

Abstract
Approximately 7,000 rare diseases affect millions of individuals in the United States. Although rare diseases taken together have an enormous impact, there is a significant gap between basic research and clinical interventions. Opportunities now exist to accelerate drug development for the treatment of rare diseases. Disease foundations and research centers worldwide focus on better understanding rare disorders. Here, the state-of-the-art drug discovery strategies for small molecules and biological approaches for orphan diseases are reviewed. Rare diseases are usually genetic diseases; hence, employing pharmacogenetics to develop treatments and using whole genome sequencing to identify the etiologies for such diseases are appropriate strategies to exploit. Beginning with high throughput screening of small molecules, the benefits and challenges of target-based and phenotypic screens are discussed. Explanations and examples of drug repurposing are given; drug repurposing as an approach to quickly move programs to clinical trials is evaluated. Consideration is given to the category of biologics which include gene therapy, recombinant proteins, and autologous transplants. Disease models, including animal models and induced pluripotent stem cells (iPSCs) derived from patients, are surveyed. Finally, the role of biomarkers in drug discovery and development, as well as clinical trials, is elucidated.
Keywords: Drug discovery, drug development, rare diseases, orphan diseases, genetic disorders, small molecules, biologics, high throughput screen, phenotypic screen, target-based screen, drug repurposing, drug repositioning, gene therapy, induced pluripotent stem cells (iPSC), recombinant proteins, biomarkers
INTRODUCTION
Translational research and drug discovery for rare genetic diseases have grown at a rapid pace. A PubMed search conducted in April 2016, using ‘rare diseases’ and ‘orphan diseases’ as keywords, showed that publications related to rare diseases or orphan diseases have significantly increased over the past two decades (Fig. 1a). Advances in rare disease diagnostics and pharmacogenomics have allowed better characterizations of rare diseases, especially those that are monogenic. Approximately 7,000 rare diseases have been identified and many have a known etiology (https://rarediseases.info.nih.gov/). Although a rare disease in the United States (US) is defined as one that affects fewer than 200,000 individuals in the US, a staggering 20 to 30 million Americans currently live with a rare disease [Schieppati et al., 2008], representing a significant collective burden.
Fig. 1a. Publications for rare and orphan disease research indexed in MEDLINE from 1996 to 2015.
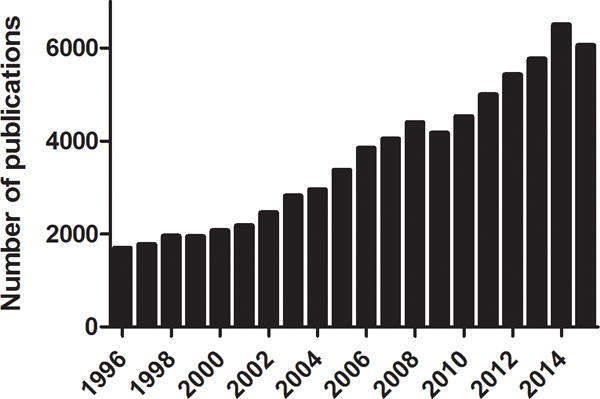
The search was conducted in April 2016 in PubMed using “rare diseases” and “orphan diseases” as keywords.
In 1983, the Orphan Drug Act (ODA) [Sanders 1983] was passed in the US to promote the development of treatments for rare diseases. In the decade before the ODA, only ten drugs for rare diseases had received the US Food and Drug Administration (FDA) approval, compared to more than 300 orphan drug approvals in the subsequent 25 years [Haffner 2006]. Based on the nature of molecular and developmental processes, a drug can be approved either as a new molecular entity (NME) or through a biologics license application (BLA). A NME is a drug that has not been marketed in the US in any form. BLA refers to the submission process that contains specific information on the manufacturing processes, chemistry, pharmacology, clinical pharmacology, and the medical effects of a biological product. If the information provided meets FDA requirements, the application is approved and a license is issued, allowing the company to market the new product. The number of NMEs and BLAs approved by the Center for Drug Evaluation and Research (CDER), a division of the FDA, increased from 5 in 2006 to 21 in 2015 for rare diseases (Fig. 1b). Orphan drugs, a term describing medications used to treat rare diseases, may offer several potential advantages, including shorter development timelines, lower cost of research and development, and less generic competition [Melnikova 2012]. In recent years, more than 35% of FDA-approved new drugs have been for the treatment of rare diseases. In turn, commercial activity in this sector has gained momentum. In 2015, two pharmaceutical companies bought rare disease assets in lucrative deals [Micklus and Muntner 2016]. Although the overall rare disease markets are notable, cost of treatment per patient may be high due to the limited number of patients suffering from each individual rare disease.
Fig. 1b. Number of new molecular entities (NMEs) and Biologics License Applications (BLAs) approved by the Center for Drug Evaluation and Research (CDER) from 2006 to 2015.
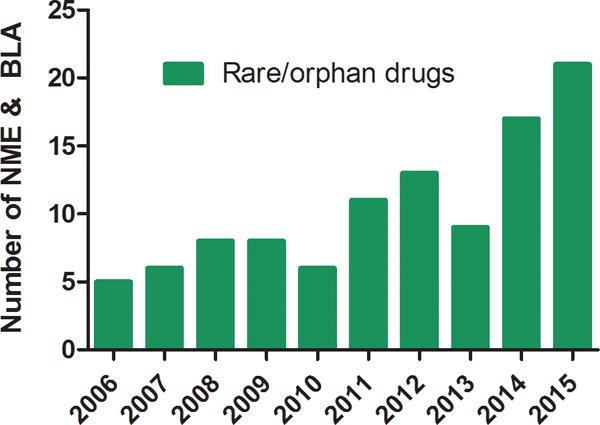
Data are from the FDA website (http://www.accessdata.fda.gov/scripts/cder/daf/).
Several factors have hindered therapeutic development for rare diseases. For example, heterogeneity in disease pathophysiology can cause large variations in drug response. Progression of many rare diseases is poorly understood due to limited natural history studies. Inadequate numbers of patients recruited for clinical trials lead to outcomes lacking statistical significance. Absence of biomarkers to measure disease also contributes to the ambiguity of clinical studies of rare diseases.
The heterogeneity of rare diseases increases the challenges that are faced in developing effective treatments. For example, Niemann–Pick disease type C (NPC) has over 200 missense mutations in the NPC1 gene that all result in a similar disease phenotype [Runz et al., 2008]. Congenital ichthyosis, a scaly skin disease, has more than 30 known subtypes with overlapping clinical phenotypes associated with different gene mutations [Dunoyer 2011]. The same drug will inevitably result in variable degrees of efficacy due to the different mutations that they carry, even though the patients are diagnosed as having the same disease. This also brings a unique opportunity for researchers and clinicians to move into the pharmacogenomics era.
The lack of natural history studies for most rare diseases renders patient ascertainment and recruitment more challenging than common diseases. For some ultra-rare diseases, there are fewer than 100 patients worldwide. Farber disease represents one such extreme example, with about 80 patients reported around the world.
FUNDING FOR RARE DISEASE DRUG DEVELOPMENT
Although the issue of commercial return on investment has been partially addressed with legislation and with some for-profit companies having robust business models, lack of funding for rare disease drug development remains, especially for early preclinical research. There are several efforts to address this disparity. The National Organization for Rare Disorders (NORD) (http://rarediseases.org/) has been successful in the past at lobbying the US Congress for improvements to the ODA [Brooks et al., 2016]. NORD provides referrals to more than 2,000 different organizations representing specific rare diseases. The organization has also provided funds through small grant programs to help develop drugs/treatments for rare diseases, which can help the collection of pilot data in order to apply for larger financial support through the NIH or other mechanisms. The NIH has programs to fund rare disease research, and the NIH official policy is to not consider the number of patients affected for a given disease when considering whether to fund an application. This policy helps to balance the funding for rare disease research.
Another alternative for funding of rare disease research resides in patient advocacy groups and foundations, which focus on a particular rare disease and often bring the disease researchers together. In recent years, while the regular funding sources have become more competitive, the funding for rare diseases appears to have remained steady. The FDA Office of Orphan Products Development (OOPD) is a major funding source for clinical grants that helps bridge the gap between basic research, clinical development, and marketing approval [Dunoyer 2011]. These grants only cover portions of phases I, II, and III clinical trials and do not fund preclinical development. OOPD grants are especially useful for academic researchers. These academically-derived early assets can in turn be licensed to pharmaceutical/biotech companies for further development or commercialization.
ORGANIZATIONS SUPPORTING RARE DISEASE DRUG DEVELOPMENT
Disease foundations
Because each rare disease individually affects a small population, the corresponding drug’s market is by definition relatively small. The price of treatment per patient is usually high because the cost of development for such therapy is shared by fewer patients. A smaller drug market typically attracts less interest from the pharmaceutical industry, especially in the context of therapeutic development for one disease at a time, and hence umbrella-type rare disease organizations can play an important role in the development of rare disease therapeutics. For example, NORD is a nonprofit patient advocacy organization dedicated to individuals with rare diseases and the organizations that serve them. NORD has more than 230 patient organization members. The programs at NORD include education, advocacy, research, and patient services. Together, they are committed to the identification, treatment, and cure of rare diseases. Many specific disease foundations and patient advocacy groups are also available to support patients and research.
Rare disease research centers
In the last few years, several translational research centers have been established worldwide to support various stages of research and development, such as the National Center for Advancing Translational Sciences (NCATS) at the NIH in the US, the International Rare Diseases Research Consortium (IRDiRC), the Translational Research Institute in Australia, and the Translational Research Informatics Center in Japan. Academic research centers for rare diseases have emerged in recent years, for example, the Center for Orphan Disease Research at the University of Pennsylvania, the Boler-Parseghian Center for Rare and Neglected Diseases at the University of Notre Dame, the Manton Center for Orphan Disease Research at Boston Children’s Hospital, the Center for Orphan Drug Research at the University of Minnesota, the Telethon Institute of Genetics and Medicine in Italy, Center for Therapeutic Innovation at the University of Miami, the Center for Rare Disease Therapies at the Keck Graduate Institute of Applied Life Sciences, the Moser Center for Leukodystrophies at the Kennedy Krieger Institute, and Institute for Advancing Medical Innovation at Kansas University Medical Center. Some of these centers and foundations distribute research funds as grants to support rare disease drug discovery, while others carry out basic research and drug development for rare diseases internally.
The NCATS and the Clinical Trials Transformation Initiative (CTTI) (https://www.ctti-clinicaltrials.org/) have led efforts to improve the quality and efficiency of clinical trials. Members of the CTTI, which is a public-private partnership involving governmental agencies, industry sponsors, patient advocacy group, academic institutions, and patients that was established in 2007 by the FDA and Duke University, contribute ideas, participate in studies, and disseminate new treatments. Engaging patients, their families, and caregivers in the process of drug development is a crucial component of the overall effort.
Approximately 50% of rare disease patients are children; thus, after the 2013 National Pediatric Research Network Act was passed, the NIH began to establish multiple consortia focused on pediatric research. Furthermore, the Clinical and Translational Science Awards program at the NCATS exists as a national network and resource established to catalyze the speed of clinical and translational research. To improve the drug development and clinical study of rare diseases, the FDA itself offers specific incentives and expedited programs, such as orphan drug designation and exclusivity, a pediatric rare disease priority review voucher, fast track, and priority review.
SMALL MOLECULE DRUG DEVELOPMENT FOR RARE DISEASES
The drug development process for rare diseases is similar to that for common diseases, which requires significant resources and typically lasts 10 to 12 years. Drugs from small molecules represent approximately 80–90% of the marketed therapeutics and have a number of advantages, including well-defined structures, relatively easy manufacturing, oral administration, and mostly non-immunogenic profiles. In addition, many of them can cross the blood-brain barrier to reach the central nervous system. From 2000 to 2008, 22% of NMEs approved by the FDA were orphan drugs, the majority of which were small molecules [Sun et al., 2016].
The drug discovery process has been revolutionized in the last two decades, with transition from low throughput animal model-based tests to high throughput molecular target-based screens. The modern process of discovery and development for a drug includes target identification, assay development, high throughput screening of small molecule libraries for hit identification, lead discovery and optimization, preclinical development, investigational new drug (IND) filing, clinical trials, and filing for final FDA approval [Paul et al., 2010] (Fig. 2). For any IND application, a clinical hold may be imposed by the FDA on the sponsors if the FDA determines that testing of the experimental drug is not safe in humans. Once an IND has been approved by the FDA, phase I clinical trial of the experimental drug can be initiated. Phase II and phase III trials have to be conducted before the drug is approved for marketing.
Fig. 2.
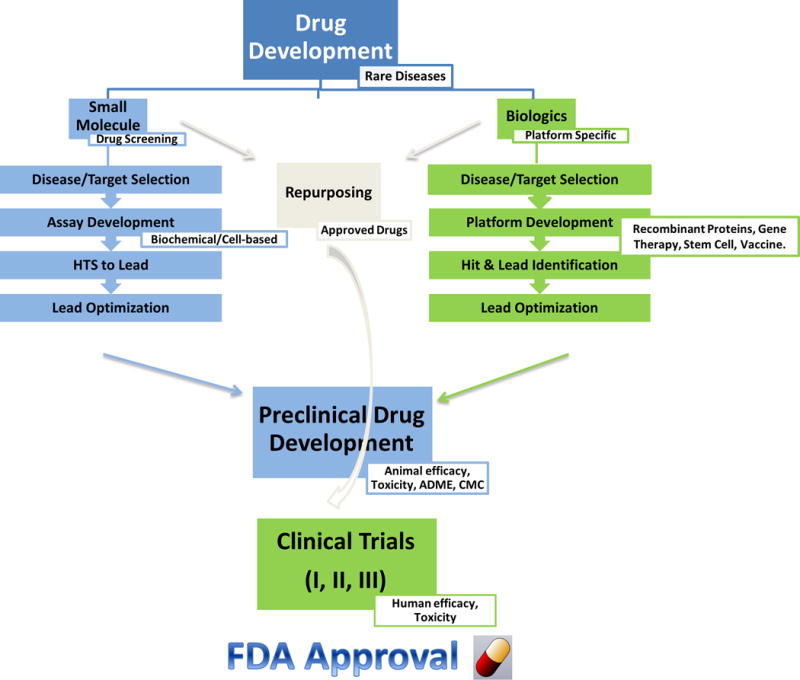
The process of drug discovery and development for rare diseases: small molecules, biologics, and repurposing approaches
Target identification
With advancements in molecular biology and the recent success in the identification of potential druggable genomic targets in the human genome [Aguero et al., 2008], molecular target-based drug discovery has become the predominant approach [Eder et al., 2014]. Whole genome or exome sequencing offers valuable opportunities to identify the causes of rare diseases. A protein target such as an enzyme, a receptor, or an ion channel related to a disease pathophysiology is usually first identified. Approaches in target identification such as direct biochemical methods, genetic interaction methods, and computational inference methods have been recently reviewed [Schenone et al., 2013].
Assay development
Once a disease target is identified, a specific assay needs to be developed to determine the candidate’s therapeutic activity. The Assay Guidance Manual (https://www.ncbi.nlm.nih.gov/books/NBK53196/) eBook is a useful resource for scientists who are interested in drug discovery for rare diseases. It provides guidelines for assay development, high throughput screening, and structure activity relationship (SAR) analyses as well as many other areas related to drug development. With the development of molecular biology techniques, recombinant proteins, and engineered cell lines expressing a specific protein or a reporter system, in vitro assays have become popular tools for screening compounds. Two major classes of assays are considered for screening of compounds (Table I). The first class is biochemical assays, which include measurements of enzyme activity, protein-protein interaction, and protein-DNA interaction. The proteins needed for these assays can be purified from primary tissues or expressed using recombinant systems. The other class of assays is cell-based assays using specially-engineered cell lines. For example, reporter-gene assays use a signal-generating reporter such as luciferase, beta-lactamase, and green fluorescent protein (GFP) that are typically linked to a special transcriptional promoter relevant to the disease target. Second messenger assays, such as those for G-protein coupled receptors, are designed to determine levels of cAMP and intracellular Ca2+ release in special cell lines. These screening assays, usually first developed in 96-well plate format, need to be miniaturized and optimized to 384- or 1536-well plate formats for the next step in large-scale screening of compounds. Use of higher density assay plates reduces consumption of proteins, cells, and other reagents, and increases the throughput of these screens. Before it is adopted for large-scale screening of compounds, an assay should meet certain statistical criteria for robustness, including a signal-to-basal ratio greater than 2, a coefficient of variation less than 10% (less than 15% for a cell-based assay), and Z′ factor greater than 0.5 [Inglese et al., 2007; Zhang et al., 1999]. Z′ factor indicates the assay robustness for HTS, which is calculated from the sample means and sample standard deviations (Z′ = 1 – (3*SD(total signal) + 3*SD(basal signal))/(Total signal – Basal signal)).
Table I.
Major types of biochemical and cell-based assays used in HTS for drug discovery
| Biochemical assays | Assay principle | Common examples |
|---|---|---|
| Enzyme substrate | Fluorescence change of enzyme substrate | Fluorogenic substrate of the enzyme of interest |
| Kinase | Monitor phosphorylated substrate, ATP or ADP | 33P-ATP, fluorophore-labeled substrate, ATP-dependent luciferase reaction, anti-ADP antibody |
| Protease | Monitor cleavage of substrate | Coumarin- or rhodamine-labeled peptide |
| Protein-protein, protein-DNA, protein-RNA interaction | Monitor binding of two proteins or protein-DNA/RNA | ELISA, FRET or AlphaScreen based assays |
| Cell-based assays | Assay principle | Common examples |
| Reporter | Monitor gene expression at the transcription/translation level | Green fluorescent protein (GFP) and luciferase assays |
| Second messenger | Monitor signal transduction activated by cell surface receptors | cAMP, Ca2+, and inositol phosphates |
| G-protein coupled receptor (GPCR) | Monitor GTP binding to receptor-activated G proteins | [35S]GTPγS and Eu-GTP™ |
| Cell proliferation/viability | Cell growth or death | Tetrazolium reduction (MTT), resazurin reduction (AlamarBlue), protease markers, and ATP content-based assays |
Fluorescence, luminescence, and time-resolved fluorescence resonance energy transfer (TR-FRET) are commonly used methods for detection in high throughput screening assays. TR-FRET assay combines standard FRET technology with time-resolved measurement of fluorescence, eliminating short-lived background fluorescence from sample components such as buffers, proteins, chemical compounds and cell lysates [Glickman et al., 2002]. Absorbance detection methods are less sensitive and have relatively large variations; thus, the absorbance readout is not recommended for a primary screening assay except for bacterial and fungus growth projects where the choice of assays is generally extremely limited. Various multifunction detection plate readers are available for applications in 96-, 384-, and 1536-well plate formats.
Compound library
Another important component in the small molecule lead discovery process is the collection of small molecule drug candidates, also referred to as a compound library. Original compound collections in large pharmaceutical companies had been assembled from previously synthesized compounds in-house, thus limiting the diversity of these collections. In the last two decades, the size and diversity of commercially-available chemical compound collections have grown substantially. Primary screens of half a million to 3 million compounds for lead compound identification for a single drug target have become routine in both pharmaceutical companies and academia.
High throughput screening
Once the assay has been developed and optimized, automated robotic screening of large collections of small-molecule compounds is performed. Developed in the 1990s, high throughput screening has evolved from 96-well plate format with reaction volumes of 100–200 μl/well to 384-well plates with 20–30 μl/well, and ultimately to 1536-well plates with only 2–8 μl/well. The miniaturization of this process has reduced reagent costs and made screens more practical. Automated screening platforms usually consist of liquid handlers for dispensing proteins, cells, compounds, and other reagents; incubators providing control of gas composition, temperature, and humidity; plate readers for detection of assay results; a robotic arm system; and software integrating all these components together. The automated robotic system increases compound screening throughput and improves data quality by reducing human error due to repeated handling of hundreds and thousands of assay plates. The robotic screenings had been initially developed and used in drug companies and have recently been adapted by academic researchers in screening centers within universities and research institutes. Using the automated screening system, a throughput of 500,000 to 1 million wells per day can be achieved, with a primary screen thus completed very rapidly.
Screening data analysis and hit selection
The screening data are loaded into a database and then analyzed using informatics software. The primary screening hits are typically selected using criteria such as “inhibition greater than 50%” for a single concentration screening, or “inhibitory concentration of 50% response (IC50) less than 5 μM and efficacy greater than 70%” for quantitative high throughput screening (qHTS) [Inglese et al., 2006]. The primary hits consist of true positive compounds, as well as false positives such as autofluorescent and other types of non-specific compounds. These false positive and non-specific compounds have to be recognized and eliminated in the hit confirmation stage.
Hit confirmation
The selected hit compounds are tested in secondary and tertiary assays to confirm their activity and selectivity. The same assay used in the primary screen is first used to confirm the activity of the compound in a concentration-dependent manner, typically using an independently-sourced sample. A compound cytotoxicity assay using the same compound concentration and incubation time as the primary screening assay is usually employed to eliminate the toxic compounds. A counter-screen, such as a mock transfected cell line or a non-target protein, is used to eliminate the non-specific compounds including fluorescent compounds or compounds that otherwise interfere with the assay signal.
Additional experiments are then used to further confirm compound activities found in the primary screens. Tertiary assays with different formats (e.g. a luminescence assay versus a fluorescence assay), primary cells (instead of engineered cell lines), and cell-based assay (for compounds identified from biochemical assays) are usually employed at this stage. All these efforts lead to the identification and prioritization of relatively few lead compounds.
Lead optimization
Once the lead compounds have been identified, chemistry optimization is an important next step for drug development, with the goal of improving the potency and selectivity of the small molecule. Medicinal chemists play a crucial role in the lead optimization process, not only by synthesizing newly designed compounds but also by leading the team effort in this important task, utilizing multiple types of experimental and computationally-derived information. The lead compound undergoes several rounds of extensive medicinal chemistry modifications to improve its potency, selectivity, water solubility, ADME (absorption, distribution, metabolism, and excretion), and toxicity profile. Chemoinformatic analysis assists with defining the SAR of the lead compound. In cases where structural information on the protein target is available, computational modeling of the interaction between a lead compound and its molecular target can yield new structures of chemical compounds with potentially improved binding properties. These rationally designed compounds are either synthesized by chemists or can be procured from millions of commercially available compounds. The optimized lead compound then moves to preclinical drug development.
Preclinical drug development
Preclinical development involves a team of experts including pharmacologists, chemists, drug metabolism specialists, toxicologists, process chemists, and formulation and regulatory experts. A few optimized lead compounds are first evaluated for their pharmacokinetics in small animals. Animal disease models, if available, are used to further confirm the efficacy of the compound and evaluate acute toxicity. The experimental results are used to guide another round of chemistry optimization to further improve the properties of the compound. The final optimized lead compound is further tested in large animal disease models and in additional toxicology studies before entering clinical trials (Fig. 2). The goals of preclinical drug development are to further establish drug efficacy in animal disease models, to characterize pharmacokinetics, and to ensure the drug safety of the lead compound(s). Identification and development of high quality lead compounds is critical for increasing the success rate of drug development in the later stages of clinical trials.
PHENOTYPIC SCREENING-BASED DRUG DISCOVERY FOR DISEASES WITH UNKNOWN TARGETS
For a genetic disease with known etiology and clear disease pathogenesis, molecular target-based drug discovery can be carried out as described above. However, the etiologies of many diseases are unknown, or in the cases of known genetic disorders, the cause-effect relationship between mutations and disease pathogenesis is unclear. For example, only a small fraction of patients with amyotrophic lateral sclerosis (ALS) have a genetic basis for their illness, and the disease pathogenesis in most patients is unknown [Kiernan et al., 2011] (Table 2). In Huntington disease, the mutation in the HTT gene was identified in 1993, but the function of the mutated protein is not completely understood and the pathophysiology of the disease is unclear (Table 2), thereby hindering the identification of a valid target for drug development. In such cases, a phenotypic screening approach is an alternative drug discovery strategy [Zheng et al., 2013]. This phenotypic screening approach for drug discovery, which is also called forward, or classical, pharmacology, allows for the activity of a drug to be determined without knowing its molecular mechanism and protein target [Takenaka 2001]. In modern phenotypic screening, a characteristic change associated with the disease (i.e., phenotype) is used to develop a cell-based assay. For example, filipin staining was used to identify compounds that were effective for the treatment of Niemann-Pick disease type C [Vanier et al., 2016]. A chemical library is then screened in the phenotypic assay to identify active hits that ameliorate the disease phenotype in the cell-based assay. In the typical phenotypic screening assays, active compounds will induce changes such as suppressing the viability of cancer cells and microbial organisms, morphological changes in cells, and functional changes in cells such as abnormal electrical activity. Phenotypic screening assays are usually more physiologically relevant and less artificial as a model system because disease-relevant cells, specifically primary cells and native cellular environment, are used. In turn, lead optimization of hits derived from phenotypic screening can be difficult since the molecular target is unknown. Despite this drawback, 28 NMEs were discovered and developed by phenotypic screenings between 1999 and 2008 [Swinney and Anthony 2011]. A more recent review summarized 48 oncology drugs approved by the FDA between 1999 and 2013. Four of them (lenalidomide, pomalidomide, romidepsin, and vorinostat) were discovered and developed entirely through the use of the phenotypic screening approach with unknown drug targets [Moffat et al., 2014], while fourteen relied on the phenotypic assays at some stages of the process. Therefore, the phenotypic screening is a useful strategy for genetic disease drug development.
Table II.
Selected rare genetic diseases and their pathophysiology and therapeutics
| Disease name | Pathophysiology | Current Therapeutics | Investigational Therapeutics | References |
|---|---|---|---|---|
| Angelman syndrome | Deletion of chromosome 15 | Clonazepam | Stem cell, small molecules (SM)* | [Grant 2000]; [Chamberlain et al., 2010] |
| Cystic fibrosis | Mutations of CFTR† | Lumacaftor/ivacaftor | Gene therapy, SM | [Griesenbach et al., 2004; Van Goor et al., 2011] |
| Retinal degeneration | Mutations of photoreceptors | Fenretinide | Gene therapy, stem cell, SM | [Kaewkhaw et al., 2016; Trifunovic et al., 2012] |
| Amyotrophic lateral sclerosis | Death of motor neurons | Riluzole | Stem cell, SM | [Bensimon et al., 1994; DeLoach et al., 2015] |
| Sickle cell disease | Substitution of hemoglobin | Hydroxyurea | Gene therapy, SM | [Charache et al., 1995; Hoban et al., 2016; Telen 2016] |
| Osteogenesis imperfecta | Substitution of glycine in collagen | Bisphosphonates | Growth hormone, gene therapy | [Evans 2012; Glorieux et al., 1998] |
| Hutchinson-Gilford progeria | Mutation of lamin A (LMNA) | Farnesyltransferase inhibitors | SM, Antisense oligonucleotide | [Lo Cicero and Nissan 2015; Moorthy et al., 2013] |
| Huntington disease | Mutation of Huntingtin gene (HTT) | Tetrabenazine | Stem cell, gene therapy, SM | [Chen et al., 2014; McLellan et al., 1974] |
Note:
SM – Small molecules.
CFTR – as cystic fibrosis transmembrane conductance regulator. All the above diseases have no cure, except that a small population of sickle cell patients can be cured by bone marrow transplantation. Current therapeutics may reduce symptoms or delay disease progression. Note that no gene therapy product has yet been approved by the US FDA.
DRUG REPURPOSING AND DRUG REPOSITIONING
Drug repurposing, also known as drug repositioning, is the identification and application of already approved drugs to treat new diseases. Drug repurposing screens aim to rapidly discover new indications by testing collections of approved drugs. The procedure is similar to the above-mentioned novel compound screening, except for the use of approved drug collections instead of a large and diverse collection of compounds [Sun et al., 2016]. For rare diseases, drug repurposing screening has emerged as an effective alternative approach for the rapid identification of new therapeutic compounds (Fig. 2). The need for new rare disease therapeutics has been met with a relative lack of interest from the private sector. In conjunction with novel drug discovery, drug repurposing could relieve this situation by increasing and expediting identification of effective drug candidates. Drug repositioning with FDA-approved drugs and clinical drug candidates offers several benefits over the classical new drug development process described above. These approved drug compounds have been used in patients, and their toxicity and safety are typically well-established [Huang et al., 2011]. Once a new indication has been identified for an approved drug, the molecule can be further evaluated in clinical trials quickly without prolonged preclinical development. In addition, since the targets for many of these drugs are known, the activity identified for these compounds (such as a kinase inhibitor or protease inhibitor) in a new disease may help identify a new drug target or therapeutic approach for that disease.
An interesting example of drug repurposing is the case of sildenafil (Viagra®; Pfizer), which was initially studied for the treatment of hypertension and angina pectoris in the 1980s. In 1998, it was repurposed for the treatment of erectile dysfunction, and in 2005 it was approved as an orphan drug for the treatment of pulmonary arterial hypertension (Revatio®; Pfizer). Vorinostat (Zolinza®; Merck & Co), an orphan drug for cutaneous T-cell lymphoma (CTCL), was discovered to induce myeloid differentiation and then subsequently identified as a broad spectrum histone deacetylase inhibitor [Marks and Breslow 2007]. This drug is now under investigation for the treatment of glioblastoma multiforme and non-small-cell lung cancer [Friday et al., 2012; Reguart et al., 2014]. In 2014, Kevin Eggan, Clifford J. Woolf, and co-workers discovered a cellular phenotype of reduced delayed-rectifier potassium channel in induced pluripotent stem cells (iPSCs)-differentiated motor neurons derived from amyotrophic lateral sclerosis (ALS) patients [Wainger et al., 2014]. An approved anticonvulsant, retigabine, was then shown to correct the phenotype and improve the in vitro survival of motor neurons derived from patients. Because retigabine is an approved drug, a phase II clinical trial of retigabine in ALS was rapidly initiated in 2015 (ClinicalTrials.gov identifier: NCT02450552).
The resource required for the development of new drugs for nearly 7,000 rare diseases is so huge that it will take well over a thousand years to develop effective therapeutics for all rare diseases using the classical drug development method. Addressing several rare diseases that share a common molecular etiology within a given project is especially attractive as the majority of rare diseases have an underlying genetic cause [Sun et al., 2016]. Undertaking drug repurposing screens for a variety of rare diseases that share a common molecular etiology will expedite drug discovery for these conditions. NCATS has established a robust drug repurposing program, New Therapeutic Uses (https://ncats.nih.gov/ntu), to facilitate both early-stage and late-stage repurposing by funding researchers who carry out repurposing projects using abandoned drug candidates.
BIOLOGICS
In recent years, biologics have grown in importance as effective therapeutics for many diseases. From 2000 to 2008, 31% of BLAs approved by the FDA were orphan drugs. Biologic products include vaccines, blood, tissues, cells, gene therapies, and recombinant proteins (enzymes, peptides, and antibodies) (http://www.fda.gov/BiologicsBloodVaccines/). The discovery and development of biologic products is different from that of small molecule drugs described above (Fig. 2). In contrast to small molecule drugs that are chemically synthesized with their structures known and well-defined, most biologics are complex products without defined structures. They can be isolated from many natural sources, including human tissues, animals, or microorganisms, using advanced biotechnology methods. It is often believed that biologics represent the most cutting-edge products in biomedical research and may provide the most effective therapies for the treatment of rare diseases that lack approved therapeutics. In addition to enzyme replacement therapies, therapeutic antibodies, and other proteins with disease-modulating properties, the biologics category includes other novel therapies, such as the use of gene vectors to correct phenotype-associated disease mutations. The NIH Genetic Modification Clinical Research Information System (GeMCRIS) is a comprehensive database for scientists, research participants, and others with an interest in human gene transfer research (https://www.gemcris.od.nih.gov/). GeMCRIS allows users to access information on human gene transfer trials registered with the NIH, including medical conditions under study, gene products being investigated, and a list of researchers and institutions carrying out these trials.
If a rare disease is caused by the deficiency of a protein, that protein, either isolated from an animal or recombinantly produced in cells, can potentially be delivered to the patient as a replacement therapy. The production of such proteins can be very challenging due to the unique post-translational modifications typically employed within the human cells, which are difficult to recapitulate during production in non-human cell culture. Lysosomal enzymes, for example, require glycosylation after protein translation; this modification is critical for binding to specific cell surface receptors. If the binding is not correct, the lysosomal enzymes will not be taken up by cells. Many of these modifications are accomplished by specific cell types. Another important factor to consider is the aseptic conditions that must be maintained in the manufacturing of these products as the process is more susceptible to microbial contamination, in contrast to the processes used in the production of small molecule drugs where the exclusive use of organic solvents prevents colonization.
In addition, many rare diseases affect the brain and the central nervous system (CNS). Biologics do not usually cross the blood-brain barrier and are thus ineffective for CNS symptoms. Intracerebroventricular or intrathecal injection, despite its risks, may offer an alternative route of administration for some biologics. Despite these limitations, biologics have emerged as the next generation of approaches to therapeutic development because they have the potential to correct the underlying pathophysiology by replacing protein function (enzyme/protein replacement therapy), preventing disease (vaccines), permanently correcting the disease (gene therapy), or boosting the power of the immune system (immunotherapy). Examples in this category include Factor VIII proteins for hemophilia [Aledort et al., 2014], vaccines to prevent smallpox and measles [Meyer et al., 1965], and monoclonal antibodies for cancer therapy [Reichert et al., 2005].
Enzyme replacement therapy (ERT)
ERT is considered a cornerstone in rare disease treatment and has been reviewed previously [Ortolano et al., 2014; Parenti et al., 2015]. ERT has been approved for 8 lysosomal storage diseases: Gaucher disease type 1, Fabry disease, mucopolysaccharidosis types I, II, IVA, and VI, Pompe disease, and lysosomal acid lipase deficiency [Hoffman et al., 1993; Ortolano et al., 2014; Sanford and Lo 2014]. ERT has also been used for the treatment of one form of immunodeficiency, adenosine deaminase (ADA) deficiency [Aiuti et al., 2009] and for infantile or juvenile-onset hypophosphatasia [Whyte 2017]. Development of enzyme replacement therapy starts with research on a small scale that produces recombinant proteins by yeast, bacteria, plant, and mammalian cells. Next, protein production is optimized and preclinical development of the lead protein product follows, including extensive profiling for immunogenicity. ERT usually requires intravenous administration at frequent intervals due to the relatively short half-life of enzymes in circulation, making it inconvenient for patients: for example, agalsidase beta (Fabrazyme®) has been approved for the treatment of Fabry disease since 2001 and has to be administered intravenously once every two weeks [Brady 2006; Lidove et al., 2007]. ERT has been efficacious at ameliorating the cardiac and renal complications in early-phase Fabry disease, lessening pain and improving the quality of life [Lidove et al., 2007]. However, the long-term use of ERT in advanced Fabry disease has not prevented progression towards organ failure and death [Weidemann et al., 2013]. In addition, some patients developed immune responses to the infused recombinant enzymes [Lidove et al., 2007]. The typically short half-life of enzymes and the need for repeated administration of large amounts of enzyme make ERT extremely expensive (an estimated US$200,000 per patient per year in 2012) [Rombach et al., 2013]. Therefore, other treatments are still needed for better management of Fabry disease.
Other recombinant human proteins
Hemophilia A is a genetic disorder in which blood does not clot normally due to deficiency of factor VIII. Several recombinant factor VIII products have been approved for the treatment of hemophilia A. The main limitation with these recombinant proteins is their short half-lives (8–12 hours for factor VIII), making repeated administrations necessary [Peyvandi et al., 2016]. A serious drawback of this and other replacement therapies is the development of antibodies directed against infused proteins, which reduces the efficacy of future treatment. One strategy for preventing antibody formation is to design genetically engineered proteins to better match the native proteins and to perform the intravenous infusion very slowly in order to minimize the immune reactions.
Stem cell-based therapy
Stem cell-based therapy has been largely confined to cord blood or bone marrow transplants to differentiate hematopoietic stem cells into key subpopulations. Stem cell-based therapies are now under investigation for a diverse range of rare diseases, including degenerative neurologic disorders such as Krabbe disease [Hoffman and Escolar 2006], Fanconi anemia [Kelly et al., 2007], and metabolic storage diseases such as the mucopolysaccharidoses [Sauer et al., 2004]. Since the discovery of iPSCs in 2006, cell-based therapy has moved to a new era. Cell-based therapy offers the opportunity for long-term correction. However, if unrelated donor cells are used, bone marrow transplantation can cause severe issues, such as failure of reconstitution, graft versus host disease, severe infections owing to immune suppression, and death. Strategies to reduce or eliminate these side effects are urgently needed.
Hematopoietic stem cells can be differentiated to all the other types of blood cells through hematopoiesis. Derived from the mesoderm, hematopoietic stem cells are located in the red bone marrow. In addition to bone marrow, umbilical cord blood is a rich source of hematopoietic stem cells and is an alternative source of cells for hematopoietic stem cell transplantation. Umbilical cord blood is collected from the placenta after a baby is born and the umbilical cord has been cut. Cord blood cells are then isolated, processed, and stored in a cord blood bank for future use. Hematopoietic stem cell transplantation has been used to treat many genetic diseases including lysosomal storage diseases [Hemsley and Hopwood 2009]. In 2011, the FDA approved HPC, Cord Blood (HEMACORD®; New York Blood Center) as the first licensed hematopoietic progenitor cells-cord (HPC-C) cell therapy for patients with disorders affecting the hematopoietic system [Allison 2012]. Although HPC, Cord blood has been “tested to exclude donors with sickle cell anemia, and anemias due to abnormalities in hemoglobins C, D, and E” and donors at high-risk of having specific infectious diseases are excluded, it is possible for the recipient to contract a genetic hemoglobinopathy or an infectious disease from an unsuspecting donor (http://hemacord.info/physicians/).
Recent advances in iPSC technology have enabled the conversion of patient cells such as skin fibroblasts and peripheral blood monocytes to iPSCs. Once expanded, the iPSC can then be further differentiated into mature cells, thereby reducing the adverse effects in stem cell-based therapy by using an autologous graft. However, the development of each cell product is different for each disease and the procedures are not standardized yet. Additionally, the process of iPSC generation, scaling-up, and differentiation can take up to several months, and it is presently associated with low yield, high variability, and very high costs. The FDA has not approved any stem cell-based products for use, other than hematopoietic progenitor cells-cord (HPC-C) cell therapy for patients with disorders affecting the hematopoietic system. In 2014, Takahashi and co-workers led the first iPSC clinical study in Japan. The first patient was transplanted with his/her own iPSC-derived retinal pigment epithelial cells for treatment of age-related macular degeneration [Garber 2015]. Although this patient did not suffer any serious adverse effects, the team decided to suspend the clinical trial due to the identification of mutations in the iPSCs from a second patient, as well as regulatory changes. The mutations identified in the second patient’s iPSCs included three single nucleotide variations and three copy-number variants that were not present in the patient’s original fibroblasts. Mutations and epigenetic/chromosomal changes are often acquired by iPSC in culture [Pera 2011]. It is not entirely clear whether the mutations were caused by the reprogramming process; however, these complications further argue for standardization of stem cell derivation, characterization, and differentiation protocols. Despite current limitations, cell-based therapies hold promise for rare diseases, and human clinical trials of stem cell-based therapies for rare diseases have started [Thomsen et al., 2014].
There are several advantages of using iPSCs in cell therapy. Firstly, iPSCs can be produced in virtually any amount and subsequently differentiated to any cell type in vitro compared to the limited availability of the other cell types used in cell therapy. Secondly, iPSCs provide autologous patient-derived cells, which negates the need to find a HLA-compatible cell donor and the need for immunosuppression. However, many obstacles have to be overcome before iPSC-based cell therapy can be used in humans, including the challenges of differentiation to many mature tissue types, short in vivo surviving times after injection of the cells, low integration into host tissue in vivo, and high costs. In addition, the genetic integrity and stability of iPSC has to be better controlled, a problem which contributed to the suspension of the first iPSC clinical trial based on cells differentiated from iPSCs [De Vos et al., 2016].
Gene therapy
Gene therapy involves the delivery of a normally-functioning gene as a nucleic acid polymer into the patient’s cells to substitute for a mutant or missing gene in order to treat a specific disease. Thus, gene therapy is ideal for the treatment of genetic diseases, most readily those caused by loss-of function mutations in a single gene. Currently, the lack of safe and effective methods to permanently deliver a gene to patients prevents the widespread application of gene therapy for the treatment of genetic diseases. Gene transfer efficacy is typically limited by insufficient delivery to the target tissue, negative immune response (autoantibody) to the treatment, and loss of therapeutic effect over time [Naldini 2015; Sasano et al., 2007]. The adeno-associated virus (AAV) is the most commonly used vector for the delivery of genes in gene therapy [Brooks et al., 2016]. While AAVs are currently used as carriers in gene therapy, a more efficient and safer gene delivery vector remains to be discovered and developed. To deliver nucleic acids into nuclei of cells, a number of barriers must be overcome. Extracellular barriers include inactivation by enzymatic degradation and recognition by the reticuloendothelial system (RES) [Hill et al., 2016]. After the genes penetrate the cell membrane, they encounter many intracellular barriers. Most exogenous genetic material is internalized through endocytosis pathways such as clathrin-mediated, caveolae-mediated, macropinocytosis, and phagocytosis. The associated vesicles are then trafficked from early endosomes to late endosomes to lysosomes where the nucleic acid cargo is degraded. The efficiency of the gene therapy platform is significantly reduced by vesicle trafficking. One of the strategies used to evade this endosomal entrapment is the design of carriers that release the nucleic acid cargo into the cytoplasm. Finally, the genes delivered to the cytoplasm need to traverse the nuclear membrane to enter the nucleus and integrate with a chromosome. The nuclear transport is aided by nuclear pore complexes and is often a barrier for larger nucleic acids such as plasmid DNA. A nuclear localization signal peptide is used for active transport of DNA to the nucleus and as a method of restricting transgene expression to the desired tissues.
Generally, two types of carriers (vectors) are used in gene therapy [Kay 2011]. Non-viral gene delivery carriers include lipoplex-mediated (liposome or phospholipids vesicle), polyplex-mediated (polymer), dendrimer-mediated (repetitively branched molecules), and graphene-mediated (a thin layer of pure carbon) gene delivery systems. Because naked plasmid DNA does not offer good therapeutic efficacy due to premature degradation, poor cellular uptake, and low protein expression, special carriers are needed. The advantages of non-viral carriers include low immunogenicity, low cost, ability to deliver large-sized DNA, lack of incorporation into host chromosomes, and lower mutation risk compared with viral carriers. Hydrophilic polymers like polyethylene glycol (PEG) have been conjugated to non-viral carriers to combat RES uptake and enhance the circulation time in blood. However, the gene delivery efficiency of the non-viral delivery method needs to be further improved.
The second method employs viral gene delivery carriers. Viruses offer a promising approach to deliver genes. The natural mechanisms of infection and transduction in viruses are very efficient, so two to three orders of magnitude less DNA is needed compared to non-viral carriers of similar efficacy [Ragusa et al., 2007]. Adenoviruses or AAV carriers are currently better choices for gene delivery carriers because the genes delivered are not integrated into the host chromosomes. Despite improvements, adenovirus carriers suffer from short duration of expression and immunogenicity. They evoke a mild immune response (to the viral carries), transduce a broad range of cell types, are non-pathogenic in humans, and provide significantly longer transgene expression. AAVs have different serotypes based on amino acid sequence of the capsid proteins, which confer different tropisms for different organs [Mason et al., 2015], a property that can help to reduce off-target effects. AAVs can typically only carry about 4.7 kb of DNA, making it very difficult to deliver a gene that encodes larger proteins. To overcome this limitation, it has been shown that a large gene can be fragmented into smaller pieces, each transported by their own AAV vector and co-administered, although such modifications increase the complexity in the resulting system. The disadvantages of viral carriers include the high cost and an immune response to the viral capsid proteins. The reported adverse reactions in clinical trials using viral carriers include a massive and uncontrolled immune response and the induction of new T-cell lymphomas [George and Fogarty 2016; Hacein-Bey-Abina et al., 2010]. As some patients may have pre-existing neutralizing antibodies against the specific viral capsid protein due to prior exposure to the virus in the community, individuals who are considering viral vector-mediated gene therapy will need to be screened to determine the baseline neutralizing antibody status. Additionally, once a patient has received a gene therapy product through a given viral vector, he/she will no longer be able to receive further doses of the product or other therapies using the same vector due to the inevitable induction of neutralizing antibodies, which is a long-term limitation of gene therapy.
Another issue in gene therapy is the delivery of genes across the blood-brain barrier (BBB) to the CNS. In this context, intracranial injections of viral gene carriers have been used to treat neuronal diseases such as aromatic L-amino acid decarboxylase (AADC) deficiency [Kumar et al., 2016]. Although it has been reported that some AAV serotypes (e.g. AAV9) can cross the BBB to deliver genes to the CNS [Rastall and Amalfitano 2015], new methods are still needed to increase the delivery efficiency of genes to the brain and to simplify the overall gene delivery procedures.
The challenge of expressing sufficient amounts of functional proteins in target tissue still remains. For example, injection of the factor IX (FIX) gene, F9, into hemophilia B patients using a recombinant AAV2 vector resulted in the production of FIX in these patients, but the efficacy was hampered by transgene retention in the extracellular space of skeletal muscle, limiting plasma FIX expression to 1% [Manno et al., 2003]. Nathwani and colleagues reported the first unequivocal successful gene therapy for hemophilia B using an AAV8 vector by demonstrating stable and safe transgenic protein expression, resulting in plasma FIX expression of 1.4%–7.2% after 3 years of follow-up [Nathwani et al., 2014].
There is a continual effort to improve the overall gene therapy platform. In particular, the use of gene editing technologies, including zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALEN), and most recently, clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9, is a rapidly evolving field [Gaj et al., 2013]. Tebas et al. used ZFNs to edit the CCR5 gene (encoding an HIV co-receptor) in autologous T-cells, which were then infused into patients with HIV [Tebas et al., 2014]. This procedure was shown to be safe within the limited number of subjects in the study. More recently, a few rare disorders have been corrected by the editing of the mutated genes in the patient’s hematopoietic stem cells and returning these edited cells to the patient. This approach was successful in treating X-linked severe combined immunodeficiency, but the trial was halted due to a serious adverse event in which malignant transformation of lymphocytes was found in several of the treated patients [Hacein-Bey-Abina et al., 2010].
Gene therapy has not yet yielded an approved product in the US. In 2011, the European Medicines Agency (EMA) rejected Glybera® (alipogene tiparvovec or AAV1–LPL) for the treatment for lipoprotein lipase (LPL) deficiency due to the lack of consistent long-lasting benefit in patients. Notably, a different version of the AAV vector expressing LPL was approved by EMA in 2012 for the treatment of LPL deficiency [Mullard 2011]. Recently, EMA approved the second gene therapy for the treatment of ADA-SCID [Mullard 2016]. The rapid advancement in genome editing technologies opens up the possibility of inactivating target genes or inserting therapeutic genes into the genome without the use of viral vectors. However, potential off-target effects of engineered nucleases and viral transcriptional enhancers continue to present concerns for these therapies.
DISEASE MODELS
Disease models offer major opportunities for studying the phenotype of rare diseases, identification of drug targets, and evaluation of drug efficacy and toxicity.
Cell-based models: cell lines and reporter lines
Cell-based models usually use primary human cell lines, immortalized cell lines (primary or engineered), or more recently, specific cell types differentiated from iPSCs derived from patient or normal human cells. For example, primary bronchial epithelial cells were used in electrophysiology studies for cystic fibrosis [Neuberger et al., 2011]. Reprogrammed cells from normal and tumorous lung tissue in respiratory papillomatosis patients were used for cell viability tests to evaluate drug cytotoxicity [Yuan et al., 2012]. A clonal cell line derived from a pheochromocytoma of the rat adrenal medulla (PC12) expressing engineered huntingtin gene (HTT Q103-linked to GFP) was used for the detection of protein aggregates (GFP) to identify small molecule therapeutics [Titus et al., 2012]. Engineered cell lines or immortalized primary cells are commonly used in primary screens to identify lead compounds largely because they are more accessible and can be rapidly expanded to large quantities for high throughput screens. However, primary human cells and patient-derived cells are more pathophysiologically relevant as models [Eglen and Reisine 2011; Vincent et al., 2015]. Many researchers rely on in vitro studies to test potential drugs. A standard model currently used by pharmaceutical companies and in academia utilizes skin fibroblasts isolated from patients, which are further modified to incorporate reporter moieties. However, the limited availability of primary cells has prevented their broad application in drug discovery.
Disease models using iPSC
Patient-specific iPSCs represent a promising type of novel disease model, especially for human genetic diseases. In the last several years, owing to the rapid development of iPSC technologies, significant advances have been made in the area of disease models derived from stem cells. The feasibility of generating iPSCs from a patient’s skin cells, blood, or other accessible cells allows for the establishment of scalable disease models using patient cells that have better pathophysiological relevance to human disease than traditional cell lines [Ebert and Svendsen 2010]. iPSCs derived from specific patients are capable of either self-renewal or differentiation into expandable progenitor cells that can be further differentiated to many types of mature cells such as neurons, cardiomyocytes, and hepatocytes for drug screenings [Ebert and Svendsen 2010; Eglen and Reisine 2011]. In 2012, familial dysautonomia iPSCs were screened against 6,912 small molecule compounds for candidate drugs. One small molecule was found to induce the transcription of the familial dysautonomia gene (IKBKAP) and rescue IKAP protein expression and the disease-specific loss of autonomic neuronal marker expression [Lee et al., 2012]. Another example, as discussed above, is the rapid advancement of retigabine into clinical trials upon discovery of its effect on the disease model using iPSCs from ALS patients.
Animal disease models
Although cell models have been used in drug development for the treatment of rare diseases, data derived from those models are usually insufficient for filing an IND with the FDA. Similar to drug development for common diseases, preclinical studies of drug candidates for rare diseases have to elucidate in detail the chemical properties of the candidate drug, including toxicology, pharmacokinetics, and dosage. Such studies are typically conducted in animals, particularly given the very limited number of patients in clinical trials. Fewer animals, fewer doses, and shorter exposures can be used to apply for an exploratory IND for rare diseases compared to common diseases [Vaquer et al., 2013]. In the preclinical phase, it is important to conduct proof of principle studies and establish solid pharmacokinetics/pharmacodynamics properties before thoroughly investigating a drug candidate in animal models of rare diseases. Recent technological advancements may assist in measuring drug target interactions. For example, the cellular thermal shift assay is capable of detecting drug target interactions in the context of cells lysates, live cells, and tissues [Jafari et al., 2014; Molina et al., 2013]. Compared to cell-based models, it is much more difficult to develop disease relevant animal models due to the long lead time and expertise needed to generate them [Vaquer et al., 2013]. Animals have many naturally occurring genetic diseases (e.g., hemophilia B in dogs) [Kay et al., 1994], including rare cancers, so they can be used for evaluation of drug efficacy in vivo. For other disorders that do not occur naturally in animals, various techniques can be used to generate appropriate animal models [Vaquer et al., 2013], such as in the case of Huntington disease (monkey) and cystic fibrosis (pig) [Wolfe 2009]. However, many rare genetic diseases still lack the desired animal models.
Both forward and reverse genetic manipulations are used to create mouse models. In forward genetics, specific genes are modified to reflect the pathophysiology of the disease. This is the fundamental approach to the study of the genetics and biochemistry of rare diseases, although it is expensive and time-consuming. Recent advancements in gene editing technology such as CRISPR/Cas9 may have the potential to significantly improve the process [Dow 2015]. CRISPR/Cas9 was used to generate large animal models of neurodegenerative diseases to better mimic human disease progression [Tu et al., 2015]. Reverse genetics is performed by treating animals with mutagenic agents and identifying genetic disorders by sequencing the genes of the animals. Deficiency of the Gmap-210 gene was identified in achondrogenesis mouse models using the reverse genetic approach [Smits et al., 2010]. The National Cancer Institute (http://mouse.ncifcrf.gov/) and the Jackson Laboratory (https://www.jax.org/jax-mice-and-services) offer a number of mouse models of genetic disorders.
BIOMARKERS
“A biomarker is a defined characteristic that is measured as an indicator of normal biological processes, pathogenic processes, or responses to an exposure or intervention, including therapeutic interventions.” (http://www.fda.gov/Drugs/DevelopmentApprovalProcess/DrugDevelopmentToolsQualificationProgram) Biomarkers are an emerging tool for clinical drug development. Biomarkers can be categorized into four types: surrogate, pharmacodynamic, predictive, and prognostic biomarkers (Table III). Identification of appropriate biomarkers can improve drug development in clinical studies and provide quantitative information for drug therapy, leading to reduced time and smaller sample sizes for clinical trials. As a surrogate endpoint, biomarkers are used to predict health outcomes and help in regulatory decisions [Katz 2004].
Table III.
Biomarker categories and examples
| Biomarker Category* | Definitions/Principles | Applications | Examples |
|---|---|---|---|
| Surrogate | Measure drug efficacy and serve as end points in clinical trials | Predict clinical benefit in therapeutic trials | Creatinine/cystatin C ratio for amyotrophic lateral sclerosis (ALS) [Bakkar et al., 2015; Tetsuka et al., 2013] |
| Pharmacodynamic | Detect the activity of a drug on the targeted pathway | Verify target engagement and guide dose selection | Multiple pro-fibrotic signaling pathways for idiopathic pulmonary fibrosis [Tzouvelekis et al., 2016] |
| Predictive | Reflect biology central to disease pathophysiology | Identify patient populations for a particular therapy | Variations in Fcγ receptors that affect the binding affinity for monoclonal antibody therapies [Smith and Clatworthy 2010] |
| Prognostic | Link with disease activity but may be distal from the targeted pathway | Select patients with a more rapid rate of disease progression | FDG–PET avidity for aggressive forms of disease in lung cancer, renal cell carcinoma and other cancers [Kelloff et al., 2005; Nakaigawa et al., 2016] |
Note:
Many of the biomarkers may fit into multiple categories depending on the context in which they are used.
Development and validation of biomarkers are not trivial undertakings even for common diseases. The effort for biomarker development is particularly necessary for clinical drug development for rare diseases because biomarkers can be used to evaluate the responses of the drug that are otherwise difficult to monitor. The time and energy needed to validate biomarkers necessitates a consortium-type approach. The Foundation for the National Institutes of Health has established the Biomarkers Consortium to develop biomarkers for use in research, therapeutic and diagnostic development, regulatory approval, and clinical practice (http://www.biomarkersconsortium.org/).
Due to the small patient population associated with each rare disease, conventional clinical trial endpoints are often not appropriate. Biomarkers can guide dose selection and monitor drug efficacy. In some cases, biomarkers serving as surrogate clinical endpoints could reduce the time needed to complete clinical trials and allow smaller sample sizes, while still generating statistically significant data. Biomarkers can also provide valuable information that could reduce uncertainty in regulatory decisions. For example, accumulation of the globotriaosylceramide (Gb(3)) was characterized in Fabry disease patients in various tissues and organs such as the kidney. Urinary Gb(3) levels were found to correlate well with renal function and could be measured as a biomarker to assess the efficacy of novel drugs [Whitfield et al., 2005].
SUMMARY
In summary, we have provided an overview of drug discovery and development strategies and methods, with an emphasis on rare genetic diseases. While small molecule drug discovery is still the main platform for rare genetic disease drug development, biologics such as recombinant proteins, antibody, stem cells, and gene therapy are expected to deliver major breakthrough therapies. The continued expansion of our knowledge of the biology and pathophysiology of rare diseases, combined with rapid advances in drug discovery technologies, will bring us closer to the discovery of much-needed new treatments.
Acknowledgments
This work was supported by the Intramural Research Program of the National Center for Advancing Translational Sciences at the National Institutes of Health. We thank Dr. DeeAnn Visk for reading and critiquing the manuscript.
Biographies
Wei Sun received his Ph.D. degree in pharmaceutical sciences from the University of North Carolina at Chapel Hill. Currently, Dr. Sun is a research scientist working in the Therapeutics for Rare and Neglected Diseases program at the National Center for Advancing Translational Science.
Wei Zheng received his Ph.D. degree in pharmacology from the State University of New York at Buffalo. Dr. Zheng joined the NIH in 2005 after working for 12 years in the pharmaceutical industry. Presently, he is the leader of Therapeutics for Rare and Neglected Diseases, Division of Pre-Clinical Innovation at the National Center for Advancing Translational Sciences.
Anton Simeonov received a Ph.D. degree in bioorganic chemistry from the University of Southern California and conducted postdoctoral research at The Scripps Research Institute. Presently serving as the scientific director at the National Center for Advancing Translational Science, Dr. Simeonov’s areas of expertise include assay development, lead discovery, and drug development for a wide range of diseases.
References
- Aguero F, Al-Lazikani B, Aslett M, Berriman M, Buckner FS, Campbell RK, Carmona S, Carruthers IM, Chan AW, Chen F, Crowther GJ, Doyle MA, Hertz-Fowler C, Hopkins AL, McAllister G, Nwaka S, Overington JP, Pain A, Paolini GV, Pieper U, Ralph SA, Riechers A, Roos DS, Sali A, Shanmugam D, Suzuki T, Van Voorhis WC, Verlinde CL. Genomic-scale prioritization of drug targets: the TDR Targets database. Nat Rev Drug Discov. 2008;7(11):900–907. doi: 10.1038/nrd2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiuti A, Cattaneo F, Galimberti S, Benninghoff U, Cassani B, Callegaro L, Scaramuzza S, Andolfi G, Mirolo M, Brigida I, Tabucchi A, Carlucci F, Eibl M, Aker M, Slavin S, Al-Mousa H, Al Ghonaium A, Ferster A, Duppenthaler A, Notarangelo L, Wintergerst U, Buckley RH, Bregni M, Marktel S, Valsecchi MG, Rossi P, Ciceri F, Miniero R, Bordignon C, Roncarolo M. Gene Therapy for Immunodeficiency Due to Adenosine Deaminase Deficiency. New Engl J Med. 2009;360(5):447–458. doi: 10.1056/NEJMoa0805817. [DOI] [PubMed] [Google Scholar]
- Aledort L, Ljung R, Mann K, Pipe S. Factor VIII therapy for hemophilia A: current and future issues. Expert Rev Hematol. 2014;7(3):373–385. doi: 10.1586/17474086.2014.899896. [DOI] [PubMed] [Google Scholar]
- Allison M. Hemacord approval may foreshadow regulatory creep for HSC therapies. Nat Biotechnol. 2012;30(4):304. doi: 10.1038/nbt0412-304. [DOI] [PubMed] [Google Scholar]
- Bakkar N, Boehringer A, Bowser R. Use of biomarkers in ALS drug development and clinical trials. Brain Res. 2015;1607:94–107. doi: 10.1016/j.brainres.2014.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med. 1994;330(9):585–591. doi: 10.1056/NEJM199403033300901. [DOI] [PubMed] [Google Scholar]
- Brady RO. Enzyme replacement for lysosomal diseases. Annu Rev Med. 2006;57:283–296. doi: 10.1146/annurev.med.57.110104.115650. [DOI] [PubMed] [Google Scholar]
- Brooks PJ, Yang NN, Austin CP. Gene Therapy: The View from NCATS. Hum Gene Ther. 2016;27(1):7–13. doi: 10.1089/hum.2016.29018.pjb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain SJ, Chen PF, Ng KY, Bourgois-Rocha F, Lemtiri-Chlieh F, Levine ES, Lalande M. Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader-Willi syndromes. P Natl Acad Sci USA. 2010;107(41):17668–17673. doi: 10.1073/pnas.1004487107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charache S, Terrin ML, Moore RD, Dover GJ, Barton FB, Eckert SV, McMahon RP, Bonds DR. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med. 1995;332(20):1317–1322. doi: 10.1056/NEJM199505183322001. [DOI] [PubMed] [Google Scholar]
- Chen Y, Carter RL, Cho IK, Chan AW. Cell-based therapies for Huntington’s disease. Drug Discov Today. 2014;19(7):980–984. doi: 10.1016/j.drudis.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos J, Bouckenheimer J, Sansac C, Lemaitre JM, Assou S. Human induced pluripotent stem cells: A disruptive innovation. Curr Res Transl Med. 2016;64(2):91–96. doi: 10.1016/j.retram.2016.04.001. [DOI] [PubMed] [Google Scholar]
- DeLoach A, Cozart M, Kiaei A, Kiaei M. A retrospective review of the progress in amyotrophic lateral sclerosis drug discovery over the last decade and a look at the latest strategies. Expert Opin Drug Discov. 2015;10(10):1099–1118. doi: 10.1517/17460441.2015.1067197. [DOI] [PubMed] [Google Scholar]
- Dow LE. Modeling Disease In Vivo With CRISPR/Cas9. Trends Mol Med. 2015;21(10):609–621. doi: 10.1016/j.molmed.2015.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunoyer M. Accelerating access to treatments for rare diseases. Nat Rev Drug Discov. 2011;10(7):475–476. doi: 10.1038/nrd3493. [DOI] [PubMed] [Google Scholar]
- Ebert AD, Svendsen CN. Human stem cells and drug screening: opportunities and challenges. Nat Rev Drug Discov. 2010;9(5):367–372. doi: 10.1038/nrd3000. [DOI] [PubMed] [Google Scholar]
- Eder J, Sedrani R, Wiesmann C. The discovery of first-in-class drugs: origins and evolution. Nat Rev Drug Discov. 2014;13(8):577–587. doi: 10.1038/nrd4336. [DOI] [PubMed] [Google Scholar]
- Eglen R, Reisine T. Primary Cells and Stem Cells in Drug Discovery: Emerging Tools for High-Throughput Screening. Assay Drug Dev Techn. 2011;9(2):108–124. doi: 10.1089/adt.2010.0305. [DOI] [PubMed] [Google Scholar]
- Evans CH. Gene delivery to bone. Adv Drug Deliv Rev. 2012;64(12):1331–1340. doi: 10.1016/j.addr.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friday BB, Anderson SK, Buckner J, Yu C, Giannini C, Geoffroy F, Schwerkoske J, Mazurczak M, Gross H, Pajon E, Jaeckle K, Galanis E. Phase II trial of vorinostat in combination with bortezomib in recurrent glioblastoma: a north central cancer treatment group study. Neuro Oncol. 2012;14(2):215–221. doi: 10.1093/neuonc/nor198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaj T, Gersbach CA, Barbas CF. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31(7):397–405. doi: 10.1016/j.tibtech.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber K. RIKEN suspends first clinical trial involving induced pluripotent stem cells. Nat Biotechnol. 2015;33(9):890–891. doi: 10.1038/nbt0915-890. [DOI] [PubMed] [Google Scholar]
- George LA, Fogarty PF. Gene therapy for hemophilia: past, present and future. Semin Hematol. 2016;53(1):46–54. doi: 10.1053/j.seminhematol.2015.10.002. [DOI] [PubMed] [Google Scholar]
- Glickman JF, Wu X, Mercuri R, Illy C, Bowen BR, He Y, Sills M. A comparison of ALPHAScreen, TR-FRET, and TRF as assay methods for FXR nuclear receptors. J Biomol Screen. 2002;7(1):3–10. doi: 10.1177/108705710200700102. [DOI] [PubMed] [Google Scholar]
- Glorieux FH, Bishop NJ, Plotkin H, Chabot G, Lanoue G, Travers R. Cyclic administration of pamidronate in children with severe osteogenesis imperfecta. N Engl J Med. 1998;339(14):947–952. doi: 10.1056/NEJM199810013391402. [DOI] [PubMed] [Google Scholar]
- Grant SGN. Molecular mechanisms of cognition: Genetics of mouse learning and memory as a route to human psychiatry. Am J Med Genet. 2000;96(4):455–455. [Google Scholar]
- Griesenbach U, Geddes DM, Alton EW. Gene therapy for cystic fibrosis: an example for lung gene therapy. Gene Ther. 2004;11(Suppl 1):S43–50. doi: 10.1038/sj.gt.3302368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, Hauer J, Lim A, Picard C, Wang GP, Berry CC, Martinache C, Rieux-Laucat F, Latour S, Belohradsky BH, Leiva L, Sorensen R, Debre M, Casanova JL, Blanche S, Durandy A, Bushman FD, Fischer A, Cavazzana-Calvo M. Efficacy of gene therapy for X-linked severe combined immunodeficiency. N Engl J Med. 2010;363(4):355–364. doi: 10.1056/NEJMoa1000164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haffner ME. Adopting orphan drugs–two dozen years of treating rare diseases. N Engl J Med. 2006;354(5):445–447. doi: 10.1056/NEJMp058317. [DOI] [PubMed] [Google Scholar]
- Hemsley KM, Hopwood JJ. Delivery of recombinant proteins via the cerebrospinal fluid as a therapy option for neurodegenerative lysosomal storage diseases. Int J Clin Pharm Th. 2009;47:S118–S123. doi: 10.5414/cpp47118. [DOI] [PubMed] [Google Scholar]
- Hill AB, Chen M, Chen CK, Pfeifer BA, Jones CH. Overcoming Gene-Delivery Hurdles: Physiological Considerations for Nonviral Vectors. Trends Biotechnol. 2016;34(2):91–105. doi: 10.1016/j.tibtech.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoban MD, Orkin SH, Bauer DE. Genetic treatment of a molecular disorder: gene therapy approaches to sickle cell disease. Blood. 2016;127(7):839–848. doi: 10.1182/blood-2015-09-618587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman EP, Barr ML, Giovanni MA, Murray MF. Lysosomal Acid Lipase Deficiency. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Ledbetter N, Mefford HC, Smith RJH, Stephens K, editors. GeneReviews(R) Seattle, WA: University of Washington; 1993. Seattle, NBK305870. [Google Scholar]
- Hoffman EP, Escolar D. Translating mighty mice into neuromuscular therapeutics - Is bigger muscle better? Am J Pathol. 2006;168(6):1775–1778. doi: 10.2353/ajpath.2006.060270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R, Southall N, Wang Y, Yasgar A, Shinn P, Jadhav A, Nguyen DT, Austin CP. The NCGC pharmaceutical collection: a comprehensive resource of clinically approved drugs enabling repurposing and chemical genomics. Sci Transl Med. 2011;3(80):80ps16. doi: 10.1126/scitranslmed.3001862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inglese J, Auld DS, Jadhav A, Johnson RL, Simeonov A, Yasgar A, Zheng W, Austin CP. Quantitative high-throughput screening: a titration-based approach that efficiently identifies biological activities in large chemical libraries. Proc Natl Acad Sci U S A. 2006;103(31):11473–11478. doi: 10.1073/pnas.0604348103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inglese J, Johnson RL, Simeonov A, Xia M, Zheng W, Austin CP, Auld DS. High-throughput screening assays for the identification of chemical probes. Nat Chem Biol. 2007;3(8):466–479. doi: 10.1038/nchembio.2007.17. [DOI] [PubMed] [Google Scholar]
- Jafari R, Almqvist H, Axelsson H, Ignatushchenko M, Lundback T, Nordlund P, Martinez Molina D. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat Protoc. 2014;9(9):2100–2122. doi: 10.1038/nprot.2014.138. [DOI] [PubMed] [Google Scholar]
- Kaewkhaw R, Swaroop M, Homma K, Nakamura J, Brooks M, Kaya KD, Chaitankar V, Michael S, Tawa G, Zou J, Rao M, Zheng W, Cogliati T, Swaroop A. Treatment Paradigms for Retinal and Macular Diseases Using 3-D Retina Cultures Derived From Human Reporter Pluripotent Stem Cell Lines. Invest Ophthalmol Vis Sci. 2016;57(5):ORSFl1–ORSFl11. doi: 10.1167/iovs.15-17639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz R. Biomarkers and surrogate markers: an FDA perspective. NeuroRx. 2004;1(2):189–195. doi: 10.1602/neurorx.1.2.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay MA. State-of-the-art gene-based therapies: the road ahead. Nat Rev Genet. 2011;12(5):316–328. doi: 10.1038/nrg2971. [DOI] [PubMed] [Google Scholar]
- Kay MA, Landen CN, Rothenberg SR, Taylor LA, Leland F, Wiehle S, Fang B, Bellinger D, Finegold M, Thompson AR, et al. In vivo hepatic gene therapy: complete albeit transient correction of factor IX deficiency in hemophilia B dogs. P Natl Acad Sci USA. 1994;91(6):2353–2357. doi: 10.1073/pnas.91.6.2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelloff GJ, Hoffman JM, Johnson B, Scher HI, Siegel BA, Cheng EY, Cheson BD, O’Shaughnessy J, Guyton KZ, Mankoff DA, Shankar L, Larson SM, Sigman CC, Schilsky RL, Sullivan DC. Progress and promise of FDG-PET imaging for cancer patient management and oncologic drug development. Clin Cancer Res. 2005;11(8):2785–2808. doi: 10.1158/1078-0432.CCR-04-2626. [DOI] [PubMed] [Google Scholar]
- Kelly PF, Radtke S, von Kalle C, Balcik B, Bohn K, Mueller R, Schuesler T, Haren M, Reeves L, Cancelas JA, Leemhuis T, Harris R, Auerbach AD, Smith FO, Davies SM, Williams DA. Stem cell collection and gene transfer in Fanconi anemia. Mol Ther. 2007;15(1):211–219. doi: 10.1038/sj.mt.6300033. [DOI] [PubMed] [Google Scholar]
- Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, Burrell JR, Zoing MC. Amyotrophic lateral sclerosis. Lancet. 2011;377(9769):942–955. doi: 10.1016/S0140-6736(10)61156-7. [DOI] [PubMed] [Google Scholar]
- Kumar SR, Markusic DM, Biswas M, High KA, Herzog RW. Clinical development of gene therapy: results and lessons from recent successes. Mol Ther Methods Clin Dev. 2016;3:16034. doi: 10.1038/mtm.2016.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G, Ramirez CN, Kim H, Zeltner N, Liu B, Radu C, Bhinder B, Kim YJ, Choi IY, Mukherjee-Clavin B, Djaballah H, Studer L. Large-scale screening using familial dysautonomia induced pluripotent stem cells identifies compounds that rescue IKBKAP expression. Nat Biotechnol. 2012;30(12):1244–1248. doi: 10.1038/nbt.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lidove O, Joly D, Barbey F, Bekri S, Alexandra JF, Peigne V, Jaussaud R, Papo T. Clinical results of enzyme replacement therapy in Fabry disease: a comprehensive review of literature. Int J Clin Pract. 2007;61(2):293–302. doi: 10.1111/j.1742-1241.2006.01237.x. [DOI] [PubMed] [Google Scholar]
- Lo Cicero A, Nissan X. Pluripotent stem cells to model Hutchinson-Gilford progeria syndrome (HGPS): Current trends and future perspectives for drug discovery. Ageing Res Rev. 2015;24(Pt B):343–348. doi: 10.1016/j.arr.2015.10.002. [DOI] [PubMed] [Google Scholar]
- Manno CS, Chew AJ, Hutchison S, Larson PJ, Herzog RW, Arruda VR, Tai SJ, Ragni MV, Thompson A, Ozelo M, Couto LB, Leonard DG, Johnson FA, McClelland A, Scallan C, Skarsgard E, Flake AW, Kay MA, High KA, Glader B. AAV-mediated factor IX gene transfer to skeletal muscle in patients with severe hemophilia B. Blood. 2003;101(8):2963–2972. doi: 10.1182/blood-2002-10-3296. [DOI] [PubMed] [Google Scholar]
- Marks PA, Breslow R. Dimethyl sulfoxide to vorinostat: Development of this histone deacetylase inhibitor as an anticancer drug. Nat Biotechnol. 2007;25(1):84–90. doi: 10.1038/nbt1272. [DOI] [PubMed] [Google Scholar]
- Mason D, Chen YZ, Krishnan HV, Sant S. Cardiac gene therapy: Recent advances and future directions. J Control Release. 2015;215:101–111. doi: 10.1016/j.jconrel.2015.08.001. [DOI] [PubMed] [Google Scholar]
- McLellan DL, Chalmers RJ, Johnson RH. A double-blind trial of tetrabenazine, thiopropazate, and placebo in patients with chorea. Lancet. 1974;1(7848):104–107. doi: 10.1016/s0140-6736(74)92338-1. [DOI] [PubMed] [Google Scholar]
- Melnikova I. Rare diseases and orphan drugs. Nat Rev Drug Discov. 2012;11(4):267–268. doi: 10.1038/nrd3654. [DOI] [PubMed] [Google Scholar]
- Merkens LS, Wassif C, Healy K, Pappu AS, DeBarber AE, Penfield JA, Lindsay RA, Roullet JB, Porter FD, Steiner RD. Smith-Lemli-Opitz syndrome and inborn errors of cholesterol synthesis: summary of the 2007 SLO/RSH Foundation scientific conference sponsored by the National Institutes of Health. Genet Med. 2009;11(5):359–364. doi: 10.1097/GIM.0b013e31819b246e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer HM, Jr, Bernheim BC, Rogers NG. Titration of Live Measles and Smallpox Vaccines by Jet Inoculation of Susceptible Children. Proc Soc Exp Biol Med. 1965;118:53–57. doi: 10.3181/00379727-118-29753. [DOI] [PubMed] [Google Scholar]
- Micklus A, Muntner S. Deal watch: Biopharma deal-making in 2015: changing the pharma landscape. Nat Rev Drug Discov. 2016;15(2):78–79. doi: 10.1038/nrd.2016.10. [DOI] [PubMed] [Google Scholar]
- Moffat JG, Rudolph J, Bailey D. Phenotypic screening in cancer drug discovery - past, present and future. Nat Rev Drug Discov. 2014;13(8):588–602. doi: 10.1038/nrd4366. [DOI] [PubMed] [Google Scholar]
- Molina DM, Jafari R, Ignatushchenko M, Seki T, Larsson EA, Dan C, Sreekumar L, Cao YH, Nordlund P. Monitoring Drug Target Engagement in Cells and Tissues Using the Cellular Thermal Shift Assay. Science. 2013;341(6141):84–87. doi: 10.1126/science.1233606. [DOI] [PubMed] [Google Scholar]
- Moorthy NS, Sousa SF, Ramos MJ, Fernandes PA. Farnesyltransferase inhibitors: a comprehensive review based on quantitative structural analysis. Curr Med Chem. 2013;20(38):4888–4923. doi: 10.2174/09298673113206660262. [DOI] [PubMed] [Google Scholar]
- Mullard A. Gene therapies advance towards finish line. Nat Rev Drug Discov. 2011;10(10):719–720. doi: 10.1038/nrd3572. [DOI] [PubMed] [Google Scholar]
- Mullard A. EMA greenlights second gene therapy. Nat Rev Drug Discov. 2016;15(5):299. doi: 10.1038/nrd.2016.93. [DOI] [PubMed] [Google Scholar]
- Nakaigawa N, Kondo K, Tateishi U, Minamimoto R, Kaneta T, Namura K, Ueno D, Kobayashi K, Kishida T, Ikeda I, Hasumi H, Makiyama K, Kubota Y, Inoue T, Yao M. FDG PET/CT as a prognostic biomarker in the era of molecular-targeting therapies: max SUVmax predicts survival of patients with advanced renal cell carcinoma. Bmc Cancer. 2016;16 doi: 10.1186/s12885-016-2097-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naldini L. Gene therapy returns to centre stage. Nature. 2015;526(7573):351–360. doi: 10.1038/nature15818. [DOI] [PubMed] [Google Scholar]
- Nathwani AC, Reiss UM, Tuddenham EG, Rosales C, Chowdary P, McIntosh J, Della Peruta M, Lheriteau E, Patel N, Raj D, Riddell A, Pie J, Rangarajan S, Bevan D, Recht M, Shen YM, Halka KG, Basner-Tschakarjan E, Mingozzi F, High KA, Allay J, Kay MA, Ng CY, Zhou J, Cancio M, Morton CL, Gray JT, Srivastava D, Nienhuis AW, Davidoff AM. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J Med. 2014;371(21):1994–2004. doi: 10.1056/NEJMoa1407309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuberger T, Burton B, Clark H, Van Goor F. Use of primary cultures of human bronchial epithelial cells isolated from cystic fibrosis patients for the pre-clinical testing of CFTR modulators. Methods Mol Biol. 2011;741:39–54. doi: 10.1007/978-1-61779-117-8_4. [DOI] [PubMed] [Google Scholar]
- Ortolano S, Vieitez I, Navarro C, Spuch C. Treatment of lysosomal storage diseases: recent patents and future strategies. Recent Pat Endocr Metab Immune Drug Discov. 2014;8(1):9–25. doi: 10.2174/1872214808666140115111350. [DOI] [PubMed] [Google Scholar]
- Parenti G, Andria G, Ballabio A. Lysosomal storage diseases: from pathophysiology to therapy. Annu Rev Med. 2015;66:471–486. doi: 10.1146/annurev-med-122313-085916. [DOI] [PubMed] [Google Scholar]
- Paul SM, Mytelka DS, Dunwiddie CT, Persinger CC, Munos BH, Lindborg SR, Schacht AL. How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nat Rev Drug Discov. 2010;9(3):203–214. doi: 10.1038/nrd3078. [DOI] [PubMed] [Google Scholar]
- Pera MF. Stem cells: The dark side of induced pluripotency. Nature. 2011;471(7336):46–47. doi: 10.1038/471046a. [DOI] [PubMed] [Google Scholar]
- Peyvandi F, Garagiola I, Young G. The past and future of haemophilia: diagnosis, treatments, and its complications. Lancet. 2016;388(10040):187–197. doi: 10.1016/S0140-6736(15)01123-X. [DOI] [PubMed] [Google Scholar]
- Ragusa A, Garcia I, Penades S. Nanoparticles as nonviral gene delivery vectors. IEEE Trans Nanobioscience. 2007;6(4):319–330. doi: 10.1109/tnb.2007.908996. [DOI] [PubMed] [Google Scholar]
- Rastall DP, Amalfitano A. Recent advances in gene therapy for lysosomal storage disorders. Appl Clin Genet. 2015;8:157–169. doi: 10.2147/TACG.S57682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reguart N, Rosell R, Cardenal F, Cardona AF, Isla D, Palmero R, Moran T, Rolfo C, Pallares MC, Insa A, Carcereny E, Majem M, De Castro J, Queralt C, Molina MA, Taron M. Phase I/II trial of vorinostat (SAHA) and erlotinib for non-small cell lung cancer (NSCLC) patients with epidermal growth factor receptor (EGFR) mutations after erlotinib progression. Lung Cancer. 2014;84(2):161–167. doi: 10.1016/j.lungcan.2014.02.011. [DOI] [PubMed] [Google Scholar]
- Reichert JM, Rosensweig CJ, Faden LB, Dewitz MC. Monoclonal antibody successes in the clinic. Nat Biotechnol. 2005;23(9):1073–1078. doi: 10.1038/nbt0905-1073. [DOI] [PubMed] [Google Scholar]
- Rombach SM, Hollak CEM, Linthorst GE, Dijkgraaf MGW. Cost-effectiveness of enzyme replacement therapy for Fabry disease. Orphanet J Rare Dis. 2013;8 doi: 10.1186/1750-1172-8-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runz H, Dolle D, Schlitter AM, Zschocke J. NPC-db, a Niemann-Pick type C disease gene variation database. Hum Mutat. 2008;29(3):345–350. doi: 10.1002/humu.20636. [DOI] [PubMed] [Google Scholar]
- Sanders TI. The Orphan Drug Act. Prog Clin Biol Res. 1983;127:207–215. [PubMed] [Google Scholar]
- Sanford M, Lo JH. Elosulfase alfa: first global approval. Drugs. 2014;74(6):713–718. doi: 10.1007/s40265-014-0210-z. [DOI] [PubMed] [Google Scholar]
- Sasano T, Kikuchi K, McDonald AD, Lai S, Donahue JK. Targeted high-efficiency, homogeneous myocardial gene transfer. J Mol Cell Cardiol. 2007;42(5):954–961. doi: 10.1016/j.yjmcc.2007.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer M, Grewal S, Peters C. Hematopoietic stem cell transplantation for mucopolysaccharidoses and leukodystrophies. Klin Padiatr. 2004;216(3):163–168. doi: 10.1055/s-2004-822629. [DOI] [PubMed] [Google Scholar]
- Schenone M, Dancik V, Wagner BK, Clemons PA. Target identification and mechanism of action in chemical biology and drug discovery. Nat Chem Biol. 2013;9(4):232–240. doi: 10.1038/nchembio.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schieppati A, Henter JI, Daina E, Aperia A. Why rare diseases are an important medical and social issue. Lancet. 2008;371(9629):2039–2041. doi: 10.1016/S0140-6736(08)60872-7. [DOI] [PubMed] [Google Scholar]
- Smith KG, Clatworthy MR. FcgammaRIIB in autoimmunity and infection: evolutionary and therapeutic implications. Nat Rev Immunol. 2010;10(5):328–343. doi: 10.1038/nri2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits P, Bolton AD, Funari V, Hong M, Boyden ED, Lu L, Manning DK, Dwyer ND, Moran JL, Prysak M, Merriman B, Nelson SF, Bonafe L, Superti-Furga A, Ikegawa S, Krakow D, Cohn DH, Kirchhausen T, Warman ML, Beier DR. Lethal skeletal dysplasia in mice and humans lacking the golgin GMAP-210. N Engl J Med. 2010;362(3):206–216. doi: 10.1056/NEJMoa0900158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Sanderson P, Zheng W. Drug combination therapy increases successful drug repositioning. Drug Discov Today. 2016 doi: 10.1016/j.drudis.2016.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinney DC, Anthony J. How were new medicines discovered? Nat Rev Drug Discov. 2011;10(7):507–519. doi: 10.1038/nrd3480. [DOI] [PubMed] [Google Scholar]
- Takenaka T. Classical vs reverse pharmacology in drug discovery. BJU Int. 2001;88(Suppl 2):7–10. doi: 10.1111/j.1464-410x.2001.00112.x. discussion 49–50. [DOI] [PubMed] [Google Scholar]
- Tebas P, Stein D, Tang WW, Frank I, Wang SQ, Lee G, Spratt SK, Surosky RT, Giedlin MA, Nichol G, Holmes MC, Gregory PD, Ando DG, Kalos M, Collman RG, Binder-Scholl G, Plesa G, Hwang WT, Levine BL, June CH. Gene Editing of CCR5 in Autologous CD4 T Cells of Persons Infected with HIV. New Engl J Med. 2014;370(10):901–910. doi: 10.1056/NEJMoa1300662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telen MJ. Beyond hydroxyurea: new and old drugs in the pipeline for sickle cell disease. Blood. 2016;127(7):810–819. doi: 10.1182/blood-2015-09-618553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetsuka S, Morita M, Ikeguchi K, Nakano I. Utility of cystatin C for renal function in amyotrophic lateral sclerosis. Acta Neurol Scand. 2013;128(6):386–390. doi: 10.1111/ane.12134. [DOI] [PubMed] [Google Scholar]
- Thomsen GM, Gowing G, Svendsen S, Svendsen CN. The past, present and future of stem cell clinical trials for ALS. Exp Neurol. 2014;262:127–137. doi: 10.1016/j.expneurol.2014.02.021. [DOI] [PubMed] [Google Scholar]
- Titus SA, Southall N, Marugan J, Austin CP, Zheng W. High-Throughput Multiplexed Quantitation of Protein Aggregation and Cytotoxicity in a Huntington’s Disease Model. Curr Chem Genomics. 2012;6:79–86. doi: 10.2174/1875397301206010079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifunovic D, Sahaboglu A, Kaur J, Mencl S, Zrenner E, Ueffing M, Arango-Gonzalez B, Paquet-Durand F. Neuroprotective strategies for the treatment of inherited photoreceptor degeneration. Curr Mol Med. 2012;12(5):598–612. doi: 10.2174/156652412800620048. [DOI] [PubMed] [Google Scholar]
- Tu Z, Yang W, Yan S, Guo X, Li XJ. CRISPR/Cas9: a powerful genetic engineering tool for establishing large animal models of neurodegenerative diseases. Mol Neurodegener. 2015;10:35. doi: 10.1186/s13024-015-0031-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzouvelekis A, Herazo-Maya J, Sakamoto K, Bouros D. Biomarkers in the Evaluation and Management of Idiopathic Pulmonary Fibrosis. Curr Top Med Chem. 2016;16(14):1587–1598. doi: 10.2174/1568026616666150930120959. [DOI] [PubMed] [Google Scholar]
- Van Goor F, Hadida S, Grootenhuis PD, Burton B, Stack JH, Straley KS, Decker CJ, Miller M, McCartney J, Olson ER, Wine JJ, Frizzell RA, Ashlock M, Negulescu PA. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. P Natl Acad Sci USA. 2011;108(46):18843–18848. doi: 10.1073/pnas.1105787108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanier MT, Gissen P, Bauer P, Coll MJ, Burlina A, Hendriksz CJ, Latour P, Goizet C, Welford RW, Marquardt T, Kolb SA. Diagnostic tests for Niemann-Pick disease type C (NP-C): A critical review. Mol Genet Metab. 2016;118(4):244–254. doi: 10.1016/j.ymgme.2016.06.004. [DOI] [PubMed] [Google Scholar]
- Vaquer G, Riviere F, Mavris M, Bignami F, Llinares-Garcia J, Westermark K, Sepodes B. Animal models for metabolic, neuromuscular and ophthalmological rare diseases. Nat Rev Drug Discov. 2013;12(4):287–305. doi: 10.1038/nrd3831. [DOI] [PubMed] [Google Scholar]
- Vincent F, Loria P, Pregel M, Stanton R, Kitching L, Nocka K, Doyonnas R, Steppan C, Gilbert A, Schroeter T, Peakman MC. Developing predictive assays: the phenotypic screening “rule of 3”. Sci Transl Med. 2015;7(293):293ps215. doi: 10.1126/scitranslmed.aab1201. [DOI] [PubMed] [Google Scholar]
- Wainger BJ, Kiskinis E, Mellin C, Wiskow O, Han SS, Sandoe J, Perez NP, Williams LA, Lee S, Boulting G, Berry JD, Brown RH, Jr, Cudkowicz ME, Bean BP, Eggan K, Woolf CJ. Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep. 2014;7(1):1–11. doi: 10.1016/j.celrep.2014.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidemann F, Niemann M, Stork S, Breunig F, Beer M, Sommer C, Herrmann S, Ertl G, Wanner C. Long-term outcome of enzyme-replacement therapy in advanced Fabry disease: evidence for disease progression towards serious complications. J Intern Med. 2013;274(4):331–341. doi: 10.1111/joim.12077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield PD, Calvin J, Hogg S, O’Driscoll E, Halsall D, Burling K, Maguire G, Wright N, Cox TM, Meikle PJ, Deegan PB. Monitoring enzyme replacement therapy in Fabry disease - Role of urine globotriaosylceramide. J Inherit Metab Dis. 2005;28(1):21–33. doi: 10.1007/s10545-005-4415-x. [DOI] [PubMed] [Google Scholar]
- Whyte MP. Hypophosphatasia: Enzyme Replacement Therapy Brings New Opportunities and New Challenges. J Bone Miner Res Jan. 2017;13 doi: 10.1002/jbmr.3075. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Wolfe JH. Gene therapy in large animal models of human genetic diseases. Introduction Ilar J. 2009;50(2):107–111. doi: 10.1093/ilar.50.2.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, Myers S, Wang JG, Zhou D, Woo JA, Kallakury B, Ju A, Bazylewicz M, Carter YM, Albanese C, Grant N, Shad A, Dritschilo A, Liu XF, Schlegel R. Use of Reprogrammed Cells to Identify Therapy for Respiratory Papillomatosis. New Engl J Med. 2012;367(13):1220–1227. doi: 10.1056/NEJMoa1203055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4(2):67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- Zheng W, Thorne N, McKew JC. Phenotypic screens as a renewed approach for drug discovery. Drug Discov Today. 2013;18(21–22):1067–1073. doi: 10.1016/j.drudis.2013.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]