

Abstract
Overexpression of the amyloid precursor protein (APP)gene on chromosome 21 in Down syndrome (DS) has been linked to increased brain amyloid levels and early-onset Alzheimer’s disease (AD). An elderly man with phenotypic DS and partial trisomy of chromosome 21 (PT21) lacked triplication of APP affording an opportunity to study the role of this gene in the pathogenesis of dementia. Multidisciplinary studies between ages 66–72 years comprised neuropsychological testing, independent neurological exams, amyloid PET imaging with 11C-Pittsburgh compound-B (PiB), plasma Amyloid-β(Aβ)measurements and a brain autopsy examination. The clinical phenotype was typical for DS and his intellectual disability was mild in severity. His serial neuropsychological test scores showed less than a 3% decline as compared to high functioning individuals with DS who developed dementia wherein the scores declined 17–28% per year. No dementia was detected on neurological examinations. On both PiB-PET scans, the patient with PT21 had lower PiB standard uptake values than controls with typical DS or sporadic AD. Plasma Aβ42 was lower than values for demented or non -demented adults with DS. Neuropathological findings showed only a single neuritic plaque and neurofibrillary degeneration consistent with normal aging but not AD. Taken together the findings in this rare patient with PT21 confirm the obligatory role of APPin the clinical, biochemical and neuropathological findings of AD in DS.
Keywords: Down syndrome, Trisomy 21, Partial Trisomy 21, Alzheimer’s disease, Dementia, Amyloid beta-Protein Precursor, amyloid-β, APP, PiB-PET
INTRODUCTION
By age 40 years and beyond, individuals with Down syndrome (DS) show the characteristic neuropathology of Alzheimer’s disease (AD) including brain deposition of amyloid-β(Aβ) protein in neuritic plaques and neurofibrillary tangles. The neuropathological changes seen in DS closely resemble those found in AD within the general population [1]. Despite the ubiquitous neuropathological changes in DS, the prevalence of dementia is variable ranging from 30–75% at age 65 [2]. As summarized by Schupf and Sergievsky[3], the most typical onset of dementia in DS occurs around age 50–55 years.
DS is most typically the result of trisomy 21 and increased dosage of specific genes on chromosome 21 may be responsible for features of the phenotype including AD [4]. In DS, triplication of the Amyloid Precursor Protein (APP) gene results in increased brain levels of Aβ based upon evidence from human studies, mouse models, and induced human pluripotent stem cells [5]. In rare instances of familial early-onset AD in the general population, there is genomic duplication of the APP locus, suggesting that the increased copy number of APP results in an AD dementia phenotype in the absence of clinical findings of DS [6 –11]. An increased brain concentration of amyloid in DS is reflected in positron emission tomography (PET) scans that have shown widespread cortical binding by age 50 years [12 –17]. The over dosage of APPin DS is linked to fetal brain accumulation of Aβ 42 [18]. Yet there are many other factors that might mitigate the effects of increased APP dosage in DS including cerebrovascular pathology, mitochondrial abnormalities, and immune system dysfunction [5, 19]. Thus it would be helpful to more directly clarify the role of APPin the pathogenesis of AD associated with DS.
Such an opportunity is afforded by studying a rare patient with DS resulting from a segmental or partial trisomy 21(PT21) in which APP is not included in the trisomic segment, thus present in the typical 2 copies rather than three as is most common in DS. A singular case study was reported by Prasher and colleagues [20]describing a 78 -year-old woman with DS resulting from PT21 involving the long arm of chromosome 21 with the karyotype [46,XX,rec(2l)dup q,inv(21)(p12q22.1)]. Southern blot and FISH analysis demonstrated that the gene sequence for APP was present in only two copies. This individual had no evidence of AD by neuropsychological, magnetic resonance imaging and neuropathological assessment.
A new case of PT21 (our present case), was included in a high-resolution genetic map of rare segmental/partial trisomies of chromosome 21 and various DS phenotypes [21]. Our patient was 65-years-old at the time and a standard -resolution karyotype on whole-blood cultures revealed a neuploidy with an unknown marker chromosome and a low-level mosaicism (10%) for a diploid cell line with a karyotype 47,XY,+mar[18]/46,XY[2]. FISH analysis characterized the marker as chromosome 21 with the proximal boundary at q11.2 and the distal boundary at q22.1 with no other chromosome showing positive hybridization. Additional molecular analysis characterized the PT21 as duplication of 18.8 MB (28.12-telo)of chromosome 21 which comprised the entirety of band q22. Excluded from the duplication was a 1.95 Mb region encompassing APP (26.17 Mb).
At the time of the 2009 report, relatively little clinical information was available on this patient. In order to more fully establish the phenotype associated with his rare PT21, we followed him for an additional 7 years. Multidisciplinary data is now presented through longitudinal neuropsychological testing, two sequential amyloid imaging scans, plasma Aβ level and ultimate neuropathological examination. This data is intended to help elucidate the role of APP in the pathogenesis of AD in DS.
Materials and Methods
Clinical examination
The dysmorphology examination was carried out according to published specifications for people with DS [22]. The neurological examination was consistent with the Unified Data Set developed for Alzheimer’s Disease Research Centers [23]. The independent neurological assessments for the diagnosis of possible dementia were carried out without examiner knowledge of any other testing results.
Genetic analysis
A standard-resolution karyotype was carried out on whole -blood cultures. Fluorescence in situ hybridization (FISH) analysis with locus specific and whole chromosome paint probe was utilized to characterize the partial chromosome 21. The methods used for molecular mapping of the PT21 have been previously described [21]. Apolipoprotein E (ApoE) genotyping was carried out according to Ossendorf and Prellwitz [24].
Neuropsychological measures
We used cognitive functioning measures previously shown to be sensitive to dementia in adults with DS [25, 26] including the Brief Praxis Test (BPT), the Dementia Questionnaire for Mentally Retarded Persons (DMR), the Severe Impairment Battery (SIB)and the Rapid Assessment for Developmental Disabilities (RADD). This battery is comprised of informant and direct measures. IQ measurement was carried out by the Wechsler Adult Intelligence Scale -Third Edition (WAIS III) [27]. Adaptive functioning was assessed by the Vineland Adaptive Behavior Scales, Second Edition (VABS-II) [28].
Amyloid brain imaging
PET with 11C-labeled Pittsburgh Compound-B (PiB) [29] scans were obtained on the patient with PT21 at the University of California, Irvine (site #1)in 2009 and the University of California, Los Angeles (site #2)in 2012. Technical issues required the 2nd scan to be carried out at site #2. The non-DS controls for the neuroimaging procedures were diagnosed as either normal or sporadic AD according to the criteria utilized in the Alzheimer’s Disease Neuroimaging Initiative [30].
At site #1 the subject was injected with 12.5±3.1 mCi of PiB and asked to rest comfortably in a dimly lit room with his eyes open. The acquisition protocol consisted of collecting four five-minute frames 51±2 minutes post PiB-injection. The subject was scanned using the High Resolution Research Tomograph (HRRT, Siemen’s Medical Systems) [31, 32]. PET reconstructions were performed using 3D ordinary Poisson ordered subset expectation maximization (3D OP-OSEM) algorithm with attenuation correction using maximum apriori transmission reconstruction (MAP-TR) and 4mm FWHM gaussian smoothing. The resulting image matrix was 256×256×207 with a voxel size of 2.5mm3.
At site #2 the subject was injected with 9.9±1.3 mCi of Pi B and scanned after 15 minute uptake time for 45 minutes covering time points from 15 minutes to 60 minutes post -injection. The resulting scan was binned into nine 5 minute frames. Scanning using the Siemens PETsyngo CT scanner. PET reconstructions were performed using filtered back-projection with CT based attenuation and scatter corrections and 3mm FWHM gaussian smoothing. The resulting image matrix was 256×256×109 with voxel sizes of 0.89×0.89×2.0mm.
Comparisons were made on 3 typical subjects with DS (TDS) at site #1 and 4 subjects with TDS at site #2. All TDS subjects had full trisomy 21 confirmed by blood karyotype. The PET-PiB scans for the TDS subjects at site #1 and site #2 were acquired with the same protocol and scanner as the PT21 subject scanned at the respective site. Because the patient with PT21 had a cardiac pacemaker, MRI scanning was precluded; instead, anatomical parcellations available in The Automated Anatomical Labeling Atlas (AAL) were used to define regions of interest (ROIs) [33]. We chose to evaluate large lobular-based ROIs in the frontal, parietal, and temporal lobes. To avoid bias from registration mismatches with the ROI template, we used the average of all cerebellum ROIs as the reference region. To align the PET scans with the AAL atlas used to define ROIs, the T1-weighted MRIs from 19 subjects with TDS acquired by Haier and colleagues [34] were used. Each PET scan was coregistered to each MRI scan using a 12 -dof transformation and normalized mutual information cost function as implemented in FMRIB’s Linear Registration Tool (FLIRT) package (www.fmrib.ox.ac.uk/fsl) [35–37]. For each PET scan, the best matching MRI was chosen based on the normalized cross correlation (average best correlations 0.78±.04) and used to spatially align the PET scans to the AAL atlas. After automated registration, each scan was visually checked for registration accuracy and manually adjusted to give the best fit to the template.
To evaluate the similarity of the patient with PT21 to controls, patient values were compared with each sample mean based on the Crawford-Howell test [38] with one tailed p-values as direction was hypothesized. This aim of the test sare to assess the degree to which the singular case may plausibly be seen as deriving from the underlying study population of controls. The Crawford-Howell test adjusts for the small sample size and the uncertainty associated with estimating variability among patients, and it has been shown to control type-Ierrors better than commonly used alternatives such as Z scores. T he t-value measured the scaled difference with the mean of the controls, and the p -value measured the degree of extremity in the reference distribution [39].
Plasma amyloid-β measurements
Plasma Aβ40 and Aβ42 were measured by a sandwich ELISA as previously described [40]. The samples used from this previous experiment have been described in detail in Head et al. Plasma from the patient with PT21 was included in this larger experiment though not reported previously.
Neuropathology examination
A standardized neuropathological protocol was utilized to carry out the autopsy brain examination [41, 42].
Ethics statement
The research protocols were approved by the Institutional Review Boards at sites #1 and #2. All experiments were done in accord with the Helsinki Declaration of 1975. For all participants with DS written informed consent for participation was obtained from the participants Legally Authorized Representative. In each case, written assent from the participant was obtained according to each protocol.
RESULTS
Clinical findings
This patient was first examined at age 65 years, having received a clinical diagnosis of DS at age 4 years. His mother was age 20 years and father 21 years at the time of his birth. With a WAIS -III full scale IQ measured at 69 and a VABS -II composite score age-equivalent of 10.5 years, he functioned within the upper range of abilities for people with DS. By example, he resided in his own apartment with minimal supervision, looked after all of his daily living needs, took a bus to his appointments and managed the cash box accurately at his day program. His general medical problems included mitral valve insufficiency, arthritis of the shoulders requiring surgery and lower spine problems resulting in multiple surgeries and an inability to walk at age 68. He wore hearing aids that corrected his sensory -neural hearing loss to the normal range. He also had a history of hypertension, EKG findings for a non-acute anteroseptal myocardial infarct, and a first degree AV-block. Because of syncopal episodes, a pacemaker was placed. The patient had a history of behavioral disturbances with some hallucinations and delusions beginning at age 66 and he was given several different psychotropic medications by his treating physicians. The mother and 2 maternal aunts were reported to have dementia of undetermined type.
On examination, the following features of the DS phenotype were present: brachycephaly, flat occiput, malar hypoplasia, down-turned corners of the mouth, flat nasal bridge, anteverted nostrils, upslanted palpebral fissures, Brushfield spots, underdeveloped philtrum, highly vaulted palate, furrowed tongue, and short neck. He did not have epicanthal folds or excess skin. He had normal hand measurements although his hands were broad with slight interphalangeal webbing but normal palmar creases. Head circumference was 54.5 cm (65th percentile according to measurements for DS [43]). Systolic blood pressure was normal in the sitting position but slightly elevated at 150 mm Hg in the standing position. His speech was characterized by dysarthria and erratic rhythm (cluttering). Prior to his back surgeries, his gait was normal without dyspraxia. Tendon reflexes were normal and there were no pathological reflexes. Multiple independent neurological examinations throughout the course of this study showed no findings for dementia up to his death at age 72 years. His death was related to pneumonia as a consequence of his medical problems. ApoE genotype indicated that the patient was homozygous for the ε3 allele (ApoEε 3/ε3).
Genetic and molecular findings
Molecular analysis revealed increase in copy number from 28.1Mb-qter of chromosome 21 [21]. The final karyotype from this high -resolution mapping of the genetic abnormality was 47,XY,+del(21)(q11.2q22.1)[18]/46,XY[2].
In Figure 1, the duplicated segments of the present case of PT21 is compared to the findings of Prasher [20], and cases of APP locus duplication without the DS phenotype [6–10]. In the present case and that of Prasher, APP was present in two copies, not three The present case spans approximately 19 Mb and gene as opposed to the Prasher case which spans approximately 12 Mb. In the cases of AD without DS [6 –10] a small chromosome duplication of chromosome 21 included the APP gene and was sufficient to result in early-onset AD but was insufficient to result in a phenotype of DS.
Figure 1.
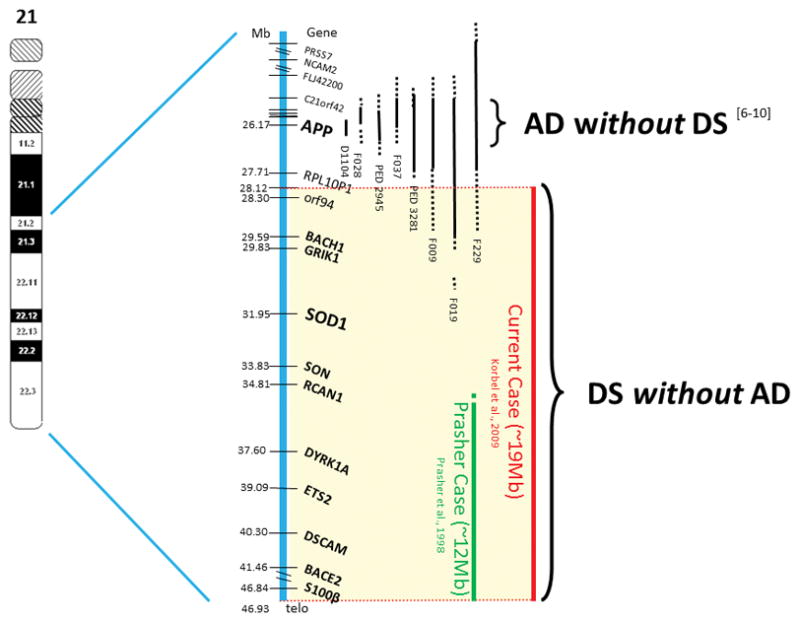
Cases of AD without DS and of DS without AD. On the left side of the figure – chromosome 21. The vertical thick blue bar adjacent to the chromosome, indicates the distal ~25Mb of chromosome 21 genes and markers with their position in Mb. The first 6 vertical thin black bars indicate six families with APP locus duplication with AD but without DS [6–10]. The next vertical green bar represents a 78-year-old partial trisomy 21 individual with DS without AD [20]. The right red bar represents the current case. Vertical solid bars – duplicated regions. Vertical dotted bars – regions of uncertainty.
Neuropsychological findings
The patient with PT21 showed less than a 3% decline in the battery of neuropsychological measures over 7 years of testing, strongly suggesting that dementia was absent. As shown in Table 1, the score change of the patient with PT21 was similar to the minor longitudinal score change of similarly high-functioning adults with DS who were not demented (n=6; age range 46–59 years; 5–9 years of follow-up). By contrast, a group of high functioning adults with DS who developed dementia (n=6; age range 45 –54 years; 2 years of follow-up) showed a decline in scores ranging from 17 –28% per year during a two-year follow-up. An independent neurological exam at each time epoch confirmed the absence of dementia.
Table 1.
Percent score change in neuropsychological assessments over 7 years of follow-up in the patient with PT21 compared to DS controls without AD dementia (5–9 years of follow-up; age range 46–59 years) and DS controls with AD dementia (2 years of follow-up; age range 45–54 years). DMR SCS scores were reversed from positive to negative to aid in interpretation of score change with the other assessments.
| Percent Score Change from Baseline over 7 Years of Observation | ||||
|---|---|---|---|---|
| Sample | BPT | DMR-SCS | SIB | RADD |
| Case (7 years) | +0.07% | +0.15% | +0.05% | −2.52% |
| DS Controls n=6 (5–9 years) | +0.20% | −0.09% | −0.15% | −0.98% |
| DS+AD Controls n=6 (2 years) | −17.02% | −27.30% | −21.12% | −28.20% |
Abbreviations: BPT; Brief Praxis Test, DMR-SCS; Dementia Questionnaire for Mentally Retarded Persons-Sum of Cognitive Scores, SIB; Severe Impairment Battery and RADD; Rapid Assessment for Developmental Disabilities.
Brain imaging findings
The PiB-standard uptake value ratios (SUVr) for the subject with PT21 were compared with the average of the TDS subjects (site #1:n=3; site #2:n=4), sporadic AD subjects (site #1: n=4; Age=82 ±9.4, site#2: n=3; Age =78.0±10.4), and healthy volunteer (NC) subjects (site #1: n=2; Age=75.5±4.9) acquired as part of ADNI (Fig 2). To evaluate the similarity of the subject with PT21 to controls, values were compared with each sample mean using the Crawford-Howell test [39] for single case comparisons as shown in Tables 2A and 2B. The Crawford-Howell test adjusted for the single-case comparison with small samples of controls, where the t-value measured the scaled difference with the mean of the controls, and the p-value measured the degree of extremity in the reference distribution. Corrected p-values were computed using the Holm method [44]. All comparisons were carried out using the R statistical package [45]. The subject with PT21 had consistently lower PiB-SUVr in all brain regions, lower than those of TDS and AD subjects at both site #1 and site #2 suggesting the PT21 subject fall sin the lower end of the TDS and AD reference distributions, and similar or lower to that observed in NCs (negative t values in Tables 2A and 2B). While statistical significance was not consistently reached, nor were the results significant after correcting for multiple comparisons, there was a clear predominance of pattern for lower SUVrs compared to all controls (AD, TDS and NC) for the patient with PT21, consistent with the neuropathological and neuropsychological results.
Figure 2.
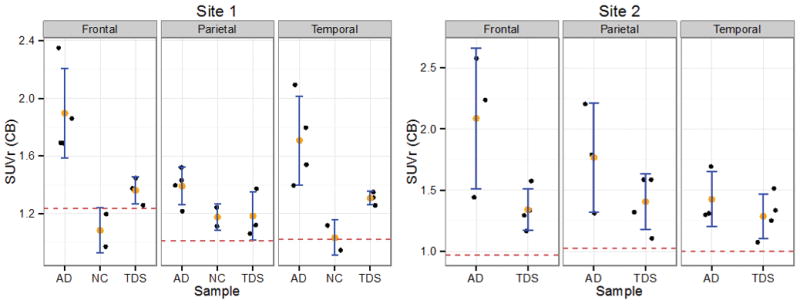
Distribution of PiB-SUVr by sample type and region. Error bars are shown for average ± one standard deviation for the sample. The patient with PT21 is shown by the red dashed horizontal line. AD: Alzheimer’s disease. NC: Normal controls. TDS: Typical Down syndrome. Left panel: Site #1 data. Right panel: Site #2 data.
Table 2A.
SUVr PiB comparisons of the patient with PT21 based on the Crawford -Howell t-test and one-tailed p-values for the site #1 data. T-value and associated p-values are tabulated for each sample and brain region, separately. P-values, shown in parentheses, were adjusted for multiple comparisons using Holm’s method [44]. AD: Alzheimer’s disease. NC: Normal controls. TDS: Typical Down syndrome.
| Region | t-TDS | p-TDS (adj) | t-NC | p-NC (adj) | t-AD | p-AD (adj) |
|---|---|---|---|---|---|---|
| Frontal | −1.14 | 0.19 (1.0) | 0.79 | 0.29 (1.0) | −1.90 | 0.08 (0.92) |
| Parietal | −0.88 | 0.24 (1.0) | −1.45 | 0.19 (1.0) | −2.61 | 0.04 (0.56) |
| Temporal | −5.32 | 0.02 (0.25) | −0.08 | 0.48 (1.0) | −1.99 | 0.07 (0.92) |
Table 2B.
SUVr PiB comparisons of the patient with PT21 based on the Crawford-Howell t-test and one-tailed p-values for the site #2 data. T-value and associated p-values are tabulated for each sample and brain region, separately. P-values, shown in parentheses, were adjusted for multiple comparisons using Holm’s method [44]. AD: Alzheimer’s disease. TDS: Typical Down syndrome.
| Region | t-TDS | p-TDS (adj) | t-AD | p-AD (adj) |
|---|---|---|---|---|
| Frontal | −1.96 | 0.07 (0.92) | −1.69 | 0.12 (1.0) |
| Parietal | −1.48 | 0.12 (1.0) | −1.44 | 0.15 (1.0) |
| Temporal | −1.39 | 0.13 (1.0) | −1.64 | 0.12 (1.0) |
Plasma Aβ determination
Figure 3 shows that the patient with PT21 was within normal levels of plasma Aβ 40 relative to both nondemented and demented adult males with DS (Figure 3A). However, plasma Aβ42 was virtually undetectable in the patient and was lower than that observed both in demented and nondemented individuals with DS (Figure 3B).
Figure 3.
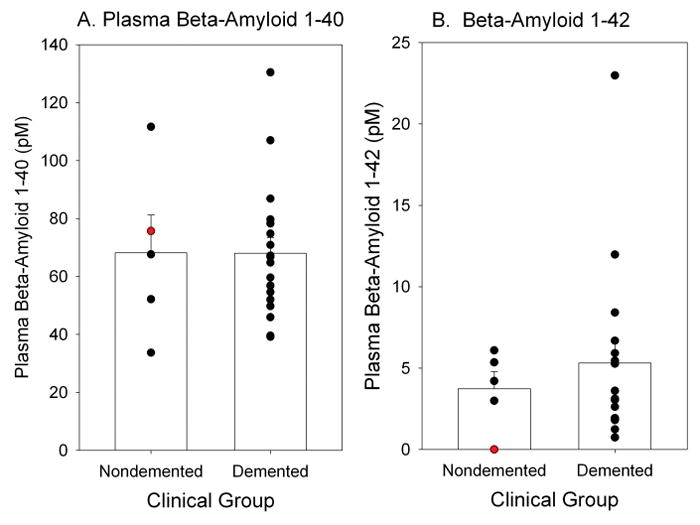
Individual plasma Aβ40 (A) and Aβ42 (B) are plotted as a function of dementia status in adult males with DS. The patient with PT21 (red symbol) is shown relative to comparison cases. Bars represent group means and error bars are standard errors of the mean.
Neuropathological findings
The brain weighed 1087g (normal weight 1300–1400g). The cerebellum was comparatively small. There was mild convolutional atrophy in the Sylvian region but the cortical ribbon appeared normal otherwise. The lateral ventricles were slightly enlarged with some reduction of periventricular white matter. No atherosclerosis was seen at the Circle of Willis.
Although diffuse plaque formation was present in a mild to moderate in degree at a number of sites, neuritic plaque formation was entirely absent except for a trace amount in CA1. Neurofibrillary tangle (NFT) formation, which was judged to be of stage III intensity by Braak and Braak criteria [42], was not observed anywhere within cerebral neocortex; it was moderate within CA1 and entorhinal-transentorhinal region, mild within amygdala, and absent within subiculum. No microinfarcts or cerebral amyloid angiopathy (CAA)were seen. The concentration of neuromelanin-bearing neurons was normal within both substantia nigra and locus ceruleus. Granulovacular degeneration was noted only in CA1 No Lewy bodies, Pick bodies or achromasic neurons were observed. In Figure 4, the middle frontal gyrus stained for Aβ is compared between the patient with PT21 and a patient with full trisomy 21 diagnosed with AD dementia who died at age 43 years.
Figure 4.

Aβ immunocytochemistry (20x objective) middle frontal gyrus; left figure shows paucity of amyloid plaques staining in the patient with PT21; right figure shows staining of a patient with full trisomy 21 and AD dementia. Sections are counterstained with cresyl violet.
DISCUSSION
Using multiple approaches, this data has shown that the lack of triplication of APP in a patient with DS is associated with the absence of dementia, very low brain amyloid levels by functional imaging, low plasma Aβ42, and the absence of AD neuropathology. These findings highlight the critical role for increased APP copy number in the development of AD in DS. Our findings are consistent with observations in the general population that mutations and duplications of APP are associated with early-onset AD [11]. However, our results do not provide evidence that lack of an extra copy of APP is the only mechanism by which dementia was prevented in the patient with PT21. Many other genes on chromosome 21 were included in the trisomic segment in the present case. Other factors responsible for the phenotypic manifestations of DS have been reviewed by Wiseman et al. [5] and include generalized transcriptional dysregulation, interaction of other genes with APP, disruption of endosomal systems, mitochondrial dysfunction and immune abnormalities. Nonetheless, the central role for APP in dementia pathogenesis for adults with DS would seem well supported by the present results.
Our patient was homozygous for the ε3 allele, the most common form of the lipoprotein [46]. In the general population, individuals with ApoEε3 have less of a risk for AD than those with the ApoEε4 allele although it is not clear that the differential risk is related to brain deposition of Aβ [47]. However, in DS ApoEε3 is considered to neither decrease norincrease the risk of dementia [48].
The phenotypic appearance of our patient was consistent with the clinical criteria for DS [49]. Overlapping features of our case with Prasher et al. [20] included short stature, Brushfield spots, mid-facial hypoplasia, hearing loss and ligamentous laxity affecting the cervical vertebrae. However, the present case of PT21 did not have clinodacyly or epicanthal folds. The difference in phenotypic appearance between the 2 cases remains of uncertain significance since phenotypic variability is characteristic of the syndrome [50]. Our patient with PT21 had a history of hypertension, cardiac rhythm disturbance and a remote myocardial infarction, which was striking since the clinical complications of atherosclerosis in adults with DS is considered to be infrequent [51, 52]. Despite his clinical findings no evidence of cerebral atherosclerotic complications was noted at autopsy.
The independent clinical evaluations by a neurologist and neuropsychologist both agreed on the absence of dementia. The multiple longitudinal measures of cognition and praxis used to follow the patient with PT21 fulfilled the criteria proposed for the diagnosis of dementia in DS (Nieuwenhuis-Mark, 2009}. The DMR has been recommended as an informant measure for dementia in DS particularly when used in longitudinal measures [53]. We have shown that the RADD correlates highly with the other measures for dementia utilized in this study [26]. Taken together the clinical and neuropsychological observations in the patient with PT21 confirm the absence of dementia through age 72 years. The patient had a history of auditory hallucinations and delusions consistent with psychotic disorder NOS specified in adults with DS [54, 55] and in adolescents [56]. However, these symptoms occurred late in his life when he was unhappily confined to a nursing home, raising the question as to whether the behaviors observed were situational in origin.
Two PiB-PET scans separated by 3 years involving independent institutional protocols and scanners were similar, showing low to absent PiB uptake over background. Despite the small sample sizes, the elevated TDS PiB-SUVr frontal and parietal values compared to normal controls were consistent with published results for DS [15]. The PiB-SUVr were also consistent with the PiB(−) aged controls and considerably lower than the PiB(+) controls, MCI, and AD subjects from the general population [57]. In fact, the subject with PT21 showed PiB-SUVr values that were lower than all of these published results. In DS without dementia there is a positive correlation of PiB retention with age [58]. Our subject had unexpectedly low PiB levels despite being in his late 60’s at the time of scanning. These imaging results do provide data suggesting that the lack of an extra copy of APPin DS may be associated with very low, to absent, uptake of PiB in brain. Despite the lack of significant differences after multiple comparison corrections of PiB retention in the PT21 subject compared to the TDS, AD, and NC groups in this case study, the low uptake of PiB in the brain is consistent with the neuropathological and neurpsychological findings and provides additional data about the in-vivo beta amyloid and neurofibrillary tangle distributions. However, they do not address the absolute affinity of PiB for amyloid beta peptide [59]. Future studies in PT21 and DS might include other ligands for amyloid and tau, such as the use of [F-18]FDDNP to identify the progression of AD symptoms in DS [13].
The plasma level of Aβ42 was dramatically low (below detectable levels) in our patient with PT21 relative to other adults with DS. This finding is consistent with the lack of APP triplication and the absence of dementia across the broad array of our study measurements as well as the neuropathological findings. Typically, plasma Aβ levels are higher in people with DS [60, 61].
The stage III neurofibrillary degeneration observed in our patient seems most consistent with “primary age-related tauopathy” (PART) that has been described as part of the normal aging process in the general population [62]. Patients with PART are described as having either no or minimal amnestic changes with only a minority having findings for dementia. Pathologically, PART is described as being characterized by the absence of Aβ-containing neuritic plaques. As in our case, the distribution of NFTs in PART tends to be restricted to the entorhinal-transentorhinal region, and amygdala, with little or no cerebral neocortical involvement. This is undoubtedly the reason that individuals with PART are usually either cognitively normal or show amnestic cognitive changes only. According to proposed criteria [42], our case, with Braak and Braak stage III NFT formation and Thal phase 0 Aβdeposition, would qualify as “primary age-related tauopathy (PART), definite, Braak stage III.”PART has not been defined for DS but the lack of convincing AD neuropathology in the present patient with PT21 would favor brain changes reflective of normal aging. It is interesting that amyloid excess appears to be necessary for the cortical spread of tau [63] and is consistent with our formulation that the neuropathology in the PT21 case is reflective of normal aging. No CAA was observed in the PT21 case, a distinguishing feature from full trisomic cases of DS [1] and the duplication-APP cases [64].
Our case differs from that of Prasher et al. [20] in the cytogenetic presentation. The present case involves a trisomy 21 with one copy of the chromosome showing a deleted segment which includes APP. The Prasher case was a disomy for chromosome 21 with an inverted duplication. However, the mechanism underlying the presentation of chromosome pathology would not appear to be relevant to the issue of AD and amyloid deposition since both cases lack an extra copy of APP.
The cases of early -onset AD with APP duplications cited in Figure 1 support the conclusions from the present case of PT21 that APP is critical to the pathogenesis of AD in DS.
Most patients with DS have been reported to have mild to moderate degrees of intellectual disability. The measured IQ of the patient with PT21 patient was close to the borderline range, yet below normal. A 26-year-old individual with PT21 (without APP triplication)and a 7q deletion had a mild DS phenotype and borderline intellectual impairment (IQ of 75) [65]. These findings suggest that lack of an extra copy of APP does not necessarily normalize intellectual functioning in PT21 and that the intellectual disability in DS most likely depends on other triplicated genes or perhaps other processes related to genomic instability [66]. The non-contributory role of APP in DS associated intellectual disability is further clarified by the 5 families, including 21 individuals, with duplication-APPin which normal cognitive function prior to the onset of AD dementia has been reported [6, 9].
A 10% mosaicism for a normal diploid cell line in the peripheral blood lymphocyte cultures was observed in the present case, consistent with advancing age in DS where acquired low-level mosaicism for a euploid cell line has been observed [67]. The significance of the small percentage of diploid cells in the present case is unknown but is unlikely to be relevant to the absence of AD in this patient. This conclusion is further supported by the observation that fibroblast cultures from this patient with PT21 showed duplication of the identical regions determined from the lymphocyte cultures with no evidence for mosaicism (Written communication with Julie R. Korenberg, PhD, MD, April 02, 2008). It is interesting that the converse may not be true since a low-level mosaicism for trisomy 21 appears sufficient to cause early-onset AD in the general population with little or no features of DS [68].
The prevalence of APP disomy in patients with DS resulting from PT21 appears to be very rare on the basis of only 2 cases, including the present report. However, unless otherwise excluded by former karyotype, PT21 without APP triplication should be considered in elderly individuals with DS who are high functioning and who exhibit no clinical signs of dementia.
Acknowledgments
The authors gratefully acknowledge our patient and his family for their contribution to the study. The US National Institutes of Health supported this study through the following grants: P50AG16573 (E.D., M.J.P., R.K., M.T., I.T.L.); R01AG 21912 (E.D., I.T.L.); R01HD065160 (E.D., I.T.L.); U01AG051412 (E.D., D.K., I.T.L.); 5U24RR021992 (D.K., S.G.P.); 5U01MH097435 (D.K., S.G.P.); 1P20RR020837 (D.K., S.G.P.); R01HD064993 (E.H.); R01AG033015 (J.R.B., J.W.S.). The National Center for Research Resources and the National Center for Advancing Translational Sciences supported this study through Grant UL1 TR000153. We acknowledge the assistance of Moyra Smith, MD, PhD and Pamela Flodman, MSc, MS for their input into the genetics discussion.
References
- 1.Mann DM. Alzheimer’s disease and Down’s syndrome. Histopathology. 1988;13:125–137. doi: 10.1111/j.1365-2559.1988.tb02018.x. [DOI] [PubMed] [Google Scholar]
- 2.Zigman W, Schupf N, Haveman M, Silverman W. The epidemiology of Alzheimer disease in intellectual disability: results and recommendations from an international conference. J Intellect Disabil Res. 1997;41(Pt 1):76–80. doi: 10.1111/j.1365-2788.1997.tb00679.x. [DOI] [PubMed] [Google Scholar]
- 3.Schupf N, Sergievsky GH. Genetic and host fact ors for dementia in Down’s syndrome. Br J Psychiatry. 2002;180:405–410. doi: 10.1192/bjp.180.5.405. [DOI] [PubMed] [Google Scholar]
- 4.Korenberg JR, Kawashima H, Pulst SM, Ikeuchi T, Ogasawara N, Yamamoto K, Schonberg SA, West R, Allen L, Magenis E. Molecular definition of a region of chromosome 21 that causes features of the Down syndrome phenotype. Am J Hum Genet. 1990;47:236–246. [PMC free article] [PubMed] [Google Scholar]
- 5.Wiseman FK, Al-Janabi T, Hardy J, Karmiloff-Smith A, Nizetic D, Tybulewicz VL, Fisher EM, Strydom A. A genetic cause of Alzheimer disease: mechanistic insights from Down syndrome. Nat Rev Neurosci. 2015;16:564–574. doi: 10.1038/nrn3983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rovelet-Lecrux A, Hannequin D, Raux G, Le Meur N, Laquerrière A, Vital A, Dumanchin C, Feuillette S, Brice A, Vercelletto M, Dubas F, Frebourg T, Campion D. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet. 2006;38:24–26. doi: 10.1038/ng1718. [DOI] [PubMed] [Google Scholar]
- 7.Sleegers K, Brouwers N, Gijselinck I, Theuns J, Goossens D, Wauters J, Del-Favero J, Cruts M, van Duijn CM, Van Broeckhoven C. APP duplication is sufficient to cause early onset Alzheimer’s dementia with cerebral amyloid angiopathy. Brain. 2006;129:2977–2983. doi: 10.1093/brain/awl203. [DOI] [PubMed] [Google Scholar]
- 8.Rovelet-Lecrux A, Frebourg T, Tuominen H, Majamaa K, Campion D, Remes AM. APP locus duplication in a Finnish family with dementia and intracerebral haemorrhage. J Neurol Neurosurg Psychiatry. 2007;78:1158–1159. doi: 10.1136/jnnp.2006.113514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cabrejo L, Guyant-Maréchal L, Laquerrière A, Vercelletto M, De la Fournière F, Thomas-Antérion C, Verny C, Letournel F, Pasquier F, Vital A, Checler F, Frebourg T, Campion D, Hannequin D. Phenotype associated with APP duplication in five families. Brain. 2006;129:2966–2976. doi: 10.1093/brain/awl237. [DOI] [PubMed] [Google Scholar]
- 10.Kasuga K, Shimohata T, Nishimura A, Shiga A, Mizuguchi T, Tokunaga J, Ohno T, Miyashita A, Kuwano R, Matsumoto N, Onodera O, Nishizawa M, Ikeuchi T. Identification of independent APP locus duplication in Japanese patients with early-onset Alzheimer disease. J Neurol Neurosurg Psychiatry. 2009;80:1050–1052. doi: 10.1136/jnnp.2008.161703. [DOI] [PubMed] [Google Scholar]
- 11.Rosenberg RN, Lambracht-Washington D, Yu G, Xia W. Genomics of Alzheimer Disease: A Review. JAMA Neurol. 2016;73:867–874. doi: 10.1001/jamaneurol.2016.0301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Landt J, D’Abrera JC, Holland AJ, Aigbirhio FI, Fryer TD, Canales R, Hong YT, Menon DK, Baron JC, Zaman SH. Using positron emission tomography and Carbon 11 -labeled Pittsburgh Compound B to image Brain Fibrillar β-amyloid in adults with down syndrome: safety, acceptability, and feasibility. Arch Neurol. 2011;68:890–896. doi: 10.1001/archneurol.2011.36. [DOI] [PubMed] [Google Scholar]
- 13.Nelson LD, Siddarth P, Kepe V, Scheibel KE, Huang SC, Barrio JR, Small GW. Positron emission tomography of brain β-amyloid and τ levels in adults with Down syndrome. Arch Neurol. 2011;68:768–774. doi: 10.1001/archneurol.2011.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sabbagh MN, Fleisher A, Chen K, Rogers J, Berk C, Reiman E, Pontecorvo M, Mintun M, Skovronsky D, Jacobson SA, Sue LI, Liebsack C, Charney AS, Cole L, Belden C, Beach TG. Positron emission tomography and neuropathologic estimates of fibrillar amyloid-β in a patient with Down syndrome and Alzheimer disease. Arch Neurol. 2011;68:1461–1466. doi: 10.1001/archneurol.2011.535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Handen BL, Cohen AD, Channamalappa U, Bulova P, Cannon SA, Cohen WI, Mathis CA, Price JC, Klunk WE. Imaging brain amyloid in nondemented young adults with Down syndrome using Pittsburgh compound B. Alzheimers Dement. 2012;8:496–501. doi: 10.1016/j.jalz.2011.09.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hartley SL, Handen BL, Devenny DA, Hardison R, Mihaila I, Price JC, Cohen AD, Klunk WE, Mailick MR, Johnson SC, Christian BT. Cognitive functioning in relation to brain amyloid-β in healthy adults with Down syndrome. Brain. 2014;137:2556–2563. doi: 10.1093/brain/awu173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jennings D, Seibyl J, Sabbagh M, Lai F, Hopkins W, Bullich S, Gimenez M, Reininger C, Putz B, Stephens A, Catafau AM, Marek K. Age dependence of brain β-amyloid deposition in Down syndrome: An [18F] florbetaben PET study. Neurology. 2015;84:500–507. doi: 10.1212/WNL.0000000000001212. [DOI] [PubMed] [Google Scholar]
- 18.Teller JK, Russo C, DeBusk LM, Angelini G, Zaccheo D, Dagna-Bricarelli F, Scartezzini P, Bertolini S, Mann DM, Tabaton M, Gambetti P. Presence of soluble amyloid beta -peptide precedes amyloid plaque formation in Down’s syndrome. Nat Med. 1996;2:93–95. doi: 10.1038/nm0196-93. [DOI] [PubMed] [Google Scholar]
- 19.Wilcock DM, Schmitt FA, Head E. Cerebrovascular contributions to aging and Alzheimer’s disease in Down syndrome. Biochim Biophys Acta. 2016;1862:909–914. doi: 10.1016/j.bbadis.2015.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prasher VP, Farrer MJ, Kessling AM, Fisher EM, West RJ, Barber PC, Butler AC. Molecular mapping of Alzheimer-type dementia in Down’s syndrome. Ann Neurol. 1998;43:380–383. doi: 10.1002/ana.410430316. [DOI] [PubMed] [Google Scholar]
- 21.Korbel JO, Tirosh-Wagner T, Urban AE, Chen XN, Kasowski M, Dai L, Grubert F, Erdman C, Gao MC, Lange K, Sobel EM, Barlow GM, Aylsworth AS, Carpenter NJ, Clark RD, Cohen MY, Doran E, Falik-Zaccai T, Lewin SO, Lott IT, McGillivray BC, Moeschler JB, Pettenati MJ, Pueschel SM, Rao KW, Shaffer LG, Shohat M, Van Riper AJ, Warburton D, Weissman S, Gerstein MB, Snyder M, Korenberg JR. The genetic architecture of Down syndrome phenotypes revealed by high-resolution analysis of human segmental trisomies. Proc Natl Acad Sci USA. 2009;106:12031–12036. doi: 10.1073/pnas.0813248106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Devlin L, Morrison PJ. Accuracy of the clinical diagnosis of Down syndrome. Ulster Med J. 2004;73:4–12. [PMC free article] [PubMed] [Google Scholar]
- 23.Morris JC, Weintraub S, Chui HC, Cummings J, Decarli C, Ferris S, Foster NL, Galasko D, Graff-Radford N, Peskind ER, Beekly D, Ramos EM, Kukull WA. The Uniform Data Set (UDS): clinical and cognitive variables and descriptive data from Alzheimer Disease Centers. Alzheimer Dis Assoc Disord. 2006;20:210–216. doi: 10.1097/01.wad.0000213865.09806.92. [DOI] [PubMed] [Google Scholar]
- 24.Ossendorf M, Prellwitz W. Rapid and easy apolipoprotein E genotyping using an improved PCR-RFLP technique. Qiagen News. 2000;1:11–13. [Google Scholar]
- 25.Lott IT, Doran E, Nguyen VQ, Tournay A, Head E, Gillen DL. Down syndrome and dementia: a randomized, controlled trial of antioxidant supplementation. Am J Med Genet A. 2011;155A:1939–1948. doi: 10.1002/ajmg.a.34114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walsh DM, Doran E, Silverman W, Tournay A, Movsesyan N, Lott IT. Rapid assessment of cognitive function in down syndrome across intellectual level and dementia status. J Intellect Disabil Res. 2015;59:1071–1079. doi: 10.1111/jir.12200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wechsler D. WAIS-III administration and scoring manual. The Psychological Corporation; San Antonio, TX: 1997. [Google Scholar]
- 28.Sparrow S, Cicchetti D, Balla D. Vineland adaptive behavior scales: (Vineland II), survey interview form/caregiver rating form. Livonia, MN: Pearson Assessments; 2005. [Google Scholar]
- 29.Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergström M, Savitcheva I, Huang GF, Estrada S, Ausén B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA, Långström B. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 30.Weiner MW, Veitch DP, Hayes J, Neylan T, Grafman J, Aisen PS, Petersen RC, Jack C, Jagust W, Trojanowski JQ, Shaw LM, Saykin AJ, Green RC, Harvey D, Toga AW, Friedl KE, Pacifico A, Sheline Y, Yaffe K, Mohlenoff B Initiative DoDAsDN. Effects of traumatic brain injury and posttraumatic stress disorder on Alzheimer’s disease in veterans, using the Alzheimer’s Disease Neuroimaging Initiative. Alzheimers Dement. 2014;1:S226–235. doi: 10.1016/j.jalz.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wienhard K, Eriksson L, Eriksson M, Casey M, Knoess C, Bruckbauer T, Hamill J, Schmand M, Gremillion T, Lenox A, Conti M, Bendriem B, Heiss W, Nutt R, Seibert J. The ECAT HRRT: NEMA NEC evaluation of the HRRT system, the new high resolution research tomograph. 2001 Ieee Nuclear Science Symposium, Conference Records. 2002;1–4:1227–1230. [Google Scholar]
- 32.de Jong HW, van Velden FH, Kloet RW, Buijs FL, Boellaard R, Lammertsma AA. Performance evaluation of the ECAT HRRT: an LSO-LYSO double layer high resolution, high sensitivity scanner. Phys Med Biol. 2007;52:1505–1526. doi: 10.1088/0031-9155/52/5/019. [DOI] [PubMed] [Google Scholar]
- 33.Tzourio-Mazoyer N, Landeau B, Papathanassiou D, Crivello F, Etard O, Delcroix N, Mazoyer B, Joliot M. Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single-subject brain. Neuroimage. 2002;15:273–289. doi: 10.1006/nimg.2001.0978. [DOI] [PubMed] [Google Scholar]
- 34.Haier RJ, Head K, Head E, Lott IT. Neuroimaging of individuals with Down’s syndrome at-risk for dementia: evidence for possible compensatory events. Neuroimage. 2008;39:1324–1332. doi: 10.1016/j.neuroimage.2007.09.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jenkinson M, Smith S. A global optimisation method for robust affine registration of brain images. Med Image Anal. 2001;5:143–156. doi: 10.1016/s1361-8415(01)00036-6. [DOI] [PubMed] [Google Scholar]
- 36.Jenkinson M, Bannister P, Brady M, Smith S. Improved optimization for the robust and accurate linear registration and motion correction of brain images. Neuroimage. 2002;17:825–841. doi: 10.1016/s1053-8119(02)91132-8. [DOI] [PubMed] [Google Scholar]
- 37.Jenkinson M, Beckmann CF, Behrens TE, Woolrich MW, Smith SM. FSL. Neuroimage. 2012;62:782–790. doi: 10.1016/j.neuroimage.2011.09.015. [DOI] [PubMed] [Google Scholar]
- 38.Crawford JR, Garthwaite PH, Howell DC. On comparing a single case with a control sample: an alternative perspective. Neuropsychologia. 2009;47:2690–2695. doi: 10.1016/j.neuropsychologia.2009.04.011. [DOI] [PubMed] [Google Scholar]
- 39.Crawford JR, Garthwaite PH. Single -case research in neuropsychology: a comparison of five forms of t-test for comparing a case to controls. Cortex. 2012;48:1009–1016. doi: 10.1016/j.cortex.2011.06.021. [DOI] [PubMed] [Google Scholar]
- 40.Head E, Doran E, Nistor M, Hill M, Schmitt FA, Haier RJ, Lott IT. Plasma amyloid - β as a function of age, level of intellectual disability, and presence of dementia in Down syndrome. J Alzheimers Dis. 2011;23:399–409. doi: 10.3233/JAD-2010-101335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Trojanowski JQ, Vinters HV, Hyman BT Aging NIo, Association As. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 2012;123:1–11. doi: 10.1007/s00401-011-0910-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Braak H, Braak E. Neuropathological stageing of Alzheimer -related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 43.Zemel BS, Pipan M, Stallings VA, Hall W, Schadt K, Freedman DS, Thorpe P. Growth Charts for Children With Down Syndrome in the United States. Pediatrics. 2015;136:e1204–1211. doi: 10.1542/peds.2015-1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.HOLM S. A simple sequentially rejective multiple test procedure. Scandinavian Journal of Statistics. 1979;6:65–70. [Google Scholar]
- 45.R Core Team. A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; Vienna, Austria: 2014. [Google Scholar]
- 46.Huang Y, Mahley RW. Apolipoprotein E: structure and function in lipid metabolism, neurobiology, and Alzheimer’s diseases. Neurobiol Dis. 2014;72(Pt A):3–12. doi: 10.1016/j.nbd.2014.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9:106–118. doi: 10.1038/nrneurol.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rohn TT, McCarty KL, Love JE, Head E. Is Apolipoprotein E4 an Important Risk Factor for Dementia in Persons with Down Syndrome? J Parkinsons Dis Alzheimers Dis. 2014;1(1):7. doi: 10.13188/2376-922x.1000004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Epstein CJ, Korenberg JR, Annerén G, Antonarakis SE, Aymé S, Courchesne E, Epstein LB, Fowler A, Groner Y, Huret JL. Protocols to establish genotype -phenotype correlations in Down syndrome. Am J Hum Genet. 1991;49:207–235. [PMC free article] [PubMed] [Google Scholar]
- 50.Lee LG, Jackson JF. Diagnosis of Down’s syndrome: clinical vs. laboratory. Clin Pediatr (Phila) 1972;11:353–356. doi: 10.1177/000992287201100610. [DOI] [PubMed] [Google Scholar]
- 51.Licastro F, Marocchi A, Penco S, Porcellini E, Lio D, Dogliotti G, Corsi MM. Does Down’s syndrome support the homocysteine theory of atherogenesis? Experience in elderly subjects with trisomy 21. Arch Gerontol Geriatr. 2006;43:381–387. doi: 10.1016/j.archger.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 52.Englund A, Jonsson B, Zander CS, Gustafsson J, Annerén G. Changes in mortality and causes of death in the Swedish Down syndrome population. Am J Med Genet A. 2013;161A:642–649. doi: 10.1002/ajmg.a.35706. [DOI] [PubMed] [Google Scholar]
- 53.Strydom A, Hassiotis A. Diagnostic instruments for dementia in older people with intellectual disability in clinical practice. Aging Ment Health. 2003;7:431–437. doi: 10.1080/13607860310001594682. [DOI] [PubMed] [Google Scholar]
- 54.Urv TK, Zigman WB, Silverman W. Psychiatric symptoms in adults with Down syndrome and Alzheimer’s disease. Am J Intellect Dev Disabil. 2010;115:265–276. doi: 10.1352/1944-7558-115.4.265. [DOI] [PubMed] [Google Scholar]
- 55.Myers BA, Pueschel SM. Psychiatric disorders in persons with Down syndrome. J Nerv Ment Dis. 1991;179:609–613. doi: 10.1097/00005053-199110000-00004. [DOI] [PubMed] [Google Scholar]
- 56.Dykens EM, Shah B, Davis B, Baker C, Fife T, Fitzpatrick J. Psychiatric disorders in adolescents and young adults with Down syndrome and other intellectual disabilities. J Neurodev Disord. 2015;7(1):9. doi: 10.1186/s11689-015-9101-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jagust WJ, Bandy D, Chen K, Foster NL, Landau SM, Mathis CA, Price JC, Reiman EM, Skovronsky D, Koeppe RA Initiative AsDN. The Alzheimer’s Disease Neuroimaging Initiative positron emission tomography core. Alzheimers Dement. 2010;6:221–229. doi: 10.1016/j.jalz.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lao PJ, Betthauser TJ, Hillmer AT, Price JC, Klunk WE, Mihaila I, Higgins AT, Bulova PD, Hartley SL, Hardison R, Tumuluru RV, Murali D, Mathis CA, Cohen AD, Barnhart TE, Devenny DA, Mailick MR, Johnson SC, Handen BL, Christian BT. The effects of normal aging on amyloid-β deposition in nondemented adults with Down syndrome as imaged by carbon 11-labeled Pittsburgh compound B. Alzheimers Dement. 2016;12:380–390. doi: 10.1016/j.jalz.2015.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lockhart A, Lamb JR, Osredkar T, Sue LI, Joyce JN, Ye L, Libri V, Leppert D, Beach TG. PIB is a non -specific imaging marker of amyloid-beta (Abeta) peptide-related cerebral amyloidosis. Brain. 2007;130:2607–2615. doi: 10.1093/brain/awm191. [DOI] [PubMed] [Google Scholar]
- 60.Mehta PD, Dalton AJ, Mehta SP, Kim KS, Sersen EA, Wisniewski HM. Increased plasma amyloid beta protein 1–42 levels in Down syndrome. Neurosci Lett. 1998;241:13–16. doi: 10.1016/s0304-3940(97)00966-x. [DOI] [PubMed] [Google Scholar]
- 61.Tokuda T, Fukushima T, Ikeda S, Sekijima Y, Shoji S, Yanagisawa N, Tamaoka A. Plasma levels of amyloid beta proteins Abeta 1–40 and Abeta 1–42(43) are elevated in Down’s syndrome. Ann Neurol. 1997;41:271–273. doi: 10.1002/ana.410410220. [DOI] [PubMed] [Google Scholar]
- 62.Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I, Arnold SE, Attems J, Beach TG, Bigio EH, Cairns NJ, Dickson DW, Gearing M, Grinberg LT, Hof PR, Hyman BT, Jellinger K, Jicha GA, Kovacs GG, Knopman DS, Kofler J, Kukull WA, Mackenzie IR, Masliah E, McKee A, Montine TJ, Murray ME, Neltner JH, Santa-Maria I, Seeley WW, Serrano-Pozo A, Shelanski ML, Stein T, Takao M, Thal DR, Toledo JB, Troncoso JC, Vonsattel JP, White CL, Wisniewski T, Woltjer RL, Yamada M, Nelson PT. Primary age -related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol. 2014;128:755–766. doi: 10.1007/s00401-014-1349-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang L, Benzinger TL, Su Y, Christensen J, Friedrichsen K, Aldea P, McConathy J, Cairns NJ, Fagan AM, Morris JC, Ances BM. Evaluation of Tau Imaging in Staging Alzheimer Disease and Revealing Interactions Between β-Amyloid and Tauopathy. JAMA Neurol. 2016;73:1070–1077. doi: 10.1001/jamaneurol.2016.2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Buss L, Fisher E, Hardy J, Nizetic D, Groet J, Pulford L, Strydom A. Intracerebral haemorrhage in Down syndrome: protected or predisposed? F1000Res. 2016:5. doi: 10.12688/f1000research.7819.1. Faculty Rev-876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Papoulidis I, Papageorgiou E, Siomou E, Oikonomidou E, Thomaidis L, Vetro A, Zuffardi O, Liehr T, Manolakos E, Vassilis P. A patient with partial trisomy 21 and 7q deletion expresses mild Down syndrome phenotype. Gene. 2014;536:441–443. doi: 10.1016/j.gene.2013.11.078. [DOI] [PubMed] [Google Scholar]
- 66.Shapiro BL. Whither Down syndrome critical regions? Hum Genet. 1997;99:421–423. doi: 10.1007/s004390050383. [DOI] [PubMed] [Google Scholar]
- 67.Jenkins EC, Schupf N, Genovese M, Ye LL, Kapell D, Canto B, Harris M, Devenny D, Lee JH, Brown WT. Increased low-level chromosome 21 mosaicism in older individuals with Down syndrome. Am J Med Genet. 1997;68:147–151. doi: 10.1002/(sici)1096-8628(19970120)68:2<147::aid-ajmg5>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 68.Ringman JM, Rao PN, Lu PH, Cederbaum S. Mosaicism for trisomy 21 in a patient with young-onset dementia: a case report and brief literature review. Arch Neurol. 2008;65:412–415. doi: 10.1001/archneur.65.3.412. [DOI] [PubMed] [Google Scholar]