

Abstract
Non-alcoholic fatty liver disease (NAFLD) is a common liver disease in Western populations. Non-alcoholic steatohepatitis (NASH) is a more debilitating form of NAFLD characterized by hepatocellular injury and inflammation, which significantly increase the risk of end-stage liver and cardiovascular diseases. Unfortunately, there are no available drug therapies for NASH. Bile acids are physiological detergent molecules that are synthesized from cholesterol exclusively in the hepatocytes. Bile acids circulate between the liver and intestine, where they are required for cholesterol solubilization in the bile and dietary fat emulsification in the gut. Bile acids also act as signaling molecules that regulate metabolic homeostasis and inflammatory processes. Many of these effects are mediated by the bile acid-activated nuclear receptor farnesoid X receptor (FXR) and the G protein-coupled receptor TGR5. Nutrient signaling regulates hepatic bile acid synthesis and circulating plasma bile acid concentrations, which in turn control metabolic homeostasis. The FXR agonist obeticholic acid has had beneficial effects on NASH in recent clinical trials. Preclinical studies have suggested that the TGR5 agonist and the FXR/TGR5 dual agonist are also potential therapies for metabolic liver diseases. Extensive studies in the past few decades have significantly improved our understanding of the metabolic regulatory function of bile acids, which has provided the molecular basis for developing promising bile acid-based therapeutic agents for NASH treatment.
Keywords: Nuclear receptor, TGR5, Metabolic syndromes, Fatty liver disease
1. Introduction
Nonalcoholic fatty liver disease (NAFLD) is characterized by excessive hepatic fat accumulation (steatosis) that is not caused by alcohol consumption (1,2). NAFLD is a common liver disease and affects ~30% of the Western population (1,2). Currently, simple hepatic steatosis does not require clinical treatment. However, in some patients it may progress to nonalcoholic steatohepatitis (NASH), which is a more debilitating form of NAFLD characterized by the presence of hepatocellular injury and inflammation. Patients with NASH have a significantly increased risk of developing fibrosis, cirrhosis, liver cancer and liver failure. In addition, patients with NASH have a significantly higher risk of cardiovascular diseases (CVD), which are the leading cause of morbidity and mortality among these patients (3,4). NASH pathogenesis is incompletely understood and is considered a result of complex interactions between genetic and environmental factors. Patients with NASH are often obese and diabetic and commonly possess other features of metabolic syndrome, such as insulin resistance, dyslipidemia and hypertension. It is thought that over-nutrition and obesity cause adipocyte stress and dysfunction, leading to inflammatory infiltration and adipose insulin resistance. As a result, uncontrolled lipolysis causes increased free fatty acid release and fatty acid lipotoxicity in non-conventional fat storage tissues, such as the skeletal muscle, pancreas and liver (2,5). Adipose-derived fatty acids serve as a major source of hepatic fat in NAFLD (2,5). Increased hepatic fat accumulation in the presence of insulin resistance further promotes hepatic triglyceride overproduction, which is a key contributing factor to dyslipidemia and higher CVD risk. Unfortunately, there are no approved therapies for NASH. Patients who develop end-stage liver disease require a liver transplantation. New therapeutic interventions that treat both liver-related and cardiovascular-related complications in patients with NASH are still needed.
Bile acids are cholesterol derivatives produced only in the hepatocytes of the liver (6). Hepatic bile acid synthesis represents the only quantitatively significant route for cholesterol elimination. Bile acids are amphipathic physiological detergents that facilitate dietary cholesterol, lipid and fat-soluble vitamin absorption in the small intestine (6,7). Studies over the past couple decades have demonstrated that bile acids act as signaling molecules that regulate intracellular signaling pathways. Many of these pathways are critically involved in the regulation of lipid, glucose and energy metabolism. In addition, bile acid synthesis and plasma bile acid concentrations are sensitive to circadian and nutrient regulation, which suggests that bile acid signaling integrates nutrient sensing to the maintenance of metabolic homeostasis. Drugs targeting bile acid metabolism and signaling have been used clinically to treat patients with hypercholesterolemia and hyperglycemia (6). New therapies targeting bile acid signaling pathways are currently being developed to treat fatty liver diseases. In this review, we will summarize the current knowledge of bile acid synthesis regulation and the mechanisms underlying bile acid signaling regulation of metabolic homeostasis. These comprise the molecular basis for the development of bile acid-based therapies for fatty liver disease.
2. A brief introduction on bile acid metabolism
Cholesterol conversion into bile acids involves several enzymatic and non-enzymatic reactions (Fig. 1A). Hepatocytes are the only cell type that expresses all the required bile acid synthetic enzymes, which are located in different intracellular compartments, including the endothelium reticulum (ER), cytosol, mitochondria and peroxisomes. Cholesterol 7α-hydroxylase (CYP7A1), which is a cytochrome P450 (CYP) enzyme residing in the ER, catalyzes the first and rate-limiting step in the classic bile acid synthesis pathway to convert cholesterol to 7α-hydroxycholesterol. 7α-hydroxycholesterol is subsequently converted to two primary bile acids, chenodeoxycholic acid (CDCA) and cholic acid (CA) (Fig. 1A). CA synthesis involves the C-12 hydroxylation of 7α-hydroxy-4-choesten-3-one to 7α, 12α-dihydroxy-4-cholesten-3-one, which is catalyzed by another cytochrome p450 enzyme sterol 12α-hydroxylase (CYP8B1) that is located in the ER (Fig. 1B). As displayed in Fig 1A, CDCA can also be produced via the alternative bile acid biosynthesis pathway, in which cholesterol is first converted to 27-hydroxycholesterol by the mitochondrial sterol 27-hydroxylase (CYP27A1). Newly synthesized bile acids are conjugated to the amino acids glycine or taurine on the side chain to form N-acyl amidates, a process that is catalyzed sequentially by two enzymes, bile acid-CoA ligase and bile acid-CoA:amino acid N-acyltransferase (8,9). Conjugation of bile acids increases bile acid water-solubility under physiological pH and decreases bile acid toxicity. The bile of human patients with defective bile acid-conjugating enzymes contains high levels of unconjugated bile acids, which causes fat-soluble vitamin malabsorption, growth retardation and liver injury (10,11). Human bile contains glycine- and taurine-conjugated bile acids in a roughly 3:1 ratio, while mouse bile contains predominantly taurine-conjugated bile acids. In this review, the terms CDCA, CA and other bile acid species mentioned later will be used to refer to both the conjugated and unconjugated forms unless specified.
Figure 1. Bile acid synthesis.

A. Primary bile acids can be synthesized by two pathways in hepatocytes. The classic bile acid synthesis pathway is considered the primary pathway in humans. In this pathway, cholesterol is first converted to 7α-hydroxycholesterol (7α-HOC) by the rate-limiting enzyme cholesterol 7α-hydroxylase (CYP7A1) in the endoplasmic reticulum. 7α-hydroxy-4 cholesten-3-one (C4) is a common precursor for chenodeoxycholic acid(CDCA) and cholic acid (CA). Sterol 12α-hydroxylase (CYP8B1) catalyzes the C-12 hydroxylation of C4, which leads to the synthesis of CA. In the alternative pathway, cholesterol is first converted to 27-hydroxycholesterol (27-HOC) by mitochondrial sterol 27-hydroxylase (CYP27A1). Oxysterol 7α-hydroxylase (CYP7B1) then catalyzes C-7 hydroxylation. The alternative pathway only produces CDCA. B. Cholesterol and cholic acid molecular structures.
Bile acids circulate between the liver and the intestine in a process called enterohepatic circulation of the bile, which is stimulated by nutrient intake and occurs a few times a day in humans (Fig. 2). Bile acids are secreted across the apical membrane of hepatocytes and stored in the gallbladder. Bile acid secretion into the bile against the concentration gradient is mediated by ATP binding cassette transporter B11 (also called the bile salt export pump [BSEP]) (12). Cholesterol and phospholipids are two other major constituents in the bile, and their efflux is mediated by ATP-binding cassette (ABC) transporters, the ABCG5/ABCG8 heterodimer (13) and canalicular phospholipid floppase multi-drug resistance 3, respectively (14). Some bile acids can be absorbed by cholangiocytes after they are secreted by hepatocytes. Cholangiocytes take up unconjugated bile acids via passive diffusion and conjugated bile acids via the apical sodium-dependent bile salt transporter (ASBT) (15). Bile acids in cholangiocytes are then secreted into the peribiliary plexus via organic solute transporter (OST) α and OSTβ, which form a heterodimer, and subsequently taken up by hepatocytes (15,16). Meal intake stimulates the release of bile acids into the intestinal tract where bile acids help emulsify dietary lipids and thus facilitate dietary lipid and fat-soluble vitamin absorption. In the small intestine, some colonized bacteria express bile salt hydrolases, which convert some of the conjugated bile acids to unconjugated bile acids. These unconjugated bile acids can then serve as substrates for bacterial 7α-dehydroxylase, which mediates the C-7 dehydroxylation reaction of primary bile acids to produce secondary bile acids deoxycholic acid (DCA) from CA and lithocholic acid (LCA) from CDCA. The majority of bile acids are efficiently reabsorbed in the ileum and transported back to the liver via portal circulation. The daily fecal loss of bile acids is approximately 5%, which is replenished by de novo bile acid biosynthesis in the hepatocytes to maintain a relatively constant bile acid pool. The ileum has high expression of bile acid transporters. ASBT mediates bile acid uptake into enterocytes, and OSTα/OSTβ heterodimers at the basolateral membrane of the enterocyte mediate bile acid efflux into the portal circulation. Conjugated bile acids in the portal circulation are taken up primarily by the sodium-dependent taurocholate transporter (17). Because the first pass extraction rate of portal bile acids by the liver is approximately 90% (18), bile acid concentrations in the systemic circulation are significantly lower than those of the portal blood under normal physiology. Under normal conditions, bile acid excretion through the kidney is minimal. However, in cholestasis, where bile acid excretion via the biliary route is impaired, more bile acids are secreted across the basolateral side of hepatocytes into the systemic circulation (19–21). Basolateral bile acid efflux from the hepatocyte can be mediated by multidrug resistance-related protein (MRP) 3, MRP4 and OSTα/OSTβ heterodimer (16,21,22). Increased systemic bile acid concentrations lead to renal bile acid excretion.
Figure 2. Bile acid transport in the enterohepatic circulation.
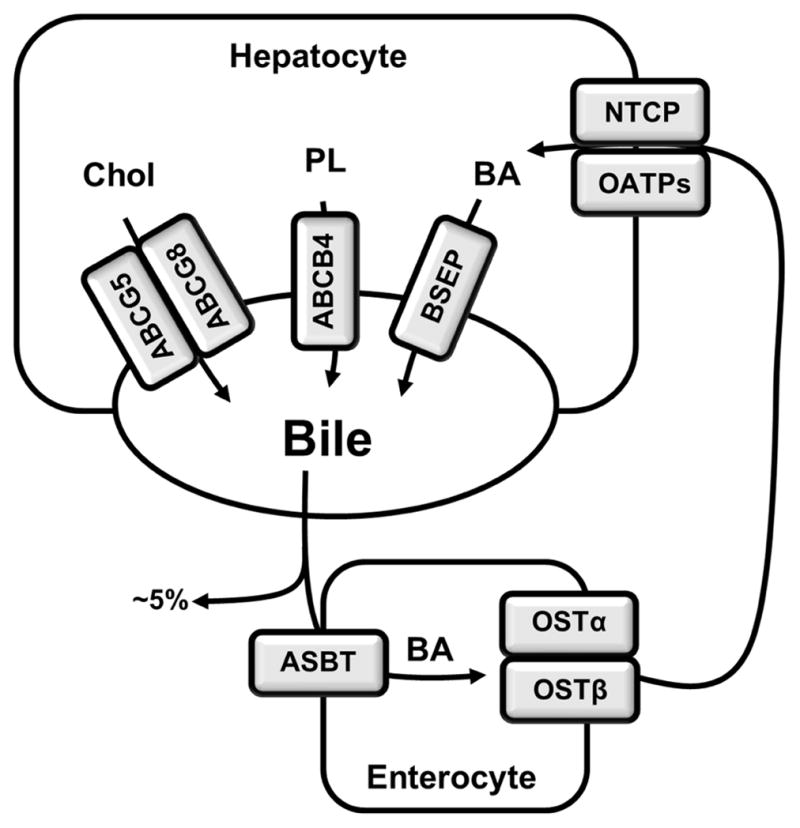
In the enterohepatic circulation, bile acids are secreted from the canalicular side of hepatocytes into the bile and stored in the gallbladder. Meal intake stimulates gallbladder bile release into the small intestine, where ~95% of bile acids can be re-absorbed in the ileum and secreted into the portal circulation. Approximately 5% of the total bile acids are lost in feces. Bile acids are taken up by the hepatocytes from the basolateral side and re-secreted into the bile. Bile acid uptake and secretion are mediated by transporters. Bile salt export pump (BSEP) is the primary canalicular bile acid efflux transporter in hepatocytes. Cholesterol is secreted into the bile by the ATP binding cassette (ABC)G5/ABCG8 heterodimer, and phospholipids are secreted by ABCB4 into the bile. Cholesterol, bile acids and phospholipids are the major constituents in the bile. They form mixed micelles for cholesterol solubilization and to prevent bile acid damage to the biliary system. In the small intestine, bile acids are taken up into the enterocytes by apical sodium-dependent bile salt transporter (ASBT) and are secreted into the portal circulation by the organic solute transporter (OST)α/OSTβ heterodimer. Sodium–dependent taurocholate transporter(NTCP) is the major basolateral conjugated bile acid uptake transporter on hepatocytes, while OATP isoforms can also mediate uptake of primarily unconjugated bile acids.
In humans, the two primary bile acids CA and CDCA make up approximately 70%–80% of the total bile acid pool. DCA is the major secondary bile acid, which may account for approximately 20% of the total bile acid pool. Among all bile acid species, the secondary bile acid LCA is highly toxic. LCA is efficiently detoxified, and only trace amounts of LCA are found in the bile. In mice and rats, CDCA and UDCA are converted to α-muricholic acids (MCA) and βMCA, respectively. MCAs have a hydroxyl group at the C-6 position and are more hydrophilic than CDCA. In C57BL6J mice, the bile acid pool contains roughly 50% MCA, ~40% CA and ~10% CDCA (23). A recent study revealed that murine Cyp2C70 was involved in MCA formation in mice (24). Humans do not appear to have a homolog of murine Cyp2c70, which may explain the different bile acid composition between mice and humans.
3. Bile acid receptors in the regulation of metabolism and inflammation
Bile acid exerts its regulatory function by activating a number of nuclear receptors and signal transduction pathways in hepatic and extrahepatic tissues. Nuclear receptors are intracellular ligand-activated transcription factors (25). A typical nuclear receptor consists of a DNA-binding domain and a ligand-binding domain. Most nuclear receptors bind to the consensus DNA sequences in their target gene promoters and regulate gene transcription in a ligand-dependent manner. Bile acids are endogenous ligands of three nuclear receptors: the farnesoid X receptor (FXR) (26–28), the pregnane X receptor (29) and the vitamin D receptor (30). These bile acid receptors sense bile acid concentrations in the enterohepatic system and in turn regulate bile acid homeostasis and detoxification mechanisms. Pregnane X and vitamin D receptor activation induces many phase-I cytochrome p450s, phase II conjugating enzymes and phase-III transporters that are involved in bile acid and drug detoxification in the hepatocyte and the intestine (reviewed in (6)). In this review, we will primarily focus our discussion on the role of FXR in the regulation of bile acid, lipid and glucose metabolism. Bile acids also activate cell surface receptors, such as the G protein-coupled receptor TGR5 and the sphingosine-1-phosphate receptor 2. TGR5 is expressed in the intestine and colon as well as metabolically active tissues, including skeletal muscle and brown adipose tissue, and is involved in the regulation of glucose and energy metabolism. Finally, bile acid-activated FXR induces the growth hormone fibroblast growth factor (FGF) 15 in mice and its ortholog FGF19 in humans. Recent studies revealed a novel role of FGF15/19 in the regulation of postprandial glucose metabolism in the liver.
3.1. FXR regulation of bile acid synthesis and transport
In the enterohepatic system, FXR is expressed in hepatocytes and enterocytes that are routinely exposed to high bile acid concentrations. Both unconjugated and conjugated bile acids, including CDCA, CA, DCA and LCA, can activate FXR (26,27). The most potent FXR ligand among all endogenous bile acid species is CDCA, which activates human FXR with an EC50 of ~10 μmol/L and murine FXR with an EC50 of ~50 μmol/L. In mice, the hydrophilic bile acid MCA does not activate FXR.
The role of FXR in the regulation of bile acid homeostasis has been extensively investigated. De novo bile acid biosynthesis is subject to bile acid feedback inhibition, which helps maintain bile acid homeostasis. Extensive studies over the past two decades support the conclusion that FXR plays a central role in mediating bile acid feedback inhibition of bile acid synthesis (6). Bile acids and FXR repress several bile acid biosynthetic genes, including CYP7A1, CYP8B1 and CYP27A1. However, major research focus has been placed on understanding the mechanisms underlying the transcriptional regulation of the rate-limiting gene CYP7A1 in the classic bile acid biosynthetic pathway. FXR can sense elevated bile acid concentrations in either the hepatocyte or the enterocyte and in turn decreases hepatic bile acid synthesis. In hepatocytes, FXR transcriptionally induces an atypical nuclear receptor called small heterodimer partner (SHP). Unlike other nuclear receptors, SHP does not have a DNA-binding domain, and it primarily interacts with other transcriptional factors as a transcriptional co-repressor. Two nuclear receptors, hepatocyte nuclear factor 4α and liver receptor homolog 1, bind to the CYP7A1 gene promoter and play a key role in maintaining basal CYP7A1 gene transcription. SHP interacts with both hepatocyte nuclear factor 4α and liver receptor homolog 1 as a co-repressor, and FXR-induction of SHP results in CYP7A1 inhibition (31,32). When bile acid levels increase in the intestine, FXR induces FGF15, which acts as an endocrine hormone to inhibit hepatic CYP7A1 gene transcription after binding to FGF receptor 4 (FGFR4) on hepatocytes (33,34). In mice, Fgfr4 deletion resulted in significantly higher hepatic CYP7A1 expression, increased bile acid synthesis and enlarged bile acid pool size, while active FGFR4 over-expression reduced hepatic CYP7A1 expression and bile acid pool size (35). Furthermore, studies have demonstrated that FGFR4 forms a plasma membrane signaling complex with β-Klotho (36), cytoplasmic tyrosine phosphatase SHP-2 (37) and FGF receptor substrate 2 (38). Additionally, ligand binding to FGFR4 activates extracellular signal-regulated kinase 1/2 signaling to cause transcriptional repression of the CYP7A1 gene (33,39,40). Humans do not express FGF15. In humans, FGF19 shares ~51% amino acid sequence homology with mouse FGF15. In contrast to mouse FGF15, which is expressed in enterocytes but not hepatocytes, human FGF19 is expressed in both hepatocytes and enterocytes and is transcriptionally induced by FXR and bile acids. Bile acids and FXR agonists have been shown to directly induce FGF19 mRNA expression in human hepatocytes (39,41). In addition, plasma FGF19 levels were elevated in patients with obstructive cholestasis, which was presumably a result of hepatic FGF19 induction (42). Studies have revealed that human FGF19 also inhibits human CYP7A1 gene transcription via the FGFR4/extracellular signal-regulated kinase signaling cascade in human hepatocytes, suggesting that FXR/FGF15/19 regulation of CYP7A1 is a conserved mechanism (39,41). Although FGF15 is not expressed in mouse hepatocytes or gallbladder, it has been reported to regulate gallbladder refilling in mice (43). Mice lacking Fgf15 had almost empty gallbladders, which was restored by recombinant FGF15 administration. In contrast, FGF19 mRNA expression could be detected in human gallbladders (39,42). Interestingly, human gallbladders contain high FGF19 levels that are approximately one hundred-fold higher than those in the blood circulation (44). Gallbladder epithelial cells express the FGF19 receptor FGFR4 and β-Klotho (44). However, FGF19 function in the human gallbladder has not been well defined.
In addition to the inhibition of de novo bile acid biosynthesis, FXR activation also reduced intracellular bile acid accumulation by modulating bile acid transporters in hepatocytes and enterocytes. When bile acid levels increase in hepatocytes, bile acids activate FXR to induce BSEP to promote biliary bile acid secretion (45) and simultaneously inhibit the sodium-dependent taurocholate transporter and basolateral bile acid uptake (46). Similarly, intestinal bile acid accumulation activated FXR to induce basolateral bile acid efflux transporters OSTα and OSTβ (47) and repressed the apical bile acid uptake transporter ASBT (48). This thereby inhibited intestinal bile acid re-absorption and promoted bile acid secretion into the portal blood. In the kidney, bile acids are excreted via MRP2 and MRP4 into the urine. In addition, ASBT and OSTα/β are involved in re-absorption of bile acids in the renal proximal tubules. FXR repression of ASBT in the kidney may presumably increase renal bile acid excretion. A recent report revealed that FXR was highly expressed in the kidney, and its activation by bile acids induced renal aquaporin 2 and decreased urine volume in mice (49).
3.2. Bile acid signaling links nutrient sensing and metabolic homeostasis
Almost all major metabolic pathways respond to changes in nutrient availability. The metabolic switch from catabolism to anabolism during the fasting-to-fed transition is subjected to complex regulation by pancreatic hormones, incretins, adipokines and growth factors. After food intake, insulin promotes glucose uptake into the muscle and adipose to prevent prolonged postprandial hyperglycemia. Insulin also induces hepatic lipogenesis and thus couples postprandial glycolysis to fatty acid synthesis in the liver. Furthermore, insulin inhibits hepatic glycogenolysis, gluconeogenesis and hepatic very low density lipoprotein(VLDL) secretion. During fasting, attenuated insulin signaling and elevated glucagon action in the liver and adipose tissue stimulate hepatic glucose production and adipose lipolysis. Fatty acids are transported to the liver to be used for fatty acid oxidation, ketogenesis and VLDL assembly. The fasting-to-fed transition is often impaired in individuals with obesity and peripheral insulin resistance. In obesity and diabetes, uncontrolled adipose fatty acid release is considered a major contributor to hepatic steatosis and hyperlipidemia. Free fatty acid deposition in the skeletal muscle is also an initial trigger of muscle insulin resistance. In the liver, increased fatty acid influx into the hepatocytes causes lipotoxicity, which is thought to be an underlying cause of NASH pathogenesis.
Both hepatic bile acid synthesis and plasma bile acid concentrations increase in response to food intake (50,51). Postprandial metabolism is highly active. Studies suggest that bile acid signaling plays an important role in the regulation of postprandial lipid, glucose and energy metabolism. After a meal, cholecystokinin stimulates gallbladder contraction and bile acid release into the small intestine, which thus increases the trans-flux of bile acids across enterocytes, resulting in postprandial elevation of plasma bile acid concentrations in humans (50,51). In mice, plasma bile acid concentrations were lower during the light cycle and increased at onset of the dark cycle as well as after refeeding in mice (23). Hepatic CYP7A1 activity and bile acid synthesis also exhibited a strong diurnal rhythm (50). In humans, plasma 7α-hydroxy-4-cholesten-3-one, which is a surrogate marker for hepatic CYP7A1 activity, increased during postprandial periods (50). In mice, refeeding after fasting also caused marked induction of hepatic Cyp7a1 mRNA expression and hepatic CYP7A1 activity (23). The mechanism underlying the diurnal control of hepatic CYP7A1 expression is a result of complex regulation by clock genes and insulin and glucagon signaling, which is reviewed elsewhere (6). The following sections will summarize current knowledge regarding how bile acids act as metabolic regulators that integrate nutrient signaling with the regulation of metabolic homeostasis.
3.3. FXR regulation of fatty acid metabolism
Early studies demonstrated that using CDCA for gallstone dissolution in humans also decreased plasma triglyceride levels (52). Bile acid sequestrants are a class of cholesterol-lowering drugs that bind bile acids in the intestine and prevent bile acid reabsorption and return to the liver. Removal of bile acids by bile acid sequestrants induces hepatic CYP7A1 expression and cholesterol catabolism, which achieves cholesterol-lowering effects. It was found that treating hypercholesterolemia with bile acid sequestrants often raised plasma triglyceride levels in many human patients (53,54). These observations collectively suggest that bile acids may have triglyceride-lowering effects. Later studies in mice revealed that when challenged with a high fat diet, Fxr knockout mice displayed worsened hepatic lipid accumulation and hyperlipidemia compared with wild type control mice. In contrast, pharmacological activation of FXR by a semisynthetic FXR agonist attenuated diet-induced hepatic steatosis and hyperlipidemia in mice (55). These studies suggest that FXR plays a role in mediating the triglyceride-lowering effects of bile acids. Studies to date have provided extensive molecular insights on the mechanisms through which FXR regulates fatty acid and triglyceride metabolism. These bile acid- and FXR-regulated pathways are summarized below.
3.3.1. De novo lipogenesis
As mentioned earlier, obesity and insulin resistance cause increased de novo lipogenesis in the liver, which is a major contributor to fat accumulation and VLDL overproduction (56). Adipose-derived free fatty acids are the primary source of hepatic triglyceride synthesis in NAFLD (5). In addition, excess carbohydrates are also converted to fatty acids in the liver. Hepatic de novo lipogenesis is regulated by two transcriptional factors: sterol-regulatory element-binding protein-1 (SREBP-1) and carbohydrate-responsive element-binding protein (ChREBP). Studies have demonstrated that FXR activation represses both SREBP-1 and ChREBP and their target genes in de novo lipogenesis in the liver (57,58). SREBPs are a group of basic helix-loop-helix leucine zipper transcription factors involved in the regulation of fatty acid and cholesterol metabolism primarily in hepatocytes but also in some extrahepatic tissues (59). SREBPs are synthesized as precursors that are sequestered in the ER by forming a protein complex with SREBP cleavage activation protein and insulin-induced genes (Insig1 and Insig2). SREBP activation involves a sterol-sensing mechanism, whereby decreased oxysterol levels in the ER promote SREBP/SREBP cleavage activation protein complex translocation to the Golgi apparatus. In the Golgi apparatus, SREBPs are cleaved by two serine proteases S1P and S2P, releasing an N-terminal transcriptionally active form of SREBP that enters the nucleus to activate target gene transcription. There are two SREBP isoforms: SREBP-2, which primarily induces genes involved in cholesterol metabolism, and SREBP-1, which is a master inducer of lipogenic genes. In addition to proteolytic cleavage activation, hepatic SREBP-1 gene transcription is also highly induced in fatty livers (60). Srebp-1 transgenic mice displayed elevated hepatic lipogenic gene expression and developed hepatic steatosis on a chow diet (61). Currently identified SREBP-1-induced lipogenic genes include acetyl-CoA carboxylase, fatty acid synthase, acetyl-CoA synthase, ATP-citrate lyase, malic enzyme, glucose-6-phosphate dehydrogenase, 6-phosphogluconate dehydrogenase and stearoyl-CoA desaturase-1 (59,62). FXR activation by either bile acids or a synthetic FXR agonist decreased hepatic SREBP-1 expression. Mechanistically, FXR-induced SHP repressed LXRα activation of the SREBP-1 gene promoter (58). Both high dietary sugar intake and postprandial hyperglycemia cause increased carbohydrate influx into the glycolysis pathway in hepatocytes. Pyruvate, the end product of glycolysis generated by liver pyruvate kinase, is converted to acetyl-CoA, which enters the TCA cycle. In the presence of high levels of NADH and ATP during the postprandial state, acetyl-CoA-derived citrate is preferentially shuttled back to the cytosol and used for fatty acid synthesis and triglyceride production. In response to carbohydrate influx, glucose induces ChREBP nuclear translocation and transcriptional activation of a set of lipogenic genes, including liver pyruvate kinase, fatty acid synthase and acetyl-CoA carboxylase, to facilitate fatty acid synthesis (63,64). Studies have demonstrated that ChREBP inhibition alleviated hepatic fat accumulation in mice (65,66).
3.3.2. Fatty acid oxidation
Recently, several genes involved in hepatic fatty acid oxidation have been identified as novel FXR targets, suggesting that bile acids also promote hepatic fatty acid oxidation via FXR activation. It was first shown that bile acids induced peroxisome proliferator-activated receptor α (PPARα) expression in human hepatocytes but not in mouse hepatocytes because of the lack of a conserved FXR binding site in the mouse Pparα gene promoter (67). PPARα is a nuclear receptor that can be activated by endogenous free fatty acids as well as fibrates, a class of lipid-lowering drugs. Activation of PPARα transcriptionally induces hepatic fatty acid oxidation genes, including the rate-limiting gene carnitine palmitoyltransferase 1A. Therefore, increased cellular free fatty acid levels can feedforward to promote fatty acid oxidation via PPARα activation. More recently, it was shown that FXR also transcriptionally induced liver carboxylesterase 1 (CES1), and over-expression of CES1 in mouse liver decreased hepatic triglyceride accumulation (68). CES1 is a neutral cholesterol ester and triglyceride hydrolase that is highly expressed in hepatocytes and macrophages. Previous studies have established a protective role of CES1 in both NAFLD and atherosclerosis (68,69). Hepatocyte CES1 mobilized intracellularly-stored cholesterol ester in the lipid droplets and simultaneously promoted cholesterol conversion into bile acids in mice (70). In macrophages, CES1 stimulated cholesterol efflux and prevented foam cell formation and inflammatory macrophage activation (69). Xu et al. demonstrated that CES1 also converted triglycerides to free fatty acids, which led to the feedforward activation of PPARα (68). As a result, FXR induction of CES1 simultaneously promoted hepatic fatty acid oxidation driven by both increased substrate availability and induction of fatty acid oxidation genes. FGF21 is another catabolic hormone induced by PPARα in the hepatocytes. In addition, a previous study demonstrated that FXR also induced hepatic expression and secretion of FGF21 (71). In the liver, FGF21 has broad metabolic actions, including stimulation of hepatic lipid oxidation, ketogenesis and gluconeogenesis, and as a result, reduces triglyceride levels and improves insulin sensitivity (72,73). The mechanism of action of FGF21 in various metabolic tissues is reviewed in detail elsewhere (74).
3.4. FXR regulation of glucose metabolism
In type 2 diabetes, peripheral insulin resistance, especially in the skeletal muscle, impairs glucose clearance resulting in postprandial hyperglycemia. In addition, hepatic gluconeogenesis is increased because of the attenuated suppressive effects of insulin, which leads to fasting hyperglycemia. Feeding mice a CA-containing diet or administration of an FXR agonist reduced fasting plasma glucose levels and improved insulin sensitivity in obese and diabetic mice (55,75). The glucose-lowering effects of bile acids and FXR may be primarily attributed to the reduction in hepatic glucose production via transcriptional repression of key gluconeogenic genes, including the genes encoding phosphoenolpyruvate carboxykinase, PEPCK, and glucose 6-phosphatase, G6Pase. To date, FXR-induced SHP has been shown to interact with and inhibit PEPCK and G6Pase transactivation through cAMP response element-binding protein (76), forkhead transcription factor O1 (77) and glucocorticoid receptor (78). Elevation of intracellular glucose concentrations can induce FXR expression and enhance FXR activity via O-Glc-N-acylation (79,80), suggesting that FXR can also sense intracellular glucose levels and consequently control glucose metabolism. In humans, circulating plasma FGF19 concentrations exhibited a pronounced diurnal rhythm and peaked during the postprandial period (51). Recently, bile acid/FXR-induced gut FGF15 has been suggested to act as a postprandial hormone, in addition to insulin, that regulates hepatic glucose metabolism (81,82). FGF15/19 has also been shown to inhibit hepatic lipogenesis (83) and increase metabolic rate (84). Fgf19 transgenic mice were resistant to diet-induced obesity and insulin resistance (84). Human patients with fatty liver disease and insulin resistance had decreased fasting FGF19 levels or an impaired hepatic response to FGF19 (85,86).
3.5. Bile acid regulation of autophagy
Autophagy is a well-conserved cellular self-degradation process that helps eliminate protein aggregates and damaged organelles to maintain cellular integrity (87). Autophagy is also a catabolic process that produces nutrients and energy by degrading macromolecules in response to nutrient deprivation (88). Cellular autophagy is under the reciprocal control of nutrient-sensing mechanistic target of rapamycin and AMP-activated protein kinase signaling in response to changes in nutrient availability and growth hormone signaling (88). As a result, autophagy is normally induced after fasting/starvation and repressed during the postprandial period. Recent studies revealed that autophagy transported intracellularly stored lipids to the lysosomes for mobilization, a process termed “lipophagy” (89). Several recent studies have linked bile acid signaling to the regulation of hepatic autophagic activity. Whole genome chromatin binding assays revealed that FXR bound to many autophagy genes, and FXR activation caused transcriptional repression of autophagy gene expression and decreased hepatic autophagic activity (90,91). In addition, Manley et al. reported that hepatic autophagic flux was inhibited in cultured primary hepatocytes treated with bile acids and in the liver of Fxr knockout mice because of increased bile acid concentrations (92). This study suggested that bile acids impaired hepatic autophagic flux by inhibiting autophagosome-lysosome fusion. Consistent with the repressive effect of bile acids on autophagic activity, administration of the bile acid sequestrant cholestyramine induced hepatic autophagy in mice, which was likely a combined result of reduced hepatic mechanistic target of rapamycin signaling and bile acid concentrations (93). The novel role of bile acid signaling regulation of hepatic autophagy further supports the general notion that bile acids act as nutrient sensors to regulate hepatic metabolism during the fasting-to-fed transition.
3.6. Bile acid-activated G protein-coupled receptors
The Gαs protein-coupled receptor TGR5 is a bile acid-activated membrane receptor (94,95). TGR5 signals through the adenylate cyclase/cAMP cascade, resulting in protein kinase A activation. TGR5 can be activated by both conjugated bile acids and free bile acids, including CDCA, CA, DCA and LCA. LCA is the most potent TGR5 agonist with an EC50 of ~0.5 μM, while DCA, CDCA and CA activate TGR5 with EC50 values ranging between 1 and 10 μM. In the enterohepatic system, TGR5 is expressed in the intestine, with relatively higher expression found in the terminal ileum and colon (94). In the liver, TGR5 is not expressed in hepatocytes but is expressed in sinusoidal endothelial cells (96), gallbladder epithelial cells and Kupffer cells (97). In other extrahepatic tissues, TGR5 is expressed in the white and brown adipose tissue, spleen, kidney, pancreas, lung, macrophages and central nervous system (94).
3.6.1. TGR5 in energy expenditure and glucose metabolism
TGR5 was first reported to mediate the anti-obesity effect of bile acids (98). Feeding mice a CA-containing diet activated TGR5 in the brown adipose tissue and skeletal muscle. TGR5 subsequently induced type 2 deiodinase and the conversion of thyroxine to the active 3,5,3′-tri-iodothyronine and thus increased energy expenditure. A similar anti-obesity effect was also observed when mice were administered a potent synthetic TGR5 agonist INT-777 (99). In contrast to the clear anti-obesity effects of pharmacological TGR5 activation, TGR5 deficiency did not appear to have a profound impact on obesity development in mice. One study revealed higher weight gain in female but not male TGR5 knockout mice (100). However, another study demonstrated that TGR5 knockout mice did not gain more weight than wild type controls under chow-fed conditions or after a high fat diet challenge (101). As mentioned earlier, systemic bile acid concentrations are usually very low because of the efficient first pass extraction rate of bile acids from the portal blood by the liver. Therefore, TGR5 in the muscle and adipose may not be highly activated by physiological concentrations of systemic bile acids.
TGR5 has also been shown to exhibit hypoglycemic effects by stimulating glucagon-like peptide-1 (GLP-1) production. GLP-1 is a gut incretin secreted by the enteroendocrine L cells in the ileum and colon. During the postprandial period, dietary carbohydrates and fats stimulate GLP-1 secretion, which enhances insulin secretion from the pancreatic β cells, inhibits glucagon production from the α cells (102), slows gastric emptying and promotes satiety (103,104). These metabolic effects are thought to collectively contribute to the glucose-lowering effect of GLP-1. It was first reported that bile acids can induce GLP-1 production in the enteroendocrine cell line STC-1 via TGR5 activation (105). It was later confirmed in mice that administration of the TGR5 agonist INT-777 enhanced GLP-1 secretion and improved glucose homeostasis (99). Currently, GLP-1 mimetics and inhibitors of dipeptidyl peptidase-4, the enzyme that rapidly inactivates GLP-1 in the circulation, are clinically used as novel type 2 diabetes therapies. Recently, the bile acid sequestrant colesevelam, which has been used as a cholesterol-lowering drug, was found to also exhibit glucose-lowering effects when given to patients with type 2 diabetes mellitus (106). This effect has been attributed, at least in part, to GLP-1 induction in the gut (107). It is thought that colesevelam, by preventing bile acid re-absorption in the ileum, may increase the amount of bile acids that reach the colon, where TGR5 is highly expressed. Furthermore, bile acid sequestrants may also delay dietary fat solubilization and absorption, which allows a higher concentration of dietary fatty acids to reach the distal ileum and induce GLP-1 secretion (108). As such, bile acid sequestrants may synergistically enhance the stimulatory effect of dietary nutrients and bile acids on gut GLP-1 production after a meal. More recently, studies have suggested the involvement of TGR5 signaling in mediating the beneficial effects of gastric bypass surgery (109,110). Many of the metabolic improvements, particularly in glucose homeostasis, were attenuated in TGR5 knockout mice after vertical sleeve gastrectomy. Such effects may be partially attributed to altered bile acid composition and action in TGR5 target tissues, which is consistent with altered bile acid metabolism in TGR5 knockout mice (100,111).
3.6.2 The anti-inflammatory role of TGR5 in macrophages
TGR5 is functionally expressed in macrophages and plays an anti-inflammatory role. To date, TGR5 activation has been shown to attenuate inflammation in various experimental models, including diabetes, NAFLD, atherosclerosis, cholestasis and inflammatory bowel disease (94,97,112–116). Recently, several new studies have revealed novel mechanistic insights on the anti-inflammatory effects of TGR5 signaling in macrophages. TGR5 activation was first shown to antagonize nuclear factor kappa(NF)-κB signaling in macrophages, leading to reduced cytokine expression upon lipopolysaccharide challenge (112). TGR5 activation has also been shown to decrease chemokine expression, which, under obese conditions, reduced adipose macrophage infiltration and improved insulin sensitivity (117). In addition to antagonizing cellular pathways that induce cytokine synthesis at the transcriptional level, a new study reported that TGR5 activation could reduce cytokine maturation and secretion by inhibiting Nod-like receptor pyrin domain-containing protein 3 (NLRP3) inflammasome activation (118). The inflammasome is a pattern-recognition receptor-containing multi-protein oligomer that recognizes pathogen-derived pathogen-associated molecular patterns and endogenously produced danger-associated molecular patterns (119). Inflammasome activation can in turn activate caspase-1, which cleaves the pro-inflammatory cytokines Interleukin (IL)-1β and IL-18 to their bioactive forms. High fatty acid and glucose levels and intracellular accumulation of cholesterol crystals have all been shown to activate the NLRP3 inflammasome, which thus links metabolic stress and intracellular organelle damage to tissue inflammation (120–123). The study by Guo et al. demonstrated that TGR5 activation caused protein kinase A-dependent phosphorylation and ubiquitination of NLRP3, resulting in decreased NLRP3 inflammasome activation (118).
4. Conclusions
Extensive studies over the past two decades have significantly expanded our knowledge of the physiological and pathophysiological functions of bile acid metabolism and signaling. Bile acids are no longer considered merely physiological detergent molecules that facilitate nutrient absorption, but are also important regulators of various cellular processes in lipid, glucose and energy metabolism and immune responses. Under normal physiology, bile acid signaling plays a role in integrating nutrient sensing to the regulation of metabolic homeostasis. Dysregulation of bile acid homeostasis may underlie the pathogenesis of many human diseases. Importantly, new mechanistic understanding of bile acid signaling action in the liver and extrahepatic tissues to date has laid the groundwork for the development of promising bile acid-based drug therapies for the treatment of liver and metabolic diseases. Indeed, bile acid sequestrants have been used for a long time as a cholesterol-lowering therapy in addition to statins. More recently, the bile acid sequestrant colesevelam has been approved to be used in combination therapies to improve glycemic control in type 2 diabetes because of its effects on inducing gut GLP-1 production (53,106,124). Additionally, the FXR agonist obeticholic acid has been approved for primary biliary cholangitis treatment (125,126). Obeticholic acid treatment has also been shown to significantly improve fibrosis and NAFLD Activity Score in a completed Phase 2b FLINT trial (127). Other bile acid-based therapies, such as the selective TGR5 agonist and the FXR and TGR5 dual agonist, are being tested in preclinical studies and phase 1 trials as potential therapies for metabolic and inflammatory diseases (113,128–131).
Acknowledgments
Financial Support: This work was supported in part by an American Diabetes Association Junior Faculty Award, NIH grants1R01DK102487-01 and P20GM103549 & P30GM118247.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ratziu V, Goodman Z, Sanyal A. Current efforts and trends in the treatment of NASH. J Hepato. 2015;62:S65–S75. doi: 10.1016/j.jhep.2015.02.041. [DOI] [PubMed] [Google Scholar]
- 2.Byrne CD, Targher G. NAFLD: a multisystem disease. J Hepatol. 2015;62:S47–S64. doi: 10.1016/j.jhep.2014.12.012. [DOI] [PubMed] [Google Scholar]
- 3.Anstee QM, Targher G, Day CP. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nature reviews Gastroenterology & hepatology. 2013;10:330–344. doi: 10.1038/nrgastro.2013.41. [DOI] [PubMed] [Google Scholar]
- 4.Lonardo A, Sookoian S, Chonchol M, Loria P, Targher G. Cardiovascular and systemic risk in nonalcoholic fatty liver disease-atherosclerosis as a major player in the natural course of NAFLD. Curr Pharm Des. 2013;19:5177–5192. [PubMed] [Google Scholar]
- 5.Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science. 2011;332:1519–1523. doi: 10.1126/science.1204265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li T, Chiang JY. Bile acid signaling in metabolic disease and drug therapy. Pharmacol Rev. 2014;66:948–983. doi: 10.1124/pr.113.008201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chiang JY. Bile acid regulation of gene expression: roles of nuclear hormone receptors. Endocr Rev. 2002;23:443–463. doi: 10.1210/er.2000-0035. [DOI] [PubMed] [Google Scholar]
- 8.Wheeler JB, Shaw DR, Barnes S. Purification and characterization of a rat liver bile acid coenzyme A ligase from rat liver microsomes. Arch Biochem Biophys. 1997;348:15–24. doi: 10.1006/abbi.1997.0391. [DOI] [PubMed] [Google Scholar]
- 9.Falany CN, Fortinberry H, Leiter EH, Barnes S. Cloning, expression, and chromosomal localization of mouse liver bile acid CoA:amino acid N-acyltransferase. J Lipid Res. 1997;38:1139–1148. [PubMed] [Google Scholar]
- 10.Setchell KD, Heubi JE, Shah S, Lavine JE, Suskind D, Al-Edreesi M, et al. Genetic defects in bile acid conjugation cause fat-soluble vitamin deficiency. Gastroenterology. 2013;144:945–955. e946. doi: 10.1053/j.gastro.2013.02.004. quiz e914–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carlton VE, Harris BZ, Puffenberger EG, Batta AK, Knisely AS, Robinson DL, et al. Complex inheritance of familial hypercholanemia with associated mutations in TJP2 and BAAT. Nat Genet. 2003;34:91–96. doi: 10.1038/ng1147. [DOI] [PubMed] [Google Scholar]
- 12.Childs S, Yeh RL, Georges E, Ling V. Identification of a sister gene to P-glycoprotein. Cancer Res. 1995;55:2029–2034. [PubMed] [Google Scholar]
- 13.Berge KE, Tian H, Graf GA, Yu L, Grishin NV, Schultz J, et al. Accumulation of dietary cholesterol in sitosterolemia caused by mutations in adjacent ABC transporters. Science. 2000;290:1771–1775. doi: 10.1126/science.290.5497.1771. [DOI] [PubMed] [Google Scholar]
- 14.Smit JJ, Schinkel AH, Oude Elferink RP, Groen AK, Wagenaar E, van Deemter L, et al. Homozygous disruption of the murine mdr2 P-glycoprotein gene leads to a complete absence of phospholipid from bile and to liver disease. Cell. 1993;75:451–462. doi: 10.1016/0092-8674(93)90380-9. [DOI] [PubMed] [Google Scholar]
- 15.Xia X, Francis H, Glaser S, Alpini G, LeSage G. Bile acid interactions with cholangiocytes. World J Gastroenterol. 2006;12:3553–3563. doi: 10.3748/wjg.v12.i22.3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kullak-Ublick GA, Stieger B, Hagenbuch B, Meier PJ. Hepatic transport of bile salts. Semin Liver Dis. 2000;20:273–292. doi: 10.1055/s-2000-9426. [DOI] [PubMed] [Google Scholar]
- 17.Hagenbuch B, Stieger B, Foguet M, Lubbert H, Meier PJ. Functional expression cloning and characterization of the hepatocyte Na+/bile acid cotransport system. Proc Natl Acad Sci U S A. 1991;88:10629–10633. doi: 10.1073/pnas.88.23.10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meier PJ. Molecular mechanisms of hepatic bile salt transport from sinusoidal blood into bile. Am J Physiol. 1995;269:G801–G812. doi: 10.1152/ajpgi.1995.269.6.G801. [DOI] [PubMed] [Google Scholar]
- 19.Boyer JL, Trauner M, Mennone A, Soroka CJ, Cai SY, Moustafa T, et al. Upregulation of a basolateral FXR-dependent bile acid efflux transporter OSTalpha-OSTbeta in cholestasis in humans and rodents. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1124–G1130. doi: 10.1152/ajpgi.00539.2005. [DOI] [PubMed] [Google Scholar]
- 20.Cui YJ, Aleksunes LM, Tanaka Y, Goedken MJ, Klaassen CD. Compensatory induction of liver efflux transporters in response to ANIT-induced liver injury is impaired in FXR-null mice. Toxicol Sci. 2009;110:47–60. doi: 10.1093/toxsci/kfp094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ballatori N, Christian WV, Lee JY, Dawson PA, Soroka CJ, Boyer JL, et al. OST alpha-OSTbeta: a major basolateral bile acid and steroid transporter in human intestinal, renal, and biliary epithelia. Hepatology. 2005;42:1270–1279. doi: 10.1002/hep.20961. [DOI] [PubMed] [Google Scholar]
- 22.Trauner M, Boyer JL. Bile salt transporters: molecular characterization, function, and regulation. Physiol Rev. 2003;83:633–671. doi: 10.1152/physrev.00027.2002. [DOI] [PubMed] [Google Scholar]
- 23.Li T, Francl JM, Boehme S, Ochoa A, Zhang Y, Klaassen CD, et al. Glucose and insulin induction of bile acid synthesis: mechanisms and implication in diabetes and obesity. J Biol Chem. 2012;287:1861–1873. doi: 10.1074/jbc.M111.305789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takahashi S, Fukami T, Masuo Y, Brocker CN, Xie C, Krausz KW, et al. Cyp2c70 is responsible for the species difference in bile acid metabolism between mice and humans. J Lipid Res. 2016;57:2130–2137. doi: 10.1194/jlr.M071183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. 1995;83:841–850. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- 26.Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, et al. Identification of a nuclear receptor for bile acids. Science. 1999;284:1362–1365. doi: 10.1126/science.284.5418.1362. [DOI] [PubMed] [Google Scholar]
- 27.Parks DJ, Blanchard SG, Bledsoe RK, Chandra G, Consler TG, Kliewer SA, et al. Bile acids: natural ligands for an orphan nuclear receptor. Science. 1999;284:1365–1368. doi: 10.1126/science.284.5418.1365. [DOI] [PubMed] [Google Scholar]
- 28.Forman BM, Goode E, Chen J, Oro AE, Bradley DJ, Perlmann T, et al. Identification of a nuclear receptor that is activated by farnesol metabolites. Cell. 1995;81:687–693. doi: 10.1016/0092-8674(95)90530-8. [DOI] [PubMed] [Google Scholar]
- 29.Xie W, Radominska-Pandya A, Shi Y, Simon CM, Nelson MC, Ong ES, et al. An essential role for nuclear receptors SXR/PXR in detoxification of cholestatic bile acids. Proc Natl Acad Sci U S A. 2001;98:3375–3380. doi: 10.1073/pnas.051014398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Makishima M, Lu TT, Xie W, Whitfield GK, Domoto H, Evans RM, et al. Vitamin D receptor as an intestinal bile acid sensor. Science. 2002;296:1313–1316. doi: 10.1126/science.1070477. [DOI] [PubMed] [Google Scholar]
- 31.Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell. 2000;6:517–526. doi: 10.1016/s1097-2765(00)00051-4. [DOI] [PubMed] [Google Scholar]
- 32.Lu TT, Makishima M, Repa JJ, Schoonjans K, Kerr TA, Auwerx J, et al. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol Cell. 2000;6:507–515. doi: 10.1016/s1097-2765(00)00050-2. [DOI] [PubMed] [Google Scholar]
- 33.Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2:217–225. doi: 10.1016/j.cmet.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 34.Katafuchi T, Esterhazy D, Lemoff A, Ding X, Sondhi V, Kliewer SA, et al. Detection of FGF15 in plasma by stable isotope standards and capture by anti-peptide antibodies and targeted mass spectrometry. Cell Metab. 2015;21:898–904. doi: 10.1016/j.cmet.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yu C, Wang F, Kan M, Jin C, Jones RB, Weinstein M, et al. Elevated cholesterol metabolism and bile acid synthesis in mice lacking membrane tyrosine kinase receptor FGFR4. J Biol Chem. 2000;275:15482–15489. doi: 10.1074/jbc.275.20.15482. [DOI] [PubMed] [Google Scholar]
- 36.Ito S, Fujimori T, Furuya A, Satoh J, Nabeshima Y, Nabeshima Y. Impaired negative feedback suppression of bile acid synthesis in mice lacking betaKlotho. J Clin Invest. 2005;115:2202–2208. doi: 10.1172/JCI23076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li S, Hsu DD, Li B, Luo X, Alderson N, Qiao L, et al. Cytoplasmic tyrosine phosphatase Shp2 coordinates hepatic regulation of bile acid and FGF15/19 signaling to repress bile acid synthesis. Cell Metab. 2014;20:320–332. doi: 10.1016/j.cmet.2014.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang C, Yang C, Chang JY, You P, Li Y, Jin C, et al. Hepatocyte FRS2α is essential for the endocrine fibroblast growth factor to limit the amplitude ofbile acid production induce d by prandial activity. Curr Mol Med. 2014;14:703–711. doi: 10.2174/1566524014666140724095112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song KH, Li T, Owsley E, Strom S, Chiang JY. Bile acids activate fibroblast growth factor 19 signaling in human hepatocytes to inhibitcholesterol 7alpha-hydroxylase gene expression. Hepatology. 2009;49:297–305. doi: 10.1002/hep.22627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kong B, Wang L, Chiang JY, Zhang Y, Klaassen CD, Guo GL. Mechanism of tissue-specific farnesoid X receptor in suppressing the expression of genes in bile-acid synthesis in mice. Hepatology. 2012;56:1034–1043. doi: 10.1002/hep.25740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Holt JA, Luo G, Billin AN, Bisi J, McNeill YY, Kozarsky KF, et al. Definition of a novel growth factor-dependent signal cascade for the suppression of bile acid biosynthesis. Genes Dev. 2003;17:1581–1591. doi: 10.1101/gad.1083503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schaap FG, van der Gaag NA, Gouma DJ, Jansen PL. High expression of the bile salt-homeostatic hormone fibroblast growth factor 19 in the liver of patients with extrahepatic cholestasis. Hepatology. 2009;49:1228–1235. doi: 10.1002/hep.22771. [DOI] [PubMed] [Google Scholar]
- 43.Choi M, Moschetta A, Bookout AL, Peng L, Umetani M, Holmstrom SR, et al. Identification of a hormonal basis for gallbladder filling. Nat Med. 2006;12:1253–1255. doi: 10.1038/nm1501. [DOI] [PubMed] [Google Scholar]
- 44.Zweers SJ, Booij KA, Komuta M, Roskams T, Gouma DJ, Jansen PL, et al. The human gallbladder secretes fibroblast growth factor 19 into bile: towards defining the role of fibroblast growth factor 19 in the enterobiliary tract. Hepatology. 2012;55:575–583. doi: 10.1002/hep.24702. [DOI] [PubMed] [Google Scholar]
- 45.Ananthanarayanan M, Balasubramanian N, Makishima M, Mangelsdorf DJ, Suchy FJ. Human bile salt export pump promoter is transactivated by the farnesoid X receptor/bile acid receptor. J Biol Chem. 2001;276:28857–28865. doi: 10.1074/jbc.M011610200. [DOI] [PubMed] [Google Scholar]
- 46.Denson LA, Sturm E, Echevarria W, Zimmerman TL, Makishima M, Mangelsdorf DJ, et al. The orphan nuclear receptor, shp, mediates bile acid-induced inhibition of the rat bile acid transporter, ntcp. Gastroenterology. 2001;121:140–147. doi: 10.1053/gast.2001.25503. [DOI] [PubMed] [Google Scholar]
- 47.Frankenberg T, Rao A, Chen F, Haywood J, Shneider BL, Dawson PA. Regulation of the mouse organic solute transporter alpha-beta, Ostalpha-Ostbeta, by bile acids. Am J Physiol Gastrointest Liver Physiol. 2006;290:G912–G922. doi: 10.1152/ajpgi.00479.2005. [DOI] [PubMed] [Google Scholar]
- 48.Chen F, Ma L, Dawson PA, Sinal CJ, Sehayek E, Gonzalez FJ, Breslow J, et al. Liver receptor homologue-1 mediates species- and cell line-specific bile acid-dependent negative feedback regulation of the apical sodium-dependent bile acid transporter. J Biol Chem. 2003;278:19909–19916. doi: 10.1074/jbc.M207903200. [DOI] [PubMed] [Google Scholar]
- 49.Zhang X, Huang S, Gao M, Liu J, Jia X, Han Q, et al. Farnesoid X receptor (FXR) gene deficiency impairs urine concentration in mice. Proc Natl Acad Sci U S A. 2014;111:2277–2282. doi: 10.1073/pnas.1323977111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Galman C, Angelin B, Rudling M. Bile acid synthesis in humans has a rapid diurnal variation that is asynchronous with cholesterol synthesis. Gastroenterology. 2005;129:1445–1453. doi: 10.1053/j.gastro.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 51.Lundasen T, Galman C, Angelin B, Rudling M. Circulating intestinal fibroblast growth factor 19 has a pronounced diurnal variation and modulates hepatic bile acid synthesis in man. J Intern Med. 2006;260:530–536. doi: 10.1111/j.1365-2796.2006.01731.x. [DOI] [PubMed] [Google Scholar]
- 52.Schoenfield LJ, Lachin JM. Chenodiol (chenodeoxycholic acid) for dissolution of gallstones: the National Cooperative Gallstone Study. A controlled trial of efficacy and safety. Ann Intern Med. 1981;95:257–282. doi: 10.7326/0003-4819-95-3-257. [DOI] [PubMed] [Google Scholar]
- 53.Garg A, Grundy SM. Cholestyramine therapy for dyslipidemia in non-insulin-dependent diabetes mellitus. A short-term, double-blind, crossover trial. Ann Intern Med. 1994;121:416–422. doi: 10.7326/0003-4819-121-6-199409150-00004. [DOI] [PubMed] [Google Scholar]
- 54.Angelin B, Einarsson K, Hellstrom K, Leijd B. Effects of cholestyramine and chenodeoxycholic acid on the metabolism of endogenous triglyceride in hyperlipoproteinemia. J Lipid Res. 1978;19:1017–1024. [PubMed] [Google Scholar]
- 55.Zhang Y, Lee FY, Barrera G, Lee H, Vales C, Gonzalez FJ, et al. Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc Natl Acad Sci U S A. 2006;103:1006–1011. doi: 10.1073/pnas.0506982103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Howard BV, Howard WJ. Dyslipidemia in non-insulin-dependent diabetes mellitus. Endocr Rev. 1994;15:263–274. doi: 10.1210/edrv-15-3-263. [DOI] [PubMed] [Google Scholar]
- 57.Caron S, Huaman Samanez C, Dehondt H, Ploton M, Briand O, Lien F, et al. Farnesoid X receptor inhibits the transcriptional activity of carbohydrate response element binding protein in human hepatocytes. Mol Cell Biol. 2013;33:2202–2211. doi: 10.1128/MCB.01004-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Watanabe M, Houten SM, Wang L, Moschetta A, Mangelsdorf DJ, Heyman RA, et al. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J Clin Invest. 2004;113:1408–1418. doi: 10.1172/JCI21025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89:331–340. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- 60.Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JM, Shimomura I, et al. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev. 2000;14:2819–2830. doi: 10.1101/gad.844900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shimano H, Horton JD, Hammer RE, Shimomura I, Brown MS, Goldstein JL. Overproduction of cholesterol and fatty acids causes massive liver enlargement in transgenic mice expressing truncated SREBP-1a. J Clin Invest. 1996;98:1575–1584. doi: 10.1172/JCI118951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Uyeda K, Yamashita H, Kawaguchi T. Carbohydrate responsive element-binding protein (ChREBP): a key regulator of glucose metabolism and fat storage. Biochem Pharmacol. 2002;63:2075–2080. doi: 10.1016/s0006-2952(02)01012-2. [DOI] [PubMed] [Google Scholar]
- 64.Postic C, Dentin R, Denechaud PD, Girard J. ChREBP, a transcriptional regulator of glucose and lipid metabolism. Annu Rev Nutr. 2007;27:179–192. doi: 10.1146/annurev.nutr.27.061406.093618. [DOI] [PubMed] [Google Scholar]
- 65.Iizuka K, Bruick RK, Liang G, Horton JD, Uyeda K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc Natl Acad Sci U S A. 2004;101:7281–7286. doi: 10.1073/pnas.0401516101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dentin R, Pegorier JP, Benhamed F, Foufelle F, Ferre P, Fauveau V, et al. Hepatic glucokinase is required for the synergistic action of ChREBP and SREBP-1c on glycolytic and lipogenic gene expression. J Biol Chem. 2004;279:20314–20326. doi: 10.1074/jbc.M312475200. [DOI] [PubMed] [Google Scholar]
- 67.Pineda Torra I, Claudel T, Duval C, Kosykh V, Fruchart JC, Staels B. Bile acids induce the expression of the human peroxisome proliferator-activated receptor alpha gene via activation of the farnesoid X receptor. Mol Endocrinol. 2003;17:259–272. doi: 10.1210/me.2002-0120. [DOI] [PubMed] [Google Scholar]
- 68.Xu J, Li Y, Chen WD, Xu Y, Yin L, Ge X, et al. Hepatic carboxylesterase 1 is essential for both normal and farnesoid X receptor-controlled lipid homeostasis. Hepatology. 2014;59:1761–1771. doi: 10.1002/hep.26714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhao B, Song J, Chow WN, St Clair RW, Rudel LL, Ghosh S. Macrophage-specific transgenic expression of cholesteryl ester hydrolase significantly reduces atherosclerosis and lesion necrosis in Ldlr mice. J Clin Invest. 2007;117:2983–2992. doi: 10.1172/JCI30485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhao B, Song J, Ghosh S. Hepatic overexpression of cholesteryl ester hydrolase enhances cholesterol elimination and in vivo reverse cholesterol transport. J Lipid Res. 2008;49:2212–2217. doi: 10.1194/jlr.M800277-JLR200. [DOI] [PubMed] [Google Scholar]
- 71.Cyphert HA, Ge X, Kohan AB, Salati LM, Zhang Y, Hillgartner FB. Activation of the farnesoid X receptor induces hepatic expression and secretion of fibroblast growth factor 21. J Biol Chem. 2012;287:25123–25138. doi: 10.1074/jbc.M112.375907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Inagaki T, Dutchak P, Zhao G, Ding X, Gautron L, Parameswara V, et al. Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metab. 2007;5:415–425. doi: 10.1016/j.cmet.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 73.Badman MK, Pissios P, Kennedy AR, Koukos G, Flier JS, Maratos-Flier E. Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab. 2007;5:426–437. doi: 10.1016/j.cmet.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 74.Kliewer SA, Mangelsdorf DJ. Fibroblast growth factor 21: from pharmacology to physiology. Am J Clin Nutr. 2010;91:254S–257S. doi: 10.3945/ajcn.2009.28449B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ma K, Saha PK, Chan L, Moore DD. Farnesoid X receptor is essential for normal glucose homeostasis. J Clin Invest. 2006;116:1102–1109. doi: 10.1172/JCI25604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee JM, Seo WY, Song KH, Chanda D, Kim YD, Kim DK, et al. AMPK-dependent repression of hepatic gluconeogenesis via disruption of CREB. CRTC2 complex by orphan nuclear receptor small heterodimer partner. J Biol Chem. 2010;285:32182–32191. doi: 10.1074/jbc.M110.134890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yamagata K, Daitoku H, Shimamoto Y, Matsuzaki H, Hirota K, Ishida J, et al. Bile acids regulate gluconeogenic gene expression via small heterodimer partner-mediated repression of hepatocyte nuclear factor 4 and Foxo1. J Biol Chem. 2004;279:23158–23165. doi: 10.1074/jbc.M314322200. [DOI] [PubMed] [Google Scholar]
- 78.Borgius LJ, Steffensen KR, Gustafsson JA, Treuter E. Glucocorticoid signaling is perturbed by the atypical orphan receptor and corepressor SHP. J Biol Chem. 2002;277:49761–49766. doi: 10.1074/jbc.M205641200. [DOI] [PubMed] [Google Scholar]
- 79.Duran-Sandoval D, Cariou B, Percevault F, Hennuyer N, Grefhorst A, van Dijk TH, et al. The farnesoid X receptor modulates hepatic carbohydrate metabolism during the fasting-refeeding transition. J Biol Chem. 2005;280:29971–29979. doi: 10.1074/jbc.M501931200. [DOI] [PubMed] [Google Scholar]
- 80.Berrabah W, Aumercier P, Gheeraert C, Dehondt H, Bouchaert E, Alexandre J, et al. The glucose sensing O-GlcNacylation pathway regulates the nuclear bile acid receptor FXR. Hepatology. 2014;59(5):2022–2033. doi: 10.1002/hep.26710. [DOI] [PubMed] [Google Scholar]
- 81.Potthoff MJ, Boney-Montoya J, Choi M, He T, Sunny NE, Satapati S, et al. FGF15/19 Regulates Hepatic Glucose Metabolism by Inhibiting the CREB-PGC-1alpha Pathway. Cell Metab. 2011;13:729–738. doi: 10.1016/j.cmet.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kir S, Beddow SA, Samuel VT, Miller P, Previs SF, Suino-Powell K, et al. FGF19 as a postprandial, insulin-independent activator of hepatic protein and glycogen synthesis. Science. 2011;331:1621–1624. doi: 10.1126/science.1198363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bhatnagar S, Damron HA, Hillgartner FB. Fibroblast growth factor-19, a novel factor that inhibits hepatic fatty acid synthesis. J Biol Chem. 2009;284:10023–10033. doi: 10.1074/jbc.M808818200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tomlinson E, Fu L, John L, Hultgren B, Huang X, Renz M, et al. Transgenic mice expressing human fibroblast growth factor-19 display increased metabolic rate and decreased adiposity. Endocrinology. 2002;143:1741–1747. doi: 10.1210/endo.143.5.8850. [DOI] [PubMed] [Google Scholar]
- 85.Wojcik M, Janus D, Dolezal-Oltarzewska K, Kalicka-Kasperczyk A, Poplawska K, Drozdz D, et al. A decrease in fasting FGF19 levels is associated with the development of non-alcoholic fatty liver disease in obese adolescents. Journal of pediatric endocrinology & metabolism: JPEM. 2012;25:1089–1093. doi: 10.1515/jpem-2012-0253. [DOI] [PubMed] [Google Scholar]
- 86.Schreuder TC, Marsman HA, Lenicek M, van Werven JR, Nederveen AJ, Jansen PL, et al. The hepatic response to FGF19 is impaired in patients with nonalcoholic fatty liver disease and insulin resistance. Am J Physiol Gastrointest Liver Physiol. 2010;298:G440–G445. doi: 10.1152/ajpgi.00322.2009. [DOI] [PubMed] [Google Scholar]
- 87.Mehrpour M, Esclatine A, Beau I, Codogno P. Autophagy in health and disease. 1. Regulation and significance of autophagy: an overview. American journal of physiology Cell physiology. 2010;298:C776–C785. doi: 10.1152/ajpcell.00507.2009. [DOI] [PubMed] [Google Scholar]
- 88.Russell RC, Yuan HX, Guan KL. Autophagy regulation by nutrient signaling. Cell research. 2014;24:42–57. doi: 10.1038/cr.2013.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Seok S, Fu T, Choi SE, Li Y, Zhu R, Kumar S, et al. Transcriptional regulation of autophagy by an FXR-CREB axis. Nature. 2014;516:108–111. doi: 10.1038/nature13949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lee JM, Wagner M, Xiao R, Kim KH, Feng D, Lazar MA, et al. Nutrient-sensing nuclear receptors coordinate autophagy. Nature. 2014;516:112–115. doi: 10.1038/nature13961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Manley S, Ni HM, Kong B, Apte U, Guo G, Ding WX. Suppression of autophagic flux by bile acids in hepatocytes. Toxicol Sci. 2014;137:478–490. doi: 10.1093/toxsci/kft246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang Y, Ding Y, Li J, Chavan H, Matye D, Ni HM, Chiang JY, et al. Targeting the Enterohepatic Bile Acid Signaling Induces Hepatic Autophagy via a CYP7A1–AKT–mTOR Axis in Mice. Cell Mol Gastroenterol Hepatol. 2016;3(2):245–260. doi: 10.1016/j.jcmgh.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, et al. A G protein-coupled receptor responsive to bile acids. J Biol Che. 2003;278:9435–9440. doi: 10.1074/jbc.M209706200. [DOI] [PubMed] [Google Scholar]
- 95.Maruyama T, Miyamoto Y, Nakamura T, Tamai Y, Okada H, Sugiyama E, et al. Identification of membrane-type receptor for bile acids (M-BAR) Biochem Biophys Res Commun. 2002;298:714–719. doi: 10.1016/s0006-291x(02)02550-0. [DOI] [PubMed] [Google Scholar]
- 96.Keitel V, Reinehr R, Gatsios P, Rupprecht C, Gorg B, Selbach O, et al. The G-protein coupled bile salt receptor TGR5 is expressed in liver sinusoidal endothelial cells. Hepatology. 2007;45:695–704. doi: 10.1002/hep.21458. [DOI] [PubMed] [Google Scholar]
- 97.Keitel V, Donner M, Winandy S, Kubitz R, Haussinger D. Expression and function of the bile acid receptor TGR5 in Kupffer cells. Biochem Biophys Res Commun. 2008;372:78–84. doi: 10.1016/j.bbrc.2008.04.171. [DOI] [PubMed] [Google Scholar]
- 98.Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, Sato H, et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature. 2006;439:484–489. doi: 10.1038/nature04330. [DOI] [PubMed] [Google Scholar]
- 99.Thomas C, Gioiello A, Noriega L, Strehle A, Oury J, Rizzo G, et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009;10:167–177. doi: 10.1016/j.cmet.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Maruyama T, Tanaka K, Suzuki J, Miyoshi H, Harada N, Nakamura T, et al. Targeted disruption of G protein-coupled bile acid receptor 1 (Gpbar1/M-Bar) in mice. J Endocrinol. 2006;191:197–205. doi: 10.1677/joe.1.06546. [DOI] [PubMed] [Google Scholar]
- 101.Vassileva G, Hu W, Hoos L, Tetzloff G, Yang S, Liu L, et al. Gender-dependent effect of Gpbar1 genetic deletion on the metabolic profiles of diet-induced obese mice. J Endocrinol. 2010;205:225–232. doi: 10.1677/JOE-10-0009. [DOI] [PubMed] [Google Scholar]
- 102.Holst JJ. The physiology of glucagon-like peptide 1. Physiol Rev. 2007;87:1409–1439. doi: 10.1152/physrev.00034.2006. [DOI] [PubMed] [Google Scholar]
- 103.Nauck MA, Niedereichholz U, Ettler R, Holst JJ, Orskov C, Ritzel R, et al. Glucagon-like peptide 1 inhibition of gastric emptying outweighs its insulinotropic effects in healthy humans. Am J Physiol. 1997;273:E981–E988. doi: 10.1152/ajpendo.1997.273.5.E981. [DOI] [PubMed] [Google Scholar]
- 104.Tang-Christensen M, Vrang N, Larsen PJ. Glucagon-like peptide 1(7–36) amide’s central inhibition of feeding and peripheral inhibition of drinking are abolished by neonatal monosodium glutamate treatment. Diabetes. 1998;47:530–537. doi: 10.2337/diabetes.47.4.530. [DOI] [PubMed] [Google Scholar]
- 105.Katsuma S, Hirasawa A, Tsujimoto G. Bile acids promote glucagon-like peptide-1 secretion through TGR5 in a murine enteroendocrine cell line STC-1. Biochem Biophys Res Commun. 2005;329:386–390. doi: 10.1016/j.bbrc.2005.01.139. [DOI] [PubMed] [Google Scholar]
- 106.Staels B, Kuipers F. Bile acid sequestrants and the treatment of type 2 diabetes mellitus. Drugs. 2007;67:1383–1392. doi: 10.2165/00003495-200767100-00001. [DOI] [PubMed] [Google Scholar]
- 107.Sonne DP, Hansen M, Knop FK. Bile acid sequestrants in type 2 diabetes: potential effects on GLP1 secretion. European journal of endocrinology/European Federation of Endocrine Societies. 2014;171:R47–R65. doi: 10.1530/EJE-14-0154. [DOI] [PubMed] [Google Scholar]
- 108.Hofmann AF. Bile acid sequestrants improve glycemic control in type 2 diabetes: a proposed mechanism implicating glucagon-like peptide 1 release. Hepatology. 2011;53:1784. doi: 10.1002/hep.24100. [DOI] [PubMed] [Google Scholar]
- 109.Ding L, Sousa KM, Jin L, Dong B, Kim BW, Ramirez R, et al. Vertical sleeve gastrectomy activates GPBAR-1/TGR5 to sustain weight loss, improve fatty liver, and remit insulin resistance in mice. Hepatology. 2016;64:760–773. doi: 10.1002/hep.28689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.McGavigan AK, Garibay D, Henseler ZM, Chen J, Bettaieb A, Haj FG, et al. TGR5 contributes to glucoregulatory improvements after vertical sleeve gastrectomy in mice. Gut. 2017;66:226–234. doi: 10.1136/gutjnl-2015-309871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Donepudi AC, Boehme S, Li F, Chiang JY. G-protein-coupled bile acid receptor plays a key role in bile acid metabolism and fasting-induced hepatic steatosis in mice. Hepatology. 2017;65:813–827. doi: 10.1002/hep.28707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wang YD, Chen WD, Yu D, Forman BM, Huang W. The G-Protein-coupled bile acid receptor, Gpbar1 (TGR5), negatively regulates hepatic inflammatory response through antagonizing nuclear factor kappa light-chain enhancer of activated B cells (NF-kappaB) in mice. Hepatology. 2011;54:1421–1432. doi: 10.1002/hep.24525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.McMahan RH, Wang XX, Cheng LL, Krisko T, Smith M, El Kasmi K, et al. Bile acid receptor activation modulates hepatic monocyte activity and improves nonalcoholic fatty liver disease. J Biol Chem. 2013;288:11761–11770. doi: 10.1074/jbc.M112.446575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Pean N, Doignon I, Garcin I, Besnard A, Julien B, Liu B, et al. The receptor TGR5 protects the liver from bile acid overload during liver regeneration in mice. Hepatology. 2013;58:1451–1460. doi: 10.1002/hep.26463. [DOI] [PubMed] [Google Scholar]
- 115.Yoneno K, Hisamatsu T, Shimamura K, Kamada N, Ichikawa R, Kitazume MT, et al. GR5 signalling inhibits the production of pro-inflammatory cytokines by in vitro differentiated inflammatory and intestinal macrophages in Crohn’s disease. Immunology. 2013;139:19–29. doi: 10.1111/imm.12045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cipriani S, Mencarelli A, Chini MG, Distrutti E, Renga B, Bifulco G, et al. The bile acid receptor GPBAR-1 (TGR5) modulates integrity of intestinal barrier and immune response to experimental colitis. PLoS One. 2011;6:e25637. doi: 10.1371/journal.pone.0025637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Perino A, Pols TW, Nomura M, Stein S, Pellicciari R, Schoonjans K. TGR5 reduces macrophage migration through mTOR-induced C/EBPbeta differential translation. J Clin Invest. 2014;124:5424–5436. doi: 10.1172/JCI76289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Guo C, Xie S, Chi Z, Zhang J, Liu Y, Zhang L, et al. Bile Acids Control Inflammation and Metabolic Disorder through Inhibition of NLRP3 Inflammasome. Immunity. 2016;45:802–816. doi: 10.1016/j.immuni.2016.09.008. [DOI] [PubMed] [Google Scholar]
- 119.Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21:677–687. doi: 10.1038/nm.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rajamaki K, Lappalainen J, Oorni K, Valimaki E, Matikainen S, Kovanen PT, et al. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS One. 2010;5:e11765. doi: 10.1371/journal.pone.0011765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nature immunology. 2011;12:408–415. doi: 10.1038/ni.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, Sharp FA, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nature immunology. 2010;11:897–904. doi: 10.1038/ni.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Goldberg RB, Fonseca VA, Truitt KE, Jones MR. Efficacy and safety of colesevelam in patients with type 2 diabetes mellitus and inadequate glycemic control receiving insulin-based therapy. Arch Intern Med. 2008;168:1531–1540. doi: 10.1001/archinte.168.14.1531. [DOI] [PubMed] [Google Scholar]
- 125.Pellicciari R, Fiorucci S, Camaioni E, Clerici C, Costantino G, Maloney PR, et al. 6alpha-ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J Med Chem. 2002;45:3569–3572. doi: 10.1021/jm025529g. [DOI] [PubMed] [Google Scholar]
- 126.Hirschfield GM, Mason A, Luketic V, Lindor K, Gordon SC, Mayo M, et al. Efficacy of obeticholic acid in patients with primary biliary cirrhosis and inadequate response to ursodeoxycholic acid. Gastroenterology. 2015;148:751–761. e758. doi: 10.1053/j.gastro.2014.12.005. [DOI] [PubMed] [Google Scholar]
- 127.Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2015;385:956–965. doi: 10.1016/S0140-6736(14)61933-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Tiwari A, Maiti P. TGR5: an emerging bile acid G-protein-coupled receptor target for the potential treatment of metabolic disorders. Drug Discov Today. 2009;14:523–530. doi: 10.1016/j.drudis.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 129.Rizzo G, Passeri D, De Franco F, Ciaccioli G, Donadio L, Rizzo G, et al. Functional characterization of the semisynthetic bile acid derivative INT-767, a dual farnesoid X receptor and TGR5 agonist. Mol Pharmaco. 2010;78:617–630. doi: 10.1124/mol.110.064501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Baghdasaryan A, Claudel T, Gumhold J, Silbert D, Adorini L, Roda A, et al. Dual farnesoid X receptor/TGR5 agonist INT-767 reduces liver injury in the Mdr2−/− (Abcb4−/−) mouse cholangiopathy model by promoting biliary HCO(−)(3) output. Hepatology. 2011;54:1303–1312. doi: 10.1002/hep.24537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Miyazaki-Anzai S, Masuda M, Levi M, Keenan AL, Miyazaki M. Dual activation of the bile acid nuclear receptor FXR and G-protein-coupled receptor TGR5 protects mice against atherosclerosis. PLoS One. 2014;9:e108270. doi: 10.1371/journal.pone.0108270. [DOI] [PMC free article] [PubMed] [Google Scholar]