

Shear stress applied on the apical side of polarizing intestinal cells induces vacuole formation via the autophagy machinery. This response is relayed through apical microvilli that act as mechanosensors linking the physical environment to the intracellular trafficking pathways.
Abstract
Flow of fluids through the gut, such as milk from a neonatal diet, generates a shear stress on the unilaminar epithelium lining the lumen. We report that exposure to physiological levels of fluid shear stress leads to the formation of large vacuoles, containing extracellular contents within polarizing intestinal epithelial cell monolayers. These observations lead to two questions: how can cells lacking primary cilia transduce shear stress, and what molecular pathways support the formation of vacuoles that can exceed 80% of the cell volume? We find that shear forces are sensed by actin-rich microvilli that eventually generate the apical brush border, providing evidence that these structures possess mechanosensing ability. Importantly, we identified the molecular pathway that regulates large vacuole formation downstream from mechanostimulation to involve central components of the autophagy pathway, including ATG5 and LC3, but not Beclin. Together our results establish a novel link between the actin-rich microvilli, the macroscopic transport of fluids across cells, and the noncanonical autophagy pathway in organized epithelial monolayers.
INTRODUCTION
Macroautophagy, herein referred to as autophagy, is a set of essential trafficking processes that reorganizes cellular organelles in response to physiological needs (Shintani and Klionsky, 2004). Autophagy was originally discovered in unicellular organisms as the “self-eating” response to starvation conditions that can restore energy balance through cellular organelle digestion (Tooze et al., 2014). Since then, researchers have found that defects in the autophagy pathway in mammalian systems contribute to intestinal pathophysiology such inflammatory bowel disease (Kaser et al., 2008; Abraham and Cho, 2009; Barrett et al., 2009; Fritz et al., 2011; Kaser and Blumberg, 2011; Khor et al., 2011; Jostins et al., 2012; Adolph et al., 2013; Vandussen et al., 2014). The autophagy pathway is critically important to the neonatal gut, as short-term nutrient deprivation is a hallmark of birth (Kuma et al., 2004). In addition, the digestive requirements of the neonatal liquid milk diet differ from those of the adult solid diet (Henning, 1981). These differences are reflected in cellular structures observed in the neonatal intestinal epithelium that are not found in the mature gut, including subapical tubulovesicular elements and large vacuoles within absorptive enterocytes (Baintner, 1994; Skrzypek et al., 2007). Here we implicate a mechanistic link between mechanical stimulation of cells by liquid flow, the autophagy machinery, and macroscopic vesicular structures in the intestinal epithelium.
A large body of literature focuses on the role of nutrient signaling, such as AMPK and mTOR, on the induction of autophagy (Chen and Klionsky, 2011). Less work has been done on other potential autophagy modulators, such as mechanical stress acting through the cytoskeleton. Microtubules are known to transport various pre- and postautophagy vesicles (Mackeh et al., 2013), as well as regulate microtubule-associated protein 1A/1B-light chain 3 (LC3), a central component of the autophagy machinery. Drugs that modify microtubules, such as Flubendazole, have recently been found to be potent autophagy inducers (Chauhan et al., 2015). While illuminating the cellular mechanisms that transduce mechanical stress remains an active field of research, the microtubule-rich primary cilia found in many epithelial cells are known mechanotransducers. A recent study reported that shear stress transduced by the primary cilium can activate canonical autophagy in kidney epithelial cells (Orhon et al., 2016). These observations, taken collectively, support a hypothesis that mechanical inputs into the cytoskeleton, such as actin interaction with microtubules, can modulate the autophagy machinery.
Components of autophagy are known to play expansive roles in intestinal epithelial cells not solely limited to canonical autophagy associated with energy balance. For instance, molecular components of the autophagy pathway are involved in the digestion and transport of lipids across the intestinal epithelium (Khaldoun et al., 2014), the secretion of cargo from specialized cell types (Dupont et al., 2011; Cleyrat et al., 2014; Vandussen et al., 2014; Bel et al., 2017; Kimura et al., 2017; Liu et al., 2017), and microbial containment and clearance (Wild et al., 2011; LaRock et al., 2015; Schwerd et al., 2016). These additional trafficking events are collectively known as “noncanonical autophagy,” and include LC3-associated phagocytosis and entosis (Overholtzer et al., 2007; Codogno et al., 2011; Florey et al., 2015; Martinez et al., 2016; Schaaf et al., 2016).
We find that intestinal epithelial cells, when organized as a monolayer, respond to shear stress by increasing flux through a vesicular trafficking pathway controlled by autophagy components. As intestinal epithelial cells lack a primary cilium, we aimed to discover another mechanical sensor of shear stress. We find that actin-rich microvillar protrusions are required to trigger the shear stress response. The intermicrovillar adhesion complex (IMAC) linking adjacent microvilli resembles the complex that links stereocilia in the inner hair cells of cochlear (Hinkel et al., 2012; Crawley et al., 2016; Li et al., 2016), which mechanically respond to sound waves (Hudspeth, 2014). Our findings provide the first evidence of a mechanosensing role of microvilli and propose a novel mechanically controlled mechanism for inducing the autophagy machinery in intestinal epithelial cells.
RESULTS
Fluid shear stress applied to the apical surface of a confluent, intestinal epithelial monolayer induces formation of vacuoles
To investigate whether intestinal cell monolayers, which lack primary cilia, respond to apical shear stress, a monolayer composed of Caco-2BBE cells was grown in a microfluidic device in the presence and absence of a fluid flow across the apical surface. Caco-2BBE cells can be directed toward enterocyte differentiation at high cell densities (Peterson and Mooseker, 1992; Crawley et al., 2014). A fluid shear stress of 0.025 dyne/cm2 mimicking physiological luminal stress on the intestinal epithelium was applied overnight (Lentle and Janssen, 2008; Ishikawa et al., 2011). Under shear stress, the Caco-2BBE monolayer height increased significantly compared with static control (Supplemental Figure S1, A–C), corroborating earlier reports on the effects of shear flow on epithelial cell polarization (Kim et al., 2012; Chi et al., 2015). Surprisingly, previously uncharacterized vacuole-like structures (termed “vacuoles” herein) appeared after 3 h of exposure to fluid shear stress and increased in number and size until plateauing after 6 h (Figure 1, A and B, and Supplemental Video 1). About 20% of cells responded by forming vacuoles in the experimental time frame, probably due to heterogeneous polarization of cells in the monolayer (Supplemental Figure S1D). Intracellular vacuoles were observed under static control conditions, but with significantly less frequency and at much smaller sizes. Vacuoles can be indicative of cellular stress preceding cellular senescence or cell death (Dakik and Titorenko, 2016). However, Caco-2BBE cells proliferated normally under shear stress despite vacuole formation, as shown by the fluorescence ubiquitination cell cycle indicator (FUCCI) reporter (Sakaue-Sawano et al., 2008), and cell numbers after overnight culturing were not different between shear and control conditions (Supplemental Figure S2, A and B). In line with this observation, proliferative signaling pathways p-ERK, and p-AKT and p-mTOR downstream from class I PI3K did not demonstrate significant changes in response to shear stress (Supplemental Figure S2, C–F). Cell death observed by TUNEL staining was not increased by shear stress compared with control (Supplemental Figure S2G). These results demonstrated that the vacuole formation process is not indicative of imminent cellular senescence or cell death.
FIGURE 1:

Shear stress induces vacuole formation in dense Caco-2BBE monolayers. (A) Representative differential interference contrast (DIC) images of vacuole formation induced by shear overnight compared with static control. Vacuoles are marked by hand in red. (B) Image processing algorithm-based quantification of the number of vacuoles formed over time as a time course of shear induction compared with static control. Five fields of view tracked and quantified per experiment. Bands represent SEM from n = 3 independent experiments. Data scales are normalized values derived from mean centering and variance scaling of each set of time course experiments. Statistical analysis was done by two-way analysis of variance (ANOVA). (C) Representative DIC/fluorescence image depicting 10 kDa fluorescently labeled dextran included in vacuoles (red) and dextran-negative vacuoles (green), comparing shear overnight to static control. (D) Quantification of the number of dextran-positive vacuoles formed as a time course of shear induction compared with static control. Quantitative data were obtained by automatically counting the number of dextran particles exceeding a size threshold over the entire movie from maximum Z-projections. Data scales, error bars, and statistical analysis are the same as B.
Next we evaluated the identity of the vacuolar structures. To determine whether shear-induced vacuoles are glycogen storage granules (Sinadinos et al., 2014), we used periodic acid–Schiff (PAS) to stain for polysaccharides in cells exposed to shear stress overnight and discovered that the vacuoles were PAS negative (Supplemental Figure S3A). In contrast, PAS-positive puncta were observed in control cells (Supplemental Figure S3A), but these puncta disappeared upon shear stress induction. This experiment also excluded the possibility that vacuolated cells are goblet cells, because PAS also detects acidic mucins produced by goblet cells. Real-time PCR (RT-PCR) also showed that expression of MUC2, LYZ, and SI, markers for goblet cells, Paneth cells, and enterocytes, respectively, was not significantly altered by shear compared with control (Supplemental Figure S3B). In addition, negative oil red staining indicated that vacuoles were not large liposomes (Supplemental Figure S3C).
We investigated whether these intracellular vacuolar structures could be identified as vacuolar apical compartments (VACs) (Gilbert and Rodriguez-Boulan, 1991; Utech et al., 2005). VACs result from macroengulfment and subsequent invagination of apical membranes that contain actin-rich microvilli. To determine the possible apical origin of vacuolar membranes, we biotinylated the apical membrane after a monolayer with a tight barrier had formed, before the start of shear flow (Supplemental Figure S4A). The vacuoles formed after exposure to shear stress were only partially decorated with biotin (Supplemental Figure S4B), unlike VACs that are completely labeled by biotin. Furthermore, the majority of vacuoles were not lined with actin-rich microvilli (Supplemental Figure S4C), another hallmark of VACs. These observations excluded the possibility that shear-induced vacuoles identify with VACs and other commonly observed, large cellular structures.
Vacuoles are enriched with extracellular fluid from altered trafficking
To determine whether shear-induced vacuoles contain extracellular material, we supplied fluorescently labeled dextran to the culture media in our shear experiments. Ten-kilodalton dextran accumulated in some vacuoles (Figure 1C and Supplemental Video 2), while 70 kDa dextran was excluded (Supplemental Figure S5A). This observation indicated that the contents of vacuoles can originate from extracellular sources, but there is size- or molecular-based selection. In addition, we confirmed that shear-induced vacuoles were intracellular by confocal microscopy, using a live-actin reporter to delineate cell borders (GFP-UtrCH) (Burkel et al., 2007) while observing the localization of dextran-labeled vacuoles (Supplemental Figure S5B).
Increased inclusion of extracellular material into vacuoles can result from distinct but related mechanisms: 1) increased uptake of extracellular material into the cell through endocytosis, and 2) prolonged residence of material within the cell through altered trafficking. We sought to determine which of these two possibilities most contributes to shear-induced vacuoles. We quantified the number of shear-induced vacuoles containing a detectable concentration of dextran compared with the total number of vacuoles over time. We found that on average (over a 3–6 h time course), ∼30% of vacuoles were dextran positive, while ∼70% were dextran negative, indicating that most shear-induced vacuoles did not incorporate extracellular material (Figure 1C). Interestingly, dextran inclusion lagged vacuole formation, indicating that vacuoles were initially formed from material already in the cell (Figure 1D). Shear-induced vacuoles only sparsely incorporated apical plasma membrane in our biotinylation experiments (Supplemental Figure S4B), supporting a small extracellular contribution to vacuoles. Because our dextran experiments suggested that extracellular uptake operated through a size-selection process, we considered whether shear stress induced fluid-phase endocytosis. Surprisingly, immunofluorescence staining of clathrin-, dynamin- and caveolin-labeled vesicles did not reveal substantial differences between shear and control conditions (Supplemental Figure S6). We verified the staining procedure with epidermal growth factor (EGF) stimulation, which showed a substantial increase in endocytic vesicles (Supplemental Figure S7; Sorkin and Von Zastrow, 2002). Activity-dependent bulk endocytosis, as marked by the appearance of VAMP4+ vesicles (Nicholson-Fish et al., 2015), was not changed in response to shear (Supplemental Figure S8). In addition, the early endosomal marker EEA1 decreased under shear compared with control (Figure 2, A and B). Taken together, these data indicate that, although endocytic uptake can contribute to shear-induced vacuoles, the majority of the contribution may come from an alternate source.
FIGURE 2:

Shear-induced vacuoles are associated with increased autophagy, but not endocytosis. (A) Representative immunofluorescence (IF) images (as a maximum Z-projection) of early endosome antigen 1 (EEA1: red) positive endosomes induced by shear overnight compared with static control. (B) Quantification of EEA1+ particles by total particle area in A. (C) Representative IF images (as a maximum Z-projection) of LC3 (green) expression induced by shear overnight compared with static control. (D) Quantification of LC3+ particles in C. (E) Representative DIC/IF images of LC3 (green) and vacuoles in one confocal image section. (F) Representative IF images (as a maximum Z-projection) of lysosomes marked by LAMP1 (red) induced by shear overnight compared with static control. (G) Quantification of the average size of individual LAMP1+ puncta in F. (H) Representative IF images of LC3 and LAMP1 colocalization around shear-induced vacuoles. Error bars represent SEM from n = 3 independent experiments. Data scales are normalized values derived from mean centering and variance scaling of each set of experiments. **P < 0.01, ****P < 0.0001 by t test.
Shear-induced vacuoles are formed by flux into a trafficking pathway associated with the autophagy machinery
Internalized material can be trafficked to multiple downstream routes, including recycling back to the plasma membrane through the recycling endosome, sorting via early endosomes into multivesicular bodies, vesicular fusion with the Golgi complex, delivery to lysosomes for degradation, and repackaging for transcytotic or exosomal release (Conibear and Stevens, 1998). Redirection among these trafficking routes by increasing flux in and decreasing flux out of certain pathways can result in the accumulation of extracellular material in vacuoles. Recent studies found that both plasma and early endosomal membranes are sources of membrane for autophagosome formation, suggesting that a possible downstream fate of endosomes is the autophagy machinery (Razi et al., 2009; Ravikumar et al., 2010). Consistent with these studies, shear-induced vacuoles partially incorporated biotinylated plasma membranes (Supplemental Figure 4B) and EEA1+ early endosomal membranes (Supplemental Figure S9A). Hence we considered whether shear-induced vacuoles result from a change of flux into an autophagic trafficking pathway. To establish an association between shear-induced vacuole formation and the autophagy machinery, we stained for LC3, a cytosolic protein that is lipidated and recruited to autophagic membranes. LC3+ puncta were markedly up-regulated throughout the epithelial monolayer under shear stress (Figure 2, C and D). Strikingly, LC3 labeled the membranes of all vacuoles almost in their entireties, in contrast to partial punctate labeling of EEA1 and biotin, suggesting a direct involvement of the autophagy machinery in the vacuolation process (Figure 2E and Supplemental Figure S9B).
Autophagosomes and their contents are known to be degraded by the cell via fusion with lysosomes (Mauvezin et al., 2015). To investigate the involvement of lysosomes in vacuole formation, cells were stained with the lysosome marker LAMP1. Although exposure to shear stress did not increase the number of LAMP1+ puncta (Supplemental Figure S9C), the sizes of individual LAMP1+ puncta were significantly increased, signifying altered trafficking and fusion activities (Figure 2, F and G). In addition to LAMP1+ puncta that represented lysosomes, LAMP1 also decorated vacuoles entirely under shear stress, similar to LC3 (Figure 2H and Supplemental Figure S10, A and B). In addition, vacuoles containing detectable dextran colocalized with lysotracker, a fluorescent acidotropic probe for tracking lysosomes in living cells (Supplemental Figure S10C). Taken together, these results demonstrated that shear-induced vacuole formation is associated with the autophagy machinery downstream from extracellular uptake.
Shear-induced vacuoles are large vesicular compartments that sometimes take up 80% of a cell’s volume, which led us to question the source of membranes that make up these large compartments. While the contributions from the plasma (Supplemental Figure 4B) and endosomal membranes (Supplemental Figure S9A) were minimal, the autophagy machinery is known to recruit membranes from the endoplasmic reticulum (ER), Golgi, and mitochondria to generate autophagosomes (Tooze and Yoshimori, 2010; Lamb et al., 2013). The ER represents a major reservoir of membranes in intestinal epithelial cells, evidenced by the staining of ER “strand” morphologies by the marker calreticulin throughout the cytoplasm at homeostasis (Figure 3A). Shear-induced vacuoles were decorated with calreticulin similar to LC3 and LAMP1, indicating that vacuolar membranes are predominantly composed of ER membranes (Figure 3A). More importantly, calreticulin-positive structures within the cell decreased upon shear induction, and this observation was coupled to the appearance of calreticulin-positive particles inside the vacuoles (Figure 3, A–C), indicating degradation. The Golgi marker golgin-97 stained stack-like structures near the nucleus whose volumes were also decreased by shear (Figure 3, C and D). However, golgin-97 did not decorate vacuoles like calreticulin, suggesting that the Golgi is not a major membrane source (Figure 3C). The observed decrease in Golgi volume under fluid shear stress is consistent with the redirection of ER membranes to vacuoles, as the Golgi structure is constructed via membrane trafficking from the ER. These results demonstrated that shear stress triggers the autophagy machinery to traffic ER membranes to form vacuoles, in parallel to reticulophagy in yeast (Lipatova and Segev, 2015).
FIGURE 3:

Shear-induced vacuolar membranes originate from the endoplasmic reticulum. (A) Representative IF images the ER marker, calreticulin (CALR) distributed in the cytoplasm or around vacuoles of cell monolayers induced by shear overnight compared with static control. (B) Three-dimensional reconstruction of cell volumes depicting CALR. (C) Quantification of the mean intensity of CALR in cell volumes exposed to overnight shear compared with control. (D) Representative IF images of the Golgi marker, Golgin-97 (GOLGA1), in cell monolayers induced by shear overnight compared with static control. (E) Three-dimensional reconstruction of cell volumes depicting GOLGA1 exposed to overnight shear compared with control. (F) Quantification of GOLGA1+ particles by total particle area. Error bars represent SEM from n = 3 independent experiments. Data scales are normalized values derived from mean centering and variance scaling of each set of experiments. ****P < 0.0001 by t test.
To further test the association between shear stress and the autophagy machinery, we pharmacologically perturbed different steps of the autophagy pathway and measured the impact on shear-induced vacuole formation. Treatment with chloroquine (CQ), a well-known lysosomotropic agent that inhibits autophagy, suppressed shear-induced vacuole formation, implying a causal role of the autophagy pathway in this phenomenon (Figure 4, A and B). CQ inhibits steps downstream from autophagosome formation, resulting in the accumulation of LC3 puncta that cannot turn over (Kimura et al., 2007; Mizushima et al., 2010; Lamoureux et al., 2013; Chauhan et al., 2015), as we also observed in the context of shear stress (Supplemental Figure S11, A and B). To corroborate that shear stress induces the autophagy machinery, we performed Western Blot analysis to evaluate the expression of LC3 and its conversion from LC3 I to the lipidated LC3 II form. Shear induced expression of LC3, with a specific increase in the LC3 II form (Figure 4, C–E). However, shear also led to an increase in p62 (Figure 4, C and F); this result was corroborated by increased p62-positive puncta (Supplemental Figure S11C). p62 is an autophagy adaptor that should be degraded during canonical autophagy, suggesting that vacuole formation involves a noncanonical process. Because both the activation and inhibition of the autophagy machinery will result in the accumulation of LC3, we also performed LC3 analysis when the downstream fusion events were blocked by CQ to clarify if shear induced or inhibited the pathway. As expected, CQ treatment increased LC3 by blocking downstream processing (Figure 4, C–E). Application of shear led to a further increase in the LC3 II/LC3 I ratio (Figure 4G). These results supported the increase of LC3 expression and lipidation by shear through a noncanonical autophagy process, which may compete with the canonical autophagy pathway to result in p62 accumulation.
FIGURE 4:

Shear stress induces the autophagy machinery to form vacuoles. (A) Representative DIC images of vacuole formation induced in cell monolayers by shear overnight in vehicle, chloroquine (CQ), or 3-methyladenine (3-MA)-treated cell monolayers. Vacuoles are marked in red. (B) Quantification of the number of vacuoles formed resulting from shear overnight or static control, comparing monolayers treated with vehicle or CQ. Data scales are normalized values derived from mean centering and variance scaling of each set of experiments. **P < 0.01 by t test. (C) Representative quantitative immunoblot of LC3B, p62, and α-tubulin (loading control) under conditions of shear stress overnight compared with static control on cell monolayers, with vehicle or CQ. Quantification of the (D) LC3B II, (E) LC3B I, and (F) p62 bands normalized to a loading control expressed as a fold difference to static condition with vehicle. *P < 0.05, **P < 0.01 by one sample t test compared with 1 (unchanged: dotted line). (G) LC3B II/LC3B I ratio comparing control and shear conditions under CQ. **P < 0.01 by t test. (H) Quantification of the number of vacuoles formed resulting from shear induction overnight or static control, comparing monolayers treated with vehicle and 3-MA. Data scales are normalized values derived from mean centering and variance scaling of each set of experiments. *P < 0.05, ****P < 0.0001 by t test. All error bars represent SEM from n = 3 independent experiments.
Treatment with 3-methyladenine (3-MA), a PI3K inhibitor that targets Vps34’s ability to modify membranes with PI(3)P to form autophagosomes, suppressed shear-induced vacuoles (Figure 4, A and H), further supporting the role of the autophagy machinery in this phenomenon. Treatment with bafilomycin A1 (BafA), a downstream lysosomal inhibitor, did not impact shear-induced vacuole number (Figure 5, A and B), but instead led to significantly larger vacuole sizes (Figure 5, A and C). CQ and BafA are thought to inhibit the same step in the canonical autophagy pathway—the fusion between autophagosomes and lysosomes. However, a recent study of a noncanonical autophagic flux in activated germinal center B cells depicted a discrepancy in response between CQ and BafA treatment (Martinez-Martin et al., 2017). Because shear-induced vacuoles are much larger than autophagosomes, we expect the vesicular fusion that forms the vacuoles and the lysosomal fusion that degrades the contents of the vacuoles to be separate events in this noncanonical process. Indeed, while CQ is a general lysosomotropic agent that can affect acidification and hence fusion of all endosomes as well as perturb the Golgi (Oda and Ikehara, 1985), BafA inhibits V-ATPase and acidification of only lysosomes and vacuoles (Yoshimori et al., 1991). The consequent increase in pH due to BafA may render acidic hydrolases nonfunctional, leading to accumulation of vacuolar contents and larger vacuole sizes. Consistent with this notion, colocalization of dextran (marking vacuolar content) and lysotracker (marking acidic pH) was decreased by BafA in the context of shear stress (Supplemental Figure S11, D and E). In addition, we observed a slight decrease in LC3 puncta under BafA treatment (Supplemental Figure S11F), due to its trafficking into and subsequent dilution by vacuoles. These results further supported that shear-induced vacuoles resulted from a noncanonical autophagy process that have separate vesicular fusion events that are differentially inhibited by small molecules.
FIGURE 5:
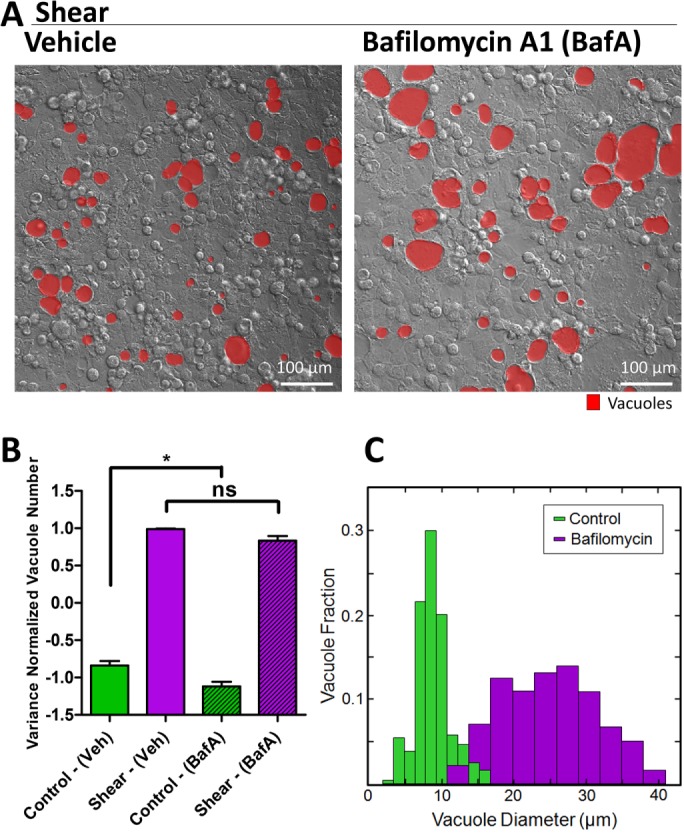
Bafilomycin A1, a V-ATPase inhibitor, blocks vacuolar acidification downstream of their formation. (A) Representative DIC images of vacuole formation induced by shear overnight, comparing BafA with vehicle-treated cell monolayers. (B) Quantification of the number of vacuoles formed resulting from shear induction overnight or static control, comparing monolayers treated with vehicle and BafA. Error bars represent SEM from n = 3 independent experiments. Data scales are normalized values derived from mean centering and variance scaling of each set of experiment. (C) Distribution of vacuole diameter measured by image analysis of experiments in A. Data represent combination of all vacuoles from n = 3 experiments. **P < 0.05 by t test.
To directly perturb the autophagy machinery, we used RNA interference (RNAi) to knock down key autophagy components, including LC3, ATG5, and Beclin. LC3 and ATG5 play important roles in both canonical and noncanonical autophagy (Dupont et al., 2011; Cleyrat et al., 2014; Martinez et al., 2016; Schaaf et al., 2016), but Beclin is specific to canonical autophagy (Naydenov et al., 2012). Knockdown of LC3, as confirmed by RT-PCR (Supplemental Figure S12A), reduced vacuole formation in response to shear (Figure 6, A and B). This result was confirmed with a second knockdown, a knockdown of another LC3 isoform, as well as a combination knockdown of multiple isoforms (Supplemental Figure S12, B–E). ATG5 forms an ATG7-mediated complex with ATG12 to regulate the conjugation of LC3 (Cheong et al., 2012). As expected, knockdown of ATG5 using two separate RNAi constructs, as confirmed by RT-PCR (Supplemental Figure S13A), down-regulated LC3-positive puncta (Supplemental Figure S13, B–D). However, it should be noted that shear-induced LC3 puncta was not suppressed completely (Supplemental Figure S13, C and D), due to either incomplete ATG5 knockdown or the existence of an alternative pathway to LC3 induction by shear. Consistent with the role of ATG5 in the autophagy machinery, knockdown of ATG5 reduced vacuole formation (Figure 6, C and D), specifically reducing the sensitivity to shear forces due to elevated basal amounts of vacuoles (Figure 6E). All of these results were repeated with a different RNAi construct (Supplemental Figure S13, E–G). The results of incomplete suppression of vacuole formation were consistent with incomplete LC3 suppression by ATG5 knockdown, although it should be noted that the remaining LC3+ puncta in ATG5 knockdown cells were distributed around vacuoles instead of the cytoplasm (Supplemental Figure S13H). RNAi knockdown of Beclin (two constructs, confirmed by RT-PCR in Supplemental Figure S13I) showed no statistical impact on shear-induced vacuole formation (Figure 6, F and G, and Supplemental Figure S13J). On the basis of these results, we conclude that shear-induced vacuole formation can be altered by perturbations in LC3 and ATG5, but not Beclin, placing it in the emerging class of noncanonical autophagic processes.
FIGURE 6:

The autophagy machinery is required for shear-induced vacuole formation. (A) Representative DIC images of vacuole formation induced by shear overnight in cell monolayers with LC3B knockdown (KD) compared with scramble control. (B) Quantification of the number of vacuoles formed under scramble or LC3B knockdown in static or shear conditions. (C) Representative DIC images of vacuole formation induced by shear overnight in cell monolayers with ATG5 knockdown compared with scramble control. (D) Quantification of the number of vacuoles formed under scramble or ATG5 knockdown in static or shear conditions. (E) Fold vacuoles induced by shear normalized to static control, comparing scramble to ATG5 knockdown. (F) Representative DIC images of vacuole formation induced by shear overnight in cell monolayers with BECN1 knockdown compared with scramble control. (G) Quantification of the number of vacuoles formed under scramble or BECN1 knockdown in static or shear conditions. Error bars represent SEM from n = 3 independent experiments. Data scales are normalized values derived from mean centering and variance scaling of each set of experiments. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by t test.
Apical shear mechanosensation depends on microvillar protrusions
We next turned our attention to the mechanism of mechanosensation by intestinal epithelial cells, which lack the well-known mechanosensor, the primary cilium. Enterocytes in the gut epithelium develop microvilli, actin-rich protrusions located at the apical surface. Caco-2BBE cells are known to adopt a differentiated enterocyte-like state in high density monolayer cultures, especially on permeable substrates. The IMAC of the stereocilia of the inner ear plays a critical role in the mechanosensation of sound waves and is implicated in deafness in Usher syndrome (Crawley et al., 2016; Li et al., 2016). Because the IMAC of microvilli has remarkable similarity to that of the stereocilia, we hypothesized that the microvilli act as the primary sensor of fluid shear stress upstream of vacuole formation. Using a live-actin reporter to label actin-rich structures, we confirmed that Caco-2BBE cells formed microvillar protrusions in high density cultures both in the absence and presence of shear stress (Supplemental Figure S14). In contrast, trophoblasts do not form microvilli without shear (Miura et al., 2015).
To investigate the potential for microvilli as a mechanosensor, we pursued three complementary perturbation strategies. First, we examined the shear stress response of the parental Caco-2 line, which is less efficient at forming microvilli than the Caco-2BBE line primarily used in this study (Figure 7A and Supplemental Figure S15, A and B; Peterson and Mooseker, 1992; Peterson et al., 1993). Parental Caco-2 cells under shear stress formed significantly fewer vacuoles compared with Caco-2 BBE cells (Figure 7, B and C). Dextran+ particles in the parental line were also decreased, correlating with the number of vacuoles (Figure 7D). Second, we evaluated the shear stress response of Caco-2BBE cells in less densely packed monolayers, where microvilli protrusions do not form due to a poor polarizing environment (Figure 7A and Supplemental Figure S15A; Peterson et al., 1993). In contrast to high density plating in the control condition, we plated cells at intermediate density where cells still formed confluent monolayers with all-around cell–cell contacts. Vacuole numbers and dextran+ particles were significantly diminished in confluent monolayers that are less densely populated (Figure 7, E–G). Third, we specifically knocked down protocadherin-24 (CDHR2, also known as PCDH24) in the Caco-2BBE line. CDHR2 is an essential adhesion molecule of the IMAC complex that bundles microvilli. CDHR2 down-regulation is known to elicit an aberrantly sparse brush border in Caco-2BBE cells (Crawley et al., 2014). We confirmed that CDHR2 expression was down-regulated in our knockdown cell line (Supplemental Figure S16A), which was accompanied by a defective brush border with few microvilli (Figure 8A and Supplemental Figure S16, B and C). Exposure of shear stress to CDHR2 knockdown cells generated significantly fewer vacuoles and dextran+ particles compared with the scramble control under shear (Figure 8, B–D). We evaluated whether shear-induced LC3 up-regulation is dependent on microvilli, and found that shear-induced LC3+ puncta were decreased with CDHR2 knockdown compared with scramble control (Figure 8, E and F). Finally, we sought to determine the expression levels of CDHR2 and LC3 in our cell lines. Both genes were detectable in the Caco-2 parental and BBE cell lines, suggesting intact machinery. The Caco-2 parental line, which has less well-formed microvilli in the experimental time frame, surprisingly expressed more CDHR2 at baseline than Caco-2BBE (Supplemental Figure 16D). Although CDHR2 expression is necessary for forming apical actin protrusions, it is not sufficient, as other molecules, such as MLPCDH, Myo7b, and ANKS4B, are also required for the formation and transport of the IMAC (Crawley et al., 2014, 2016; Weck et al., 2016). LC3 expression in this cell line (both isoforms) was also elevated compared with BBE (Supplemental Figure 16, E and F). Taken together, these results supported a necessary role of intestinal microvilli in the transduction of apical shear stress signals to induce vacuole formation.
FIGURE 7:

Microvilli are required for shear-induced vacuole formation. (A) Representative scanning electron micrographs of the apical surfaces of cell monolayers cultured overnight, comparing densely plated Caco-2BBE, Caco-2 parental cells, and less densely (but confluently) plated Caco-2BBE cells. Representative DIC images of shear-induced vacuole formation overnight (B, E), comparing (B) Caco-2BBE (control) with Caco-2 parental and (E) dense (control) with less dense (confluent) plating. Vacuoles are marked in red. (C, F) Quantification of vacuole number as a time course of shear stress or static control, comparing conditions outlined in B and E. Statistical analysis was performed by two-way ANOVA. (D, G) Quantification of the number of dextran-positive vacuoles resulting from overnight shear induction or static control, comparing conditions outlined in B and E. Error bars represent SEM from n = 3 independent experiments. Data scales are normalized values derived from mean centering and variance scaling of each set of experiments. **P < 0.01, ***P < 0.001 by t test.
FIGURE 8:
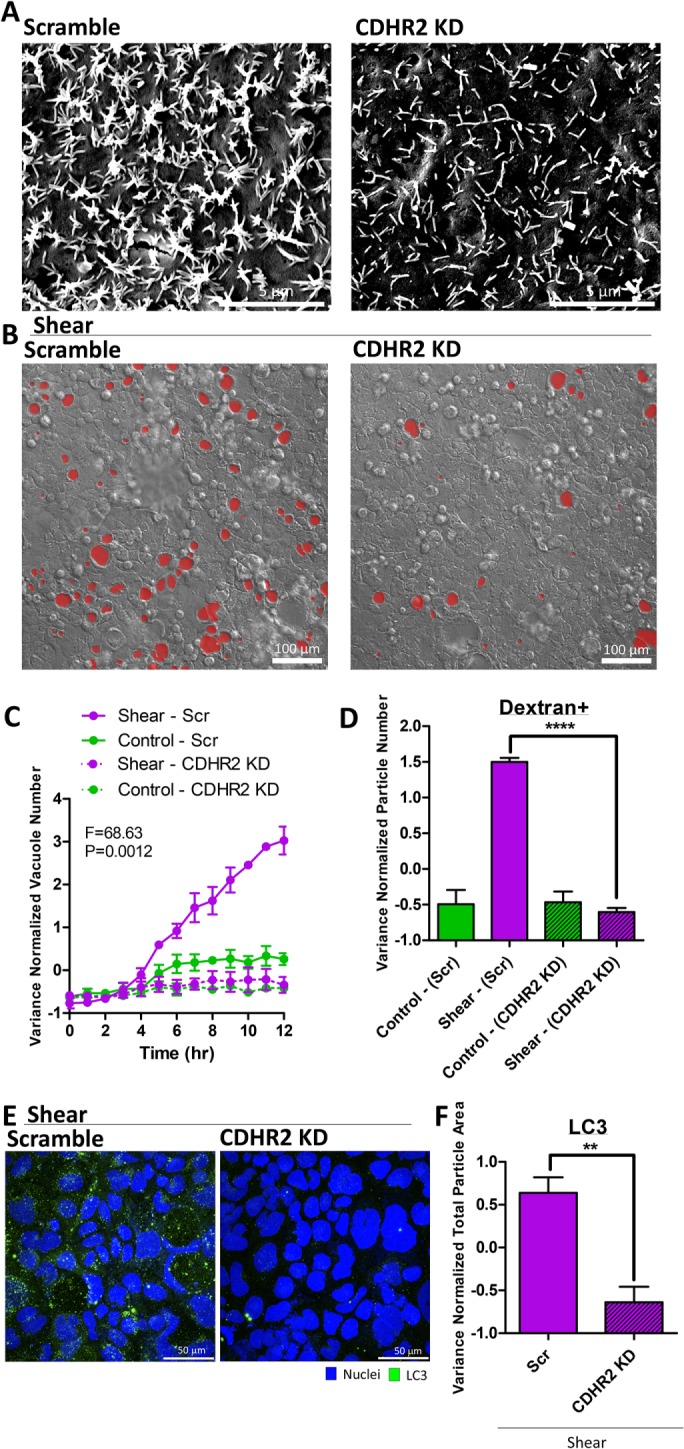
Perturbing microvilli by CDHR2 knockdown suppressed vacuole formation and autophagic trafficking. (A) Representative scanning electron micrographs of the apical surfaces of cell monolayers cultured overnight, comparing CDHR2 knockdown with scramble control. (B) Representative DIC images of shear-induced vacuole formation overnight, comparing CDHR2 knockdown with scramble control. (C) Quantification of vacuole number as a time course of shear stress or static control, comparing conditions outlined in B. Statistical analysis was performed by two-way ANOVA. (D) Quantification of the number of dextran-positive vacuoles resulting from overnight shear induction or static control, comparing conditions outlined in B. (E) Representative IF images (as a maximum Z-projection) of LC3 (green) expression induced by shear overnight, comparing CDHR2 knockdown with scramble control. (F) Quantification of LC3+ particles in E. Error bars represent SEM from n = 3 independent experiments. Data scales are normalized values derived from mean centering and variance scaling of each set of experiments. **P < 0.01, ****P < 0.0001 by t test.
DISCUSSION
In this study, we report that intestinal epithelial cell monolayers transduce apical shear stress by mechanosensitive microvillar protrusions. Unlike trophoblasts, which respond to shear stress by forming microvilli (Miura et al., 2015), our results suggest that epithelial monolayers use microvilli as mechanosensors. Exposure to shear stress induces intracellular vacuole formation (Figure 9). Although endocytosis contributes to this event (Tooze et al., 2014), our data support that shear stress mostly redirects intracellular trafficking flux, including endosomes, into an autophagic trafficking pathway. Vacuole formation depends on central autophagy components such as LC3 and ATG5, and is related to degradative processes such as LAMP1+ lysosomal recruitment and fusion. Glycogen-containing granules are reduced by shear, consistent with studies reporting autophagy to be a central pathway for degrading glycogen stores, specifically in glycogen storage diseases (Nascimbeni et al., 2012; Sedwick, 2013). Taking the results together, we have uncovered a novel mechanism in epithelial monolayers that links mechanical forces to the autophagy machinery.
FIGURE 9:

Shear induces vacuole formation via a noncanonical autophagic pathway. Microvilli on the apical side of relatively immature epithelial cells are formed by a CDHR2-dependent clustering mechanism. Shear stress on loosely packed and motile microvilli stimulates the autophagy machinery in a noncanonical pathway that is distinct from Beclin-dependent canonical autophagy. This noncanonical process triggers distinct trafficking events originating from the endoplasmic reticulum that are modulated differently by the small molecule inhibitors CQ and BafA, resulting in the formation of large LC3 and LAMP1-decorated degradative vacuoles.
Although shear-induced vacuole formation shares components of the canonical autophagy pathway, our data suggest that it is a distinct process that is broadly classified as noncanonical autophagy (Figure 9). Canonical autophagy begins with phagopore formation, maturation into autophagosomes, and ends with lysosomal fusion into autolysosomes where degradation of vesicular contents occurs (Shintani and Klionsky, 2004). Shear-induced vacuole formation does not end with autolysosomes that are observed as small puncta in the cell. Instead, previously uncharacterized, large vacuolar structures with acidic pH are formed. These structures are distinct from swelled autolysosomes that form when downstream degradation is blocked (Mauvezin et al., 2015). The differences between shear-induced vacuole formation and canonical autophagy are further highlighted by our results. Although canonical autophagy consists of four steps that are regulated by specific molecules—1) initiation by ULK1; 2) nucleation by Beclin, WIPI, and others; 3) elongation by ATG5, ATG7, and LC3; and 4) recycling by ATG9—noncanonical autophagic processes have been shown to bypass certain steps. In our studies, shear-induced vacuole formation partially depends on ATG5 but is independent of Beclin, bypassing the canonical nucleation step. In addition, the canonical autophagy adaptor molecule p62 is not degraded by shear-induced vacuole formation, as also observed in recently identified noncanonical autophagic processes (Florey et al., 2015; Martinez-Martin et al., 2017). Furthermore, CQ and BafA treatment showed distinct phenotypes, again mirroring the phenotype in the Martinez-Martin et al. report. Here the CQ phenotype is consistent with inhibition of a vesicular fusion step to form vacuoles, while the BafA phenotype is consistent with inhibition of downstream lysosomal fusion and degradation. Importantly, our observations in cilia-deficient but microvilli-rich intestinal epithelial cells are distinct from canonical autophagy triggered by shear forces acting on the primary cilia of kidney epithelial cells (Orhon et al., 2016).
We surmise that clues to the cellular function of these vacuoles can be gleaned from their identities. Based on our data, vacuoles do not correspond to well-known cellular structures, such as mucin granules, lipid droplets, or VACs. However, we determined that the substantial membrane requirement for vacuole formation is provided by the ER. ER-labeled membrane components were observed inside vacuoles, suggesting that the ER was turned over in a retinophagy-like process (Lipatova and Segev, 2015). Moreover, image mass spectrometry showed that phosphatidylethanolamine, the conjugating lipid for LC3, was significantly reduced (data not shown). The ER is a major site for secretory trafficking. Recent studies have demonstrated that misfolded proteins induce redirected trafficking from the ER–Golgi–plasma membrane secretory pathway to unconventional ERAD I (retrograde translocation from the ER into the cytosol) or ERAD II (autophagosome–lysosome pathway) (Gee et al., 2011). Furthermore, a noncanonical secretory pathway has also been shown to utilize the autophagy machinery, including ATG5 (Dupont et al., 2011; Cleyrat et al., 2014). Because intestinal enterocytes are responsible for the transport of nutrients from the luminal to the basal side, we speculate that macronutrients encountered at different developmental time points may require professional degradation–secretory pathways that may involve the autophagy machinery.
Clues to the physiological relevance of shear-induced autophagic trafficking can be gleaned from studies of the neonatal intestine. In the developing gut, introduction of a liquid diet from milk induces profound changes in duodenal enterocytes, including the formation of subapical tubulovesicular elements that feed intracellular vacuoles (Moxey and Trier, 1979; Henning, 1981). These changes occur when epithelial cells are immature with less organized and rigid microvillar protrusions and minimal mucus secretion, resembling the state of Caco-2BBE monolayers (Supplemental Figure S17; Muncan et al., 2011; Tourneur and Chassin, 2013). We surmise that a nonrigid brush border is required for mechanoresponse. Other studies have indeed revealed that polarized Caco-2 cells have characteristics of neonatal enterocytes (Schnabl et al., 2009). Dietary nutrients and immunoglobulins (Ig) traverse the intestinal mucosa by bulk endocytosis through enterocytes, and defects in the initialization, processing, and recycling of vesicles result in neonatal diarrheal diseases such as microvillus inclusion disease (Knowles et al., 2014; Weis et al., 2016). Although there are many possible stimulating factors in milk, such as nutrients, microbes, and endocrine inputs, the actual mechanism for inducing these profound epithelial changes remains to be elucidated. We speculate that mechanical shear forces from fluid flow may contribute to downstream processing of vacuoles and their contents through the autophagy machinery (Supplemental Figure S18). Enterocytes of the proximal small intestine of P0 mouse neonates before suckling exhibit no apparent subapical tubular network, vacuoles, or LC3 staining. Upon suckling, subapical tubulovesicular elements form in enterocytes in association with LC3 vesicles (P2), followed by the appearance of LC3-decorated vacuoles (P5). These structures, as well as lysosomal proteases and β-galactosidase for digesting milk proteins, disappear over time into adulthood (Muncan et al., 2011). Investigation of the contribution of mechanical stimuli to intracellular trafficking is a relatively unexplored cell biological topic that may have a significant impact on the understanding of luminal sensing in the gut.
MATERIALS AND METHODS
Cell culture
Caco-2BBE, Caco-2, and HEK293FT cells were cultured in DMEM (4.5 g/l glucose; Corning) in 37°C incubator supplied by 5% CO2, supplemented with 20%, 10%, and 10% fetal bovine serum (Sigma), respectively. All media were supplemented with 1% penicillin/streptomycin and 2 mM l-glutamine (Hyclone).
Microfluidic device fabrication
Microfluidic devices are constructed from polydimethylsiloxane (PDMS) to the specifications below. Designs were produced in Adobe illustrator and made into photolithography masks (Front Range Photomask). Photolithography was performed on a 3-in. (76.2-mm) silicon wafer according to standard procedure (MicroChem Procedures). The masters were then treated with Trichloro(1H,1H,2H,2H-perfluorooctyl)silane (Sigma) to create nonstick surfaces. PDMS (Corning) was then poured onto the wafers in 10:1 base to curing agent, desiccated, and baked at 60°C for 4 h. After removing the PDMS from the master, holes for tubing were punched in the inlets and outlets. The chambers were then sealed to a four-well Nunc Lab-Tek chamber (Nunc) slide according to standard procedure (Bhattacharya et al., 2005). The chambers were then coated with a 50 μM collagen (Sigma), 300 μM Matrigel (Sigma) mixture in DMEM (Sigma). After 1 h under UV light, the devices were rinsed with DMEM, covered with phosphate-buffered saline (PBS), and refrigerated until used.
Live cell imaging with microfluidic control
For shear experiments, 25 mM HEPES was added in the media when cells were seeded in the microfluidic device. Cells were seeded at a density of 3 × 107 cells/ml to obtain dense confluency and 7.5 × 106 cells/ml to obtain confluency at a lower cell density for microvilli perturbation experiments. After 5 h of incubation to allow monolayer formation, unattached cells were washed off before the start of overnight shear experiments. Live cell imaging was performed with a Nikon A1R (Nikon) or a LSM 710 (Zeiss) fluorescence confocal microscope using a 20× objective in 30-min intervals. At the end of the experiment, cells were fixed with 4% paraformaldehyde (PFA), and then the devices were dissembled for further cellular analysis.
To visualize the cytoskeleton and microvillar changes, Caco-2BBE cells were transfected with a GFP-UtrCH plasmid, containing the actin-binding domain of utrophin (Burkel et al., 2007). To study cell proliferation and cell cycle, cells were transfected with a FUCCI plasmid (Sakaue-Sawano et al., 2008). For dextran uptake assay, FITC-dextran (70 kDa; Molecular Probes) at a final concentration of 5 mg/ml, RFP-dextran (10 kDa; Molecular Probes) at a final concentration of 10 mg/ml, and/or LysoTracker (Molecular Probes) at a final concentration of 50 nM were supplied continuously to the microfluidic devices for the duration of the experiments. Live cell imaging was conducted and dextran-positive particle count (as determined by manual thresholding) was conducted by scripts in ImageJ (National Institutes of Health [NIH]).
Immunofluorescence microscopy
Fixed cells were permeabilized with 0.1% Triton X and 1% bovine serum albumin (BSA; Sigma) in PBS, and then blocked with 5% BSA and 5% goat serum (Jackson ImmunoResearch) in PBS for 1 h at room temperature. Cells were stained with primary antibodies diluted in the blocking buffer for 1.5–2 h, and then with fluorescent secondary antibodies (Life Technologies), Hoechst (Life Technologies), and phalloidin (Life Technologies) for 1 h. Images were taken using 20× or 60× objectives on a Nikon A1R (Nikon) or a LSM 710 (Zeiss) confocal microscope. Antibodies against CAV1 (BD Biosciences), clathrin (Cell Signaling Technology), dynamin II (BD Biosciences), EEA1 (BD Biosciences), LAMP1 (BD Biosciences), LC3 (MBL, Novus), CALR (Abcam), GOLGA1 (Golgin97; Molecular Probes) were used.
The TUNEL assay was performed according to the manufacturer’s specifications (Roche). Positive control for TUNEL was Caco-2BBE cells induced to undergo cell death with sodium butyrate (100 mM) for 48 h. For quantification, sizes of single particles were measured using custom scripts in ImageJ after thresholding out excessively large particle clusters. Numbers of particles were estimated by the total area occupied by positively stained regions as determined by manual thresholding in ImageJ. Mean intensities were calculated on whole fields of view. Whole cell volume quantification was performed by Z-maximal projection.
Apical surface biotinylation
Caco-2BBE cells were seeded in the microfluidic device for 1 d to ensure monolayer establishment, and then were washed with ice-cold PBS supplemented with 0.1 mM CaCl2 and 1.0 mM MgCl2 (PBS-CM). EZ-Link Sulfo-NHS-LC-Biotin (0.5 mg/ml) (ThermoScientific) was freshly prepared in ice-cold PBS-CM and applied to the apical surface of the monolayer in the microfluidic chamber at 4°C for two consecutive 20-min periods, after which Stretavidin-Cy3 (Sigma) was applied for 1 h at 4°C. Additional biotin was quenched with 100 mM glycine and washed before the start of microfluidic experiments.
Histochemical staining
Samples were fixed with 4% PFA for 1 h at room temperature, and then dehydrated with 60% isopropanol for 5 min. Samples were then stained with oil red O solution for 10 min and imaged with brightfield microscopy (EVOS). Periodic acid–Schiff staining was performed to manufacturer’s specifications (Abcam).
Small molecule studies
Bafilomycin A1 (BafA; 100 nM; Sigma) and 50 μM CQ (Enzo Life Sciences) were prepared in culture media from a dimethyl sulfoxide (DMSO)-based stock solution. 3-MA (5 mM; Sigma) was dissolved directly in media. Small molecules were supplied to the microfluidic devices continuously for the duration of the experiments. Control cells were treated with the DMSO vehicle.
RNA knockdown
Small interfering RNA against LC3A and LC3B (Santa Cruz Biotechnology), and BECN1 (Qiagen) were transfected into Caco-2BBE cells with PolyJet to manufacturer’s specifications (SignaGen Laboratory). Control cells were treated with scramble RNAi. CDHR2 (also known as protocadherin-24 PCDH24) shRNA (Sigma) and nontargeting scramble shRNA expressed from the pLKO.1 vector (Addgene) were transduced into Caco-2BBE cells as previously described (Crawley et al., 2014). In addition, LC3B shRNAs (Sigma) and ATG5 shRNAs (Sigma) were used for loss-of-function studies. Cells were used 48 h later for experiments.
Real-time RT-PCR
RNA was extracted using RNeasy kit (Qiagen), and reverse transcribed using the Quantitect reverse transcription kit (Qiagen) to the manufacturer’s specifications. Real-time PCR was performed using a StepOne system (ABI) or a CFX96 (BioRad) with commercially available primers (reatimepcr.com or Sigma), and relative quantities were calculated from Ct values using GAPDH expression as a housekeeping gene.
Electron microscopy
Samples were prepared by washing with 0.1 M sodium cacodylate and fixed with 2.5% glutaldehyde in sodium cacodylate for 1 h at room temperature, followed by postfixation in 1% osmium tertroxide for 1 h in sodium cacodylate. After washing with 0.1 M sodium cacodylate, samples were dehydrated with a serial dilution of ethanol, then a 50:50 mixture of ethanol and propylene oxide, and finally propylene oxide. Electron micrographs were taken by 250 ESEM (Quanta).
Automated image analysis of vacuole count and size, and monolayer height
Because vacuoles tend to have uniform intensity, while cells are full of contrasting features, images were first passed through a variance filter in ImageJ, followed by further analysis in Matlab (MathWorks). Images were binarized, and morphologically transformed using “bottom hat” (dilation and erosion). Vacuolar boundaries were filtered by area and circularity, yielding outlines of vacuoles while ignoring other cell features. Vacuolar diameters were estimated by the diameters of circles with the same area as each object to negate the noise of boundary identification. Vacuole number and size quantified by this automated algorithm were initially compared with results measured manually.
To determine the height of cell monolayers, we used the highest actin signal below the dextran level as the apical boundary of the monolayer. The basal boundary was determined as the highest actin signal above the substrate, which was nonfluorescent. The difference between the top and bottom was recorded at every pixel, with errors removed and the surface smoothed.
Immunoblot
Cells were lysed in RIPA buffer (Sigma) supplemented with protease inhibitor cocktail (Roche) and phosphotase inhibitor cocktail (Roche). Sample lysates were boiled with Laemmli sample buffer and loaded in gel for electrophoresis. After blot transfer, polyvinylidene difluoride membranes were blocked with blocking buffer (LI-COR) and incubated with primary antibodies overnight. After 3× washing with PBS, secondary antibodies were applied for 1 h and then washed with TBST (0.05% Tween 20) before imaging. Blots were scanned by the Odyssey imager (LI-COR), and integrated intensities using the top and bottom of the bands as background were quantified by the Odyssey software. Primary antibodies such as LC3 (Novus), p-ERK (Cell Signaling), p-AKT (Cell Signaling), p62 (SQSTM1) (Abnova), and GAPDH (Origene) were used for this experiment.
Mouse experiments
All animal experiments were performed under protocols approved by the Vanderbilt University Animal Care and Use Committee and in accordance with NIH guidelines. Postnatal duodenal tissues were harvested, fixed in 4% PFA on ice for 1 h, embedded in OCT, and then frozen at −80°C. Tissues were sectioned at 5 µm thickness and then stained with phalloidin (Life Technologies) or an antibody against LC3 (Cell Signaling) for 1.5–2 h. After secondary antibody (Life Technologies) and Hoescht (Life Technologies) staining for 1 h, sections were imaged on the Nikon A1R confocal microscope as above.
Variance normalization of data
Because vacuole numbers vary from experiment to experiment mainly due to extrinsic factors affecting the maturation state of the apical brush border of individual cells, we normalized each set of experiments by mean centering and variance scaling. Briefly, the mean of the whole data set (consisting of control conditions and perturbations) was set as zero, giving values less than the mean a negative and values greater than the mean a positive value. This value was then converted to the number of SDs away from the mean. This type of normalization has been used routinely in large-scale multivariate analysis for multicondition comparisons (Lau et al., 2012, 2013; Simmons et al., 2015).
Supplementary Material
Acknowledgments
We are grateful to Nick Ferrell, Anne Kenworthy (Vanderbilt), and Sabrina Spencer (University of Colorado Boulder) for reagents, Janice Williams in the Cell Imaging Shared Resource for her help with electron microscopy, and Alan Simmons, Alina Starchenko, Andres Guillen, Chehronai Fozil, Won Jae Huh, Jeffrey Spraggins, Eunyoung Choi, Bhuminder Singh, Cao Zheng, James Higginbotham, Bob Coffey, and members of the Epithelial Biology Center and Center for Quantitative Systems Biology, as well as Mark Donowitz at the Johns Hopkins School of Medicine for stimulating discussions. K.S.L is funded by R01DK103831, VICTR (2ULTR000445), and pilot project grants from P30DK058404 and P50CA095103 from the NIH, an Innovator Award from the AACR-Landon Foundation (15-20-27-LAUK), and a Crohn’s and Colitis Foundation CDA (308221). S.W.K. and Y.S.O. are funded by R01DK103831 and VICTR (UL1TR000445) from the NIH. E.C.R. holds a Career Award from the Burroughs Wellcome Fund. J.R.G. is supported by R01DK48370 and R01DK70856 from the NIH. S.W.C. and M.J.T. are funded by R01DK075555 and R01DK095811 from the NIH.
Abbreviations used:
- AKT
protein kinase B
- ATG5
autophagy related 5
- BafA
bafilomycin A1
- BECN1
beclin 1
- CALR
calreticulin
- Cav1
caveolin 1
- CDHR2
cadherin-related family member 2
- CQ
chloroquine
- DMSO
dimethyl sulfoxide
- EEA1
early endosome antigen 1
- EGF
epidermal growth factor
- ER
endoplasmic reticulum
- ERK
extracellular signal-regulated kinases
- FUCCI
fluorescence ubiquitination cell cycle indicator
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GOLGA1
golgin A1
- IMAC
intermicrovillar adhesion complex
- LAMP1
lysosomal-associated membrane protein 1
- LC3
microtubule-associated protein 1A/1B-light chain 3
- Lyz
lysozyme
- 3-MA
3-methyladenine
- mTOR
mammalian target of rapamycin
- Muc2
mucin 2
- OCT
optimum cutting temperature (compound)
- PAS
periodic acid–Schiff
- PCDH24
protocadherin 24
- PDMS
polydimethylsiloxane
- PFA
paraformaldehyde
- PI3K
phosphatidylinositol-3-kinase
- SI
sucrase-isomaltase
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
- Utr-CH
calponin homology domain of utrophin
- VACs
vacuolar apical compartments
- VAMP4
vesicle-associated membrane protein 4
- Vps34
vacuolar protein sorting 34.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E17-01-0021) on August 30, 2017.
REFERENCES
- Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med. 2009;361:2066–2078. doi: 10.1056/NEJMra0804647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adolph TE, Tomczak MF, Niederreiter L, Ko H-J, Böck J, Martinez-Naves E, Glickman JN, Tschurtschenthaler M, Hartwig J, Hosomi S, et al. Paneth cells as a site of origin for intestinal inflammation. Nature. 2013;503:272–276. doi: 10.1038/nature12599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baintner K. Demonstration of acidity in intestinal vacuoles of the suckling rat and pig. J Histochem Cytochem. 1994;42:231–238. doi: 10.1177/42.2.7507141. [DOI] [PubMed] [Google Scholar]
- Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, Brant SR, Silverberg MS, Taylor KD, Barmada MM, et al. Genome-wide association defines more than thirty distinct susceptibility loci for Crohn’s disease. Nat Genet. 2009;40:955–962. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bel S, Pendse M, Wang Y, Li Y, Ruhn KA, Hassell B, Leal T, Winter SE, Xavier RJ, Hooper LV. Paneth cells secrete lysozyme via secretory autophagy during bacterial infection of the intestine. Science. 2017;357:1047–1052. doi: 10.1126/science.aal4677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya S, Datta A, Berg JM, Gangopadhyay S. Studies on surface wettability of poly(dimethyl) siloxane (PDMS) and glass under oxygen-plasma treatment and correlation with bond strength. J Microelectromechanical Syst. 2005;14:590–597. [Google Scholar]
- Burkel BM, Von Dassow G, Bement WM. Versatile fluorescent probes for actin filaments based on the actin-binding domain of utrophin. Cell Motil Cytoskeleton. 2007;64:822–832. doi: 10.1002/cm.20226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan S, Ahmed Z, Bradfute SB, Arko-Mensah J, Mandell MA, Won Choi S, Kimura T, Blanchet F, Waller A, Mudd MH, et al. Pharmaceutical screen identifies novel target processes for activation of autophagy with a broad translational potential. Nat Commun. 2015;6:8620. doi: 10.1038/ncomms9620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Klionsky DJ. The regulation of autophagy—unanswered questions. J Cell Sci. 2011;124:161–170. doi: 10.1242/jcs.064576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong H, Lu C, Lindsten T, Thompson CB. Therapeutic targets in cancer cell metabolism and autophagy. Nat Biotechnol. 2012;30:671–678. doi: 10.1038/nbt.2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi M, Yi B, Oh S, Park D-J, Sung JH, Park S. A microfluidic cell culture device (μFCCD) to culture epithelial cells with physiological and morphological properties that mimic those of the human intestine. Biomed Microdevices. 2015;17:9966. doi: 10.1007/s10544-015-9966-5. [DOI] [PubMed] [Google Scholar]
- Cleyrat C, Darehshouri A, Steinkamp MP, Vilaine M, Boassa D, Ellisman MH, Hermouet S, Wilson BS. Mpl traffics to the cell surface through conventional and unconventional routes. Traffic. 2014;15:961–982. doi: 10.1111/tra.12185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codogno P, Mehrpour M, Proikas-Cezanne T. Canonical and non-canonical autophagy: variations on a common theme of self-eating? Nat Rev Mol Cell Biol. 2011;13:7–12. doi: 10.1038/nrm3249. [DOI] [PubMed] [Google Scholar]
- Conibear E, Stevens TH. Multiple sorting pathways between the late Golgi and the vacuole in yeast. Biochim Biophys Acta Mol Cell Res. 1998;1404:211–230. doi: 10.1016/s0167-4889(98)00058-5. [DOI] [PubMed] [Google Scholar]
- Crawley SW, Shifrin DA, Jr, Grega-Larson NE, McConnell RE, Benesh AE, Mao S, Zheng Y, Zheng QY, Nam KT, Millis BA, et al. Intestinal brush border assembly driven by protocadherin-based intermicrovillar adhesion. Cell. 2014;157:433–446. doi: 10.1016/j.cell.2014.01.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawley SW, Weck ML, Grega-Larson NE, Shifrin DA, Tyska MJ. ANKS4B is essential for intermicrovillar adhesion complex formation. Dev Cell. 2016;36:190–200. doi: 10.1016/j.devcel.2015.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dakik P, Titorenko VI. Communications between mitochondria, the nucleus, vacuoles, peroxisomes, the endoplasmic reticulum, the plasma membrane, lipid droplets, and the cytosol during yeast chronological aging. Front Genet. 2016;7:177. doi: 10.3389/fgene.2016.00177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J. 2011;30:4701–4711. doi: 10.1038/emboj.2011.398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florey O, Gammoh N, Kim SE, Jiang X, Overholtzer M. V-ATPase and osmotic imbalances activate endolysosomal LC3 lipidation. Autophagy. 2015;11:88–99. doi: 10.4161/15548627.2014.984277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz T, Niederreiter L, Adolph T, Blumberg RS, Kaser A. Crohn’s disease: NOD2, autophagy and ER stress converge. Gut. 2011;60:1580–1588. doi: 10.1136/gut.2009.206466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee HY, Noh SH, Tang BL, Kim KH, Lee MG. Rescue of ΔF508-CFTR trafficking via a GRASP-dependent unconventional secretion pathway. Cell. 2011;146:746–760. doi: 10.1016/j.cell.2011.07.021. [DOI] [PubMed] [Google Scholar]
- Gilbert T, Rodriguez-Boulan E. Induction of vacuolar apical compartments in the Caco-2 intestinal epithelial cell line. J Cell Sci. 1991;100(Pt 3):), 451–458. doi: 10.1242/jcs.100.3.451. [DOI] [PubMed] [Google Scholar]
- Henning SJ. Postnatal development: coordination of feeding, digestion, and metabolism. Am J Physiol. 1981;241:G199–G214. doi: 10.1152/ajpgi.1981.241.3.G199. [DOI] [PubMed] [Google Scholar]
- Hinkel I, Duluc I, Martin E, Guenot D, Freund J-N, Gross I. Cdx2 controls expression of the protocadherin Mucdhl, an inhibitor of growth and β-catenin activity in colon cancer cells. Gastroenterology. 2012;142:875–885.e3. doi: 10.1053/j.gastro.2011.12.037. [DOI] [PubMed] [Google Scholar]
- Hudspeth AJ. Integrating the active process of hair cells with cochlear function. Nat Rev Neurosci. 2014;15:600–614. doi: 10.1038/nrn3786. [DOI] [PubMed] [Google Scholar]
- Ishikawa T, Sato T, Mohit G, Imai Y, Yamaguchi T. Transport phenomena of microbial flora in the small intestine with peristalsis. J Theor Biol. 2011;279:63–73. doi: 10.1016/j.jtbi.2011.03.026. [DOI] [PubMed] [Google Scholar]
- Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaser A, Blumberg RS. Autophagy, microbial sensing, endoplasmic reticulum stress, and epithelial function in inflammatory bowel disease. Gastroenterology. 2011;140:1738–1747. doi: 10.1053/j.gastro.2011.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaser A, Lee A, Franke A, Glickman JN, Tilg H, Nieuwenhuis EES, Higgins DE, Glimcher LH, Blumberg RS. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell. 2008;134:743–756. doi: 10.1016/j.cell.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaldoun SA, Emond-Boisjoly M-A, Chateau D, Carrière V, Lacasa M, Rousset M, Demignot S, Morel E. Autophagosomes contribute to intracellular lipid distribution in enterocytes. Mol Biol Cell. 2014;25:118–132. doi: 10.1091/mbc.E13-06-0324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;474:307–317. doi: 10.1038/nature10209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Huh D, Hamilton G, Ingber DE. Human gut-on-a-chip inhabited by microbial flora that experiences intestinal peristalsis-like motions and flow. Lab Chip. 2012;12:2165–2174. doi: 10.1039/c2lc40074j. [DOI] [PubMed] [Google Scholar]
- Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3:452–460. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- Kimura T, Jia J, Kumar S, Choi SW, Gu Y, Mudd M, Dupont N, Jiang S, Peters R, Farzam F, et al. Dedicated SNAREs and specialized TRIM cargo receptors mediate secretory autophagy. EMBO J. 2017;36:42–60. doi: 10.15252/embj.201695081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles BC, Roland JT, Krishnan M, Tyska MJ, Lapierre LA, Dickman PS, Goldenring JR, Shub MD. Myosin Vb uncoupling from RAB8A and RAB11A elicits microvillus inclusion disease. J Clin Invest. 2014;124:2947–2962. doi: 10.1172/JCI71651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- Lamb CA, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol. 2013;14:759–774. doi: 10.1038/nrm3696. [DOI] [PubMed] [Google Scholar]
- Lamoureux F, Thomas C, Crafter C, Kumano M, Zhang F, Davies BR, Gleave ME, Zoubeidi A. Blocked autophagy using lysosomotropic agents sensitizes resistant prostate tumor cells to the novel Akt inhibitor AZD5363. Clin Cancer Res. 2013;19:833–844. doi: 10.1158/1078-0432.CCR-12-3114. [DOI] [PubMed] [Google Scholar]
- LaRock DL, Chaudhary A, Miller SI. Salmonellae interactions with host processes. Nat Rev Microbiol. 2015;13:191–205. doi: 10.1038/nrmicro3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau KS, Cortez-Retamozo V, Philips SR, Pittet MJ, Lauffenburger DA, Haigis KM. Multi-scale in vivo systems analysis reveals the influence of immune cells on TNF-α-induced apoptosis in the intestinal epithelium. PLoS Biol. 2012;10:e1001393. doi: 10.1371/journal.pbio.1001393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau KS, Schrier SB, Gierut J, Lyons J, Lauffenburger DA, Haigis KM. Network analysis of differential Ras isoform mutation effects on intestinal epithelial responses to TNF-α. Integr Biol (Camb) 2013;5:1355–1365. doi: 10.1039/c3ib40062j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lentle RG, Janssen PWM. Physical characteristics of digesta and their influence on flow and mixing in the mammalian intestine: a review. J Comp Physiol B. 2008;178:673–690. doi: 10.1007/s00360-008-0264-x. [DOI] [PubMed] [Google Scholar]
- Li J, He Y, Lu Q, Zhang M. Mechanistic basis of organization of the Harmonin/USH1C-mediated brush border microvilli tip-link complex. Dev Cell. 2016;36:179–189. doi: 10.1016/j.devcel.2015.12.020. [DOI] [PubMed] [Google Scholar]
- Lipatova Z, Segev N. A role for macro-ER-phagy in ER quality control. PLoS Genet. 2015;11:e1005390. doi: 10.1371/journal.pgen.1005390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T-C, Naito T, Liu Z, Van Dussen KL, Haritunians T, Li D, Endo K, Kawai Y, Nagasaki M, Kinouchi Y, et al. LRRK2 but not ATG16L1 is associated with Paneth cell defect in Japanese Crohn’s disease patients. JCI Insight. 2017;2:e91917. doi: 10.1172/jci.insight.91917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackeh R, Perdiz D, Lorin S, Codogno P, Poüs C. Autophagy and microtubules—new story, old players. J Cell Sci. 2013;126:1071–1080. doi: 10.1242/jcs.115626. [DOI] [PubMed] [Google Scholar]
- Martinez J, Cunha LD, Park S, Yang M, Lu Q, Orchard R, Li QZ, Yan M, Janke L, Guy C, et al. Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature. 2016;533:115–119. doi: 10.1038/nature17950. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Martinez-Martin N, Maldonado P, Gasparrini F, Frederico B, Aggarwal S, Gaya M, Tsui C, Burbage M, Keppler SJ, Montaner B, et al. A switch from canonical to noncanonical autophagy shapes B cell responses. Science. 2017;355:641–647. doi: 10.1126/science.aal3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauvezin C, Nagy P, Juhasz G, Neufeld TP. Autophagosome-lysosome fusion is independent of V-ATPase-mediated acidification. Nat Commun. 2015;6:7007. doi: 10.1038/ncomms8007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura S, Sato K, Kato-Negishi M, Teshima T, Takeuchi S. Fluid shear triggers microvilli formation via mechanosensitive activation of TRPV6. Nat Commun. 2015;6:8871. doi: 10.1038/ncomms9871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moxey PC, Trier JS. Development of villus absorptive cells in the human fetal small intestine: a morphological and morphometric study. Anat Rec. 1979;195:463–482. doi: 10.1002/ar.1091950307. [DOI] [PubMed] [Google Scholar]
- Muncan V, Heijmans J, Krasinski SD, Büller NV, Wildenberg ME, Meisner S, Radonjic M, Stapleton KA, Lamers WH, Biemond I, et al. Blimp1 regulates the transition of neonatal to adult intestinal epithelium. Nat Commun. 2011;2:452. doi: 10.1038/ncomms1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nascimbeni AC, Fanin M, Masiero E, Angelini C, Sandri M. The role of autophagy in the pathogenesis of glycogen storage disease type II (GSDII) Cell Death Differ. 2012;19:1698–1708. doi: 10.1038/cdd.2012.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naydenov NG, Harris G, Morales V, Ivanov AI. Loss of a membrane trafficking protein αSNAP induces non-canonical autophagy in human epithelia. Cell Cycle. 2012;11:4613–4625. doi: 10.4161/cc.22885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson-Fish JC, Kokotos AC, Gillingwater TH, Smillie KJ, Cousin MA. VAMP4 is an essential cargo molecule for activity-dependent bulk endocytosis. Neuron. 2015;88:973–984. doi: 10.1016/j.neuron.2015.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda K, Ikehara Y. Weakly basic amines inhibit the proteolytic conversion of proalbumin to serum albumin in cultured rat hepatocytes. Eur J Biochem. 1985;152:605–609. doi: 10.1111/j.1432-1033.1985.tb09238.x. [DOI] [PubMed] [Google Scholar]
- Orhon I, Dupont N, Zaidan M, Boitez V, Burtin M, Schmitt A, Capiod T, Viau A, Beau I, Kuehn EW, et al. Primary-cilium-dependent autophagy controls epithelial cell volume in response to fluid flow. Nat Cell Biol. 2016;18:657–667. doi: 10.1038/ncb3360. [DOI] [PubMed] [Google Scholar]
- Overholtzer M, Mailleux AA, Mouneimne G, Normand G, Schnitt SJ, King RW, Cibas ES, Brugge JS. A nonapoptotic cell death process, entosis, that occurs by cell-in-cell invasion. Cell. 2007;131:966–979. doi: 10.1016/j.cell.2007.10.040. [DOI] [PubMed] [Google Scholar]
- Peterson MD, Bement WM, Mooseker MS. An in vitro model for the analysis of intestinal brush border assembly. II. Changes in expression and localization of brush border proteins during cell contact-induced brush border assembly in Caco-2BBe cells. J Cell Sci. 1993;105(Pt 2):461–472. doi: 10.1242/jcs.105.2.461. [DOI] [PubMed] [Google Scholar]
- Peterson MD, Mooseker MS. Characterization of the enterocyte-like brush border cytoskeieton of the C2 BBe clones of the human intestinal cell line. Caco-2. 1992;600:581–600. doi: 10.1242/jcs.102.3.581. [DOI] [PubMed] [Google Scholar]
- Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol. 2010;12:747–757. doi: 10.1038/ncb2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razi M, Chan EYW, Tooze SA. Early endosomes and endosomal coatomer are required for autophagy. J Cell Biol. 2009;185:305–321. doi: 10.1083/jcb.200810098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 2008;132:487–498. doi: 10.1016/j.cell.2007.12.033. [DOI] [PubMed] [Google Scholar]
- Schaaf MBE, Keulers TG, Vooijs MA, Rouschop KMA. LC3/GABARAP family proteins: autophagy-(un)related functions. FASEB J. 2016;30:3961–3978. doi: 10.1096/fj.201600698R. [DOI] [PubMed] [Google Scholar]
- Schnabl KL, Field C, Clandinin MT. Ganglioside composition of differentiated Caco-2 cells resembles human colostrum and neonatal rat intestine. Br J Nutr. 2009;101:694–700. doi: 10.1017/S0007114508048289. [DOI] [PubMed] [Google Scholar]
- Schwerd T, Pandey S, Yang HT, Bagola K, Jameson E, Jung J, Lachmann RH, Shah N, Patel SY, Booth C, et al. Impaired antibacterial autophagy links granulomatous intestinal inflammation in Niemann-Pick disease type C1 and XIAP deficiency with NOD2 variants in Crohn’s disease. Gut. 2016;66:1060–1073. doi: 10.1136/gutjnl-2015-310382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedwick C. The fly muscles in on glycogen autophagy. PLoS Biol. 2013;11:1–2. doi: 10.1371/journal.pbio.1001709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990–995. doi: 10.1126/science.1099993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons AJ, Banerjee A, McKinley ET, Scurrah CR, Herring CA, Gewin LS, Masuzaki R, Karp SJ, Franklin JL, Gerdes MJ, et al. Cytometry-based single-cell analysis of intact epithelial signaling reveals MAPK activation divergent from TNF-α-induced apoptosis in vivo. Mol Syst Biol. 2015;11:835. doi: 10.15252/msb.20156282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinadinos C, Valles-Ortega J, Boulan L, Solsona E, Tevy MF, Marquez M, Duran J, Lopez-Iglesias C, Calbó, Blasco E, et al. Neuronal glycogen synthesis contributes to physiological aging. Aging Cell. 2014;13:935–945. doi: 10.1111/acel.12254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skrzypek T, Valverde Piedra JL, Skrzypek H, Kazimierczak W, Biernat M, Zabielski R. Gradual disappearance of vacuolated enterocytes in the small intestine of neonatal piglets. J Physiol Pharmacol. 2007;58(Suppl 3):87–95. [PubMed] [Google Scholar]
- Sorkin A, Von Zastrow M. Signal transduction and endocytosis: close encounters of many kinds. Nat Rev Mol Cell Biol. 2002;3:600–614. doi: 10.1038/nrm883. [DOI] [PubMed] [Google Scholar]
- Tooze SA, Abada A, Elazar Z. Endocytosis and autophagy: exploitation or cooperation? Cold Spring Harb Perspect Biol. 2014;6:a018358. doi: 10.1101/cshperspect.a018358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tooze SA, Yoshimori T. The origin of the autophagosomal membrane. Nat Cell Biol. 2010;12:831–835. doi: 10.1038/ncb0910-831. [DOI] [PubMed] [Google Scholar]
- Tourneur E, Chassin C. Neonatal immune adaptation of the gut and its role during infections. Clin Dev Immunol. 2013;2013 doi: 10.1155/2013/270301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utech M, Ivanov AI, Samarin SN, Bruewer M, Turner JR, Mrsny RJ, Parkos CA, Nusrat A. Mechanism of IFN-γ-induced endocytosis of tight junction proteins: myosin II-dependent vacuolarization of the apical plasma membrane. Mol Biol Cell. 2005;16:5040–5052. doi: 10.1091/mbc.E05-03-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandussen KL, Liu T-C, Li D, Towfic F, Modiano N, Winter R, Haritunians T, Taylor KD, Dhall D, Targan SR, et al. Genetic variants synthesize to produce paneth cell phenotypes that define subtypes of Crohn’s disease. Gastroenterology. 2014;146:200–209. doi: 10.1053/j.gastro.2013.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weck ML, Crawley SW, Stone CR, Tyska MJ. Myosin-7b promotes distal tip localization of the intermicrovillar adhesion complex. Curr Biol. 2016;26:2717–2728. doi: 10.1016/j.cub.2016.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis GV, Knowles BC, Choi E, Goldstein AE, Williams JA, Manning EH, Roland JT, Lapierre LA, Goldenring JR. Loss of MYO5B in mice recapitulates Microvillus Inclusion Disease and reveals an apical trafficking pathway distinct to neonatal duodenum. Cell Mol Gastroenterol Hepatol. 2016;2:131–157. doi: 10.1016/j.jcmgh.2015.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, Richter B, Korac J, Waidmann O, Choudhary C, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011;333:228–233. doi: 10.1126/science.1205405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimori T, Yamamoto A, Moriyama Y, Futai M, Tashiro Y. Bafilomycin A1, a specific inhibitor of vacuolar-type H+-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J Biol Chem. 1991;266:17707–17712. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.