

Abstract
Although toxic when inhaled in high concentrations, the gas carbon monoxide (CO) is endogenously produced in mammals, and various beneficial effects are reported. For potential medicinal applications and studying the molecular processes underlying the pharmacological action of CO, so-called CO-releasing molecules (CORMs), such as tricabonyldichlororuthenium(II) dimer (CORM-2), have been developed and widely used. Yet, it is not readily discriminated whether an observed effect of a CORM is caused by the released CO gas, the CORM itself, or any of its intermediate or final breakdown products. Focusing on Ca2+- and voltage-dependent K+ channels (KCa1.1) and voltage-gated K+ channels (Kv1.5, Kv11.1) relevant for cardiac safety pharmacology, we demonstrate that, in most cases, the functional impacts of CORM-2 on these channels are not mediated by CO. Instead, when dissolved in aqueous solutions, CORM-2 has the propensity of forming Ru(CO)2 adducts, preferentially to histidine residues, as demonstrated with synthetic peptides using mass-spectrometry analysis. For KCa1.1 channels we show that H365 and H394 in the cytosolic gating ring structure are affected by CORM-2. For Kv11.1 channels (hERG1) the extracellularly accessible histidines H578 and H587 are CORM-2 targets. The strong CO-independent action of CORM-2 on Kv11.1 and Kv1.5 channels can be completely abolished when CORM-2 is applied in the presence of an excess of free histidine or human serum albumin; cysteine and methionine are further potential targets. Off-site effects similar to those reported here for CORM-2 are found for CORM-3, another ruthenium-based CORM, but are diminished when using iron-based CORM-S1 and absent for manganese-based CORM-EDE1.
Keywords: Potassium channel, K+ channel, Patch clamp, Carbon monoxide, Hemeoxygenase, Metal carbonyl
Graphical abstract
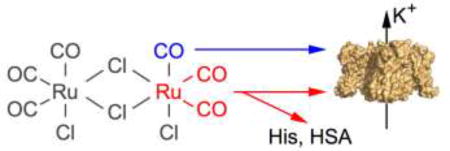
1. Introduction
Carbon monoxide (CO) is an odorless toxic gas, typically generated during incomplete combustion of organic matter. However, CO is also endogenously produced in mammals (Tenhunen et al., 1968) and affects many processes such as vascular tone regulation (Coburn, 1979) and synaptic transmission (Zhuo et al., 1993). Therefore, CO may also serve as a physiological gaseous messenger with therapeutic potential. This aspect spurred the development of CO-releasing molecules (CORMs) to be used as drugs, circumventing complications during therapeutic inhalation of CO gas.
Among several different CORMs synthesized, tricarbonyldichlororuthenium(II) dimer (CORM-2; Motterlini et al., 2002), most likely because of its commercial availability, has been used extensively in in-vitro studies. Compared with application of CO itself, CORMs are safer and easier to use in experimental settings; however, a drawback of using CORMs is the potential problem of eliciting molecular reactions that are unrelated to CO itself but originate from other by-products. Unfortunately, such CORM-mediated side effects have not been studied systematically.
Studies utilizing CORMs have implicated numerous molecular effectors of CO (reviewed in e.g. Gullotta et al. 2012; Wegiel et al., 2013). For example, it is generally accepted that activation of large-conductance, Ca2+- and voltage-activated K+ (KCa1.1) channels contributes to the CO-dependent vasorelaxation (Wang et al., 1997; Williams et al., 2004). However, vasorelaxation induced by CO gas and CORM-2 apparently involves different molecular mechanisms (Decaluwé et al., 2012). Furthermore, CO-mediated activation of KCa1.1 channels in human umbilical vein endothelial cells is not mimicked by CORM-2 (Dong et al., 2008).
The tetrameric KCa1.1 channels are composed of a transmembrane central pore domain surrounded by four voltage-sensing domains, similar to voltage-gated K+ (Kv) channels. Two large cytosolic C-terminal domains (RCK1 and RCK2), which are absent in Kv channels, form a gating ring structure. The channel open probability is controlled by transmembrane voltage and the conformation of the gating ring, which changes upon binding of intracellular Ca2+ (Hoshi et al., 2013) or a plethora of molecules, among them possibly CO (Hou et al., 2009). Activating impacts of CO gas or several CORMs have been reported, but the underlying molecular mechanisms are still under debate. Proposed molecular determinants for CO effects on KCa1.1 include extracellular histidines (Wang and Wu, 1997), channel-bound heme (Jaggar et al., 2005), H365 and H394 within RCK1 (Hou et al., 2008b), and C911 within RCK2 (Williams et al., 2008; Telezhkin et al., 2011).
Here we analyzed the mechanism by which CORM-2 – as compared to CO gas – affects KCa1.1, Kv11.1 (hERG1) and Kv1.5 channels. We present generally applicable experimental strategies for identifying and avoiding side effects originating from CORM-2 and related CO-releasing compounds.
2. Materials and methods
2.1. Expression plasmids and mutagenesis
Wild-type human K+ channels used in this study were: KCa1.1, (hSlo1, KCNMA1, U11058), Kv1.5 (KCNA5, P22460), Kv10.1 (hEAG1, KCNH1, AJ0013668), Kv11.1 (hERG1, KCNH2, NM_000238), and Kv11.3 (hERG3, KCNH7, NP_150375). Mutations were introduced by overlap extension PCR (Expand High Fidelity, Roche, Mannheim, Germany), verified by DNA sequencing.
2.2. Cell culture
HEK 293T cells (DSMZ, Braunschweig, Germany) were maintained in DMEM/F-12 (Life Technologies, Darmstadt, Germany) supplemented with 10% fetal bovine serum at 37 °C in a humidified 5% CO2 incubator. Cells were trypsinized, diluted with culture medium, and seeded on 12-mm glass coverslips. Patch-clamp experiments were performed 2–3 days after plating. Cells were transfected with the respective plasmids using the Rotifect® (Roth, Karlsruhe, Germany) transfection reagent. CD8-encoding plasmids (10–20% of total DNA) were co-transfected to allow identification of transfected cells using anti-CD8-coated beads (Dynabeads, Invitrogen, Karlsruhe, Germany).
2.3. Electrophysiological measurements
Whole-cell and inside-out voltage-clamp experiments were performed as described previously (Gessner et al., 2010; Gessner et al., 2012). Briefly, patch pipettes with resistances of 1.0–2.5 MΩ were used. The series resistance was compensated for by more than 70% to minimize voltage errors. Patch-clamp amplifier EPC9 or EPC10 was operated with PatchMaster software (both HEKA Elektronik, Lambrecht, Germany). Leak and capacitive currents were corrected with a p/n method with a leak holding voltage of −110 mV. Currents were low-pass filtered at 5 kHz and sampled at a rate of 25 kHz. All experiments were performed at 21–23 °C.
Internal solutions contained (mM): 140 KCl, 10 EGTA, 10 HEPES (pH 7.4 with KOH). For whole-cell recordings the external (bath) solution consisted of (mM): 146 NaCl, 4 KCl, 2 CaCl2, 2 MgCl2, 10 HEPES (pH 7.4 with NaOH). To allow buffering of pH between 6.5 and 8.5, MES or TRIS was used instead of HEPES when appropriate. For inside-out recordings the external (pipette) solution composed of (in mM): 140 KCl, 2 CaCl2, 2 MgCl2, 10 HEPES (pH 7.4 with KOH).
Solutions containing CO gas (1 mM) were prepared on the day of experiments by bubbling the solution for >20 min with CO gas and stored in gas-tight rubber-sealed glass vials. CORM-2 and CORM-3 aliquots were dissolved in DMSO (50 or 100 mM) vigorously mixed for 5 s and immediately diluted in the bath to the final concentration, resulting in a maximal final DMSO concentration of 0.1%. CORM-S1 and CORM-EDE1 were dissolved in DMSO (50 mM), aliquoted and stored at −20 °C in the dark for up to 2 months, a period during which no significant loss of CO-releasing activity was observed in a standard myoglobin assay. For CORM structures see Supplementary Fig. 1. CO release from CORM-S1 and CORM-EDE1 was triggered by illumination of the solution in the focus of a 40× objective with blue light from a 100-W HBO mercury lamp (400–470 nm). CORM-2 was from Sigma-Aldrich (Darmstadt, Germany); iCORM-2, CORM-3, CORM-S1 and CORM-EDE1 were synthesized according to protocols published previously (Motterlini et al., 2002; Johnson et al., 2007; Kretschmer et al., 2011; Mede et al., 2016).
2.4. Peptide synthesis
Solid-phase peptide synthesis was carried out on Rink amide MBHA resin (0.53 mmol/g, Iris) applying a standard Fmoc (9-fluorenylmethyloxycarbonyl) protocol with 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate) (HBTU) and hydroxybenzotriazole (HOBt) as coupling agents. Synthesis was performed using an automated peptide synthesizer EPS221 (Intavis, Cologne, Germany). Peptide cleavage was carried out using 1 ml of the mixture: 75 mg phenol, 25 µl ethanedithiol, 50 µl thioanisol, 50 µl water in 1 ml trifluoroacetic acid (TFA) per 100 mg of resin. The mixture was gently shaken for 3 h, and then the peptide was filtered off the resin and precipitated in cold diethyl ether. After several washing steps using diethyl ether, the crude peptides were purified by semi-preparative RP-HPLC on a Shimadzu LC-8A system equipped with a C18 column (Knauer Eurospher 100) using 0.1% TFA in water (eluent A) and 0.1% TFA in 90% acetonitrile/water (eluent B) as elution system. Aliquots were prepared, lyophilized and stored at −20 °C.
Peptides used:
| Kv11.1 wild type | H-GNMEQPHMDSRIGWLHNLGDQI-NH2 | (1) |
| Kv11.1 H578D | H-GNMEQPDMDSRIGWLHNLGDQI-NH2 | (2) |
| Kv11.1 H587Y | H-GNMEQPHMDSRIGWLYNLGDQI-NH2 | (3) |
| Kv11.1 H578D:H587Y | H-GNMEQPDMDSRIGWLYNLGDQI-NH2 | (4) |
| Control-His | H-AAAAHAAAA-NH2 | (5) |
| Control-Ala | H-AAAAAAAAA-NH2 | (6) |
| Control-Cys | H-AAAACAAAA-NH2 | (7) |
2.5. Sample preparation
Peptides or Ac-His-NHMe were dissolved in ddH2O and immediately mixed with freshly prepared CORM-2 (in ddH2O) solutions at different ratios (4:1 and 1:1). Mixtures were incubated at room temperature for different durations (15 min or 12 h), centrifuged and the supernatant was injected into the LC-ESI-MS system micrOTOF-Q III (Bruker Daltonics GmbH, Bremen, Germany) equipped with a C18 column (EC100/2 Nucleoshell RP18 Gravity 2.7 µm column, Macherey-Nagel, Düren, Germany), and detected at 220 nm. The column temperature was 25 °C. Analysis of the MS data was performed using Bruker Compass Data Analysis 4.1 software.
2.6. Amino acid analysis
Peptide concentration was determined by amino acid analysis after hydrolyzing about 500 µg peptide in 6 N HCl in an oxygen-free environment at 110 °C for 24 h. The hydrolysis product was subjected to ion exchange chromatography-based amino acid analysis in an Eppendorf Amino Acid Analyzer LC 3000 in comparison to an external standard (Laborservice Onken, Gründau, Germany). The peptide contents varied between 84.2 and 88.9%.
2.7. Mass spectrometry
The molar mass of the precursor peptides and the reaction products was measured using a Dionex UltiMate 3000 LC (ThermoScientific, Dreieich, Germany) coupled to a micrOTOF-Q III system (Bruker Daltonics). CORM-2 and peptides 1–7 or Ac-His-NHMe were dissolved in ddH2O and mixed to final concentrations of 12.5 µM and 50 µM, respectively. Samples were incubated at room temperature for ≥15 min prior to application to the LC-MS/MS device.
2.8. Electrophysiology data analysis and statistics
Electrophysiological data were analyzed with FitMaster (HEKA Elektronik) and IgorPro (WaveMetrics, Lake Oswego, OR, USA). Averaged data are presented as means ± S.E.M. (n = number of independent measurements) unless specified otherwise. Groups of data were compared with a two-sided Student’s t-test assuming unequal variances followed by Holms-Bonferroni correction. The resulting P values are specified.
3. Results
3.1. CORM-2 activates KCa1.1 channels independently of CO
Quantitative evaluation of how CORM-2 as compared to CO dissolved in the intracellular solution activates KCa1.1 channels was performed after transient expression of KCNMA1 (hSlo1) in HEK 293T cells. K+ current was recorded in the inside-out patch-clamp configuration in the virtual absence of intracellular Ca2+ during repetitive depolarizing voltage steps; the impact of 50 µM CORM-2, which should be capable of releasing up to 300 µM CO, and that of intracellular buffer with an estimated concentration of 250 µM CO gas were compared.
As shown in Fig. 1A, depolarization to 100 mV produced outward currents that were augmented up to five-fold by the application of 50 µM CORM-2 (Fig. 1A, top). Application of solution containing 250 µM CO, however, had a much weaker effect (at most two-fold). Both effects developed with a comparable time course, reaching steady state after about 2 min (Fig. 1B). RuCl2[DMSO]4, the final breakdown product of CORM-2 dissolved in DMSO (iCORM-2, see Supplementary Fig. 1), did not affect KCa1.1 currents (I/ICtrl = 0.82 ± 0.05, n = 5, P = 0.15). At this point one may conclude that CORM-2 results in a local concentration of CO higher than 250 µM or that CORM-2 itself or a transient intermediate is responsible for the current-enhancing effect. When applied to the extracellular face in the whole-cell configuration, CORM-2 had no activating but rather a slightly inhibiting effect (Supplementary Fig. 2). This result argues in favor of the molecular target being located on the cytoplasmic side and the effector not being CO alone because CO gas should readily diffuse through the membrane. Moreover, KCa1.1 activation by CORM-2 was only partially reversible upon washout (Supplementary Fig. 3A, B). Recording of current responses to voltage ramps revealed that current enhancement is voltage dependent. Analysis of the ramp currents yielded the half-maximal voltage (V0.5) of channel activation as robust parameter (Supplementary Fig. 3C); CORM-2 shifted V0.5 in the hyperpolarizing direction by up to 24 mV in a concentration-dependent manner with an EC50 of about 15 µM and a Hill coefficient close to unity (Supplementary Fig. 3D).
Fig. 1. Activation of KCa1.1 channels by CORM-2 and CO.

A) Current traces in response to the indicated pulse protocol recorded from inside-out membrane patches of HEK 293T cells expressing KCa1.1 wild type (wt) or mutant H365A:H394A (HA:HA) before (black) and 3 min after the application of either 50 µM CORM-2, 250 µM CO, or 50 µM CORM-2 in the presence of 1 mM histidine (colored). B) Time courses of the normalized steady-state current with 50 µM CORM-2 (without and with 1 mM histidine) or 250 µM CO application at time zero. Straight lines connect data points for clarity. C) Mean steady-state current increases by 50 µM CORM-2 (red) and 250 µM CO (green) for the indicated channel types and conditions. Data in B and C are means ± S.E.M. with n indicated in parentheses. Asterisks indicate difference to respective wild-type data: *P<0.05; **P<0.01; ***P<0.001.
Based on the previous report that the effect of CORM-2 in KCa1.1 channels is strongly diminished in a mutant lacking two regulatory histidine residues in the cytosolic gating ring structure (H365A:H394A; Hou et al., 2008b), we measured the impact of 50 µM CORM-2 on KCa1.1 channels in the presence of 1 mM free histidine and found that the current enhancement was reduced to the level obtained with CO solution alone (Fig. 1). Consistent with this finding, KCa1.1 mutant H365A:H394A (HA:HA) was activated by CO solution much like the wild type, while the effect of CORM-2 was strongly diminished, albeit not to the level of CO alone (Fig. 1). We thus conclude that H365 and H394 are major – but most likely not the only – determinants for KCa1.1 activation by CORM-2 but not by CO, while CO has a minor impact on the channel by means of a different mechanism.
Cysteine at position 911 in KCa1.1 was previously suggested to be a determinant for channel activation by CO (Telezhkin et al., 2011). In our assay, mutant C911A was indistinguishable from the wild type with respect to activation by CORM-2 (P = 0.97); the impact of CO was smaller than for the wild type (1.16 ± 0.10 vs. 1.47 ± 0.09) but activation by CO persisted (P = 0.028) (Fig. 1C). Further, mutation E535A, which was shown to eliminate RCK1-mediated channel activation by Ca2+ (Zhang et al., 2010), did not affect the impact of CORM-2 (P = 0.93) or CO (P = 0.87) (Fig. 1C). Elimination of RCK2-mediated channel activation by Ca2+ via mutation ΔCaBowl (D884 and D885 removed) (Schreiber and Salkoff, 1997; Hou et al., 2010) did not affect channel activation by CORM-2 (P = 0.99) but fully abolished current enhancement by CO gas (I/ICtrl = 0.78 ± 0.02, P = 0.00065); in fact, CO even diminished current through ΔCaBowl channels (P = 0.015). In summary, KCa1.1 channel activation by CO gas appears to involve structural components of RCK2 (including C911) and clearly differs from activation by CORM-2, which largely depends on histidine residues in RCK1.
3.2. CORM-2 potently inhibits Kv11.1 channels
The results shown above clearly demonstrate that CORM-2 application with the intention of delivering CO bears the potential risk of unspecific target modification, unrelated to the CO effect itself. We therefore asked whether other K+ channels are affected by CORM-2 similarly as KCa1.1. Here we focused on cardiac Kv11.1 (hERG1, KCNH2) channels. Kv11.1 is an important target in safety pharmacology because its inhibition increases the risk of fatal cardiac arrhythmia (Finlayson et al., 2004). Kv11.1 channels were assayed in the whole-cell mode after transient expression in HEK 293T cells, and CORM-2 as well as CO-containing bath solutions were applied to the extracellular face of the membrane. Kv11.1 channels were activated and subsequently inactivated by 400-ms depolarizations to 20 mV; the peak current at 0 mV, following short recovery from inactivation at −120 mV, was measured and analyzed (Fig. 2A). Application of 50 µM CORM-2 strongly inhibited the current (to 28.2 ± 2.1% within 5 min) whereas 250 µM CO was without noticeable effect (P = 0.981) (Fig. 2A, B). Neither was the current blocked by application of 100 µM RuCl3 (101 ± 5%, n = 5). Like for KCa1.1, the effect of CORM-2 was completely eliminated in the presence of 1 mM free histidine; even 100 µM histidine was sufficient to noticeably diminish the activity of CORM-2 (Fig. 2C). Alanine at 1 mM, however, did not appreciably abolish the effect of CORM-2, while 1 mM imidazole was as effective as 100 µM histidine (Fig. 2B, C), indicating that the imidazole moiety of histidine likely is a receptor for CORM-2 or one of its breakdown products. Pre-application of 30 µM human serum albumin (HSA) also fully abolished CORM-2 effects on Kv11.1 (Fig. 2C), thus suggesting that systemic application of CORM-2 is unlikely to modify Kv11.1 channels in the heart.
Fig. 2. Inhibition of Kv11.1 channels by CORM-2.

A) Whole-cell Kv11.1 currents elicited with the indicated voltage protocol before (black) and 5 min after addition of 250 µM CO or 50 µM CORM-2 in the absence and presence of 1 mM histidine. Depolarization to 20 mV lasted 400 ms (not shown). B) Maximal outward current amplitudes at 0 mV as a function of time, normalized to the level before application of 50 µM CORM-2 at time zero with 1 mM of the indicated substances in the bath solution: His, histidine; Im, imidazole; Ala, alanine. Straight lines connect data points for clarity. C) Normalized remaining current after 5 min application of 50 µM CORM-2 plus the competing compounds, as indicated (HSA, human serum albumin). Data in B and C are means ± S.E.M. with n indicated in parentheses of C. Asterisks indicate difference to control, i.e. for application of 50 µM CORM-2 only (***P<0.001).
3.3. Pore-loop histidines of Kv11.1 are molecular targets of CORM-2
Free or surface-exposed histidine, such as in HSA (see, e.g., PDB entry 1E78), may attenuate the impact of CORM-2 on Kv11.1 channels by competing as alternative molecular target with surface-exposed histidine residues of Kv11.1 proteins. To test whether histidines that were previously shown to be relevant for redox sensing of the channel (Pannaccione et al., 2002) are involved, we introduced the mutations H578D and H587Y: selection of aspartate and tyrosine to replace histidine was motivated by finding such residues at the equivalent positions in the closely related Kv10.1 (EAG1, KCNH1) channels (Fig. 3A). Elimination of these pore-loop histidines from Kv11.1 channels markedly attenuated the impact of 50 µM CORM-2 after 300 s: I / ICtrl = 55.3 ± 3.8% (n = 6) vs. 28.2 ± 2.1% for the wild type (n = 21; P < 0.001) (Fig. 3B, C). Moreover, closely related Kv11.3 (hERG3, KCNH7) channels, which lack the corresponding histidines, were only marginally inhibited by CORM-2; 72.8 ± 4.2%, n = 7 (Fig. 3B, C). In conclusion, histidine residues may serve as molecular targets for CORM-2 inhibition of K+ channels.
Fig. 3. Impact of CORM-2 on Kv11.1 mutant and Kv11.3.

A) Multiple sequence alignment of part of the S5–S6 loop of Kv11.1, Kv10.1, and Kv11.3; histidines in Kv11.1 that were mutated are highlighted. B) Representative current traces for the indicated pulse protocols for Kv11.1-HD:HY (Kv11.1-H578D:H587Y, left) and Kv11.3 (right) channels in the whole-cell configuration before (black) and 300 s after application of 50 µM CORM-2 (red). C) Mean relative remaining current for the indicated channel variants after CORM-2 application. Data are means ± S.E.M. with n indicated in parentheses; ***P<0.001 difference compared with hERG1 (wild type).
The pore-loop histidines of Kv11.1 or free or protein-bound histidines in general may serve as binding sites for CORM-2 or fragments thereof. We therefore synthesized 22mer peptides corresponding to pore-loop residues G572 to I593 of Kv11.1 (wild type (1) or histidine mutants (2–4)) and studied their interaction with CORM-2 and its degradation products. Peptides were dissolved in water, incubated with an aqueous CORM-2 solution at a peptide:CORM-2 ratio of 4:1 for 15 min, and analyzed using HPLC and LC-ESI-MS/MS. As shown in Fig. 4A (left), CORM-2 treatment of the wild-type peptide (1) decreased the original peptide peak in the elution profile while additional peaks at longer elution times appeared, thus indicating stable peptide modifications. In contrast, neither a decrease of the peak of the unmodified peptide nor additional peaks upon incubation with CORM-2 were detected for the histidine-free peptide (4) (Fig. 4A, right). LC-ESI-MS/MS analysis (Fig. 4B) revealed an increase in mass compatible with an attachment of Ru(CO)2 to peptide (1), but no modification of (4); peptides (2) and (3), i.e. those harboring only one histidine residue, were also subject to CORM-2 modification (Supplementary Fig. 4).
Fig. 4. Peptide modification by CORM-2.

A) HPLC elution profiles of 22mer peptides corresponding to the pore loop sequences of wild-type Kv11.1 channels (wt, (1)) or its double mutant H578D:H587Y (HD:HY, (4)). Colored traces are from peptides preincubated with CORM-2, black: peptides without preincubation. The elution peak for the non-modified peptides is marked with an asterisk. B) Mass spectra of wild-type and double-mutant peptides (as in (A)) before (black) and after incubation with CORM-2 (red). The labeled peaks correspond to (M+4H+)4+, (M+3H+)3+ and (M+2H+)2+ of the peptides or the peptide:Ru(CO)2 complexes.
We also assessed incubation of CORM-2 with control peptide 5 ([M+H]+ 723.39 m/z) or with Ac-His-NHMe ([M+H]+ 211.12 m/z), which resembles a histidine with free side chain but protected termini. In both cases the m/z value was increased by 157.89 m/z, pointing towards attachment of Ru(CO)2. For Ac-His-NHMe, however, a further signal ([M+H]+ 579.12 m/z) indicated an additional complex formed during the incubation, most likely a 2:1-complex of Ac-His-NHMe and Ru(CO)2. Both signals, the 1:1 and the 2:1 complex, respectively, increased according to the base peak chromatogram over time in approximately the same ratio. In contrast, a 2:1 complex of the control peptide 5 and Ru(CO)2 was not detected. This might be due to steric hindrance of the peptide backbone upon coordination to Ru(CO)2 on either coordination side.
3.4. Histidine-dependent and -independent modification of K+ channels by CORM-2
Results shown thus far indicate that pore-loop histidines are among the targets of CORM-2 that result in a functional impact on Kv11.1 channels. Kv10.1 channels, which are closely related to Kv11.1, lack pore-loop histidines (Fig. 5A). However, these channels were very sensitive to CORM-2 (I / ICtrl = 9.0 ± 1.7%, n = 6, Fig. 5B), suggesting that other histidines or other residues are involved. Free histidine (1 mM) eliminated the effect of CORM-2, and solution with 250 µM CO had no influence on channel function (Fig. 5C). An involvement of histidine residue H364 within S5 of Kv10.1 could not be tested because mutations at this position rendered the channel nonfunctional. However, mutation of H343 at the intracellular end of the voltage-sensing S4 helix to arginine significantly diminished the channel’s sensitivity to CORM-2 (I / ICtrl = 51.0 ± 4.0%, n = 6; P = 0.00002) (Fig. 5B, C).
Fig. 5. CO-independent inhibition of K+ channels by CORM-2.

A) Membrane topology of α subunits of Kv10.1 (left) and Kv1.5 (right) channels with those histidine residues that are potentially accessible from the extracellular side highlighted. B) Representative whole-cell current traces before (black) and after (red) application of 50 µM CORM-2 for wild-type channel constructs (top) and mutants in which Ala or Arg was replaced for His: Kv10.1-H343R (blue dot in the cartoon), Kv1.5-AARA. C) Mean normalized remaining current after application of 50 µM CORM-2 without (red) and with 1 mM free histidine (blue) or in 250 µM CO-containing solutions (green) for the indicated channel types. Asterisks indicate difference to the respective control (application of CORM-2 only; **P<0.01; ***P<0.001). Data are means ± S.E.M. with n indicated in parentheses.
Kv1.5 channels, which like Kv11.1 are also expressed in the heart and therefore also potential targets with relevance for safety pharmacology (Schmitt et al., 2014), were also insensitive to CO but strongly inhibited by 50 µM CORM-2 (I / ICtrl = 8.2 ± 1.5%, n = 7; Fig. 5B, C). Although 1 mM free histidine fully abolished the inhibitory influence of CORM-2 (I / ICtrl = 93.3 ± 1.4%, n = 6), histidine residues within the channel protein apparently only play a minor role because a mutant channel in which all four putative target histidines were eliminated (mutant AARA, combining mutations H284A, H289A, H416R and H463A, Fig. 5A) still responded to CORM-2 (AARA: I / ICtrl = 37.2 ± 5.6%, n = 9) (Fig. 5B, C). Taken together, the data show that various voltage-gated K+ channels are insensitive to CO gas but sensitive to CORM-2. Although the CORM-2 effects on all channels analyzed could be diminished by co-application of free histidine, histidines are not the only determinants.
3.5. pH dependence of CORM-2 modification of Kv11.1 channels
An involvement of histidines in the inhibition of Kv11.1 channels by CORM-2 suggests that this effect may depend on the pH value. Indeed, while 50 µM CORM-2 caused channel inhibition to I / ICtrl = 28% at pH 7.4, the effect was augmented at pH 6.5 (I / ICtrl = 4%) and virtually abolished at pH 8.5 (I / ICtrl = 105%) (Fig. 6A). A detailed analysis of the pH dependence revealed that CORM-2 inhibition of Kv11.1 channels is described by two pK values of 7.0 and 8.3 with 30% and 70% contribution, respectively (Fig. 6B). These pK values may correspond to the side-chain pKa values of histidine (pKa <7) and cysteine (pK 8.3) serving as targets for modification by CORM-2. This idea is further supported by the small size of this pK-7 component in Kv1.5 channels (Supplementary Fig. 5), for which elimination of the histidine residues only partially removed the CORM-2 sensitivity (Fig. 5B, C).
Fig. 6. pH dependence of Kv11.1 inhibition by CORM-2.

A) Representative traces of whole-cell Kv11.1 currents before (black) and 5 min after (red) application of 50 µM CORM-2 at pH 6.5 and pH 8.5. B) Normalized mean remaining current after CORM-2 application as a function of pH, fit with a two-component Hill function (solid line). Resulting pK values are indicated by vertical bars. Data in B are means ± S.E.M. for n = 5–21.
3.6. Involvement of residues other than histidine
The pH titration experiments suggest that only a fraction of CORM-2 effects on KV channels can be accounted for modification of histidine residues, and that cysteine might be an alternative target. We therefore systematically assessed the impact of different free amino acids (1 mM) on the potency of 50 µM CORM-2 to inhibit Kv11.1 channels. As demonstrated in Fig. 7, only cysteine and histidine fully abolished Kv11.1 inhibition by CORM-2. Nevertheless, also methionine, aspartate, and threonine had substantial impacts on CORM-2 activity, whereas alanine, asparagine, isoleucine, tryptophan, and proline were clearly without effect.
Fig. 7. Influence of free amino acids on Kv11.1 inhibition by CORM-2.

Mean remaining Kv11.1 current 5 min after co-application of 50 µM CORM-2 and 1 mM of the indicated amino acids. Asterisks indicate P values compared to application of CORM-2 only (dashed line). Data are means ± S.E.M. with n indicated in parentheses. *P<0.05; **P< 0.01; ***P<0.001 for tests versus CORM-2 alone.
We therefore also synthesized 9mer peptides with central His (5), Ala (6), or Cys (7), flanked by four Ala on each side. Incubation of these peptides (each 50 µM) with CORM-2 (12.5 µM) for 15 min and subsequent LC-MS analysis revealed that only in the His-containing peptide Ru(CO)2 adducts could be detected as inferred from a mass increase of 157.89 m/z (Supplementary Fig. 6). For an increased CORM-2:peptide ratio (1:1) and increased incubation time (12 h) histidine-independent interactions of CORM-2 with peptides, i.e. also with (4), were observed, thus indicating that CORM-2-induced peptide modifications are not specific for histidine only.
3.7. Not all metal-based CORMs cause CORM-2-like side effects
CORM-3 is ruthenium-based, where the metal ion is pre-bound to the amino acid glycine (Supplementary Fig. 1). However, this feature does not prevent inhibition of Kv11.1 and Kv1.5 channels by CORM-3, and in both cases channel inhibition was strongly diminished by the presence of 1 mM free histidine (Supplementary Fig. 7).
We also tested photo-CORMs with iron (CORM-S1; Kretschmer et al., 2011) or manganese (CORM-EDE1; Mede et al., 2016) as the metal center. Blue-light illumination of CORM-EDE1 (100 µM), which triggers release of CO, was without effect on Kv11.1 (Fig. 8A, B) and Kv1.5 (Fig. 8C, D) currents. Illumination of CORM-S1 (100 µM) also failed to affect Kv1.5 channels (Fig. 8C, D) but clearly inhibited Kv11.1 channels (Fig. 8A, B). This effect (I / ICtrl = 46.9 ± 3.9%, n = 6) was only partially attenuated by co-application of 1 mM histidine (I / ICtrl = 80.0 ± 3.1%, n = 3) and, hence, suggests a different molecular mechanism from that for CORM-2.
Fig. 8. Influence of the photo-CORMs CORM-S1 and CORM-EDE1 on Kv11.1 and Kv1.5 channels.

A) Representative whole-cell current traces of Kv11.1 channels for the pulse protocol shown on top before (black) and after blue-light illumination of 100 µM CORM-S1 (brown) or 100 µM CORM-EDE1 (blue) present in the bath solution. B) Time course of mean normalized currents as in A with an indication of the illumination protocol. C, D) As in A and B for Kv1.5 channels. Data in B and D are means ± S.E.M. with n indicated in parentheses; straight lines connect the data points for clarity.
4. Discussion
Physiologically produced CO, mainly by the catabolism of heme catalyzed by heme oxygenase, is a putative cellular messenger. However, unlike NO, which has been studied extensively and has clearly identified molecular targets, the medicinal benefits of CO and the underlying molecular mechanisms are less well defined. For possible clinical applications and for studying CO-related biology, the local release of CO by means of CORMs is desirable. However, potential confounding factors must be recognized and alternative strategies need to be devised. We show here that CORM-2 has the propensity of forming Ru(CO)2 adducts to histidine side chains. For voltage-gated K+ channels the functional consequences are diverse: while KCa1.1 channels are activated, Kv11.1, Kv10.1, and Kv1.5 are inhibited by CORM-2 in a CO-independent manner. These modulations involve the histidine residues previously identified to be important for various aspects of gating. While H365 and H394 in KCa1.1 affect channel regulation by intracellular pH (Hou et al., 2008a), the extracellular histidines in Kv11.1 (H578 and H587) are implicated in the channel’s regulation by iron-mediated oxidation (Pannaccione et al., 2002).
Ru(CO)2 adduct formation is largely independent of the local environment because it occurs with free histidine and within peptides or proteins such as HSA. In our experimental settings, co-application of 1 mM histidine or 30 µM HSA “quenched” adverse effects of CORM-2 on K+ channels. This quenching effect is consistent with the observation that CORM-2 does not elicit obvious cardiac side effects when administered systemically to rodents (Motterlini et al., 2002; Cepinskas et al., 2008). Excess HSA in the blood serving as scavenger for Ru(CO)2 adducts probably prevents CORM-2-dependent inhibition of Kv11.1 and Kv1.5 channels that otherwise could result in cardiac arrhythmia (Finlayson et al., 2004; Schmitt et al., 2014). Vascular side effects due to activation of KCa1.1 are even less likely because extracellular CORM-2 did not affect these channels (Supplementary Fig. 2). Similarly, extracellularly applied CORM-2 does not activate KCa1.1 channels in human umbilical vein endothelial cells (Dong et al., 2008). The sensitivity of KCa1.1 to intracellularly applied CORM-2 suggests that CORM-2 or its breakdown products cannot cross the membrane or that they immediately react with off-site histidines on the extracellular side of the membrane.
Despite the rapid decomposition of CORM-2 in a physiological environment some systemic functional impacts may occur distant from the systemic application site. Such effects may originate from HSA-Ru(CO)2 complexes serving as temporary CO storage molecules. Compatible with this notion, binding of Ru(CO)n fragments, with n ranging from 0 to 3, typically to histidine, has been described (Santos-Silva et al., 2001; Santos et al., 2012; Seixas et al., 2015; Tabe et al., 2015; Valensin et al., 2010). Moreover, Tabe et al. (2015) observed CO release from ruthenium carbonyl-incorporated cross-linked hen egg white lysozyme (HEWL). Loss or release of a carbonyl moiety, however, does not necessarily imply release of CO because CORM-3-mediated adduct formation to HEWL, HSA, and hemoglobin was also shown to result in the release of CO2 (Santos-Silva et al., 2011). Future studies must clarify whether physiological impacts resulting from systemically administered CORM-2 depend on the release of CO at all.
Protein modifications by CORM-2, as we have shown for KCa1.1 and Kv11.1 channels, may have been misinterpreted in previous studies as CO effect. In particular for KCa1.1 channels this resulted in contradictory models on the molecular mechanism of channel activation by CO (reviewed in Heinemann et al., 2014). For KCa1.1 channels we conclude that CO gas and CORM-2 mediate channel activation by different mechanisms, the latter requiring histidine residues H365 and H394 within the RCK1 domain. The impact of CO itself accounts for at most 20% of the channel activation observed with CORM-2. Such differences in the mode of action may also underlie the observed differences in vasorelaxing effects of CO vs. CORM-2 in vascular ring preparations (Decaluwé et al., 2012). The mechanism by which CO gas activates KCa1.1 channels, however, remains elusive, albeit the involvement of heme moieties is a likely scenario (Jaggar et al., 2005).
Conclusions from previous studies utilizing CORM-2 and related compounds to elucidate the molecular mechanism of CO action need to be revisited because iCORM-2 (Supplementary Fig. 1) and hemoglobin may not be proper controls for distinguishing between CO gas effects and formation of Ru(CO)2 adducts. Hemoglobin binds CO, but it may also simultaneously serve as an acceptor for Ru(CO)2 adducts via surface-exposed histidines. iCORM-2 may be a proper control if it exerts a similar effect as CORM-2, such as observed for the inhibition of Kv2.1 channels (Jara-Oseguera et al., 2011).
We also noticed quenching of CORM-2 effects by free cysteine and methionine (Fig. 7). Similar to sulfite species (McLean et al., 2012), these amino acids may serve as physiological triggers to initiate CO release from CORM-2. Assuming such a mechanism, the observed quenching of the CORM-2 side effect might be explained by induced decomposition of CORM-2 such that no Ru(CO)2 adducts to histidine can be formed. Alternatively, cysteine or methionine may serve as acceptors for binding of Ru(CO)n fragments. However, we did not observe Ru(CO)2 adducts for cysteine-containing peptides (Supplementary Fig. 6). This finding may be explained by either very weak binding or the necessity of a secondary amino acid that was not present in our peptide assay. The latter was experimentally observed in a ferritin / CORM-2 composite in which Ru(CO)2 adduction to cysteine required a glutamate residue (Fujita et al., 2014).
Tavares et al. (2011) suggested that CORM-2 stimulates ROS production in E. coli, which amplifies the cytotoxic effect (Nobre et al., 2007) of CO gas, an effect that can be quenched by co-application of hemoglobin or antioxidants including GSH, free cysteine, and methionine. In fact, part of the Kv11.1 response to CORM-2 in the presence of 1 mM histidine was a small but significant increase in current (Fig. 2B), which was strongly diminished by mutagenesis of extracellular histidines (mutant Kv11.1-H578D:H587Y) (Supplementary Fig. 8). This effect is reminiscent of a transient current increase when applying H2O2 to Kv11.1 channels (Kolbe et al., 2010) and, hence, indicates that oxidation or protonation of the histidine residues may also take place on CORM-2 application.
Metal- or metal-carbonyl-based side effects are not expected when using transition metalfree CORMs such as CORM-A1 (Motterlini et al., 2005; Supplementary Fig. 1) or more recently developed metal-free CORMs (e.g., Palao et al., 2016; Ji et al., 2017). Among the metal-based CORMs, metal-carbonyl adduct formation is not specific for ruthenium but was also observed for iridium (Catarino et al., 2016). We found that the iron-based CORM-S1, despite being without effect on Kv1.5, inhibits Kv11.1 channels when decomposed with blue light (Kretschmer et al., 2011). In fact, application of the expected final breakdown products CO and cysteamine alone had no effects on Kv11.1 channels, and application of Fe2+ alone inhibited Kv11.1 channels with at least 20-fold slower kinetics (Supplementary Fig. 9). Our experiments with manganese-based CORM-EDE1 (Mede et al., 2016) gave no indication for similar side effects on Kv11.1 and Kv1.5 channels (Fig. 8). Manganese-based CORMs may therefore represent attractive alternatives for physiological tests, in particular when CO release is triggered with visible light.
In conclusion, due to multiple side effects elicited by decomposing metal-based CORMs, a direct proof for the involvement of the released CO gas in functional assays is not straightforward. In future experiments utilizing CORMs as source of CO, in particular under conditions with inorganic buffers, great care regarding the derived conclusions on the impact of CO is warranted. Application of CO gas itself remains the gold standard in physiological experiments for testing whether or not CORM effects are to be associated with the action of the gaseous messenger CO.
Supplementary Material
Acknowledgments
This work was supported by the German Research Foundation (DFG, FOR 1738 and HE2993/16-1) and the National Institutes of Health (to TH, GM121375). We furthermore thank M. Keating for providing cDNA coding for KCNH2 and E.M. Franke for technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Chemical compounds studied in this article
CORM-2 (PubChem CID: 10951331); CORM-3 (PubChem CID: 91826084)
Statement of conflicts of interest
The authors declare no competing interests.
References
- Caterino M, Petruk AA, Vergara A, Ferraro G, Marasco D, Doctorovich F, Estrin DA, Merlino A. Mapping the protein-binding sites for iridium(III)-based CO-releasing molecules. Dalton Trans. 2016;45:12206–12214. doi: 10.1039/c6dt01685e. [DOI] [PubMed] [Google Scholar]
- Cepinskas G, Katada K, Bihari A, Potter RF. Carbon monoxide liberated from carbon monoxide-releasing molecule CORM-2 attenuates inflammation in the liver of septic mice. Am. J. Physiol. 2008;294:G184–G191. doi: 10.1152/ajpgi.00348.2007. [DOI] [PubMed] [Google Scholar]
- Coburn RF. Mechanisms of Carbon Monoxide Toxicity. Prev. Med. 1979;8:310–322. doi: 10.1016/0091-7435(79)90008-2. [DOI] [PubMed] [Google Scholar]
- Decaluwé K, Pauwels B, Verpoest S, Van de Voorde J. Divergent mechanisms involved in CO and CORM-2 induced vasorelaxation. Eur. J. Pharmacol. 2012;674:370–377. doi: 10.1016/j.ejphar.2011.11.004. [DOI] [PubMed] [Google Scholar]
- Dong DL, Chen C, Huang W, Chen Y, Zhang XL, Li Z, Li Y, Yang BF. Tricarbonyldichlororuthenium(II) dimer (CORM2) activates non-selective cation current in human endothelial cells independently of carbon monoxide releasing. Eur. J. Pharmacol. 2008;590:99–104. doi: 10.1016/j.ejphar.2008.05.042. [DOI] [PubMed] [Google Scholar]
- Finlayson K, Witchel HJ, McCulloch J, Sharkey J. Acquired QT interval prolongation and HERG: implications for drug discovery and development. Eur. J. Pharmacol. 2004;500:129–142. doi: 10.1016/j.ejphar.2004.07.019. [DOI] [PubMed] [Google Scholar]
- Fujita K, Tanaka Y, Sho T, Ozeki S, Abe S, Hikage T, Kuchimaru T, Kizala-Kondoh S, Ueno T. Intracellular CO Release from Composite of Ferritin and Ruthenium Carbonyl Complexes. J. Am. Chem. Soc. 2014;136:16902–16908. doi: 10.1021/ja508938f. [DOI] [PubMed] [Google Scholar]
- Gessner G, Cui Y-M, Otani Y, Ohwada T, Soom M, Hoshi T, Heinemann SH. Molecular mechanism of pharmacological activation of BK channels. Proc. Natl. Acad. Sci. USA. 2012;109:3552–3557. doi: 10.1073/pnas.1114321109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gessner G, Macianskiene R, Starkus JG, Schönherr R, Heinemann SH. The amiodarone derivative KB130015 activates hERG1 potassium channels via a novel mechanism. Eur. J. Pharmacol. 2010;632:52–59. doi: 10.1016/j.ejphar.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gullotta F, di Masi A, Coletta M, Ascenzi P. CO metabolism, sensing, and signaling. Biofactors. 2012;38:1–13. doi: 10.1002/biof.192. [DOI] [PubMed] [Google Scholar]
- Heinemann SH, Hoshi T, Westerhausen M, Schiller A. Carbon monoxide – physiology, detection and controlled release. Chem. Comm. 2014;50:3644–3660. doi: 10.1039/c3cc49196j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi T, Pantazis A, Olcese R. Transduction of voltage and Ca2+ signals by Slo1 BK channels. Physiology. 2013;28:172–189. doi: 10.1152/physiol.00055.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou S, Xu R, Heinemann SH, Hoshi T. Reciprocal regulation of the Ca2+ and H+ sensitivity in the SLO1 BK channel conferred by the RCK1 domain. Nat. Struct. Mol. Biol. 2008a;15:403–410. doi: 10.1038/nsmb.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou S, Xu R, Heinemann SH, Hoshi T. The RCK1 high-affinity Ca2+ sensor confers carbon monoxide sensitivity to Slo1 BK channels. Proc. Natl. Acad. Sci. USA. 2008b;105:4039–4043. doi: 10.1073/pnas.0800304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou S, Heinemann SH, Hoshi T. Modulation of BKCa channel gating by endogenous signaling molecules. Physiology. 2009;24:26–35. doi: 10.1152/physiol.00032.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou S, Vigeland LE, Zhang G, Xu R, Li M, Heinemann SH, Hoshi T. Zn2+ activates large conductance Ca2+-activated K+ channel via an intracellular domain. J. Biol. Chem. 2010;285:6434–6442. doi: 10.1074/jbc.M109.069211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaggar JH, Li A, Parfenova H, Liu J, Umstot ES, Dopico AM, Leffler CW. Heme is a carbon monoxide receptor for large-conductance Ca2+-activated K+ channels. Circ. Res. 2005;97:805–812. doi: 10.1161/01.RES.0000186180.47148.7b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji X, De La Cruz LKC, Pan Z, Chittavong V, Wang B. pH sensitive metal-free carbon monoxide prodrugs with tunable and predictable release rates. Chem. Comm. 2017;53:9628–9631. doi: 10.1039/c7cc04866a. [DOI] [PubMed] [Google Scholar]
- Johnson TR, Mann BE, Teasdale IP, Adams H, Foresti R, Green CJ, Motterlini R. Metal carbonyls as pharmaceuticals? [Ru(CO)3Cl(glycinate)], a CO-releasing molecule with an extensive aqueous solution chemistry. Dalton Trans. 2007;15:1500–1508. doi: 10.1039/b613629j. [DOI] [PubMed] [Google Scholar]
- Jara-Oseguera A, Ishida IG, Rangel-Yescas GE, Espinosa-Jalapa N, Pérez-Guzmán JA, Elías-Vinas D, Le Lagadec R, Rosenbaum T, Islas DL. Uncoupling charge movement from channel opening in voltage-gated potassium channels by ruthenium complexes. J. Biol. Chem. 2011;286:16414–16425. doi: 10.1074/jbc.M110.198010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolbe K, Schönherr R, Gessner G, Sahoo N, Hoshi T, Heinemann SH. Cysteine 723 in the C-linker segment confers oxidation inhibition of hERG1 potassium channels. J. Physiol. 2010;588:2999–3009. doi: 10.1113/jphysiol.2010.192468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretschmer R, Gessner G, Görls H, Heinemann SH, Westerhausen M. Dicarbonylbis(cysteamine)iron(II): A light-induced carbon monoxide releasing molecule based on iron (CORM-S1) J. Inorg. Biochem. 2011;105:6–9. doi: 10.1016/j.jinorgbio.2010.10.006. [DOI] [PubMed] [Google Scholar]
- McLean S, Mann BE, Poole RK. Sulfite species enhance carbon monoxide release from CO-releasing molecules: Implications for the deoxymyoglobin assay of activity. Anal. Biochem. 2012;427:36–40. doi: 10.1016/j.ab.2012.04.026. [DOI] [PubMed] [Google Scholar]
- Mede R, Klein M, Claus RA, Krieck S, Quickert S, Görls H, Neugebauer U, Schmitt M, Gessner G, Heinemann SH, Popp J, Bauer M, Westerhausen M. CORM-EDE1: a highly water-soluble and nontoxic manganese-based photoCORM with a biogenic ligand sphere. Inorg. Chem. 2016;55:104–113. doi: 10.1021/acs.inorgchem.5b01904. [DOI] [PubMed] [Google Scholar]
- Motterlini R, Clark JE, Foresti R, Saratchandra P, Mann BE, Green CJ. Carbon monoxide-releasing molecules: characterization of biochemical and vascular activities. Circ. Res. 2002;90:e17–e24. doi: 10.1161/hh0202.104530. [DOI] [PubMed] [Google Scholar]
- Motterlini R, Sawle P, Hammad J, Bains S, Alberto R, Foresti R, Green CJ. CORM-A1: a new pharmacologically active carbon monoxide-releasing molecule. FASEB J. 2005;19:284–286. doi: 10.1096/fj.04-2169fje. [DOI] [PubMed] [Google Scholar]
- Nobre LS, Seixas JD, Romao CC, Saraiva LM. Antimicrobial action of carbon monoxide-releasing compounds. Antimicrob. Agents Chemother. 2007;51:4303–4307. doi: 10.1128/AAC.00802-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palao E, Slanina T, Muchová L, Solomek T, Vitek L, Klán P. Transition-metal-free CO-releasing BODIPY derivatives activatable by visible to NIR light as promising bioactive molecules. J. Am. Chem. Soc. 2017;138:126–133. doi: 10.1021/jacs.5b10800. [DOI] [PubMed] [Google Scholar]
- Pannaccione A, Castaldo P, Ficker E, Annunziato L, Taglialatela M. Histidines 578 and 587 in the S5–S6-linker of the human Ether-a-gogo Related Gene-1 K+ channels confer sensitivity to reactive oxygen species. J. Biol. Chem. 2002;277:8912–8919. doi: 10.1074/jbc.M111353200. [DOI] [PubMed] [Google Scholar]
- Santos-Silva T, Mukhopadhyay A, Seixas JD, Bernardes GJL, Romao CC, Romao MJ. CORM-3 reactivity toward proteins: the crystal structure of a Ru(II) dicarbonyl-lysozyme complex. J. Am. Chem. Soc. 2011;133:1192–1195. doi: 10.1021/ja108820s. [DOI] [PubMed] [Google Scholar]
- Santos MFA, Seixas JD, Coelho AC, Mukhopadhyay A, Reis PM, Romao MJ, Romao CC, Santos-Silva T. New insights into the chemistry of fac-[Ru(CO)3]2+ fragments in biologically relevant conditions: The CO releasing activity of [Ru(CO)3Cl2(1,3-thiazole)], and the X-ray crystal structure of its adduct with lysozyme. J. Inorg. Biochem. 2012;117:285–291. doi: 10.1016/j.jinorgbio.2012.06.018. [DOI] [PubMed] [Google Scholar]
- Schreiber M, Salkoff L. A Novel Calcium-Sensing Domain in the BK Channel. Biophys. J. 1997;73:1355–1363. doi: 10.1016/S0006-3495(97)78168-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt N, Grunnet M, Olesen S-P. Cardiac potassium channel subtypes: new roles in repolarization and arrhythmia. Physiol. Rev. 2014;94:609–653. doi: 10.1152/physrev.00022.2013. [DOI] [PubMed] [Google Scholar]
- Seixas JD, Santos MFA, Mukhopadhyay A, Coelho AC, Reis PM, Veiros LF, Marques AR, Penacho N, Goncales AML, Romao MJ, Bernardes GJL, Santos-Silva T, Romao CC. A contribution to the rational design of Ru(CO)3Cl2L complexes for in vivo delivery of CO. Dalton Trans. 2015;44:5058–5075. doi: 10.1039/c4dt02966f. [DOI] [PubMed] [Google Scholar]
- Tabe H, Fujita K, Abe S, Tsujimoto M, Kuchimaru T, Kizaka-Kondoh S, Takano M, Kitagawa S, Ueno T. Preparation of a cross-linked porous protein crystal containing Ru carbonyl complexes as a CO-releasing extracellular scaffold. Inorg. Chem. 2015;54:215–220. doi: 10.1021/ic502159x. [DOI] [PubMed] [Google Scholar]
- Tavares AFN, Teixeira M, Romao CC, Seixas JD, Nobre LS, Saraiva LM. Reactive oxygen species mediate bactericidal killing elicited by carbon monoxide-releasing molecules. J. Biol. Chem. 2011;286:26708–26717. doi: 10.1074/jbc.M111.255752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telezhkin V, Brazier SP, Mears R, Müller CT, Riccardi D, Kemp PJ. Cysteine residue 911 in the C-terminal tail of human BKCa α channel subunit is crucial for its activation by carbon monoxide. Eur. J. Physiol. 2011;461:665–675. doi: 10.1007/s00424-011-0924-7. [DOI] [PubMed] [Google Scholar]
- Tenhunen R, Marver HS, Schmid R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc. Natl. Acad. Sci. USA. 1968;61:748–755. doi: 10.1073/pnas.61.2.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valensin D, Anzini P, Gaggelli E, Gaggelli N, Tamasi G, Cini R, Gabbiani C, Michelucci E, Messori L, Kozlowski H, Valensin G. fac-{Ru(CO)3}2+ selectively targets the histidine residue of the β-amyloid peptide 1–28. Implications for new Alzheimer’s disease treatments based on ruthenium complexes. Inorg. Chem. 2010;49:4720–4722. doi: 10.1021/ic902593e. [DOI] [PubMed] [Google Scholar]
- Wang R, Wu L, Wang Z. The direct effect of carbon monoxide on KCa channels in vascular smooth muscle cells. Eur. J. Physiol. 1997;434:285–291. doi: 10.1007/s004240050398. [DOI] [PubMed] [Google Scholar]
- Wang R, Wu L. The chemical modification of KCa channels by carbon monoxide in vascular smooth muscle cells. J. Biol. Chem. 1997;272:8222–8226. doi: 10.1074/jbc.272.13.8222. [DOI] [PubMed] [Google Scholar]
- Wegiel B, Douglas WH, Otterbein LE. The social network of carbon monoxide in medicine. Trends Mol. Med. 2013;19:3–11. doi: 10.1016/j.molmed.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SEJ, Wootton P, Mason HS, Bould J, Iles DE, Riccardi D, Peers C, Kemp PJ. Hemoxygenase-2 is an oxygen sensor for a calcium-sensitive potassium channel. Science. 2004;306:2093–2097. doi: 10.1126/science.1105010. [DOI] [PubMed] [Google Scholar]
- Williams SE, Brazier SP, Baban N, Telezhkin V, Müller CT, Riccardi D, Kemp PJ. A structural motif in the C-terminal tail of slo1 confers carbon monoxide sensitivity to human BKCa channels. Eur. J. Physiol. 2008;456:561–572. doi: 10.1007/s00424-007-0439-4. [DOI] [PubMed] [Google Scholar]
- Zhang G, Huang S-Y, Yang J, Shi J, Yang X, Moller A, Zou X, Cui J. Ion sensing in the RCK1 domain of BK channels. Proc. Natl. Acad. Sci. USA. 2010;107:18700–18705. doi: 10.1073/pnas.1010124107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo M, Small SA, Kandel ER, Hawkins RD. Nitric oxide and carbon monoxide produce activity-dependent long-term synaptic enhancement in hippocampus. Science. 1993;260:1946–1950. doi: 10.1126/science.8100368. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.