

Fibronectin-ligated α5β1 integrin promotes malignancy by inducing tissue tension.
Abstract
Tumors are fibrotic and characterized by abundant, remodeled, and cross-linked collagen that stiffens the extracellular matrix stroma. The stiffened collagenous stroma fosters malignant transformation of the tissue by increasing tumor cell tension to promote focal adhesion formation and potentiate growth factor receptor signaling through kinase. Importantly, collagen cross-linking requires fibronectin (FN). Fibrotic tumors contain abundant FN, and tumor cells frequently up-regulate the FN receptor α5β1 integrin. Using transgenic and xenograft models and tunable two- and three-dimensional substrates, we show that FN-bound α5β1 integrin promotes tension-dependent malignant transformation through engagement of the synergy site that enhances integrin adhesion force. We determined that ligation of the synergy site of FN permits tumor cells to engage a zyxin-stabilized, vinculin-linked scaffold that facilitates nucleation of phosphatidylinositol (3,4,5)-triphosphate at the plasma membrane to enhance phosphoinositide 3-kinase (PI3K)-dependent tumor cell invasion. The data explain why rigid collagen fibrils potentiate PI3K activation to promote malignancy and offer a perspective regarding the consistent up-regulation of α5β1 integrin and FN in many tumors and their correlation with cancer aggression.
INTRODUCTION
Tumors are highly fibrotic (Van den Hooff, 1986; Walker, 2001; Lopez-Novoa and Nieto, 2009; Arendt et al., 2010; Ewald et al., 2011; Samuel et al., 2011; Nakagawa et al., 2014). Fibrotic tumors contain abundant quantities of extracellular matrix (ECM) proteins, such as type I collagen, fibronectin (FN), tenascin, and assorted proteoglycans. Cancerous tissues also exhibit altered levels and activity of ECM receptors, including integrins, discoidin receptors, and syndecans (Ioachim et al., 2002; West et al., 2005; Desgrosellier and Cheresh, 2010; Nam et al., 2010; Ozbek et al., 2010; Gehler et al., 2013; Pickup et al., 2014; Theocharis et al., 2015). Elevated β1-integrin levels and focal adhesion kinase (FAK) activity correlate positively with high tumor grade and predict for poor patient prognosis (Recher et al., 2004; dos Santos et al., 2012; Sulzmaier et al., 2014). Inhibiting β1-integrin ligand binding represses the malignant phenotype of tumor cells in vitro and in vivo (Weaver et al., 1997; Stoeltzing et al., 2003; Huang et al., 2011; Blandin et al., 2015), and transgenic ablation of β1 integrin or FAK prevent oncogene-induced malignant transformation and metastasis (van Nimwegen et al., 2005). Function-blocking antibodies to integrins and FAK inhibitors are also under clinical consideration to improve the treatment of fibrotic tumors (Walsh et al., 2010; Stokes et al., 2011; Lagares et al., 2012; Jiang et al., 2016). These findings emphasize the interplay among tissue fibrosis, the ECM, and integrin signaling in malignancy.
Tumors are mechanically corrupted tissues with high interstitial pressure, elevated compression, increased solid stress, ECM stiffening, and increased cellular tension (Heldin et al., 2004; Paszek et al., 2005; Levental et al., 2009; Lopez et al., 2011; Samuel et al., 2011; Provenzano et al., 2012; Mouw et al., 2014; Laklai et al., 2016). Fibrotic tumors contain a stiffened ECM, which associates with cancer aggression and high patient mortality (Shimosato et al., 1980; Maeshima et al., 2002; Willis and Borok, 2007; Levental et al., 2009; Egeblad et al., 2010). Elevating tumor cell contractility (tension) or stiffening the tumor ECM through increased deposition, remodeling, and cross-linking of type 1 fibrillar collagen promotes the malignant transformation and aggressiveness of a tumor tissue in culture and in vivo (Paszek et al., 2005; Levental et al., 2009; Kraning-Rush et al., 2012; Acerbi et al., 2015; Laklai et al., 2016). Moreover, reducing cell tension or interstitial pressure or preventing matrix stiffening by inhibiting collagen cross-linking decreases tumor incidence and aggression and can improve treatment efficacy (Paszek et al., 2005; Levental et al., 2009; Samuel et al., 2011; Provenzano et al., 2012; Chauhan et al., 2013; Miller et al., 2015). Cell tension and ECM stiffness promote focal adhesion assembly, and β1-integrin ligation and signaling is necessary for tension-dependent malignant transformation and tumor metastasis (Paszek et al., 2005; Levental et al., 2009; Samuel et al., 2011; Pickup et al., 2013; Kaushik et al., 2016). What remains unclear is whether tissue mechanics promote malignancy and tumor aggression by engaging specific β1-integrin heterodimers, and if so, why? Indeed, while thick perpendicular collagen fibers associate with invasive cancers (Provenzano et al., 2006; Levental et al., 2009; Conklin et al., 2011; Carey et al., 2012), and a stiffened collagen ECM can promote malignant transformation in culture (Paszek and Weaver, 2004; Levental et al., 2009; Miroshnikova et al., 2011; Cassereau et al., 2015), the major collagen receptor, α2β1 integrin, inhibits rather than promotes malignancy in vivo (Zutter et al., 1995; Ramirez et al., 2011).
FN is frequently elevated in primary and metastatic tumors (Nam et al., 2010), and the premetastatic niche contains abundant FN (Kaplan et al., 2005). Moreover, expression of the major FN receptor α5β1 integrin is increased in many solid primary tumors, and the levels of either FN or α5β1 integrin independently correlate with poor prognosis in cancer patients (Yao et al., 2007; Dingemans et al., 2010; Nam et al., 2010). Functionally, ligated α5β1 integrin promotes tumor cell growth and survival, and blocking FN ligation by soluble tripeptide composed of l-arginine, glycine, and l-aspartic acid (RGD) sensitizes breast tumor spheroids to radiation treatment (Park et al., 2006; Nam et al., 2010; Yao et al., 2016). Importantly, FN can be coassembled with type I collagen (Sevilla et al., 2013), and lysyl oxidase (LOX)-dependent collagen cross-linking requires FN (Kubow et al., 2009; Cox et al., 2013). The binding of FN to cross-linked collagen increases the ability of fibroblasts to unfold FN to reveal cryptic binding sites, including the synergy site (Friedland et al., 2009; Seong et al., 2013), which is uniquely ligated by α5β1 integrin (Garcia et al., 2002). Cells with FN-synergy site–bound α5β1 integrin engage an adhesive bond that enables them to exert higher forces on their ECM (Friedland et al., 2009; Kong et al., 2009; Benito-Jardon et al., 2017). Consistently, tumor cells sorted for higher cell-surface α5β1 integrin are more contractile and migrate faster on a rigid FN ECM (Mierke et al., 2011). This suggests that a stiff, collagen–FN-rich ECM may favor ligation of the FN synergy site by α5β1 integrin to enhance integrin–growth factor receptor signaling that fosters malignancy. Here we used mouse models of mammary cancer, three-dimensional (3D) organotypic mammary epithelial cell (MEC) cultures, and a force reactor with quantitative analysis to explore the relationship between a stiff collagen ECM, FN ligation of the synergy site by α5β1 integrin, and tumor cell invasion.
RESULTS
Mammary tumor invasion colocalizes with collagen and FN fibers that decrease when stromal stiffening and malignancy are inhibited
We first determined whether an association exists between FN and ECM collagen stiffness–dependent mammary tumor invasion in vivo. We looked for colocalization of collagen and FN and tumor cell invasion in the mammary glands of late-stage FVB PyMT tumors that atomic force microscopy indentation previously revealed was significantly stiffer than that of a normal mammary gland (normal gland: 0.4 kPa, PyMT tumor: 3 kPa) (Lopez et al., 2011; Pickup et al., 2013). Confocal images of PyMT mammary tumor tissue immunostained for FN (Figure 1A, green) and imaged by second harmonic generation (Figure 1A, light blue) showed that a high percentage of invading tumor cells (red) could be observed on thick collagen fibers that costained with FN (Figure 1A, quantified in Figure 1B). To implicate FN in tension-dependent tumor invasion, we next analyzed mammary tissue from MMTV-Neu mice treated with (LOX-i) and without (control) the small-molecule inhibitor β-aminopropionitrile (BAPN) that inhibits LOX activity and that we previously showed prevents collagen cross-linking and ECM stiffening and reduces tumor progression (Siegel, 1974; Kagan, 2000; Kirschmann et al., 2002; Levental et al., 2009). Confocal imaging of mammary tumors from the BAPN-treated mice confirmed that the tumors had lower cell tension compared with their nontreated controls, as revealed by decreased immunostaining for myosin activity (Figure 1C, “pMLC,” green, quantified in Figure 1D), reflecting the decreased ECM stiffness of the tissue (PyMT tumor: 3 kPa; PyMT tumor + BAPN: 0.1 kPa) (Levental et al., 2009; Lopez et al., 2011). Immunofluorescence imaging analysis further indicated that the BAPN-inhibited tumors had reduced integrin mechanosignaling (Dumbauld et al., 2010), as indicated by lower levels of FAK phosphorylation at residue 397 (Figure 1C, “p397FAK,” red, quantified in Figure 1D). Intriguingly, immunostaining showed that the BAPN-treated mice also had lower α2 integrin (Figure 1C, “Integrin α2,” red) and α5 integrin (Figure 1C, “Integrin α5,” red), as well as reduced tissue FN (Figure 1C, “FN,” green, quantified in Figure 1D). These findings provide evidence of a plausible relationship among FN, collagen, and stiffness-dependent tumor cell invasion
FIGURE 1:

Mammary tumor invasion colocalizes with collagen and FN fibers that decrease when stromal stiffening and malignancy are inhibited. (A) Zoomed-in second harmonic generation images of collagen (blue), FN (green), and nuclei (red), as well as an overlay in the 7-mo-old PyMT tumors at the invasive front (left four images) evident from the zoomed-out overlay image (rightmost image). Scale bar: 20 μm. (B) Quantification of cells migrating along FN-coated vs. non–FN-coated collagen fibers. (C) Confocal immunofluorescence images of mammary tissue stained for p397FAK, pMLC, FN, and α2 integrin, αv integrin, and α5 integrin in tissue excised from 7-mo-old control (Her2/Neu) or LOX inhibitor–treated (+LOX-i) Her2/Neu transgenic mice with quantification of fluorescence intensity in D. Scale bar: 10 μm. Results are the mean ± SEM of three separate experiments. For animal studies, three animals were used per condition with three to five images per animal analyzed for quantification. *, p < 0.05; ***, p < 0.001.
A link between the malignant phenotype of mammary tumors and expression of FN and its integrin receptors
To explore the possibility of a relationship between FN and the malignant phenotype of MECs, we conducted reconstituted basement membrane (rBM) mammosphere assays using the immortalized MEC tumor progression series HMT3522 (Weaver et al., 1996, 1997). As previously reported, the nonmalignant S1 MECs in this tumor-progression series formed growth-arrested polarized acini when grown within a 3D rBM for 2 wk (Weaver et al., 1997; Wang et al., 1998; Rizki et al., 2008). The polarized acini phenotype was indicated by apical-lateral cortical actin (phalloidin, green, Figure 2A), cell–cell-localized β-catenin (β-catenin, green, Figure 2A, left) and basally localized β4 integrin (Figure 2A, left). The S1 MECs also assembled an endogenous basement membrane, as indicated by basally deposited laminin 5 (Laminin V) (Figure 2A, left). By comparison, the malignant T4-2 MECs formed continuously growing (unpublished data), tumor cell colonies, lacking apical-basal polarity and stable cell–cell adhesions (Figure 2A, middle). Flow-activated cell sorting (FACs) revealed that the HMT-3522 T4-2 cells in this series express significantly higher plasma membrane levels of the FN integrin receptors αv and α5 and β1 (Figure 2B), whose levels were confirmed by immunoblot analysis of protein isolated from 3D rBM colonies (Figure 2C). Organotypic rBM cultures also revealed that only the tumor colonies deposited FN in the 3D rBM assay (Figure 2A, middle, “Fibronectin”). Importantly, inhibiting β1-integrin ligand binding, using the ligand function–blocking antibody (AIIB2; Weaver et al., 1997) or by pharmacologically inhibiting the activity of the epidermal growth factor receptor (EGFR) by treating the cultures with tyrphostin (Wang et al., 1998), reverted the invasive colony phenotype of the T4-2 MECs toward that of the differentiated nonmalignant S1 MECs (Weaver et al., 1997; Weigelt et al., 2014). Thus the reverted T4-2 MECs formed growth-arrested (unpublished data), apically-basally polarized acini after 2 wk of growth within a 3D rBM (Figure 2A, right). Confocal images of immunofluorescently stained cultures showed that the reverted tumor colonies had apical-lateral cortical actin (Figure 2A, right, “F-actin,” green) and cell–cell-localized β-catenin (Figure 2A, right, “β-catenin,” green). Immunostaining also revealed that the reverted tumor colonies had basally localized β4 integrin (Figure 2A, right, “β4 integrin,” green) and assembled an endogenous basement membrane, as revealed by basally deposited laminin 5 (Figure 2A, right, “Laminin V,” green). Interestingly, the reverted tumor colonies no longer secreted FN into the 3D rBM (Figure 2A, right, “Fibronectin,” green) and an immunoblot of total colony protein showed a dramatic reduction of integrin α5, α2, and β1 in the reverted acini (Figure 2C). These findings establish a causal association between FN and its integrin receptors α5, αv, and β1 and expression of the malignant phenotype of mammary tissue–like structures in 3D rBM culture.
FIGURE 2:

Link between the malignant phenotype of mammary tumors and expression of FN and its integrin receptors. (A) Phase-contrast and confocal immunofluorescence images of Ki-67 (insert), phalloidin (F-actin), β-catenin, β4 integrin, laminin-5, and FN-stained colonies of nonmalignant (S-1), malignant (T4-2), and phenotypically reverted (T4 Rvt) HMT-3522 human MECs grown within an rBM for 2 wk. Scale bar: 10 μm. (B) Bar graphs of FACS analysis of membrane-localized integrins in S-1 compared with T4-2 MECs. (C) Representative immunoblot image of α5, αv, and β1 integrin in lysates from S-1, T4-2, and T4 Rvt 3D rBM colonies shown in A with corresponding quantification of signal intensity normalized to E-cadherin loading control. Fifty acini were analyzed in three separate experiments. ***, p < 0.001.
FN-ligated α5β1 integrin regulates the malignant phenotype of MECs in vitro and in vivo
We next asked whether FN ligation by either of its integrin receptors, α5 or αv, influenced the malignant phenotype of MEC HMT-3522 tumor cells. We treated the HMT-3522 T4-2 MECs, which secrete FN (see Figure 2A), with either α5- or αv-integrin function–blocking antibodies and assayed for changes in cell growth and colony morphology after 2 wk within rBM compared with the effect of blocking the activity of α2, α3, or β1 integrin. Blocking ligand binding to αv (Figure 3A, second column), α2 (Figure 3A, third column), or α3 integrin (unpublished data) had little to no effect on the growth and morphological behavior of the T4-2 MECs in a 3D rBM. However, inhibiting α5 integrin repressed the malignant phenotype of the T4-2 MECs in rBM (Figure 3A, fourth column), similar to what was observed following inhibition of β1-integrin ligand binding (Figure 3A, fifth column; see also Figure 2A). After 2 wk of culture within rBM the T4-2 MECs treated with either immunoglobulin G (IgG) isotype–matched control or inhibitory antibodies to α2 or αv integrin formed continuously growing, large, disorganized, and invasive colonies in rBM, as indicated by disorganized β-catenin (Figure 3A, top), α6 integrin (Figure 3A, middle), and actin (Actin; Figure 3A, bottom). By contrast, the T4-2 MECs treated with function-blocking antibodies against either α5 or β1 integrin assembled morphologically reverted colonies in rBM that were reminiscent of differentiated nonmalignant S1 mammary acini (Figure 2A). T4-2 MECs in which α5β1 integrins were inhibited assembled growth-arrested (unpublished data), polarized structures as indicated by cell–cell-localized β-catenin (Figure 3A; top panel), basal α6 integrin (Figure 3A, middle), and apical-lateral cortical actin (Figure 3A, bottom). The reverted T4-2 MECs also formed acini that were at least 60–70% smaller than the nonreverted colonies (Figure 3B). Moreover, preventing α5- or β1-integrin ligand binding significantly impaired the anchorage-independent growth and survival of the T4-2 MECs in soft agar (Figure 3C). These data suggest that FN-ligated α5β1 integrin regulates expression of the malignant phenotype of the T4-2 MEC in a 3D rBM.
FIGURE 3:

FN-ligated α5β1 integrin regulates the malignant phenotype of MECs in vitro and in vivo. (A) Confocal immunofluorescence images of β-catenin, α6 integrin, and actin (Phalloidin) staining of malignant (T4-2) MEC colonies grown for 2 wk in rBM in the presence of a function-blocking antibody (mAb) to αv, α2, α5, or β1 or an IgG isotype–matched control mAb. Scale bar: 30 μm. (B) Bar graph showing relative size of the T4-2 colonies shown in A. (C) Bar graph showing percentage of tumor colonies formed in soft agar (40+ microns) following treatment with function-blocking mAbs to αv, α2, α5, or β1 integrin or an IgG isotype–matched control mAbs. (D) Mean fluorescence intensity of integrin α2 and α5 expression in S-1 cells overexpressing Itga2. (E) Mean fluorescence intensity of integrin α2 and α5 expression in S-1 cells overexpressing Itga5. (F) Confocal immunofluorescence images of β-catenin, β4 integrin, and collagen IV staining of colonies of nonmalignant (S-1) vector (Ctrl) MECs and MECs expressing elevated α2 or α5 integrin grown in rBM with or without the addition of FN (+FN) for 2 wk. Scale bar: 10 μm. (G) Bar graph showing relative size of S-1 MEC colonies shown in D. (H) Bar graph showing percent Ki-67–positive S-1 MEC colonies shown in D. (I) Bright-field images of low (top) and high (bottom) magnification of H&E sections of tissue excised 2 mo following injection of malignant T4-2 MECs with IgG or a function-blocking antibody to α5 integrin and nonmalignant S-1 MECs expressing empty vector or an α5 integrin. Scale bar: 10 μm. Red arrows indicate areas of necrosis observed upon blocking of integrin a5. Red arrows indicate hyperplastic or dysplastic cellular structures. (J) Table summarizing tumor score and histological features. Scale bar: 10 μm. **, p < 0.01; ***, p < 0.001.
We next investigated whether FN-ligated α5β1 integrin could promote the malignant phenotype of nonmalignant MECs. HMT-3522 S1 nonmalignant MECs (which express negligible α5 integrin) were engineered to express either a tetracycline (Tet)-regulated enhanced green fluorescent protein (eGFP)-tagged α5 integrin or an eGFP-tagged α2 integrin. Immunoblot and FACS analysis confirmed a Tet-modulated increase in total and cell-surface α5 integrin and α2 integrin in two independent nonmalignant HMT-3522 S1 MEC lines (Figure 3, D and E) and two independent nonmalignant MCF10A MEC lines (Damiano et al., 2014). Neither ectopic expression of α5 integrin nor α2 integrin significantly altered plasma membrane levels of α1, α2, α3, α6, β1, or β4 integrin in either the HMT-3522 S1 or the MCF10A MECs (unpublished data). Furthermore, MCF10A MECs expressing elevated levels of α5 integrin showed greater adhesion to FN, and those engineered to express high α2 integrin adhered more to a type I collagen ECM (Damiano et al., 2014). The nonmalignant S1 MECs expressing either more α2 integrin or α5 integrin assembled growth-arrested (Figure 3F), polarized acini with a cleared central lumen after 10–14 d of growth within rBM. This “normal” phenotype was indicated by immunofluorescence staining, which revealed cell–cell-localized β-catenin (Figure 3F, top), basally localized β4 integrin (Figure 3F, middle), and basally deposited collagen IV (Figure 3F, bottom). Importantly, when the α5 integrin was ligated through addition of FN to the rBM, the HMT-3522 S1 MECs expressing ectopic α5 integrin formed continuously growing colonies (Figure 3, G and H) that by 2 wk were almost double the size of the colonies formed by the non–FN-ligated α5 integrin–expressing S1 MECs (Figure 3G). The FN-ligated α5 integrin–expressing S1 MEC colonies were unable to clear their lumens (Figure 3F), showed diffuse cell–cell-localized β-catenin (Figure 3D, top) and disorganized β4 integrin (Figure 3F, middle), and failed to assemble an endogenous collagen IV basement membrane (Figure 3F, bottom). In the presence of exogenous FN, nonmalignant S1 MECs expressing elevated cell-surface α5 integrin formed colonies that were 30-40% larger than the S1 MECs expressing higher α2 integrin, despite the availability of abundant collagen (Figure 3G). We observed a similar tumor-promoting phenotype following FN ligation in the nonmalignant immortalized MCF10A MECs ectopically engineered to express higher α5-integrin levels (Figure 4A) (Damiano et al., 2014) with little to no effect on tissue phenotype following collagen ligation of MCF10A MECs expressing higher α2-integrin levels (Figure 4A). These findings demonstrate that FN-ligated α5β1 integrin can promote the malignant phenotype of nontransformed MEC tissue-like structures in vitro, reminiscent of prior studies implicating FN ligation on acinar morphogenesis and growth of MCF10A (Williams et al., 2008).
FIGURE 4:

FN-ligated α5β1 integrin increases cell tension to promote mammary malignancy. (A) Phase-contrast and confocal immunofluorescence images of β1 integrin, phalloidin (F-actin), laminin V, Ki-67 and activated caspase 3 (Caspase-3), and DAPI (nuclei)-stained colonies of nonmalignant (MCF10A) human MECs expressing empty vector or elevated α2 or α5 integrin treated with or without a ROCK inhibitor and malignant cells (HMT-3522 T4-2) incubated with nonspecific IgG or function-blocking antibodies to α2 or α5 integrin or treated with or without a ROCK inhibitor grown within a collagen gel with added FN for 2 wk. Scale bar: 8 μm. (B) Bar graph showing relative size of the nonmalignant colonies shown in A. (C) Bar graph showing relative size of the malignant colonies shown in A. (D) Immunofluorescence confocal images (bottom) and phase-contrast images (top) of 3D cultures of α2/Col 1 and α5/FN nonmalignant MECs stained for the ROCK target p696MYPT (top). Scale bar: 50 μm. (E) Second harmonic generation images of nonmalignant (MCF10A) human mammary epithelial day 20 acini (green) expressing empty vector or elevated α5 integrin with or without FN embedded in collagen (blue) and installed into a 3D tension bioreactor system and subjected either to 0% (400 Pa) or 10% (4000 Pa) stretch, as described previously (Cassereau et al., 2015). Arrows in E indicate invasive cells and colony circularity (left) and area (right) are quantified in F. Results are the mean ± SEM of three to five separate experiments. **, p < 0.01; ***, p < 0.001. (Ten to 15 acini were analyzed per condition in three separate experiments.)
To further implicate FN ligation of α5β1 integrin in the malignant behavior of mammary tissues, we manipulated α5-integrin expression and function in nonmalignant and malignant MECs and assayed for effects on their malignant behavior in vivo. We inoculated nonmalignant HMT-3522 S1 MECs expressing eGFP (nonmalignant control) or α5 integrin (+α5 integrin), and HMT-3522 T4-2 malignant MECs treated with either an α5-integrin function–blocking monoclonal antibody (α5-integrin inhibited) or an isotype IgG control antibody (tumor control), into the rear flanks of BALB/c nu/nu mice. Two months following MEC inoculation, the mice were killed and the tumor phenotype was assessed. The control tumors had formed large, actively growing, invasive, and angiogenic tumor masses, as indicated by elevated proliferating cell nuclear antigen (PCNA) immunostaining, negligible activated caspase 3 immunostaining, a clearly visible vasculature (confirmed by CD34 immunostaining), and histopathological analysis that confirmed the presence of invasive cell masses (Figure 3, I and J, green arrows indicate proliferating cells, and Supplemental Figure 1C, left panels). By comparison, T4-2 cells treated with the α5-integrin function–blocking antibody formed only small, nonproliferating tumor colonies that immunostained positively for the apoptosis marker–activated caspase 3. The tumors formed by the cells treated with the α5-integrin function–blocking antibody also lacked a vasculature (Supplemental Figure 1D, top left panels), as revealed by negligible CD34 immunostaining (Supplemental Figure 1D, bottom left panels), and they showed histopathological evidence of cystic degeneration and necrosis (Figure 3, I and J, red arrows indicate dying cells, and Supplemental Figure 1C, right panels). As expected, the majority of the injected nonmalignant S1 MECs failed to survive (red arrow indicate dying cells), and those that did formed ductal-like differentiated tissue-like structures (Figure 3I, zoom, see arrows; Figure 3J). By contrast, those S1 MECs expressing high levels of α5 integrin not only survived, but they grew to form hyperplastic/dysplastic cell masses that apparently induced an angiogenic response, as indicated by a visible vasculature and positive CD34 tissue immunostaining (Figure 3, I and J, green arrows point to proliferating cells, and Supplemental Figure 1D, top and bottom right panels). These in vivo tumor studies suggested that engagement of α5β1 integrin by MECs might stimulate tumor angiogenesis, another key feature attributed to a malignant tissue (Pickup et al., 2014). To directly address this possibility, we grew the nonmalignant S-1 MECs ectopically expressing α5 integrin or α2 integrin within a 3D rBM doped with either type I collagen or FN inside of a tissue culture insert until they formed colonies (10–12 d culture). The tissue culture inserts containing the rBM/FN-embedded MEC colonies were then transferred into a Transwell in which vascular endothelial cells were grown on the bottom to semiconfluence and then overlaid with a layer of type I collagen (Supplemental Figure 1A). The growth and invasion of the vascular endothelial cells into the collagen gel was then monitored over several days, and extent of network formation was quantified (60 d). Consistent with the in vivo findings, we noted that the colonies formed by the nonmalignant MECs expressing FN-ligated α5 integrin, but not collagen-ligated α2 integrin, induced more endothelial network-like structures (Supplemental Figure 1B, top left bar graphs). We next conducted a similar endothelial network assay using the malignant HMT-3522 T4-2 MECs incubated with IgG control antibody or function-blocking antibodies against α2, αv, α5, or β1 integrin. Findings showed that only the T4-2 colonies incubated with the IgG control or the α2- or αv-integrin function–blocking antibodies were able to stimulate a significant amount of vascular network formation (Supplemental Figure 1B) compared with those tumor cells that had been phenotypically reverted by blocking either α5- or β1-integrin function (Supplemental Figure 1B, top right bar graphs). Consistent with these findings, enzyme-linked immunosorbent assay (ELISA) revealed that only the FN-ligated α5β1-integrin MECs expressed abundant vascular endothelial growth factor (VEGF), and that inhibiting α5 or β1 integrin in the malignant T4-2 MECs significantly reduced levels of tumor-secreted VEGF (Supplemental Figure 1B, bottom right and left bar graphs). These findings indicate that FN-ligated α5β1 integrin can regulate the malignant phenotype of mammary tissues both in vitro and in vivo.
FN-ligated α5β1 integrin increases cell tension to promote mammary malignancy
We next determined whether the ability of FN ligation of α5β1 integrin to promote the malignant behavior of mammary tissues depended on cell tension. We treated 3D collagen–FN cultures of the nonmalignant MCF10A MECs engineered to express higher cell-surface α5 integrin or α2 integrin, with either a ROCK inhibitor (Y27632, 10 µM) (Figure 4A) or a myosin inhibitor (blebbistatin; unpublished data). Two weeks following embedment within a 3D collagen–rBM–FN hydrogel, vector control, nonmalignant MCF10A MECs, and the MCF10A MECs ectopically expressing higher α2 integrin assembled growth-arrested (Figure 4A, bottom, “Ki-67”), tissue-like structures (Figure 4A, top, DIC phase-contrast images) that resembled acini. Confocal immunofluorescence images confirmed that both the MCF10A vector control, and α2 integrin–overexpressing MEC acini were polarized. Colony phenotype was indicated by apical-lateral cortical actin (Figure 4A, second row, “β1 integrin,” “F-actin,” and “DAPI”); deposition of an endogenous basally localized, basement membrane (Figure 4A, third row, “Laminin V”); and lumen clearance, indicated by detectable apoptotic cells in the colony core (Figure 4A, bottom row, activated “Caspase-3” and “DAPI”). By contrast, the MCF10A MECs ectopically expressing higher α5 integrin formed continuously growing, disorganized colonies lacking a central lumen (Figure 4A, third column) that were significantly larger than those formed by either the vector control or α2 integrin–overexpressing MECs (Figure 4B). Critically, treatment of the MCF10A MEC α5 integrin–overexpressing MEC colonies with ROCK inhibitor phenotypically reverted the colonies so they resembled growth-arrested, differentiated acini, with a size and organization that resembled the vector control MCF10A MEC acini (Figure 4A, compare fourth to first column, and Figure 4B). These findings suggest FN ligation of α5β1 integrin promotes a tumor phenotype in MECs by increasing cell tension. Consistent with this notion, immunostaining revealed that the FN-ligated MECs expressing higher α5 integrin, but not the collagen-ligated MECs expressing higher α2 integrin, stained positively for activity of the ROCK target, myosin phosphatase binding protein (MyPT) (p696MyPT) (Figure 4D). Similarly, treating the malignant HMT-3522 T4-2 MECs that overexpressed α5β1 integrin (Figure 2), which we previously showed had elevated RhoGTPase and ROCK activity (Paszek et al., 2005), with the same ROCK inhibitor, also reverted their tissue phenotype toward that resembling the nonmalignant MCF10A MECs or the T4-2 MECs incubated with an α5-integrin function–blocking antibody (Figure 4A, compare fifth through seventh columns with eighth column). The T4-2 MECs treated with the ROCK inhibitor assembled smaller (Figure 4C), growth-arrested (Figure 4D, bottom row), polarized acini (Figure 4A) that were similar to those generated in the presence of a function-blocking antibody to α5 integrin (Figure 4A, right). In marked contrast, the T4-2 MECs generated by the IgG isotype–treated control tumors or those in which α2 integrin was inhibited formed larger (Figure 4C), proliferative, invasive, and disorganized colonies (Figure 4A, right). These findings suggest that the ability of FN ligation of α5β1 integrin to promote the malignant behavior of mammary tissues, in vitro at least, depends on cell tension.
We next confirmed whether links between tissue tension and FN-ligated α5β1 integrin and expression of the malignant phenotype of mammary tissues existed by manipulating ECM stiffness to increase cell tension. Consistent with an enhancement of stiffness-induced induction of cell tension, we observed that MECs ectopically expressing α5 integrin, as opposed to those ectopically expressing high αvβ3 integrin, demonstrated strong directional migration (Isenberg et al., 2009; Hartman et al., 2016) in response to a gradient of FN stiffness, or those ectopically expressing α2 integrin in response to a gradient of collagen I stiffness (Supplemental Figure 3C). The impact of ECM stiffness and FN ligation of α5β1 integrin was also illustrated by a set of invasion-monitoring studies that employed the 3D tension bioreactor with and without added FN. The 3D tension bioreactor permits the rapid stiffening of the ECM from 400 to 4000 Pa through the application of a uniaxial stretch (Cassereau et al., 2015). Findings revealed that nonmalignant MCF10A control preassembled MEC colonies embedded within a nonstiffened 3D collagen–rBM remained quiescent and noninvasive for 48 h, as indicated by retention of colony circularity (Figure 4F). Upon uniaxial stretch and collagen stiffening, the nonmalignant MECs still did not invade, although they did begin to project membrane protrusions (Figure 4, E and F). MEC colonies ectopically expressing higher α5 integrin exhibited a similar phenotype to that demonstrated by the vector control and α2 integrin–overexpressing MECs when embedded within the relaxed and stretched, collagenous 3D tension bioreactor, nevertheless their protrusion activity was enhanced (Figure 4, E and F). However, and importantly, addition of FN to the 3D tension bioreactor significantly enhanced the protrusive behavior of the control mammary colonies and induced the rapid and significant invasion of the MEC colonies ectopically expressing the higher cell-surface α5 integrin (Figure 4E, compare with and without FN, see white arrows). These findings imply that FN ligation of α5β1 integrin could promote the malignant behavior of mammary tissues by enhancing tumor cell tension.
FN-ligated α5β1 integrin and not collagen-ligated α2β1 integrin increases MEC tension
We next asked whether FN-ligated α5β1 integrin induced more actomyosin tension in the nonmalignant MECs and whether this modified their integrin adhesions (Regent et al., 2011). Collagen–FN gel contraction assays revealed that both the nonmalignant HMT-3522 and MCF10A MECs ectopically expressing higher α5 integrin contracted 3D collagen–FN gels better than the MECs engineered to express higher α2 integrin (Supplemental Figure 2). These findings were verified by traction force microscopy (TFM) which directly demonstrated that the nonmalignant MECs ectopically expressing higher α5 integrin exerted higher maximum cell traction force on an FN ECM, as compared with nonmalignant MECs ectopically expressing α2 integrin plated on collagen I ECM (Figure 5, A and B). The nonmalignant MECs expressing more α5 integrin that were plated on FN also showed significantly higher levels of nuclear yes-associated protein (YAP), reflecting enhanced activity of the mechano-activated Hippo pathway (Aragona et al., 2013), as compared with the nonmalignant MECs expressing higher α2 integrin plated on collagen I (Figure 5C, quantified in Figure 5D). Indeed, the FN-ligated nonmalignant MECs expressing higher α5β1 integrin assembled more and apparently larger peripheral integrin adhesions (Figure 5E) that stained strongly with p397FAK (Figure 5E, quantified in Figure 5F). Immunostaining also showed that MECs with FN-ligated α5β1 integrin recruited more of the force-activated molecules vinculin (Figure 5G, quantified in Figure 5H) and zyxin (Figure 5I, quantified in Figure 5J) to their ECM adhesions, as compared with MECs with collagen 1-ligated α2β1 integrin (Figure 5, E–J). These findings show that FN ligation of α5β1 integrin induces a greater amount of actomyosin tension in MECs at their ECM adhesions. The higher FN-ligated α5β1-integrin adhesion force in turn permits the assembly of tension-regulated scaffolding molecules and enhances mechano-signaling through YAP.
FIGURE 5:

FN-ligated α5β1 integrin and not collagen-ligated α2β1 increases MEC cell tension. (A) Force maps of α2/Col 1 and α5/FN nonmalignant MECs and bar graphs (B) showing maximum traction generated by MECs at the cell edge. (C) Immunofluorescence images of α2/Col 1 and α5/FN nonmalignant MECs stained for YAP (arrow indicating nuclear localization of YAP). Scale bar: 15 μm. (D) Bar graphs quantifying percent nuclear YAP in MECs shown in H. Results are the mean ± SEM of three separate experiments with 10–15 acini analyzed per condition. (E) Immunofluorescence confocal images of nonmalignant MECs expressing either exogenous α2 integrin plated on type I collagen (α2/Col 1) or exogenous α5 integrin plated on FN (α5/FN) stained for p397FAK (green) or with phalloidin (F-actin; red). Scale bar: 10 μm. (F) Bar graph quantifying size of peripheral adhesions shown in E. (G) Immunofluorescence confocal images of α2/Col 1 and α5/FN nonmalignant MECs stained for vinculin (red). Scale bar: 3 μm. (H) Bar graphs showing quantification of relative amount of vinculin recruited to focal adhesions in MECs shown in G. Quantification is relative to α5-overexpressing cells on FN-coated matrices. (I) Immunofluorescence confocal images of α2/Col 1 and α5/FN nonmalignant MECs stained for zyxin (green). Scale bar: 3 μm. (J) Bar graphs showing quantification of relative zyxin recruited to focal adhesions in MECs shown in I. Quantification is relative to α5-overexpressing cells on FN-coated matrices. Arrows in C, E, G and I indicate peripheral localization of adhesion molecules. Results are the mean ± SEM of three separate experiments with 10–15 cells analyzed per each condition for each experiment. ***, p < 0.001. Scale bars: 50 μm.
FN-ligated α5β1 integrin enhances mechanotransduction in MECs
Unlike αv integrin, α5 integrin binds to both the proline-histidine-serine-arginine-asparagine (PHSRN) synergy and RGD sites of FN, and only FN-bound α5β1 integrin, but not αvβ3 integrin, exhibits a unique adhesive bond phenotype in which ligand binding is reinforced in response to force (Friedland et al., 2009; Cao et al., 2012; Seong et al., 2013). Consistently, shear-force adhesion studies showed that nonmalignant MECs engineered to ectopically express high levels of α5 integrin attached to either full-length (unpublished data) or a recombinant 9–10 domain FN, bound with much greater strength as compared with MECs plated on recombinant 9–10 domain FN in which the PHSRN domain was mutated (Figure 6A and Supplemental Figure 3B), as previously shown in fibroblasts and CHO cells (Gallant and Garcia, 2007; Friedland et al., 2009). MECs expressing higher α5 integrin bound with at least twice as much strength to a recombinant wild-type 9–10 domain of FN compared with MECs ectopically expressing αvβ3 integrin ligated to the same 9–10 domain of FN. The enhanced ligand binding strength depended on synergy-site binding by α5β1 integrin, because the strength of adhesion decreased significantly when the synergy-site region in the 9–10 domain of FN was mutated. Given these findings and prior work emphasizing differential roles of FN domains during fibrillogenesis (Sechler et al., 1997), we further explored the relevance of FN domains on cell tension generation. We first observed that the size of the integrin adhesions and the amount of p397FAK significantly decreased when the MECs expressing higher α5 integrin were plated on a recombinant 9–10 domain of FN in which the synergy site was mutated (Figure 6B, quantified in Figure 6C). Similarly, the amount of the mechano-activated molecule vinculin that appeared to be recruited to the integrin adhesions was substantially reduced when the MECs were plated on an FN lacking the synergy site (Figure 6D, quantified in Figure 6E) (Dumbauld et al., 2010). Consistently, the vinculin fluorescent resonance energy transfer (FRET) index acquired using a vinculin FRET force sensor showed a significant decrease in tension applied to the vinculin molecule in the MECs plated on the 9–10 domain of FN with the mutant synergy site as compared with the wild-type domain (Figure 6F, quantified in Figure 6G) (Grashoff et al., 2010). The amount of zyxin, which is also force activated (Hirata et al., 2008; Hoffman et al., 2012), that was recruited to actin stress fibers and localized at the integrin adhesions was similarly reduced in the MECs expressing higher α5 integrin when they were plated on the recombinant FN lacking the PHSRN synergy domain (Figure 6, H–K). The importance of the α5β1-integrin adhesive bond in enhancing mechanosignaling in MECs was illustrated by a dramatic reduction in nuclear YAP in the MECs ectopically expressing high α5 integrin plated on the 9–10 domain of FN in which the synergy site was mutated (Figure 6L, quantified in Figure 6M). These findings illustrate the importance of the FN synergy site for the engagement of the α5β1 integrin and the enhancement of MEC tension subsequent to integrin adhesion maturation and increased cellular mechanosignaling.
FIGURE 6:

The α5β1-integrin adhesive bond enhances mechanotransduction in MECs. (A) Bar graphs showing adhesion strength of nonmalignant MECs expressing αv or α5 plated on recombinant FN 9–10 (FN 9–10 WT) and α5 plated on recombinant FN 9–10 with the synergy-site mutated (FN 9–10 Syn). (B) Immunofluorescence confocal images of MECs expressing α5 integrin plated on FN 9–10 WT or FN 9–10 Syn stained for p397FAK (green) and actin with phalloidin (F-actin; red). Scale bar: 10 μm. (C) Bar graph quantifying size of peripheral adhesions shown in B. (D) Immunofluorescence confocal images of MECs expressing α5 integrin plated on FN 9–10 WT or FN 9–10 Syn stained for vinculin. Scale bar: 3 μm. (E) Bar graphs showing quantification of relative amount of vinculin recruited to focal adhesions in MECs shown in D. (F) FRET images of MECs expressing α5 integrin and the vinculin force sensor plated on polyacrylamide gels conjugated with FN 9–10 WT or FN 9–10 Syn. Scale bar: 5 μm. (G) Bar graphs showing quantification of FRET index at focal adhesions in MECs shown in F. (H) Immunofluorescence confocal images of MECs expressing α5 integrin plated on FN 9–10 WT or FN 9–10 Syn stained for zyxin. Scale bar: 3 μm. (I) Bar graphs showing quantification of zyxin recruited to focal adhesions in MECs shown in H. (J) Immunofluorescence confocal images of MECs expressing α5 integrin plated on FN 9–10 WT or FN 9–10 Syn double-stained for zyxin and F-actin (Phalloidin). Scale bar: 3 μm. (K) Bar graphs showing quantification of zyxin colocalization to actin in MECs shown in J. (L) Immunofluorescence images of MECs expressing α5 integrin plated on FN 9–10 WT or FN 9–10 Syn stained for YAP. Scale bar: 15 μm. (M) Bar graphs quantifying percent nuclear YAP in MECs shown in I. For the immunofluorescence and FRET experiments, results are represented with the mean ± SEM of three separate experiments with either 10–15 cells (immunofluorescence analysis) or 25 cells (FRET analysis) analyzed per each condition for each experiment *, p < 0.05; ***, p < 0.001.
α5β1-Integrin engagement by the RGD and synergy site of FN is required for the tension-dependent malignant behavior of MEC organoids
We next investigated the relevance of the α5β1-integrin adhesive bond in the tension-induced malignant behavior of mammary tissues in culture. MECs ectopically expressing high α5 integrin were grown within rBM gels doped with either a recombinant 9–10 domain wild-type FN or recombinant 9–10 domain FN in which the synergy site was mutated and then assayed for colony morphology after 2 wk. Consistent with the importance of synergy-site ligation and activation of the α5β1 integrin, in the absence of the synergy site, FN-ligated MECs formed small, growth-arrested acini in rBM (unpublished data) and failed to activate the hippo pathway, as revealed by lack of nuclear YAP (Figure 7A, right). By comparison, the MECs within rBM doped with a wild-type 9–10 FN grew to form large, disorganized colonies and showed abundant nuclear YAP immunostaining (Figure 7A, left). To directly test the importance of the α5β1-integrin adhesive bond in the tension-dependent malignant behavior of mammary tissues, we studied the impact of the synergy site of FN in permitting stiffness-induced MEC invasion in the 3D tension bioreactor doped with collagen–rBM–FN (see Figure 3) (Cassereau et al., 2015). Mammary colonies expressing high α5 integrin embedded within the compliant (400 Pa) nonstretched tension bioreactor remained quiescent and noninvasive (Figure 7B, left image, quantified in Figure 7C, left and right bar graphs). In response to uniaxial stretch and gel stiffening (4000 Pa), MECs from the colony invaded into the gel (Figure 7B, middle, see white arrows) and grew larger, as indicated by a significant drop in colony circularity (Figure 7C, left bar graph) and an increase in colony size (Figure 7C, right bar graph). However, inhibition of synergy-site binding, through treatment of the colonies with the small molecule ATN-161, prevented MEC invasion in response to ECM stiffness (Figure 7B, right, quantified in Figure 7C, left bar graph). The findings indicate that FN-ligated α5β1 integrin, by virtue of its unique ability to bind to the FN synergy site, enhances cell tension to induce mechanosignaling that promotes the malignant behavior of MECs.
FIGURE 7:

The α5β1-integrin adhesive bond is required for the tension-dependent malignant behavior of MEC organoids. (A) Immunofluorescence confocal images (bottom) and phase-contrast images (top) of 3D cultures of α5/FN9–10 WT and α5/FN9–10 Syn. Site mutant nonmalignant MECs stained nuclear YAP. (B) Second harmonic generation images of nonmalignant (MCF10A) human mammary epithelial day 20 acini (green), expressing empty vector or elevated α5 integrin with FN and with or without FN synergy-site inhibitor, embedded in collagen (blue) and installed into a 3D tension bioreactor system and subjected either to 0% (400 Pa) or 10% (4000 Pa) stretch. Arrows in B indicate invasive cells, and colony circularity (left) and area (right) are quantified in C. Results are the mean ± SEM of three separate experiments with at least 10 acini analyzed per each condition each time. ***, p < 0.001,one-way ANOVA. Scale bars: 50 μm.
α5β1-integrin ligation by the RGD and synergy site of FN enhances MEC tension and promotes malignancy by amplifying growth factor–stimulated PI3K signaling
We next investigated how elevated cell tension mediated by ligation of α5β1 integrin by FN promoted the malignant behavior of MECs. EGFR signaling enhances MEC growth and survival by activating PI3K and extracellular signal–regulated kinase (ERK) (Zahir et al., 2003; Yuan and Cantley, 2008), and oncogenic transformation requires PI3K and ERK signaling (Turner and Grose, 2010; Kandoth et al., 2013; Stephen et al., 2014). Consistently, we observed greatly reduced p202/204ERK and p473Akt levels in the mammary epithelium of Her2/Neu mice in which collagen cross-linking and ECM stiffening had been prevented by inhibiting LOX activity (Figure 8A) (Levental et al., 2009), and this reduction in PI3K and ERK signaling correlated with lower p397FAK, pMLC, α5-integrin, and FN expression in the tissues (Figure 1B). Culture assays also showed that FN ligation of α5β1 integrin increased the levels and duration of EGFR-stimulated p473Akt and p202/204ERK activity in nonmalignant and malignant MECs (Supplemental Figure 4). Thus FN-ligated, nonmalignant HMT-3522 S1 MECs expressing α5β1 integrin showed a 300% increase in p473Akt and a 200% increase in p202/204ERK 90 min after EGF treatment compared with control cells expressing empty vector (Figure 8B, left bar graphs). Similarly, treating malignant HMT-3522 T4-2 MECs plated on FN with a function-blocking antibody to α5 integrin significantly decreased p473Akt and p202/204ERK activation in response to EGF (Figure 8B, right bar graphs). Nonmalignant S1 MECs cultured in 3D rBM gels doped with FN formed disorganized, invasive, and nonpolarized colonies, yet inhibition of PI3K using a small-molecule inhibitor (LY294002, 5 μM) (Figure 8C, left bar graphs) or MAP kinase inhibitor (PD98059, 20 μM) (unpublished data) reverted their disorganized phenotype toward differentiated nonmalignant MEC acini (Figure 8C, left bar graphs). Inhibition of either PI3K (Figure 8D, left bar graphs) or ERK activity (unpublished data) also significantly decreased the colony size of the HMT-3522 nonmalignant S1 MECs with FN-ligated α5β1 integrin. Treatment of malignant T4-2 MECs with the same PI3K (Figure 8C, right bar graphs) or ERK inhibitor (unpublished data) also reverted the malignant phenotype to that exhibited by noninvasive, growth-arrested, polarized, nonmalignant MEC acini and significantly decreased their colony size (Figure 8D, right bar graphs). Importantly, the α5β1 integrin–mediated increase in PI3K signaling required ligation of the synergy site of FN, because nonmalignant α5β1 integrin–expressing MCF10A MECs showed a profound and sustained increase in EGF-stimulated Akt and ERK activation only when the cells were able to ligate a recombinant FN with an intact synergy site (Figure 8E).
FIGURE 8:

The FN synergy site–ligated α5β1 integrin increases MEC tension and promotes malignancy by amplifying PI3K signaling. (A) Confocal immunofluorescence images of pAkt substrate, pThr202/pTyr204ERK staining, and DAPI-stained nuclei of mammary tissue excised from 7-mo-old Her2/Neu transgenic mice (three mice/condition, three to five images per mouse used for analysis) treated with or without lysyl oxidase inhibitor (Lox). Scale bar: 10 μm. (B) Bar graphs showing level of p473Akt and pThr202/pTyr204ERK normalized to total cellular Akt and ERK in control or α5 integrin–expressing nonmalignant HMT-3522 S-1 MECs and in T4-2 malignant MECs treated with either nonspecific IgG or a function-blocking antibody to α5 integrin at 90 min following EGF treatment. (C) Confocal immunofluorescence images of β4 integrin and β-catenin in colonies of control or α5 integrin–expressing nonmalignant HMT-3522 S-1 MECs and in T4-2 colonies treated with and without the PI3K inhibitor LY294002. Scale bar: 10 μm. (D) Bar graphs showing size of nonmalignant and malignant MEC colonies in C. (E) Line graphs showing time course of EGF-stimulated pThr202/pTyr204ERK (top) and p473Akt (bottom) levels normalized to total ERK and Akt in nonmalignant MCF10A MECs expressing elevated α5 integrin plated on wild-type (WT) or synergy site–mutated (Syn) FN. (F) Confocal images of nonmalignant MEC cells expressing elevated α5 integrin, a probe for PIP3 activity (mKO2-PH-Grp1), and the focal adhesion protein vinculin plated on recombinant 9–10 FN with (WT) or without (Syn) site mutated. Scale bar: 3 μm. Line graphs showing time course of quantification of EGF-stimulated PIP3 recruited to focal adhesions in nonmalignant MECs plated on wild-type (WT) or synergy site–mutated (Syn) FN. Measurements of all pixels in adhesions were averaged over the whole cell. Results are the mean ± SEM of three separate experiments for all studies; at least 10 cells/condition were used for each experimental repeat or 15–20 colonies/condition for 3D culture studies. ***, p < 0.001.
A force-stabilized vinculin-talin-actin-zyxin scaffolding complex facilitates PI3K-mediated conversion of phosphatidylinositol (3,4)-bisphosphate (PIP2) into phosphatidylinositol (3,4,5)-triphosphate (PIP3) (Rubashkin et al., 2014). We therefore asked whether the force-mediated stabilization of the vinculin-talin-actin-zyxin scaffolding complex by FN ligation of α5β1 integrin required engagement of the FN synergy site to enrich PIP3 at integrin adhesions. Consistently, a greater amount of mKO2-PH-Grp1 (a PIP3 localization reporter; Martin-Belmonte et al., 2007) could be detected at the focal adhesions (indicated by vinculin mEmerald) following EGF stimulation in the nonmalignant MCF10A MECs overexpressing the α5 integrin that were plated on recombinant 9–10 FN domains, but only when the synergy site was intact (Figure 8F). These findings demonstrate that the engagement of an FN-ligated α5β1 integrin increases cell tension to stabilize a vinculin-talin-actin-zyxin scaffolding complex that is able to promote the malignant behavior of MECs by amplifying PI3K signaling in response to EGFR stimulation.
DISCUSSION
Here we provide evidence that the FN associated with stiffened type I collagen fibers in the tumor microenvironment could foster malignant transformation and tumor progression by promoting the engagement of α5β1 integrin to increase tumor cell mechanosignaling. The findings are consistent with previous data supporting the role of FN in mediating the cellular response to extracellular mechanical cues (Carraher et al., 2013). Our data suggest that the activation of FN-ligated α5β1 integrin provides the mechanical strength to transmit higher adhesion forces that promote the recruitment of vinculin, zyxin, and other focal adhesion proteins, which ultimately drive PIP nucleation at the plasma membrane to enhance growth factor–dependent PI3K signaling in the invading tumor cells. The data present a plausible explanation as to why a stiffened collagen ECM potentiates PI3K activation to promote tumor progression and provides insight into the relationship between the consistent up-regulation of α5β1 and FN in many tumors as well as its association with metastasis. Using two-dimensional (2D) and 3D culture assays, a 3D tension bioreactor, and mouse models, we showed that FN-ligated α5β1 integrin, by virtue of its ability to enhance cell tension through its synergy site, can promote expression of the malignant phenotype of MECs in vitro and in vivo. Our findings provide one perspective for why α5β1 integrin and its ligand FN are so frequently elevated in many solid tumors, where interstitial pressure and tension are also high, and in contractile primary and metastatic cancer cell lines and tumor cells, which have high Rho and ROCK activity (Maschler et al., 2005; Yao et al., 2007; Craig et al., 2009; Dingemans et al., 2010; Nam et al., 2010; Mierke et al., 2011; Yu et al., 2012). Our data are also consistent with prior results showing that cancer cell lines expressing abundant FN, when sorted for high membrane α5-integrin levels, migrate faster and contract collagenous matrices to a greater extent (Mierke et al., 2011), and studies showing that blocking the activity of RGD-binding receptors is critical for expression of the malignant phenotype of cultured breast cancer cells (Park et al., 2006; Nam et al., 2010). Here we not only identified α5β1 integrin as the key RGD receptor, but we also demonstrated that the ability of α5β1 integrin to promote the malignant phenotype of MECs requires binding to both the RGD and synergy site of FN, because this permits its critical adhesive bond activation, which is required to enhance cell tension and integrin mechanosignaling (Figures 3 and 4). Importantly, prior data showed that unfolding of FN reveals the otherwise hidden synergy site that permits the engagement α5β1 integrin (Friedland et al., 2009; Seong et al., 2013). Our data expand upon these observations to imply that collagen-mediated ECM stiffening could promote malignancy by fostering α5β1-integrin binding to the FN synergy site along stiff collagen fibrils that then permit the force-dependent exposure of the synergy site on FN molecules (Baneyx et al., 2002; Barker et al., 2005; Martino et al., 2009; Cao et al., 2012; Seo et al., 2015).
Tumors contain abundant quantities of type I collagen, and cross-linked, remodeled type I collagen contributes critically to the tensile strength of a tissue (Levental et al., 2009; Lopez et al., 2011). Nevertheless, we failed to quantify differences in α2 integrin in either Her2/Neu mouse mammary tissue or in the 3D organotypic cultures of malignant MECs in which the tension had been reduced (Figures 1 and 2). Instead, we established a relationship among cell tension, ECM stiffness, and elevated expression of the FN receptor α5β1 integrin in mouse mammary tissue and mammary tumor organoids. We determined that MECs with FN-ligated α5β1 integrin, but not collagen I-ligated α2β1, exerted higher traction forces and were able to contract collagen gels better (Figure 5 and Supplemental Figure 2). Our data are consistent with prior studies suggesting that α2β1 integrin represses expression of the malignant phenotype of MECs in culture and is a tumor suppressor in the mammary gland in vivo (Zutter et al., 1995) and recent data indicating that α2β1 integrin activates FAK by a force-independent mechanism (Seong et al., 2013). Interestingly, FN and collagen are often secreted and processed in tandem (Ledger et al., 1980), and an FN matrix can serve as a scaffold to guide collagen assembly (Singh et al., 2010; Kubow et al., 2015). Indeed, just as FN deposition and unfolding requires a collagen scaffold, collagen assembly and remodeling require FN (McDonald, 1988; Dallas et al., 2005, 2006; Kadler et al., 2008), during which time FN can be cross-linked to collagen (Eyre et al., 1984; Mosher, 1984). FN fibril assembly and collagen remodeling form a feedback loop, with collagen-mediated ECM stiffness and cell contractility inducing conformational changes in FN that reveal hidden binding sites required for FN matrix assembly, which then further facilitate collagen remodeling (Baneyx et al., 2002; Li et al., 2003). Thus our data are consistent with a paradigm wherein the highly cross-linked collagen forms the scaffold upon which an FN meshwork is assembled. This stiffened ECM, by virtue of its ability to modify FN, would then favor tumor progression by permitting α5β1-integrin binding to enhance tension-dependent signaling in tumors (Cao et al., 2012; Seo et al., 2015). Nevertheless, it is also feasible that FN-bound α5β1 integrin could collaborate with other transmembrane collagen receptors such as the discoidin domain receptor (DDR) or syndecans to foster malignant progression, particularly because DDRs are also tension regulated (Liebersbach and Sanderson, 1994; Shyu et al., 2005; Shintani et al., 2008; Vuoriluoto et al., 2008).
PI3K and ERK regulate cell growth, survival, and invasion, and the levels and activity of these kinases are frequently elevated in tumors (McCubrey et al., 2007; Chappell et al., 2011). Accordingly, an assortment of pharmacological inhibitors have been developed to target these kinases and their associated signaling molecules to treat various solid cancers with various degrees of clinical success (McCubrey et al., 2008; Engelman, 2009; Wong et al., 2010; Thorpe et al., 2015). Here we determined that, while the Her2/Neu tumors, which are surrounded by a stiffened ECM, have elevated ERK and PI3K activity as expected, merely preventing collagen cross-linking and stiffening by inhibiting LOX activity significantly reduced both ERK and Akt activity and did so in tandem with a reduction in α5 integrin and FN. We also observed that MECs with α5β1 integrin ligated by a wild-type, but not a synergy-site mutated FN, nucleated more vinculin-talin-actin-zyxin scaffold and recruited more PIP3 to amplify EGF-dependent activation of Akt and ERK. Inhibiting PI3K or ERK repressed the malignant behavior of FN-ligated mammary MEC 3D tissue-like structures expressing elevated α5β1 integrin (Figure 9). Our findings highlight the possibility that a stiffened tumor ECM stroma could amplify oncogenic signaling by enhancing EGFR signaling through PI3K and ERK via ligation of α5β1 integrin by the FN synergy site. The net result would be a reduced efficacy of many of the therapies targeting this pathway, as has been recently demonstrated (Hirata et al., 2015). The data thereby provide one plausible explanation for why, in some instances, targeted molecular therapies are less effective and suggest that combinatorial treatments that target both the mechanical properties of the cell or tissue and specific oncogenic signaling pathways might prove to be a better therapeutic option (Sanz-Moreno et al., 2011; Calvo et al., 2013; Sadok et al., 2015; Jiang et al., 2016; Laklai et al., 2016). Our findings are therefore clinically relevant, as they identify a tumor-specific mechanical signature, because the synergy site is only engaged in a high-force environment, such as in a wound or at the invasive front of a tumor. Thus inhibiting the ability of α5β1 integrin to ligate the synergy site of FN could prove to be a tractable cancer-specific therapy (Garcia et al., 1998; Baneyx et al., 2002). The data also suggest that new tools that are being developed to detect the FN synergy site in tumor biopsies might be useful biomarkers to identify patients with potential kinase-treatment resistance (Barker et al., 2005).
FIGURE 9:
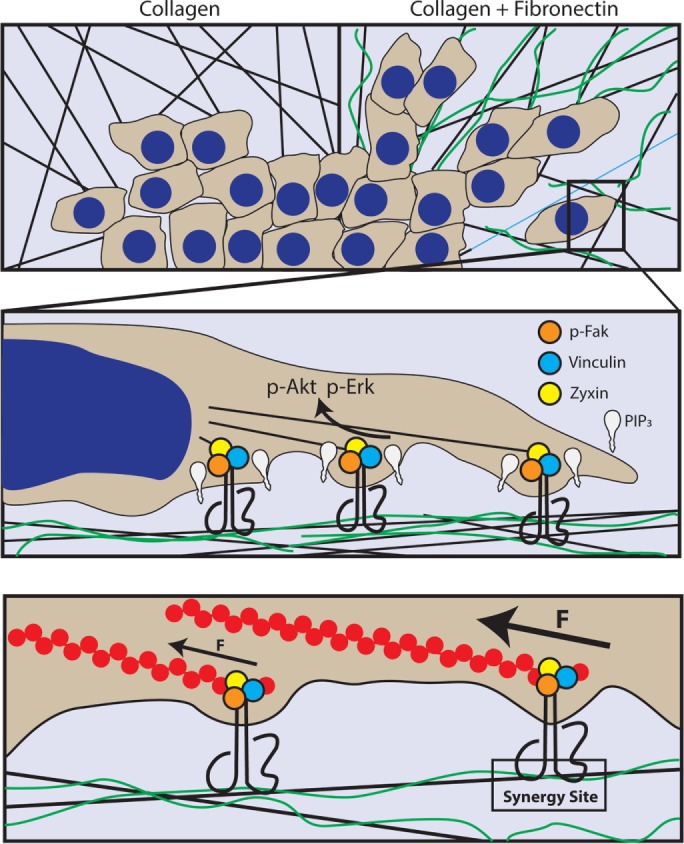
Model describing how ligation of α5β1-integrin synergy site promotes MEC malignancy. When tumor cells encounter FN-rich, stiff collagen fibers at invasive fronts, they engage the α5β1-integrin synergy site to recruit vinculin and zyxin to sites of focal adhesions in a tension-dependent manner to nucleate PIP3 at the plasma membrane and enhance PI3K–dependent tumor cell invasion.
MATERIALS AND METHODS
Antibodies and reagents
The following primary antibodies were used for our studies: collagen IV, CIV 22, PCNA, and PC10 (DAKO); laminin-5 3-chain specific, BM165 (gift from M.P. Marinkovich, Stanford University); β1 integrin, AIIB2 (gift from C.H. Damsky, Lawrence Berkeley National Laboratory); TS2/16 (American Type Culture Collection); β4 integrin 3E1, ASC-3, ASC-8; α1 integrin FB12, α2 integrin 10G11, α4 integrin P1H4, α5 integrin SAM-1 and P1D6, α6 integrin GoH3, αv integrin M9, FN, 3E3, E-cadherin, vinculin (Chemicon International); pAkt substrate, pMLCK Thr-18/Ser-19, YAP, activated caspase 3 (Cell Signaling); pMypt Thr-850 (Millipore), pFak Tyr-397 (Invitrogen); Ki-67, β-catenin, Akt and pSer-472/473/474-Akt, Erk1 (BD Transduction Laboratories); p-Erk1/2 (Thr-202/Tyr-204) (New England BioLabs); Flt-1/VEGFR1 antibody-1 (NeoMarkers). Phalloidin and anti-mouse/rabbit/rat IgG secondary antibodies, conjugated with fluorescein isothiocyanate (FITC), Texas red (Jackson Laboratories), Alexa Fluor 488/555/647 (Invitrogen), and horseradish peroxidase (HRP; Amersham Pharmacia Biotech) were used. Inhibitors used were: EGFR-specific tyrosine kinase Tyrphostin AG 1478 (200 nM), MEK1: PD98059 (20 μM) and PI3K LY 294002 (20 μM) (BIOMOL), FN synergy-site inhibitor (50 μM; gift from A.P. Mazar). In addition, the following proteins were used: FN synergy-site mutants (gift from H. P. Erickson, Duke University), human recombinant VEGF Receptor 1 Flt-1 (Oncogene), and RGD peptide (Biomol International).
Cell culture
HMT-3522, MCF10A MECs, and HDMVECs were grown and manipulated in 2D and 3D rBM (Collaborative Research) and collagen matrices. Phenotypic reversion of T4-2 cells was as previously described (Weaver et al., 1997). For inhibition of integrin function, the 3D multicellular structures were pre-incubated with anti-α2, α5, or αv integrin–blocking antibodies or IgG isotype–matched control monoclonal antibody (mAb; 20 g IgG/ml). When added to 3D cultures, FN concentration was 50 μg/ml (WT and mutant FN were gifts of H. P. Erickson [Duke University] and were purified as described in Purification of FN domains 7–10). Colony size and morphology were measured after 10–12 d in culture. Inhibition studies were performed using ROCKi Y27632 (10 µM), PI3Ki LY294002 (5 µM), MAPKi PD98059 (20 µM), EGFRi tyrphostin AG 1476 (100 mM). Adult human dermal microvascular endothelial cells (HDMVECs) were grown on collagen type I–coated flasks (Collaborative Biomedical) in EGM-2 bullet kit media (Bio-Whitaker). Angiogenesis induction by MECs was assayed by coculturing 3D rBM-generated mammary organoids in cell culture inserts (0.45-µm pore size; Biocoat; BD Labware) with HDMVECs that had been overlaid with a 1-micron layer of acellular collagen type I (BD Pharmingen). HDMECs invasion through the collagen overlay and network formation was assessed after 2 d by staining with toluidine blue. Anchorage-independent growth was assessed using a soft agar assay. Briefly, 20,000 cells in 1.0 ml 0.35% agarose with or without integrin-blocking antibodies, as indicated, were overlaid with 1.0 ml 0.5% agarose containing 1X growth media, and colonies larger than 40 μm in diameter were scored positive after 21 d.
Cell and tissue staining
The 3D rBM gels, as well as tissue sections, were prepared by mixing cultures with fresh collagen following embedment and freezing in sucrose with Tissue-Tek OCT compound (Miles Laboratories), then sectioning them in 10- to 20-µm-thick slices for analysis. All samples were incubated with primary mAbs followed directly by either FITC-, Texas red-, or Alexa Fluor–conjugated secondary antibodies and DAPI, when indicated. Nuclei were counterstained with diaminophenyl-indole (Sigma-Aldrich). Images were compared and quantified based on fluorescence intensity signal following minimal thresholding to subtract background. For mouse studies, when mice were killed, lesions were photographed, dissected, measured, macroscopically analyzed, fixed in 4% paraformaldehyde (PFA), and paraffin embedded. H&E sections were evaluated for histopathological evidence of tumor phenotype, and tissue sections were analyzed by immunofluorescence as described.
Image acquisition and analysis
All immunofluorescence images were recorded at 20–120× magnification and conventional images were recorded at 40–60× magnification. FRET images were acquired using a 60×/WI 1.2 NA Plan-Apo objective. Immunofluorescence and FRET images were acquired using an Olympus IX81 epifluorescence microscope with Spot color CCD camera, Nikon TE2000-U inverted microscope, Bio-Rad MRC 1024 laser-scanning confocal microscope attached to a Nikon Diaphot 200 microscope, and a spinning-disk/ total internal reflection fluorescence (TIRF) microscope setup with Andor’s iXon3 EMCCD camera with the Yokogawa CSU-X1 confocal scanner, a Nikon TIRF illuminator, and a MOSAIC module for fluorescence recovery after photobleaching (FRAP) and photoactivation on a motorized Nikon Ti-E inverted microscope base. Where immunofluorescence intensity was quantified, images were taken using identical image-acquisition parameters and were immunostained using the same primary and secondary antibody solutions. Images were imported into ImageJ (NIH), and positive signal was thresholded to secondary-only controls. The thresholded images are quantified from positive signal and presented as mean pixel density per group of samples.
The 3D tension bioreactor
PyMT organoids were cultured in rBM (Corning) for 3–5 d. The organoids were isolated by solubilizing the gels using phosphate-buffered saline (PBS)-EDTA. The isolated organoids were subsequently embedded in 2.5 mg/ml collagen I solution supplemented with FN (1 µg/ml). The collagen gels were stretched in a tension bioreactor with 0% or 10% strain for 72 h (Cassereau et al., 2015). Organoids were fixed with 4% PFA overnight for subsequent immunofluorescence staining.
Flow cytometry
Cells were isolated, blocked with 1% bovine serum albumin (BSA) in PBS for 60 min, incubated with saturating concentrations of primary mAb for 60 min, washed three times, and labeled with FITC- or phycoerythrin-conjugated goat IgG (Millipore). Stained cells were washed three times and immediately analyzed on a FACScan (BD Pharmingen). All manipulations were conducted at 4°C.
PIP3 localization analysis
Stable lines of MCF10A cells expressing tagged α5 integrin and transiently transfected with vinculin-mEmerald and KO2-PH-Grp1 (a fluorescent PIP3 probe courtesy of Keith Mostov) were plated on recombinant FN 9–10 or recombinant FN 9–10 with the synergy-site mutants. For analysis methods used, refer to the Supplemental Experimental Procedures.
Mouse studies
Cohorts of PyMT and FVB mice were maintained in accordance with University of California IACUC guidelines under protocol AN092125.
Her2/Neu mice were treated starting at 5 mo of age with BAPN (3 mg/kg; Spectrum) in the drinking water or a LOX function–blocking polyclonal antibody (3 mg/kg; OpenBiosystems) injected intraperitoneally twice per week (Levental et al., 2009). Mice were killed at 7–8 mo of age, at which time the fourth mammary gland was PFA fixed, paraffin sections were analyzed for histology, and parallel sections were stained as described.
PyMT mice were treated with BAPN (3 mg/kg; Spectrum) in the drinking water starting at 4 wk of age. The mice were killed at 14 wk of age with the tumors being harvested for PFA fixation and paraffin embedding for staining as described.
Four-week-old BALB/c nu/nu mice were subcutaneously injected in the rear flanks (5E6 cells/injection, together with Matrigel with or without addition of function-blocking antibody or IgG isotype control). Tumor development was monitored via external palpation, and lesions were measured and monitored biweekly for 2 mo upon detection (instant readout digital calipers; Electron Microscopy Sciences).
Retroviral and lentiviral infections and vectors
Standard retroviral and lentiviral infection procedures were used. For detailed descriptions of lentiviral and retroviral constructs and vectors, please refer to the Supplemental Experimental Procedures.
Western and ELISA procedures
Equal amounts of cell protein lysate (either RIPA or Laemmli lysate; BCA; Pierce) were separated on reducing SDS–PAGE gels, transferred to nitrocellulose or polyvinylidene fluoride membranes, and probed with primary antibody. Bands were visualized and quantified using a Fujifilm Gel Documentation system in combination with HRP-conjugated secondary antibodies and ECL-Plus system (Amersham Pharmacia). Specific activity for Akt and Erk was calculated by normalizing densitrometric values of phosphorylated to total AKT or ERK and E-cadherin. Integrin protein levels were assessed using nonreducing SDS–PAGE gels. VEGF, Il-8, and bFGF levels in the media of 10- to 12-day 3D rBM cultures of MECs were measured using sandwich ELISA (R&D Systems), according to the manufacturer’s instructions. OD measurements were performed using a Fluoroskan Ascent FL (Labsystems).
Real time-PCR analysis
Random-primed cDNA was prepared from total isolated RNA using Trizol reagent (Invitrogen), and target cDNA sequences were quantified via real-time PCR using SYBR Green I reagent (Roche) according to the manufacturer’s protocol. An Eppendorf Realplex2 quantitative PCR machine was used for all studies.
Real time-PCR primers
The following primer sequences were used: 18S: forward 5′-cggctaccacatccaaggaa-3′, reverse 5′-gctggaattaccgcggct-3′; FN 1: forward 5′-agtgggagacctcgagaag-3, reverse 5′-gtccctcggaacatcagaaa-3′, 5′-agtgggagacctcgagaag-3′, reverse 5′-gtccctcggaacatcagaaa-3′, VEGF: forward 5′-caggctgcacccatggcagaa-3′, reverse 5′-gcatcgcatcaggggcacaca-3′.; ITGB1: forward 5′-cgaggtcatggttcatgttg-3′, reverse 5′-tcccatttggcattcatttt-3′; ITGA5: forward 5′-agcctcagaaggaggaggac-3′, reverse 5′-ggttaatggggtgattggtg-3′, ITGA2 forward 5′-tgaccaaattctgcaggaca-3′, reverse 5′-ggagccaatctggtcacct-3′; ITGAV forward 5′-ccaccaagctttggctattc-3′, reverse 5′-caggcagtgagcaggtttta-3′.
Cell contractility, migration, and durotaxis assays
Collagen gel contraction was measured by imaging projected gel areas of cell-embedded collagen gels as described previously (Asaga et al., 1991). Traction-force microscopy studies were performed as described by Dembo and colleagues and processed using LIBTRC-2.0 software (Dembo and Wang, 1999; Reinhart-King et al., 2003). For durotaxis studies, bright-field images were captured every 5 min over 12 h using gradient-stiffness polyacrylamide gel (Isenberg et al., 2009). Cell migration time-course images were compiled, and cell speed, persistence, distance, and directionality were analyzed on a single-cell basis with ImageJ (NIH) and the Chemotaxis plug-in (Meijering et al., 2012).
Substrate preparation
ECM–cross-linked PA gels were prepared and mechanically analyzed as described; single stiffness substrates and mechanically gradient substrates (Lakins et al., 2012) with modifications made as described in Przybyla et al. (2016). Briefly, two droplets, each containing 12.5 μl of a soft (100 Pa) or stiff (60,000 Pa) acrylamide/bis-acrylamide mixture, were placed adjacent to each other on a large hydrophobic coverglass (no. 1, 45 mm × 50 mm; Fisher Scientific) and then covered with a small circular activated coverglass (no. 1, 18-mm diameter; Fisher Scientific) to merge the drops. By carefully maintaining the interface, a uniform gradient of 3.33Pa/µm along the length of substrate was achieved. Regions of different rigidities were distinguished by using a fluorescently labeled bis-acrylamide in the stiff solution creating a gradient of fluorescence correlated with the mechanical gradient.
Substrate elastic modulus was measured via atomic force microscopy (Asylum Research) using the Hertz model. Briefly, a silicon beaded–tip cantilever (5-μm diameter, 0.07 N/m, silicon nitride) was used to measure 90 × 90 μm elastic modulus maps down the length of the gel along the mechanical gradient while simultaneously measuring fluorescence intensity with a inverted epifluorescence microscope. We used these measurements to assess the mechanical gradient and develop an algorithm for determining the elastic modulus of the gel solely from the fluorescence intensity. This model allowed us to determine the particular surface stiffness seeded cells were adhered to when tracking cell motility.
Adhesion strength assay and generation of recombinant FN
Preparation of adhesive ligands: the previously described Promega Pinpoint vector containing the sequence for a fragment spanning the 7th to 10th type III repeat of human FN (FN7–10) was cut with NruI and ligated to yield an expression vector for a fragment spanning the 9th to 10th type III repeat of human FN (FN9–10). The synergy-site mutant PHSAN [FN9–10(PHSAN)] was generated using the Stratagene QuikChange Site-Directed Mutagenesis kit and primers 5′-GGGTGCCCCACTCTGCGAATTCCATCACCC-3′ (forward) and 5′-GGGTGATGGAATTCGCAGAGTGGGGCACCC-3′ (reverse). Constructs were verified by DNA sequencing. Proteins were expressed in JM109 cells (Promega) in the presence of d-biotin and purified by affinity chromatography. Protein concentration and purity were confirmed by Western blotting and Coomassie blue staining.
Preparation of micropatterned substrates.
Micropatterned substrates were generated by microcontact printing of self-assembled monolayers of alkanethiols on gold using a polydimethylsiloxane (PDMS) stamp (Sylgard 184/186 elastomer kit) with circular patterns (10-μm-diameter circles, 75-μm center-to-center spacing). Arrays of methyl-terminated alkanethiol [HS-(CH2)11-CH3; Sigma-Aldrich] circles were stamped onto Au-coated glass coverslips. The remaining exposed areas were functionalized with a tri(ethylene glycol)-terminated alkanethiol [HS-(CH2)11-(CH2CH2O)3-OH; ProChimia Surfaces] to generate a cell adhesive–resistant background. Patterned substrates were coated with purified adhesive ligands (20 μg/ml), blocked with 1% heat-denatured BSA, incubated in PBS (Ca2+/Mg2+), then seeded with cells at a density of 210 cells/mm2, and incubated for 16 h at 37°C.
Cell adhesion assay description.
Cell adhesion to FN-coated islands was measured using a hydrodynamic spinning-disk system. Micropatterned substrates with adherent cells were spun in PBS supplemented with 2 mM dextrose for 5 min at constant speeds. The applied shear stress (τ) is given by the formula τ = 0.8r(ρμω3)1/2, where r is the radial position and ρ, μ, and ω are the fluid density, viscosity, and rotational speed, respectively. After being spun, cells were fixed in 3.7% formaldehyde, permeabilized in 1% Triton X-100, and stained with ethidium homodimer-1 (Invitrogen). Adherent cells were counted at specific radial positions using a 10× objective lens in a Nikon TE300 microscope equipped with a Ludl motorized stage, Spot-RT camera, and an Image-Pro analysis system. A total of 61 fields (80–100 cells per field before spinning) were analyzed, and cell counts were normalized to the number of cell counts at the center of the disk. The fraction of adherent cells (f) as a function of shear stress τ (force/area) was then fitted to a sigmoid curve f = f0/[1 + exp[b (τ − τ50)]], where τ50 is the shear stress for 50% detachment, b is the inflection slope, and f0 is the y-intercept. τ50 represents the mean adhesion strength for the cell population.
Vinculin tension sensor and FRET
FRET images were collected and analyzed as previously described (Graham et al., 2001). Briefly, images were background subtracted and adjusted for spectral bleedthrough. Relative FRET index was calculated by taking the ratio of pixel intensity in the FRET image (donor excitation and acceptor emission) to pixel intensity in the donor image (donor excitation and emission). Average cell FRET index was calculated for each cell in a sample of 25–30 cells. Cell averages were then used to calculate a sample average, which was then used in Student’s t tests to evaluate whether the sampled populations had different means.
Purification of FN domains 7–10 (wild-type, double, and triple synergy-site mutants)
BL21(DE3) bacteria transformed with pET constructs expressing wild-type human FN domains 7–10 (Fn7-10) and the double (R1374A/R1379A) and triple (R1374A/P1376A/R1379A) synergy-site mutants were generous gifts of H. P. Erickson (Duke University). Bacteria were grown to an OD600 of 0.5–0.6 in 125 ml of 2× YT with 50 µg/ml ampicillin at 37°C in a shaking incubator, and expression was induced for 2–3 h by addition of IPTG to 0.4 mM. Bacteria were pelleted and resuspended on ice in 5 ml of 0.2 µg/ml lysozyme in 50 mM Tris-HCl/100 mM NaCl/1 mM EDTA, pH 8 (at 4°C). MgCl2, DNAseI, and PMSF were added to final concentrations of 10 mM, 190 U/ml, and 1 mM, respectively, and bacteria were digested for 30 min with stirring on ice. Lysis was effected by addition of Triton X-100 0.1% and sonication with two 30-s bursts. The supernatant containing soluble Fn7–10 was obtained by centrifugation for 30 min at 15,000 × g (at 4°C) and precipitated by addition of solid ammonium sulfate to 40% saturation (at 4°C). Following centrifugation for 30 min at 15,000 × g, the pelleted fraction was redissolved in 10 ml of 20 mM Tris-HCl, pH 7.9 (at 4°C). Redissolved proteins were filtered (0.22-µm pore size) and loaded at 1 ml/min onto a 1.6 cm × 10 cm Q-Sepharose HP (GE Healthcare) column equilibrated in 20 mM Tris-HCl, pH 7.9 (at 4°C) and washed in the same until OD260 returned to baseline. Bound proteins were eluted by a linear gradient of 0–0.5 M NaCl over 100 min, and 5-ml fractions were collected. Fractions containing wild-type and mutant forms of Fn7–10 were identified by SDS–PAGE and staining with Coomassie blue and eluted between 0.26 and 0.35 M NaCl. Peak fractions were pooled and desalted by exhaustive dialysis against PBS and sterilized by filtration through 0.22-µm syringe filters. Purified Fn7–10 concentrations were estimated by micro-BCA Assay (Pierce Chemical).
Doxycycline-inducible expression of α integrins
For HMT3522 cell lines S1 and T4-2, α5 and α2 integrins were cloned and expressed as doxycycline-inducible eGFP fusions in the retroviral vector pHRS puro Tet MCS. pHRS puro Tet MCS features in reverse orientation, an expression cassette for puromycin acetyltransferase back to back with a hepatamerized TetO minimal cytomegalovirus (CMV) promoter downstream of which α-integrin fusions were cloned. After transduction and selection for puromycin at 1 µg/ml, resistant cells were transduced with the recombinant retrovirus generated from MFG tTA, which constitutively expresses the TETOFF transcriptional transactivator form the 5′LTR. Transduced cells were induced by withdrawal of doxycycline for 2 d and sorted by flow cytometry for eGFP-positive cells to obtain a more homogeneous population of α5 and α2 integrin–overexpressing cells. For MCF10A cells, overexpressing lines were prepared using recombinant lentiviruses. Expression of the synthetic reverse tetracycline transactivator (TETON) rtTAs-M2 was obtained by transduction of target cells with the lentiviral vector pLV rtTAs-M2 IRES Neor. In pLV rtTAs-M2 IRES Neor, rtTAs-M2 is under control of the human EEF1α promoter and is coexpressed with neomycin phosphotransferase via a bicistronic (IRES-mediated) mRNA. Transduced cells were selected with 250 µg/ml G418. The complete coding regions (cds) of human α5 and α2 integrins were subcloned into the lentiviral vector pLV puro Tet MCS. PCR was used to introduce a Kozak concensus sequence to the 5′ ends of the cds and at the 3′ end to remove the stop codon and enable in-frame fusion to five tandem repeats of the myc epitope recognized by mAb 9E10 followed in frame by TagRFPT fluorescence protein. α-Integrin fusions were cloned downstream of the hepatamerized TetO minimal CMV promoter in pLV puro Tet MCS. pLV puro Tet MCS expresses puromycin acetyltransferase from an internal expression cassette in reverse orientation, and transduced G418r rtTAs-M2–expressing cells were selected with puromycin at 1 µg/ml. Following selection, expression of integrins was obtained by induction with doxycycline and a more homogeneous expressing population was obtained by flow sorting for TagFPT-positive cells.
Statistical analysis
Statistical analysis was performed using Prism/GraphPad Software (La Jolla, CA) at indicated p values. Unless otherwise stated, two-tailed Student’s t tests were used for significance testing to compare mean differences between two conditions, and one-way analysis of variance (ANOVA) was used to compare means of more than two conditions. Means were presented as ±SEM of at least three independent experiments. Where indicated, sample size, n, refers to either the number of animals (for animal studies with indicated number of images analyzed per animal, and unless otherwise indicated, three to five images were analyzed per animal) or the number of times each experiment was repeated (for all in vitro studies, at least 10 cells per condition were analyzed for each replica). Statistical significance was considered at p < 0.05 and was calculated only for experiments with a sample size of at least three. Adhesion-strength values were specifically analyzed via ANOVA and Tukey’s post hoc test with a p value < 0.05 considered significant.
Supplementary Material
Acknowledgments
We thank H. P. Erickson for FN mutants (Redick et al., 2000), K. Mostov for the fluorescent PIP3 probe (Martin-Belmonte et al., 2007), and M. Dembo for the LIBTRC 2.0 traction-force software. This work was supported by National Institutes of Health (NIH) grants R01CA192914 (V.M.W.), 2R01CA085482-11A1 (H. Moses and V.M.W.), 1U01CA202241 (V.M.W.); the U.S. Department of Defense Breast Cancer Research Program (DOD BCRP) grant BC122990 (V.M.W.); National Science Foundation (NSF) GRFP 1144247 and NIH/National Cancer Institute F31CA180422 (Y.A.M.); NSF GRFP 1106400 (G.O.); and NIH R01GM065918 (A.J.G.).
Abbreviations used:
- 2D
two-dimensional
- 3D
three-dimensional
- ANOVA
analysis of variance
- BAPN
β-aminopropionitrile
- BSA
bovine serum albumin
- ECM
extracellular matrix
- eGFP
enhanced green fluorescent protein
- EGFR
epidermal growth factor receptor
- ELISA
enzyme-linked immunosorbent assay
- ERK
extracellular signal–regulated kinase
- FAK
focal adhesion kinase
- FITC
fluorescein isothiocyanate
- FN
fibronectin
- FRAP
fluorescence recovery after photobleaching
- FRET
fluorescent resonance energy transfer
- HRP
horseradish peroxidase
- IgG
immunoglobulin G
- LOX
lysyl oxidase
- mAb
monoclonal antibodies
- MEC
mammary epithelial cell
- MyPT
myosin phosphatase binding protein
- PBS
phosphate-buffered saline
- PCNA
proliferating cell nuclear antigen
- PHSRN
proline-histidine-serine-arginine-asparagine
- PI3K
phosphoinositide 3-kinase
- PIP2
phosphatidylinositol (3,4)-bisphosphate
- PIP3
phosphatidylinositol (3,4,5)-triphosphate
- rBM
reconstituted basement membrane
- Tet
tetracycline
- TIRF
total internal reflection fluorescence
- VEGF
vascular endothelial growth factor
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E17-02-0126) on September 6, 2017.
REFERENCES
- Acerbi I, Cassereau L, Dean I, Shi Q, Au A, Park C, Chen YY, Liphardt J, Hwang ES, Weaver VM. Human breast cancer invasion and aggression correlates with ECM stiffening and immune cell infiltration. Integr Biol (Camb) 2015;7:1120–1134. doi: 10.1039/c5ib00040h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aragona M, Panciera T, Manfrin A, Giulitti S, Michielin F, Elvassore N, Dupont S, Piccolo S. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell. 2013;154:1047–1059. doi: 10.1016/j.cell.2013.07.042. [DOI] [PubMed] [Google Scholar]
- Arendt LM, Rudnick JA, Keller PJ, Kuperwasser C. Stroma in breast development and disease. Semin Cell Dev Biol. 2010;21:11–18. doi: 10.1016/j.semcdb.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asaga H, Kichuchi S, Yoshizato K. Collagen gel contraction by fibroblasts requires cellular fibronectin but not plasma fibronectin. Exp Cell Res. 1991;193:167–174. doi: 10.1016/0014-4827(91)90552-6. [DOI] [PubMed] [Google Scholar]
- Baneyx G, Baugh L, Vogel V. Fibronectin extension and unfolding within cell matrix fibrils controlled by cytoskeletal tension. Proc Natl Acad Sci USA. 2002;99:5139–5143. doi: 10.1073/pnas.072650799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker TH, Baneyx G, Cardo-Vila M, Workman GA, Weaver M, Menon PM, Dedhar S, Rempel SA, Arap W, Pasqualini R, et al. SPARC regulates extracellular matrix organization through its modulation of integrin-linked kinase activity. J Biol Chem. 2005;280:36483–36493. doi: 10.1074/jbc.M504663200. [DOI] [PubMed] [Google Scholar]
- Benito-Jardon M, Klapproth S, Gimeno LI, Petzold T, Bharadwaj M, Muller DJ, Zuchtriegel G, Reichel CA, Costell M. The fibronectin synergy site re-enforces cell adhesion and mediates a crosstalk between integrin classes. eLife. 2017;6:e22264. doi: 10.7554/eLife.22264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blandin AF, Renner G, Lehmann M, Lelong-Rebel I, Martin S, Dontenwill M. β1 integrins as therapeutic targets to disrupt hallmarks of cancer. Front Pharmacol. 2015;6:279. doi: 10.3389/fphar.2015.00279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo F, Ege N, Grande-Garcia A, Hooper S, Jenkins RP, Chaudhry SI, Harrington K, Williamson P, Moeendarbary E, Charras G, Sahai E. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol. 2013;15:637–646. doi: 10.1038/ncb2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Zeller MK, Fiore VF, Strane P, Bermudez H, Barker TH. Phage-based molecular probes that discriminate force-induced structural states of fibronectin in vivo. Proc Natl Acad Sci USA. 2012;109:7251–7256. doi: 10.1073/pnas.1118088109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey SP, Kraning-Rush CM, Williams RM, Reinhart-King CA. Biophysical control of invasive tumor cell behavior by extracellular matrix microarchitecture. Biomaterials. 2012;33:4157–4165. doi: 10.1016/j.biomaterials.2012.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carraher CL, Schwarzbauer JE. Regulation of matrix assembly through rigidity-dependent fibronectin conformational changes. J Biol Cheml. 2013;288:14805–14814. doi: 10.1074/jbc.M112.435271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassereau L, Miroshnikova YA, Ou G, Lakins J, Weaver VM. A 3D tension bioreactor platform to study the interplay between ECM stiffness and tumor phenotype. J Biotechnol. 2015;193:66–69. doi: 10.1016/j.jbiotec.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappell WH, Steelman LS, Long JM, Kempf RC, Abrams SL, Franklin RA, Basecke J, Stivala F, Donia M, Fagone P, et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR inhibitors: rationale and importance to inhibiting these pathways in human health. Oncotarget. 2011;2:135–164. doi: 10.18632/oncotarget.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan VP, Martin JD, Liu H, Lacorre DA, Jain SR, Kozin SV, Stylianopoulos T, Mousa AS, Han X, Adstamongkonkul P, et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat Commun. 2013;4:2516. doi: 10.1038/ncomms3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conklin MW, Eickhoff JC, Riching KM, Pehlke CA, Eliceiri KW, Provenzano PP, Friedl A, Keely PJ. Aligned collagen is a prognostic signature for survival in human breast carcinoma. Am J Pathol. 2011;178:1221–1232. doi: 10.1016/j.ajpath.2010.11.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox TR, Bird D, Baker AM, Barker HE, Ho MW, Lang G, Erler JT. LOX-mediated collagen crosslinking is responsible for fibrosis-enhanced metastasis. Cancer Res. 2013;73:1721–1732. doi: 10.1158/0008-5472.CAN-12-2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig DH, Gayer CP, Schaubert KL, Wei Y, Li J, Laouar Y, Basson MD. Increased extracellular pressure enhances cancer cell integrin-binding affinity through phosphorylation of beta1-integrin at threonine 788/789. Am J Physiol Cell Physiol. 2009;296:C193–C204. doi: 10.1152/ajpcell.00355.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallas SL, Chen Q, Sivakumar P. Dynamics of assembly and reorganization of extracellular matrix proteins. Curr Top Dev Biol. 2006;75:1–24. doi: 10.1016/S0070-2153(06)75001-3. [DOI] [PubMed] [Google Scholar]
- Dallas SL, Sivakumar P, Jones CJ, Chen Q, Peters DM, Mosher DF, Humphries MJ, Kielty CM. Fibronectin regulates latent transforming growth factor-beta (TGF beta) by controlling matrix assembly of latent TGF beta-binding protein-1. J Biol Chem. 2005;280:18871–18880. doi: 10.1074/jbc.M410762200. [DOI] [PubMed] [Google Scholar]
- Damiano L, Stewart KM, Cohet N, Mouw JK, Lakins JN, Debnath J, Reisman D, Nickerson JA, Imbalzano AN, Weaver VM. Oncogenic targeting of BRM drives malignancy through C/EBPbeta-dependent induction of alpha5 integrin. Oncogene. 2014;33:2441–2453. doi: 10.1038/onc.2013.220. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Dembo M, Wang YL. Stresses at the cell-to-substrate interface during locomotion of fibroblasts. Biophys J. 1999;76:2307–2316. doi: 10.1016/S0006-3495(99)77386-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10:9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingemans AM, van den Boogaart V, Vosse BA, van Suylen RJ, Griffioen AW, Thijssen VL. Integrin expression profiling identifies integrin alpha5 and beta1 as prognostic factors in early stage non-small cell lung cancer. Mol Cancer. 2010;9:152. doi: 10.1186/1476-4598-9-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- dos Santos PB, Zanetti JS, Ribeiro-Silva A, Beltrao EI. Beta 1 integrin predicts survival in breast cancer: a clinicopathological and immunohistochemical study. Diagn Pathol. 2012;7:104. doi: 10.1186/1746-1596-7-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumbauld DW, Michael KE, Hanks SK, Garcia AJ. Focal adhesion kinase-dependent regulation of adhesive forces involves vinculin recruitment to focal adhesions. Biol Cell. 2010;102:203–213. doi: 10.1042/BC20090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egeblad M, Rasch MG, Weaver VM. Dynamic interplay between the collagen scaffold and tumor evolution. Curr Opin Cell Biol. 2010;22:697–706. doi: 10.1016/j.ceb.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550–562. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- Ewald AJ, Werb Z, Egeblad M. Dynamic, long-term in vivo imaging of tumor-stroma interactions in mouse models of breast cancer using spinning-disk confocal microscopy. Cold Spring Harb Protoc. 2011;2011:pdb top97. doi: 10.1101/pdb.top97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyre DR, Paz MA, Gallop PM. Cross-linking in collagen and elastin. Annu Rev Biochem. 1984;53:717–748. doi: 10.1146/annurev.bi.53.070184.003441. [DOI] [PubMed] [Google Scholar]
- Friedland JC, Lee MH, Boettiger D. Mechanically activated integrin switch controls alpha5beta1 function. Science. 2009;323:642–644. doi: 10.1126/science.1168441. [DOI] [PubMed] [Google Scholar]
- Gallant ND, Garcia AJ. Quantitative analyses of cell adhesion strength. Methods Mol Biol. 2007;370:83–96. doi: 10.1007/978-1-59745-353-0_7. [DOI] [PubMed] [Google Scholar]
- Garcia AJ, Huber F, Boettiger D. Force required to break alpha5beta1 integrin-fibronectin bonds in intact adherent cells is sensitive to integrin activation state. J Biol Chem. 1998;273:10988–10993. doi: 10.1074/jbc.273.18.10988. [DOI] [PubMed] [Google Scholar]
- Garcia AJ, Schwarzbauer JE, Boettiger D. Distinct activation states of alpha5beta1 integrin show differential binding to RGD and synergy domains of fibronectin. Biochemistry. 2002;41:9063–9069. doi: 10.1021/bi025752f. [DOI] [PubMed] [Google Scholar]
- Gehler S, Ponik SM, Riching KM, Keely PJ. Bi-directional signaling: extracellular matrix and integrin regulation of breast tumor progression. Crit Rev Eukaryot Gene Expr. 2013;23:139–157. doi: 10.1615/critreveukargeneexpr.2013006647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham DL, Lowe PN, Chalk PA. A method to measure the interaction of Rac/Cdc42 with their binding partners using fluorescence resonance energy transfer between mutants of green fluorescent protein. Anal Biochem. 2001;296:208–217. doi: 10.1006/abio.2001.5306. [DOI] [PubMed] [Google Scholar]
- Grashoff C, Hoffman BD, Brenner MD, Zhou R, Parsons M, Yang MT, McLean MA, Sligar SG, Chen CS, Ha T, Schwartz MA. Measuring mechanical tension across vinculin reveals regulation of focal adhesion dynamics. Nature. 2010;466:263–266. doi: 10.1038/nature09198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman CD, Isenberg BC, Chua SG, Wong JY. Vascular smooth muscle cell durotaxis depends on extracellular matrix composition. Proc Natl Acad Sci USA. 2016;113:11190–11195. doi: 10.1073/pnas.1611324113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldin CH, Rubin K, Pietras K, Ostman A. High interstitial fluid pressure—an obstacle in cancer therapy. Nat Rev Cancer. 2004;4:806–813. doi: 10.1038/nrc1456. [DOI] [PubMed] [Google Scholar]
- Hirata E, Girotti MR, Viros A, Hooper S, Spencer-Dene B, Matsuda M, Larkin J, Marais R, Sahai E. Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin beta1/FAK signaling. Cancer Cell. 2015;27:574–588. doi: 10.1016/j.ccell.2015.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata H, Tatsumi H, Sokabe M. Mechanical forces facilitate actin polymerization at focal adhesions in a zyxin-dependent manner. J Cell Sci. 2008;121:2795–2804. doi: 10.1242/jcs.030320. [DOI] [PubMed] [Google Scholar]
- Hoffman LM, Jensen CC, Chaturvedi A, Yoshigi M, Beckerle MC. Stretch-induced actin remodeling requires targeting of zyxin to stress fibers and recruitment of actin regulators. Mol Biol Cell. 2012;23:1846–1859. doi: 10.1091/mbc.E11-12-1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Park CC, Hilsenbeck SG, Ward R, Rimawi MF, Wang YC, Shou J, Bissell MJ, Osborne CK, Schiff R. Beta1 integrin mediates an alternative survival pathway in breast cancer cells resistant to lapatinib. Breast Cancer Res. 2011;13:R84. doi: 10.1186/bcr2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioachim E, Charchanti A, Briasoulis E, Karavasilis V, Tsanou H, Arvanitis DL, Agnantis NJ, Pavlidis N. Immunohistochemical expression of extracellular matrix components tenascin, fibronectin, collagen type IV and laminin in breast cancer: their prognostic value and role in tumour invasion and progression. Eur J Cancer. 2002;38:2362–2370. doi: 10.1016/s0959-8049(02)00210-1. [DOI] [PubMed] [Google Scholar]
- Isenberg BC, Dimilla PA, Walker M, Kim S, Wong JY. Vascular smooth muscle cell durotaxis depends on substrate stiffness gradient strength. Biophys J. 2009;97:1313–1322. doi: 10.1016/j.bpj.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, Nywening TM, Hawkins WG, Shapiro IM, Weaver DT, et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med. 2016;22:851–860. doi: 10.1038/nm.4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadler KE, Hill A, Canty-Laird EG. Collagen fibrillogenesis: fibronectin, integrins, and minor collagens as organizers and nucleators. Curr Opin Cell Biol. 2008;20:495–501. doi: 10.1016/j.ceb.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan HM. Intra- and extracellular enzymes of collagen biosynthesis as biological and chemical targets in the control of fibrosis. Acta Trop. 2000;77:147–152. doi: 10.1016/s0001-706x(00)00128-5. [DOI] [PubMed] [Google Scholar]
- Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, MacDonald DD, Jin DK, Shido K, Kerns SA, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438:820–827. doi: 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik S, Pickup MW, Weaver VM. From transformation to metastasis: deconstructing the extracellular matrix in breast cancer. Cancer Metastasis Rev. 2016;35:655–667. doi: 10.1007/s10555-016-9650-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschmann DA, Seftor EA, Fong SF, Nieva DR, Sullivan CM, Edwards EM, Sommer P, Csiszar K, Hendrix MJ. A molecular role for lysyl oxidase in breast cancer invasion. Cancer Res. 2002;62:4478–4483. [PubMed] [Google Scholar]
- Kong F, Garcia AJ, Mould AP, Humphries MJ, Zhu C. Demonstration of catch bonds between an integrin and its ligand. J Cell Biol. 2009;185:1275–1284. doi: 10.1083/jcb.200810002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraning-Rush CM, Califano JP, Reinhart-King CA. Cellular traction stresses increase with increasing metastatic potential. PLoS ONE. 2012;7:e32572. doi: 10.1371/journal.pone.0032572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubow KE, Klotzsch E, Smith ML, Gourdon D, Little WC, Vogel V. Crosslinking of cell-derived 3D scaffolds up-regulates the stretching and unfolding of new extracellular matrix assembled by reseeded cells. Integr Biol (Camb) 2009;1:635–648. doi: 10.1039/b914996a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubow KE, Vukmirovic R, Zhe L, Klotzsch E, Smith ML, Gourdon D, Luna S, Vogel V. Mechanical forces regulate the interactions of fibronectin and collagen I in extracellular matrix. Nat Commun. 2015;6:8026. doi: 10.1038/ncomms9026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagares D, Busnadiego O, Garcia-Fernandez RA, Kapoor M, Liu S, Carter DE, Abraham D, Shi-Wen X, Carreira P, Fontaine BA, et al. Inhibition of focal adhesion kinase prevents experimental lung fibrosis and myofibroblast formation. Arthritis Rheum. 2012;64:1653–1664. doi: 10.1002/art.33482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakins JN, Chin AR, Weaver VM. Exploring the link between human embryonic stem cell organization and fate using tension-calibrated extracellular matrix functionalized polyacrylamide gels. Methods Mol Biol. 2012;916:317–350. doi: 10.1007/978-1-61779-980-8_24. [DOI] [PubMed] [Google Scholar]
- Laklai H, Miroshnikova YA, Pickup MW, Collisson EA, Kim GE, Barrett AS, Hill RC, Lakins JN, Schlaepfer DD, Mouw JK, et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat Med. 2016;22:497–505. doi: 10.1038/nm.4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledger PW, Uchida N, Tanzer ML. Immunocytochemical localization of procollagen and fibronectin in human fibroblasts: effects of the monovalent ionophore, monensin. J Cell Biol. 1980;87:663–671. doi: 10.1083/jcb.87.3.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SF, Csiszar K, Giaccia A, Weninger W, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139:891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Redick SD, Erickson HP, Moy VT. Force measurements of the alpha5beta1 integrin-fibronectin interaction. Biophys J. 2003;84:1252–1262. doi: 10.1016/S0006-3495(03)74940-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebersbach BF, Sanderson RD. Expression of syndecan-1 inhibits cell invasion into type I collagen. J Biol Chem. 1994;269:20013–20019. [PubMed] [Google Scholar]
- Lopez JI, Kang I, You WK, McDonald DM, Weaver VM. In situ force mapping of mammary gland transformation. Integr Biol (Camb) 2011;3:910–921. doi: 10.1039/c1ib00043h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Novoa JM, Nieto MA. Inflammation and EMT: an alliance towards organ fibrosis and cancer progression. EMBO Mol Med. 2009;1:303–314. doi: 10.1002/emmm.200900043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeshima AM, Niki T, Maeshima A, Yamada T, Kondo H, Matsuno Y. Modified scar grade: a prognostic indicator in small peripheral lung adenocarcinoma. Cancer. 2002;95:2546–2554. doi: 10.1002/cncr.11006. [DOI] [PubMed] [Google Scholar]
- Martin-Belmonte F, Gassama A, Datta A, Yu W, Rescher U, Gerke V, Mostov K. PTEN-mediated apical segregation of phosphoinositides controls epithelial morphogenesis through Cdc42. Cell. 2007;128:383–397. doi: 10.1016/j.cell.2006.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martino MM, Mochizuki M, Rothenfluh DA, Rempel SA, Hubbell JA, Barker TH. Controlling integrin specificity and stem cell differentiation in 2D and 3D environments through regulation of fibronectin domain stability. Biomaterials. 2009;30:1089–1097. doi: 10.1016/j.biomaterials.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maschler S, Wirl G, Spring H, Bredow DV, Sordat I, Beug H, Reichmann E. Tumor cell invasiveness correlates with changes in integrin expression and localization. Oncogene. 2005;24:2032–2041. doi: 10.1038/sj.onc.1208423. [DOI] [PubMed] [Google Scholar]
- McCubrey JA, Steelman LS, Abrams SL, Bertrand FE, Ludwig DE, Basecke J, Libra M, Stivala F, Milella M, Tafuri A, et al. Targeting survival cascades induced by activation of Ras/Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways for effective leukemia therapy. Leukemia. 2008;22:708–722. doi: 10.1038/leu.2008.27. [DOI] [PubMed] [Google Scholar]
- McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M, Tafuri A, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007;1773:1263–1284. doi: 10.1016/j.bbamcr.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JA. Extracellular matrix assembly. Annu Rev Cell Biol. 1988;4:183–207. doi: 10.1146/annurev.cb.04.110188.001151. [DOI] [PubMed] [Google Scholar]
- Meijering E, Dzyubachyk O, Smal I. Methods for cell and particle tracking. Methods Enzymol. 2012;504:183–200. doi: 10.1016/B978-0-12-391857-4.00009-4. [DOI] [PubMed] [Google Scholar]
- Mierke CT, Frey B, Fellner M, Herrmann M, Fabry B. Integrin alpha5beta1 facilitates cancer cell invasion through enhanced contractile forces. J Cell Sci. 2011;124:369–383. doi: 10.1242/jcs.071985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BW, Morton JP, Pinese M, Saturno G, Jamieson NB, McGhee E, Timpson P, Leach J, McGarry L, Shanks E, et al. Targeting the LOX/hypoxia axis reverses many of the features that make pancreatic cancer deadly: inhibition of LOX abrogates metastasis and enhances drug efficacy. EMBO Mol Med. 2015;7:1063–1076. doi: 10.15252/emmm.201404827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miroshnikova YA, Jorgens DM, Spirio L, Auer M, Sarang-Sieminski AL, Weaver VM. Engineering strategies to recapitulate epithelial morphogenesis within synthetic three-dimensional extracellular matrix with tunable mechanical properties. Phys Biol. 2011;8:026013. doi: 10.1088/1478-3975/8/2/026013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosher DF. Cross-linking of fibronectin to collagenous proteins. Mol Cell Biochem. 1984;58:63–68. doi: 10.1007/BF00240605. [DOI] [PubMed] [Google Scholar]
- Mouw JK, Yui Y, Damiano L, Bainer RO, Lakins JN, Acerbi I, Ou G, Wijekoon AC, Levental KR, Gilbert PM, et al. Tissue mechanics modulate microRNA-dependent PTEN expression to regulate malignant progression. Nat Med. 2014;20:360–367. doi: 10.1038/nm.3497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa H, Hikiba Y, Hirata Y, Font-Burgada J, Sakamoto K, Hayakawa Y, Taniguchi K, Umemura A, Kinoshita H, Sakitani K, et al. Loss of liver E-cadherin induces sclerosing cholangitis and promotes carcinogenesis. Proc Natl Acad Sci USA. 2014;111:1090–1095. doi: 10.1073/pnas.1322731111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam JM, Onodera Y, Bissell MJ, Park CC. Breast cancer cells in three-dimensional culture display an enhanced radioresponse after coordinate targeting of integrin alpha5beta1 and fibronectin. Cancer Res. 2010;70:5238–5248. doi: 10.1158/0008-5472.CAN-09-2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozbek S, Balasubramanian PG, Chiquet-Ehrismann R, Tucker RP, Adams JC. The evolution of extracellular matrix. Mol Biol Cell. 2010;21:4300–4305. doi: 10.1091/mbc.E10-03-0251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CC, Zhang H, Pallavicini M, Gray JW, Baehner F, Park CJ, Bissell MJ. Beta1 integrin inhibitory antibody induces apoptosis of breast cancer cells, inhibits growth, and distinguishes malignant from normal phenotype in three dimensional cultures and in vivo. Cancer Res. 2006;66:1526–1535. doi: 10.1158/0008-5472.CAN-05-3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paszek MJ, Weaver VM. The tension mounts: mechanics meets morphogenesis and malignancy. J Mammary Gland Biol Neoplasia. 2004;9:325–342. doi: 10.1007/s10911-004-1404-x. [DOI] [PubMed] [Google Scholar]
- Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, et al. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Pickup MW, Laklai H, Acerbi I, Owens P, Gorska AE, Chytil A, Aakre M, Weaver VM, Moses HL. Stromally derived lysyl oxidase promotes metastasis of transforming growth factor-beta-deficient mouse mammary carcinomas. Cancer Res. 2013;73:5336–5346. doi: 10.1158/0008-5472.CAN-13-0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickup MW, Mouw JK, Weaver VM. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014;15:1243–1253. doi: 10.15252/embr.201439246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:418–429. doi: 10.1016/j.ccr.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG, Keely PJ. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. 2006;4:38. doi: 10.1186/1741-7015-4-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przybyla L, Lakins JN, Sunyer R, Trepat X, Weaver VM. Monitoring developmental force distributions in reconstituted embryonic epithelia. Methods. 2016;94:101–113. doi: 10.1016/j.ymeth.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez NE, Zhang Z, Madamanchi A, Boyd KL, O’Rear LD, Nashabi A, Li Z, Dupont WD, Zijlstra A, Zutter MM. The α2β1 integrin is a metastasis suppressor in mouse models and human cancer. J Clin Invest. 2011;121:226–237. doi: 10.1172/JCI42328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recher C, Ysebaert L, Beyne-Rauzy O, Mansat-De Mas V, Ruidavets JB, Cariven P, Demur C, Payrastre B, Laurent G, Racaud-Sultan C. Expression of focal adhesion kinase in acute myeloid leukemia is associated with enhanced blast migration, increased cellularity, and poor prognosis. Cancer Res. 2004;64:3191–3197. doi: 10.1158/0008-5472.can-03-3005. [DOI] [PubMed] [Google Scholar]
- Redick SD, Settles DL, Briscoe G, Erickson HP. Defining fibronectin’s cell adhesion synergy site by site-directed mutagenesis. J Cell Biol. 2000;149:521–527. doi: 10.1083/jcb.149.2.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regent M, Planus E, Bouin AP, Bouvard D, Brunner M, Faurobert E, Millon-Fremillon A, Block MR, Albiges-Rizo C. Specificities of beta1 integrin signaling in the control of cell adhesion and adhesive strength. Eur J Cell Biol. 2011;90:261–269. doi: 10.1016/j.ejcb.2010.09.006. [DOI] [PubMed] [Google Scholar]
- Reinhart-King CA, Dembo M, Hammer DA. Endothelial cell traction forces on RGD-derivatized polyacrylamide substrata. Langmuir. 2003;19:1573–1579. [Google Scholar]
- Rizki A, Weaver VM, Lee SY, Rozenberg GI, Chin K, Myers CA, Bascom JL, Mott JD, Semeiks JR, Grate LR, et al. A human breast cell model of preinvasive to invasive transition. Cancer Res. 2008;68:1378–1387. doi: 10.1158/0008-5472.CAN-07-2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubashkin MG, Cassereau L, Bainer R, DuFort CC, Yui Y, Ou G, Paszek MJ, Davidson MW, Chen YY, Weaver VM. Force engages vinculin and promotes tumor progression by enhancing PI3K activation of phosphatidylinositol (3,4,5)-triphosphate. Cancer Res. 2014;74:4597–4611. doi: 10.1158/0008-5472.CAN-13-3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadok A, McCarthy A, Caldwell J, Collins I, Garrett MD, Yeo M, Hooper S, Sahai E, Kuemper S, Mardakheh FK, Marshall CJ. Rho kinase inhibitors block melanoma cell migration and inhibit metastasis. Cancer Res. 2015;75:2272–2284. doi: 10.1158/0008-5472.CAN-14-2156. [DOI] [PubMed] [Google Scholar]
- Samuel MS, Lopez JI, McGhee EJ, Croft DR, Strachan D, Timpson P, Munro J, Schroder E, Zhou J, Brunton VG, et al. Actomyosin-mediated cellular tension drives increased tissue stiffness and beta-catenin activation to induce epidermal hyperplasia and tumor growth. Cancer Cell. 2011;19:776–791. doi: 10.1016/j.ccr.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz-Moreno V, Gaggioli C, Yeo M, Albrengues J, Wallberg F, Viros A, Hooper S, Mitter R, Feral CC, Cook M, et al. ROCK and JAK1 signaling cooperate to control actomyosin contractility in tumor cells and stroma. Cancer Cell. 2011;20:229–245. doi: 10.1016/j.ccr.2011.06.018. [DOI] [PubMed] [Google Scholar]
- Sechler JL, Corbett SA, Schwarzbauer JE. Modulatory roles for integrin activation and the synergy site of fibronectin during matrix assembly. Mol Biol Cell. 1997;8:2563–2573. doi: 10.1091/mbc.8.12.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo BR, Bhardwaj P, Choi S, Gonzalez J, Andresen Eguiluz RC, Wang K, Mohanan S, Morris PG, Du B, Zhou XK, et al. Obesity-dependent changes in interstitial ECM mechanics promote breast tumorigenesis. Sci Transl Med. 2015;7:301ra130. doi: 10.1126/scitranslmed.3010467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seong J, Tajik A, Sun J, Guan JL, Humphries MJ, Craig SE, Shekaran A, Garcia AJ, Lu S, Lin MZ, et al. Distinct biophysical mechanisms of focal adhesion kinase mechanoactivation by different extracellular matrix proteins. Proc Natl Acad Sci USA. 2013;110:19372–19377. doi: 10.1073/pnas.1307405110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevilla CA, Dalecki D, Hocking DC. Regional fibronectin and collagen fibril co-assembly directs cell proliferation and microtissue morphology. PLoS ONE. 2013;8:e77316. doi: 10.1371/journal.pone.0077316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimosato Y, Suzuki A, Hashimoto T, Nishiwaki Y, Kodama T, Yoneyama T, Kameya T. Prognostic implications of fibrotic focus (scar) in small peripheral lung cancers. Am J Surg Pathol. 1980;4:365–373. doi: 10.1097/00000478-198008000-00005. [DOI] [PubMed] [Google Scholar]
- Shintani Y, Fukumoto Y, Chaika N, Svoboda R, Wheelock MJ, Johnson KR. Collagen I-mediated up-regulation of N-cadherin requires cooperative signals from integrins and discoidin domain receptor 1. J Cell Biol. 2008;180:1277–1289. doi: 10.1083/jcb.200708137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyu KG, Chao YM, Wang BW, Kuan P. Regulation of discoidin domain receptor 2 by cyclic mechanical stretch in cultured rat vascular smooth muscle cells. Hypertension. 2005;46:614–621. doi: 10.1161/01.HYP.0000175811.79863.e2. [DOI] [PubMed] [Google Scholar]
- Siegel RC. Biosynthesis of collagen crosslinks: increased activity of purified lysyl oxidase with reconstituted collagen fibrils. Proc Natl Acad Sci USA. 1974;71:4826–4830. doi: 10.1073/pnas.71.12.4826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P, Carraher C, Schwarzbauer JE. Assembly of fibronectin extracellular matrix. Annu Rev Cell Dev Biol. 2010;26:397–419. doi: 10.1146/annurev-cellbio-100109-104020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephen AG, Esposito D, Bagni RK, McCormick F. Dragging ras back in the ring. Cancer Cell. 2014;25:272–281. doi: 10.1016/j.ccr.2014.02.017. [DOI] [PubMed] [Google Scholar]
- Stoeltzing O, Liu W, Reinmuth N, Fan F, Parry GC, Parikh AA, McCarty MF, Bucana CD, Mazar AP, Ellis LM. Inhibition of integrin alpha5beta1 function with a small peptide (ATN-161) plus continuous 5-FU infusion reduces colorectal liver metastases and improves survival in mice. Int J Cancer. 2003;104:496–503. doi: 10.1002/ijc.10958. [DOI] [PubMed] [Google Scholar]
- Stokes JB, Adair SJ, Slack-Davis JK, Walters DM, Tilghman RW, Hershey ED, Lowrey B, Thomas KS, Bouton AH, Hwang RF, et al. Inhibition of focal adhesion kinase by PF-562,271 inhibits the growth and metastasis of pancreatic cancer concomitant with altering the tumor microenvironment. Mol Cancer Ther. 2011;10:2135–2145. doi: 10.1158/1535-7163.MCT-11-0261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzmaier FJ, Jean C, Schlaepfer DD. FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer. 2014;14:598–610. doi: 10.1038/nrc3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theocharis AD, Skandalis SS, Neill T, Multhaupt HA, Hubo M, Frey H, Gopal S, Gomes A, Afratis N, Lim HC, et al. Insights into the key roles of proteoglycans in breast cancer biology and translational medicine. Biochim Biophys Acta. 2015;1855:276–300. doi: 10.1016/j.bbcan.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer. 2015;15:7–24. doi: 10.1038/nrc3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010;10:116–129. doi: 10.1038/nrc2780. [DOI] [PubMed] [Google Scholar]
- Van den Hooff A. Connective tissue as an active participant in the process of malignant growth. Anticancer Res. 1986;6:775–780. [PubMed] [Google Scholar]
- van Nimwegen MJ, Verkoeijen S, van Buren L, Burg D, van de Water B. Requirement for focal adhesion kinase in the early phase of mammary adenocarcinoma lung metastasis formation. Cancer Res. 2005;65:4698–4706. doi: 10.1158/0008-5472.CAN-04-4126. [DOI] [PubMed] [Google Scholar]
- Vuoriluoto K, Jokinen J, Kallio K, Salmivirta M, Heino J, Ivaska J. Syndecan-1 supports integrin alpha2beta1-mediated adhesion to collagen. Exp Cell Res. 2008;314:3369–3381. doi: 10.1016/j.yexcr.2008.07.005. [DOI] [PubMed] [Google Scholar]
- Walker RA. The complexities of breast cancer desmoplasia. Breast Cancer Res. 2001;3:143–145. doi: 10.1186/bcr287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh C, Tanjoni I, Uryu S, Tomar A, Nam JO, Luo H, Phillips A, Patel N, Kwok C, McMahon G, et al. Oral delivery of PND-1186 FAK inhibitor decreases tumor growth and spontaneous breast to lung metastasis in pre-clinical models. Cancer Biol Ther. 2010;9:778–790. doi: 10.4161/cbt.9.10.11433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Weaver VM, Petersen OW, Larabell CA, Dedhar S, Briand P, Lupu R, Bissell MJ. Reciprocal interactions between beta1-integrin and epidermal growth factor receptor in three-dimensional basement membrane breast cultures: a different perspective in epithelial biology. Proc Natl Acad Sci USA. 1998;95:14821–14826. doi: 10.1073/pnas.95.25.14821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver VM, Fischer AH, Peterson OW, Bissell MJ. The importance of the microenvironment in breast cancer progression: recapitulation of mammary tumorigenesis using a unique human mammary epithelial cell model and a three-dimensional culture assay. Biochem Cell Biol. 1996;74:833–851. doi: 10.1139/o96-089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver VM, Petersen OW, Wang F, Larabell CA, Briand P, Damsky C, Bissell MJ. Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J Cell Biol. 1997;137:231–245. doi: 10.1083/jcb.137.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigelt B, Ghajar CM, Bissell MJ. The need for complex 3D culture models to unravel novel pathways and identify accurate biomarkers in breast cancer. Adv Drug Deliv Rev. 2014;69–70:42–51. doi: 10.1016/j.addr.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West RB, Nuyten DS, Subramanian S, Nielsen TO, Corless CL, Rubin BP, Montgomery K, Zhu S, Patel R, Hernandez-Boussard T, et al. Determination of stromal signatures in breast carcinoma. PLoS Biol. 2005;3:e187. doi: 10.1371/journal.pbio.0030187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CM, Engler AJ, Slone RD, Galante LL, Schwarzbauer JE. Fibronectin expression modulates mammary epithelial cell proliferation during acinar differentiation. Cancer Res. 2008;68:3185–3192. doi: 10.1158/0008-5472.CAN-07-2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis BC, Borok Z. TGF-beta-induced EMT: mechanisms and implications for fibrotic lung disease. Am J Physiol Lung Cell Mol Physiol. 2007;293:L525–534. doi: 10.1152/ajplung.00163.2007. [DOI] [PubMed] [Google Scholar]
- Wong KK, Engelman JA, Cantley LC. Targeting the PI3K signaling pathway in cancer. Curr Opin Genet Dev. 2010;20:87–90. doi: 10.1016/j.gde.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao ES, Zhang H, Chen YY, Lee B, Chew K, Moore D, Park C. Increased beta1 integrin is associated with decreased survival in invasive breast cancer. Cancer Res. 2007;67:659–664. doi: 10.1158/0008-5472.CAN-06-2768. [DOI] [PubMed] [Google Scholar]
- Yao H, Veine DM, Livant DL. Therapeutic inhibition of breast cancer bone metastasis progression and lung colonization: breaking the vicious cycle by targeting alpha5beta1 integrin. Breast Cancer Res Treat. 2016;157:489–501. doi: 10.1007/s10549-016-3844-6. [DOI] [PubMed] [Google Scholar]
- Yu M, Ting DT, Stott SL, Wittner BS, Ozsolak F, Paul S, Ciciliano JC, Smas ME, Winokur D, Gilman AJ, et al. RNA sequencing of pancreatic circulating tumour cells implicates WNT signalling in metastasis. Nature. 2012;487:510–513. doi: 10.1038/nature11217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497–5510. doi: 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahir N, Lakins JN, Russell A, Ming W, Chatterjee C, Rozenberg GI, Marinkovich MP, Weaver VM. Autocrine laminin-5 ligates alpha6beta4 integrin and activates RAC and NFkappaB to mediate anchorage-independent survival of mammary tumors. J Cell Biol. 2003;163:1397–1407. doi: 10.1083/jcb.200302023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zutter MM, Santoro SA, Staatz WD, Tsung YL. Re-expression of the alpha 2 beta 1 integrin abrogates the malignant phenotype of breast carcinoma cells. Proc Natl Acad Sci USA. 1995;92:7411–7415. doi: 10.1073/pnas.92.16.7411. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.