

Heterozygous mutations in caveolin-1 (CAV1) have been linked to pulmonary arterial hypertension (PAH), but their impact on caveolae is unclear. We show that a PAH-associated frameshift mutation introduces an endoplasmic reticulum retention signal in CAV1 that partially disrupts caveolae assembly and interferes with their ability to serve as membrane buffers.
Abstract
Caveolin-1 (CAV1) is an essential component of caveolae and is implicated in numerous physiological processes. Recent studies have identified heterozygous mutations in the CAV1 gene in patients with pulmonary arterial hypertension (PAH), but the mechanisms by which these mutations impact caveolae assembly and contribute to disease remain unclear. To address this question, we examined the consequences of a familial PAH-associated frameshift mutation in CAV1, P158PfsX22, on caveolae assembly and function. We show that C-terminus of the CAV1 P158 protein contains a functional ER-retention signal that inhibits ER exit and caveolae formation and accelerates CAV1 turnover in Cav1–/– MEFs. Moreover, when coexpressed with wild-type (WT) CAV1 in Cav1–/– MEFs, CAV1-P158 functions as a dominant negative by partially disrupting WT CAV1 trafficking. In patient skin fibroblasts, CAV1 and caveolar accessory protein levels are reduced, fewer caveolae are observed, and CAV1 complexes exhibit biochemical abnormalities. Patient fibroblasts also exhibit decreased resistance to a hypo-osmotic challenge, suggesting the function of caveolae as membrane reservoir is compromised. We conclude that the P158PfsX22 frameshift introduces a gain of function that gives rise to a dominant negative form of CAV1, defining a new mechanism by which disease-associated mutations in CAV1 impair caveolae assembly.
INTRODUCTION
Caveolae are specialized plasma membrane domains rich in cholesterol (Rothberg et al., 1992; Parton and del Pozo, 2013; Chaudhary et al., 2014). Found at the plasma membrane of many cell types, these 50- to 100-nm invaginations are especially highly abundant in adipocytes, smooth muscle, and endothelial cells (Ariotti and Parton, 2013). Caveolae have numerous cellular functions, including buffering cells from mechanical stress, maintaining membrane integrity, and regulating clathrin-independent endocytosis (Parton and del Pozo, 2013; Chaudhary et al., 2014). They also are thought to control a variety of signal transduction pathways by mechanisms that are currently a matter of debate (Collins et al., 2012; Parton and del Pozo, 2013; Cheng and Nichols, 2016).
The primary structural components of caveolae are a family of proteins known as caveolins. Caveolins 1, 2, and 3 are integral membrane proteins whose cytoplasmically oriented N- and C-termini are connected by a predicted hairpin (Parton et al., 2006). Caveolin-1 (CAV1) is a 178-amino-acid protein that was the first protein identified as a component of caveolae (Rothberg et al., 1992) and that also is required for caveolae formation (Galbiati et al., 2001; Razani et al., 2001; Kirkham et al., 2008). Caveolae biogenesis begins in the endoplasmic reticulum through a stepwise series of oligomerization events (Monier et al., 1995; Scheiffele et al., 1998; Hayer et al., 2010a; Busija et al., 2017). Newly synthesized caveolin becomes incorporated into complexes composed of 14–16 CAV1 monomers (Monier et al., 1995) that correspond to an 8S complex (Hayer et al., 2010a). This first oligomerization event occurs in the endoplasmic reticulum (ER) and the oligomers are quickly transported to the Golgi complex for further maturation. In the Golgi compartment, 8S complexes undergo another round of oligomerization to form 70S scaffolds of CAV1 oligomers that become enriched in cholesterol and lipids before being trafficked to the plasma membrane (Hayer et al., 2010a). Proper caveolae assembly also requires the presence of several accessory proteins, including the cavins, PACSIN2, and EHD-2 (Vinten et al., 2005; Hill et al., 2008; Liu et al., 2008; Hansen et al., 2011; Senju et al., 2011; Moren et al., 2012; Stoeber et al., 2012; Ludwig et al., 2013; Kovtun et al., 2015).
The 178-amino-acid CAV1 protein consists of an N-terminal domain, scaffolding domain, intramembrane domain, and C-terminal domain (Parton et al., 2006; Root et al., 2015). The functions of the various domains of CAV1 have been extensively studied via truncation and mutagenesis experiments (Song et al., 1997; Schlegel and Lisanti 2000). For example, the C-terminus of CAV1 (residues 135–178) is important for membrane attachment, exit from the Golgi complex and homotypic interactions between caveolin complexes to form 70S scaffolds (Song et al., 1997; Ren et al., 2004; Hayer et al., 2010a). This portion of CAV1 is also speculated to be important for proper trafficking of the protein, because C-terminal truncations and other mutant forms of CAV1 often accumulate in the Golgi compartment when ectopically expressed (Luetterforst et al., 1999; Machleidt et al., 2000; Lee et al., 2002; Ren et al., 2004).
The functions of both CAV1 and caveolae have been extensively studied using knockout mice, and work in this animal model has highlighted an importance for CAV1 in modulating proliferation, adipose and vascular homeostasis, and metabolism (Razani et al., 2001, 2002b; Cohen et al., 2003a, b, 2004). CAV1 and caveolae have been linked to a number of diseases such as lung injury/disease, myopathies, lipodystrophy, cardiovascular disease, and cancer (Razani and Lisanti, 2001; Lee et al., 2002). However, the mechanisms by which defects in CAV1 or caveolae give rise to disease are still poorly understood.
One of the many diseases in which CAV1 is implicated is pulmonary arterial hypertension (PAH) (Mathew et al., 2004, 2007; Mathew, 2011; Patel et al., 2007; Austin et al., 2012; Austin and Loyd 2014; Bakhshi et al., 2013; Garg et al., 2015; Prewitt et al., 2015; Schrauwen et al., 2015; Han, Copeland, et al., 2016). PAH is a fatal disease arising from progressive right ventricular failure that is induced by progressive increase of pulmonary vascular resistance (Hoeper et al., 2013). Most cases of familial PAH with a known etiology involve mutations in BMPR2 (Austin and Loyd, 2014), although the details of the molecular and cellular pathogenesis of the disease remain unclear. However, many of the signaling pathways disrupted in PAH are regulated by caveolin (Mathew et al., 2007; Patel et al., 2007) and decreases in the expression of CAV1 and number of caveolae have been reported in endothelial cells in clinical and experimental cases of PAH (Drab et al., 2001; Jasmin et al., 2006; Mathew, 2011; Bakhshi et al., 2013; Austin and Loyd, 2014).
In recent years, genetic analysis of PAH patients has revealed several heterozygous mutations in the C-terminus of CAV1, further implicating a critical role for both CAV1 and caveolae in this disease (Austin et al., 2012; Garg et al., 2015; Schrauwen et al., 2015; Han, Copeland, et al., 2016). One set of mutations consists of novel heterozygous frameshift mutations in CAV1 in both a family with PAH and a patient with idiopathic form of the disease (P158PfsX22 and P158HfsX22, respectively) (Austin et al., 2012). Both of these frameshift mutations are predicted to generate a mutant form of CAV1 that is one amino acid longer than the wild-type protein and contains a novel C-terminus (residues 159–179), and the predicted protein sequences of the familial and idiopathic mutations are nearly identical with the exception of the first residue (Austin et al., 2012). A second type of C-terminal mutation, discovered in a patient with both PAH and congenital generalized lipodystrophy, leads to the generation of a truncated protein missing the final 19 residues (F160X) (Garg et al., 2015; Schrauwen et al., 2015; Han, Copeland, et al., 2016).
The mechanisms by which these mutant forms of CAV1 contribute to disease are not yet clear. Interestingly, however, they appear to have distinct effects on CAV1 trafficking and caveolae assembly. For example, wild-type numbers of caveolae are observed in patient skin fibroblasts heterozygous for F160X CAV1 (Garg et al., 2015; Han, Copeland, et al., 2016), and the F160X CAV1 mutant is correctly targeted to caveolae when expressed in Cav1–/– MEFs (Han, Copeland, et al., 2016). In contrast, in skin fibroblasts isolated from patients heterozygous for the familial P158PfsX22 mutation (hereafter referred to as CAV1 P158), reduced numbers of caveolae are observed (Marsboom et al., 2017), and CAV1 P158 localizes to the endoplasmic reticulum when expressed in Cav1–/– MEFs (Marsboom et al., 2017). The basis for these differences is unknown but could potentially reflect either a loss of function or gain of function resulting from the introduction of a novel C-terminus for CAV1 P158. Here we test this idea.
RESULTS
CAV1 P158 contains a functional ER retention signal
Defects in the intracellular trafficking of CAV1 typical result in the accumulation of the protein in either the Golgi complex or aggresomes (Luetterforst et al., 1999; Machleidt et al., 2000; Lee et al., 2002; Ren et al., 2004; Tiwari et al., 2016). In contrast, the P158 mutant has been reported to be excluded from caveolae, instead localizing to the endoplasmic reticulum (ER) (Marsboom et al., 2017). Given that the frameshift introduces a novel C-terminus, we considered the possibility that the mutation may have introduced a sorting signal. C-terminal dilysine motifs such as KKXX and KXKXX function as sorting signals that cause proteins to be retained in or retrieved to the ER (Jackson et al., 1993; Cosson et al., 1998). Inspection of the amino acid sequence of the novel sequence introduced by the frameshift in CAV1 P158 revealed a putative ER retention signal at its C-terminus (amino acids 176–179) (Table 1).
TABLE 1:
C-terminal amino acid sequences of CAV1 constructs.
| Name | Amino acid sequence |
|---|---|
| Wild-type CAV1 | 158 PLFEAVGKIFSNVRINLQKEI-178 |
| CAV1-P158 | 158 PSLKLLGKYSAMSASTCRKKYK 179 |
| CAV1-P158-AAYK | 158 PSLKLLGKYSAMSASTCRAAYK 179 |
| CAV1-P158-ΔKKYK | 158 PSLKLLGKYSAMSASTCR––175 |
| CAV1-KKYK | 158 PLFEAVGKIFSNVRINLQKEIKKYK 182 |
In a previous study, Ostermeyer et al. (2001) reported that a CAV1 construct bearing a C-terminal dilysine motif (CAV1-KKSL) was sequestered in the ER and as a consequence was also targeted to lipid droplets. Consistent with this, we found that CAV1 P158, but not wild-type (WT) CAV1, was not only localized to the ER as previously reported but was also present in punctate structures that strongly colocalized with adipose differentiation related protein (ADRP), a marker of lipid droplets (Figure 1, A, B, F, and G). Unlike WT CAV1, CAV1 P158 also failed to colocalize with cavin-1 (Figure 2, A–C and G), consistent with a recent report (Marsboom et al., 2017).
FIGURE 1:

CAV1 P158 is targeted to the ER and lipid droplets as the result of the introduction of a functional ER retention signal by the frameshift mutation. (A–E) Immunofluorescence staining of WT CAV1, CAV1-P158, CAV1-P158-AAYK, CAV1-P158-ΔKKYK, and CAV1-KKYK (green) expressed in Cav1–/– MEFs. Cells were colabeled with the ER marker calreticulin (CalR, red) and the lipid droplet marker ADRP (cyan). The dashed box indicates the region shown in the zooms. Scale bars = 10 μm. (F, G) Quantification of the extent of colocalization between CAV1 constructs with CalR or ADRP, respectively, calculated by Pearson’s correlation. The data were analyzed with a nonparametric Kluskal–Wallis test and post hoc Dunn’s multiple comparisons test to calculate p values. Data are averaged over three independent experiments for the following numbers of regions of interest (ROIs): Calreticulin/WT CAV1, n = 33; calreticulin/CAV1-P158, n = 30; calreticulin/CAV1-P158-AAYK, n = 52; calreticulin/CAV1-P158-ΔKKYK, n = 43; calreticulin/CAV1-KKYK, n = 40; ADRP/WT CAV1, n = 85; ADRP/CAV1-P158, n = 73; ADRP/CAV1-P158-AAYK, n = 104; ADRP/CAV1-P158-ΔKKYK, n = 107; and ADRP/CAV1-KKYK, n = 114. Asterisks (*) and hashtags (#) indicate statistically significant differences compared with wild-type CAV1 and CAV1-P158, respectively. ***/###, p < 0.001.
FIGURE 2:
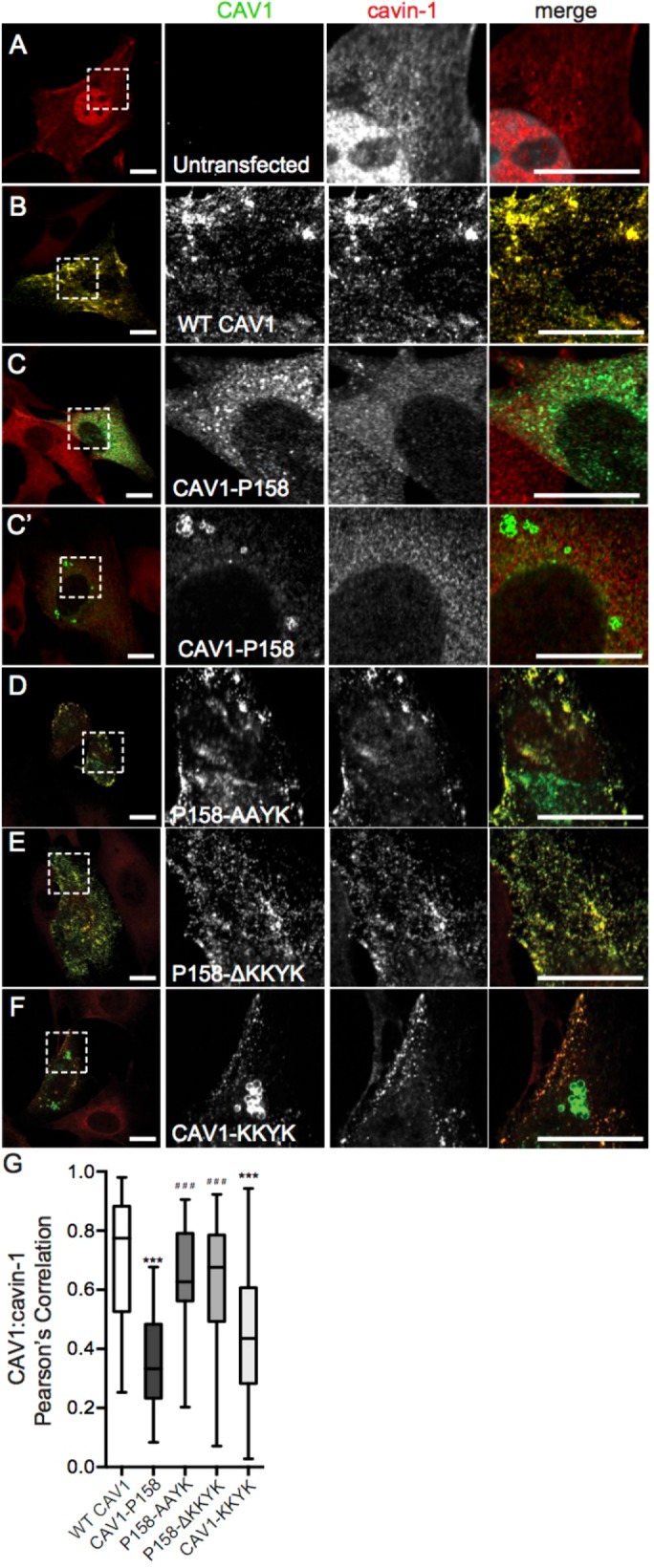
Disruption of the KKYK motif of CAV1-P158 enables the protein to traffic to caveolae. (A) Endogenous cavin-1 immunofluorescence in Cav1–/– MEFs costained with a CAV1 antibody. Note the diffuse distribution of cavin-1 in the absence of Cav1 expression. (B–F) Endogenous cavin-1 immunofluorescence in transfected Cav1–/– MEFs. (B) WT CAV1 colocalizes with endogenous cavin-1 in Cav1–/– MEFs. (C, C′) CAV1-P158 fails to colocalize with cavin-1 and is instead distributed in a diffuse/reticular pattern (C) and/or localizes to vesicular structures with open lumens (C′). (D) CAV1-P158-AAYK colocalizes with cavin-1. (E) CAV1-P158Δ-KKYK also colocalizes with cavin-1. (F) CAV1-KKYK partially colocalizes with cavin-1 but is also found in lipid droplets and/or the ER (also see Figure 1). (G) Quantification of the extent of colocalization between cavin-1 and CAV1 constructs as calculated by Pearson’s correlation. Data are averaged over two to three independent experiments for the following numbers of ROIs: WT CAV1, n = 38; CAV1-P158, n = 40; CAV1-P158-AAYK, n = 52; CAV1-P158Δ-KKYK, n = 46; and CAV1-KKYK, n = 52. Kluskal–Wallis nonparametric ANOVA and a post hoc Dunn’s multiple comparisons test were performed to determine p values. Asterisks (*) and hashtags (#) indicate statistically significant differences compared with wild-type CAV1 and CAV1-P158, respectively. ***/###, p < 0.001. Scale bars = 10 μm.
To test whether the putative dilysine motif in the C-terminus of CAV1 P158 functions as an ER-retention/retrieval signal, we performed mutagenesis to either disrupt or truncate the dilysine motif (Table 1). The constructs were then individually transfected into Cav1–/– MEFs and the cells were then fixed and stained with either ADRP and calreticulin (Figure 1, C and D) or cavin-1 (Figure 2, D and E). Wild-type CAV1 served as a control for normal trafficking and CAV1 P158 served as a control for ER retention and lipid droplet localization. Both CAV1 P158-AAYK and CAV1 P158-ΔKKYK mutants were excluded from the ER and lipid droplets (Figure 1, C, D, F, and G) and strongly colocalized with cavin1 (Figure 2, D, E, and G). This suggests that the putative dilysine motif in CAV1 P158 indeed functions as an ER-retention/retrieval signal and inhibits delivery of the mutant protein to caveolae.
We next tested whether the KKYK motif is sufficient to redirect WT CAV1 to the ER using a construct consisting of WT CAV1 appended with the CAV1 P158 dilysine motif on the C-terminus (Table 1 and Figures 1E and 2F). Similar to the results of Ostermeyer et al. (2001), we found that CAV1-KKYK was readily detectible in lipid droplets (Figure 1, E and G) but also colocalized with Cavin1 to some degree when expressed in Cav1–/– MEFs (Figure 2, F and G). This is different from the behavior of CAV1 P158, which appeared to be primarily excluded from Cavin1-positive puncta (Figure 2, C, C′, and G). Furthermore, CAV1-KKYK was largely excluded from the ER (Figure 1, E and F), whereas CAV1 P158 was at least partially localized to the ER (Figure 1, F and G). These differences suggest that both the ER retention motif and the additional residues introduced by the frameshift modulate the intracellular trafficking and localization of CAV1 P158.
Coexpression of CAV1 P158 with WT CAV1 partially inhibits delivery of WT CAV1 to caveolae
Patients harboring the CAV1 P158 mutation also have a wild-type CAV1 gene (Austin et al., 2012). Recent work suggests that WT CAV1 interacts directly with CAV1 P158 and that the two partially colocalize in caveolae upon their coexpression in Cav1–/– MEFs (Marsboom et al., 2017). However, in patient skin fibroblasts, the number of caveolae is decreased relative to controls (Marsboom et al., 2017), suggesting that not all of the CAV1 is ultimately delivered to caveolae. We therefore wondered whether expression of CAV1 P158 might also prevent the trafficking of WT CAV1 to caveolae.
To test this idea, we carried out cotransfection experiments using equivalent amounts of cDNA for each construct. Cells were costained for cavin-1 to mark caveolae, calreticulin to mark the ER, and ADRP as a lipid droplet marker. As a positive control, we cotransfected cells with Myc-tagged WT CAV1 and HA-tagged WT CAV1. As expected, both were detectible together at the plasma membrane in Cavin-1 positive structures (Figure 3, A and C) and were also excluded from both lipid droplets and the ER (Figure 3, D, F, G, and I). However, in cells cotransfected with WT CAV1 and CAV1 P158, CAV1 P158 only weakly colocalized with Cavin 1 (Figure 3, B and C) and some CAV1 P158P was still associated with the ER and lipid droplets (Figure 3, E, F, H, and I).
FIGURE 3:

Coexpressed WT CAV1 and CAV1-P158 partially colocalize in the ER and lipid droplets. Cav1–/– MEFs coexpressing wild-type CAV1 constructs (WT+WT, white bars in graphs) or wild-type and mutant constructs (WT+P158, gray bars in graphs) were costained with cavin-1 (A, B), calreticulin (D, E), or ADRP (G, H). WT HA-CAV1 or HA-CAV1-P158 staining is shown in green, WT Myc-CAV1 staining is red, and endogenous cavin-1, calreticulin, or ADRP staining is shown in cyan. Scale bars, 10 μm. (C, F, I) Quantification of the extent of colocalization between the indicated CAV1 constructs and cavin-1, calreticulin, or ADRP as calculated by Pearson’s correlation. Data were averaged over two to three independent experiments for the following numbers of cells: cavin-1, 114 WT/WT and 30/44 WT/P158 cells; CalR, 40 WT/WT and 39 WT/P158 cells; ADRP, 18 WT/WT and 26 WT/P158 cells. Ordinary one-way ANOVA and post hoc Bonferroni’s multiple comparisons test were performed to determine p values. n.s., not significant; ***, p < 0.001. Scale bars = 10 μm.
Interestingly, the trafficking of WT CAV1 to the plasma membrane was also partially impaired in the presence of the mutant protein in CAV1–/– MEFs. In particular, in cells coexpressing WT CAV1 and CAV1 P158, a significantly increased amount of WT CAV1 was observed in the ER (Figure 3, E and F) and lipid droplets (Figure 3, H and I). This suggests that the P158 mutant behaves in a dominant-negative manner by causing the partial retention of WT CAV1 in the ER and lipid droplets.
To address how exactly the CAV1 P158 mutant functions as a dominant negative against WT CAV1, we assessed the ability of CAV1 P158 and WT CAV1 to oligomerize. WT CAV1 normally undergoes a series of homo- and hetero-oligomerization events, leading to the formation of 8S and 70S complexes (Hayer et al., 2010a). To test whether these complexes form normally, we performed sucrose gradient fractionation of Cav1–/– MEFs extracted with 0.5% Triton X-100 (TX-100). As expected, when expressed individually WT CAV1 was found in both 8S and 70S complexes (Figure 4A, left panel). In contrast, individually expressed CAV1 P158 was found predominantly at the bottom of the gradient, indicating it associates with very large complexes (Figure 4A, left panel). However, when cells were cotransfected with WT CAV1 and CAV1 P158, CAV1 P158 partially redistributed to 8S and 70S complexes (Figure 4A, right panel). This strongly suggests it is capable of forming hetero-oligomeric complexes with WT CAV1 and thus may affect the behavior of WT CAV1 via direct interactions. Some high-molecular-weight complexes of CAV1 P158 were still apparent under these conditions, however, again demonstrating that the mutant was not completely rescued by coexpression with WT CAV1.
FIGURE 4:

CAV1-P158 partially cofractionates with WT CAV1 in coexpressing cells. (A, B) Cav1–/– MEFs transfected with WT CAV1, P158-CAV1, or WT CAV1 + P158-CAV1 were lysed in either (A) 0.5% TX-100 or (B) 0.2% TX-100 + 0.4% SDS at room temperature. Extracts were run through 10–40% sucrose velocity gradients, and fractions were analyzed by SDS–PAGE/Western blot. The positions of the 8S and 70S complexes for WT CAV1 are indicated. (C) DRM analysis of Cav1–/– MEFs expressing WT CAV1, CAV1-P158, or WT CAV1 + CAV1-P158. The position of the DRM fractions is indicated. For the cotransfected cells, blotting was performed for each construct individually using tag-specific antibodies as well as using a Cav1 antibody to facilitate comparison with results for patient fibroblasts, where we could not differentiate between the WT and mutant protein (see below). The higher-molecular-weight bands detected by the myc antibody in fractions 12–14 in C were not observed when the blots were probed with an anti-caveolin antibody and thus likely represent cross-reaction to an unknown antigen. Data are representative of two independent experiments.
We wondered whether the high-molecular-weight complexes of CAV1 P158 consist of 8S complexes. To test this, we extracted them under conditions previously shown to dissociate 70S complexes using a combination of TX-100 and SDS (Hayer et al., 2010a). The large CAV1-P158 complexes were solubilized to oligomers smaller than 8S in size under these conditions (Figure 4B), suggesting that they are either not organized correctly or are unstable.
Since caveolae are normally detergent resistant (Parton and Simons 2007), we next examined whether CAV1 P158 associates with detergent-resistant membranes (DRMs) and found that it fails to do so (Figure 4C, left panel). Coexpression of WT CAV1 and CAV1 P158 led to an increased amount of CAV1 P158 in DRMs, suggesting the wild-type protein may partially rescue targeting of CAV1 P158 to DRMs (Figure 4C, right panel).
CAV1 P158 has a shortened half-life compared with WT CAV1 in reconstituted MEFs
When associated properly with caveolae, endogenous CAV1 has a long half-life, up to 36 h (Hayer et al., 2010b). In contrast, mutant forms of caveolin family members often exhibit reduced half-lives (Galbiati et al., 1999, 2000). To determine whether this is also the case for CAV1 P158, we assessed the half-life of the transfected proteins to determine whether the mutant protein is turned over more rapidly that WT CAV1. For these experiments, Cav1–/– MEFs were transfected with CAV1 or CAV1 P158, cotransfected with two WT CAV1 constructs, or cotransfected with WT CAV1 and CAV1 P158. CAV1-P132L, which is known to turn over rapidly (Hanson et al., 2013), was studied as a control. After transfection, cells were treated with cycloheximide (CHX) to inhibit new protein synthesis, and the CAV1 protein levels were measured at different time points by Western blotting. Endogenous Cav1 in WT Cav1+/+ MEFs was also studied as a control.
The half-life of the WT CAV1 protein was reduced compared with endogenous CAV1 (Figure 5A). This is expected given that CAV1 turns over more rapidly on overexpression (Hayer et al., 2010b). Interestingly, the half-life of the mutant CAV1 was decreased even further, suggesting that it is even more rapidly turned over (Figure 5A). We also noted that levels of CAV1 P158 protein were substantially lower than WT CAV1, making its detection by Western blotting difficult (Figure 5A). This effect was reproducible and was not due to differences in the transfection efficiencies for the two constructs (Supplemental Figure 1) and is also not the result of differences in tagging since both were tagged with HA. We also asked whether coexpression of CAV1 P158 impacts the turnover of WT CAV1. We found that the half-life of WT CAV1 was further decreased in the presence of the mutant protein (Figure 5B). Furthermore, levels of CAV1 P158 were increased in cotransfected cells compared with cells expressing CAV1 P158 alone. These findings suggest that CAV1 P158 behaves in a dominant negative manner in part by enhancing the turnover of WT CAV1 and that CAV1 P158 is also partially rescued by WT CAV1.
FIGURE 5:

CAV1-P158 has a shortened half compared with WT CAV1 in reconstituted Cav1–/– MEFs. (A) Untransfected Cav1+/+ MEFs (“WT MEF”) or Cav1–/– MEFs transiently transfected with WT CAV1, CAV1-P158, or CAV1-P132L were incubated in the continuous presence of 100 μg/ml CHX and samples were collected at the indicated timepoints. Whole-cell lysates were analyzed by SDS–PAGE/Western blot. Densitometry was performed on blots probed with an anti-Cav1 antibody to determine the levels of endogenous or transfected CAV1 at each sampling time. (B) As in A, except Cav1–/– MEFs were cotransfected with either WT CAV1+WT CAV1 or WT CAV1+CAV1-P158. Densitometry was performed on blots probed with an anti-Cav1 antibody for cells cotransfected with WT CAV1 + WT CAV1. For cells cotransfected with HA-P158 + WT Myc-CAV1, densitometry was performed on blots probed with a myc or HA antibody. We note that in cells cotransfected with HA-CAV1 and Myc-CAV1, HA-CAV1 appeared to be preferentially expressed for unknown reasons. Blots and densitometry results are representative of two independent experiments.
Endogenous CAV1 is localized normally and caveolae are present in patient fibroblasts expressing CAV1 P158
The results of our experiments in Cav1–/– MEFs suggest that CAV1 P158 contains targeting information in its C-terminus introduced by the frameshift mutation that interferes with delivery of the mutant protein to caveolae. Some CAV1 P158 can be incorporated into caveolae when coexpressed with WT CAV1. CAV1 P158 can also function as a dominant negative by partially inhibiting the proper trafficking of WT CAV1 to the plasma membrane as well as by enhancing its turnover. However, given that these experiments were carried out under conditions where both CAV1 P158 and WT CAV1 are overexpressed, it is unclear which phenotype prevails when the proteins are expressed at physiological levels.
To address this question, we assessed the subcellular distribution of CAV1 in skin fibroblasts isolated from heterozygous CAV1 P158 patients using immunofluorescence microscopy. CAV1 was detected using N-terminally directed antibodies, which should recognize both wild-type and mutant CAV1. Numerous puncta consistent with the appearance of caveolae were detected in both the control cells and patient fibroblasts (Figure 6). These CAV1-positive puncta strongly colocalized with caveolin-2 (Figure 6, A–C) and the caveolar accessory proteins cavin1 (Figure 6, D–F) and EHD2 (Figure 6, G–I), verifying their identity as caveolae. Under normal conditions, endogenous CAV1 is undetectable in the ER and lipid droplets. In line with this, no CAV1 was detected in ADRP-positive structures in either control or patient fibroblasts (Figure 6, J–L). This suggests that when expressed at physiological levels in patient cells, little CAV1 accumulates in lipid droplets, a secondary compartment of ER-accumulated caveolins (Ostermeyer et al., 2001). We speculate that unlike transiently overexpressed CAV1, any endogenous CAV1 protein retained in the ER of patient cells is efficiently cleared from the organelle, without overwhelming the degradation machinery, thus preventing its detectible accumulation.
FIGURE 6:
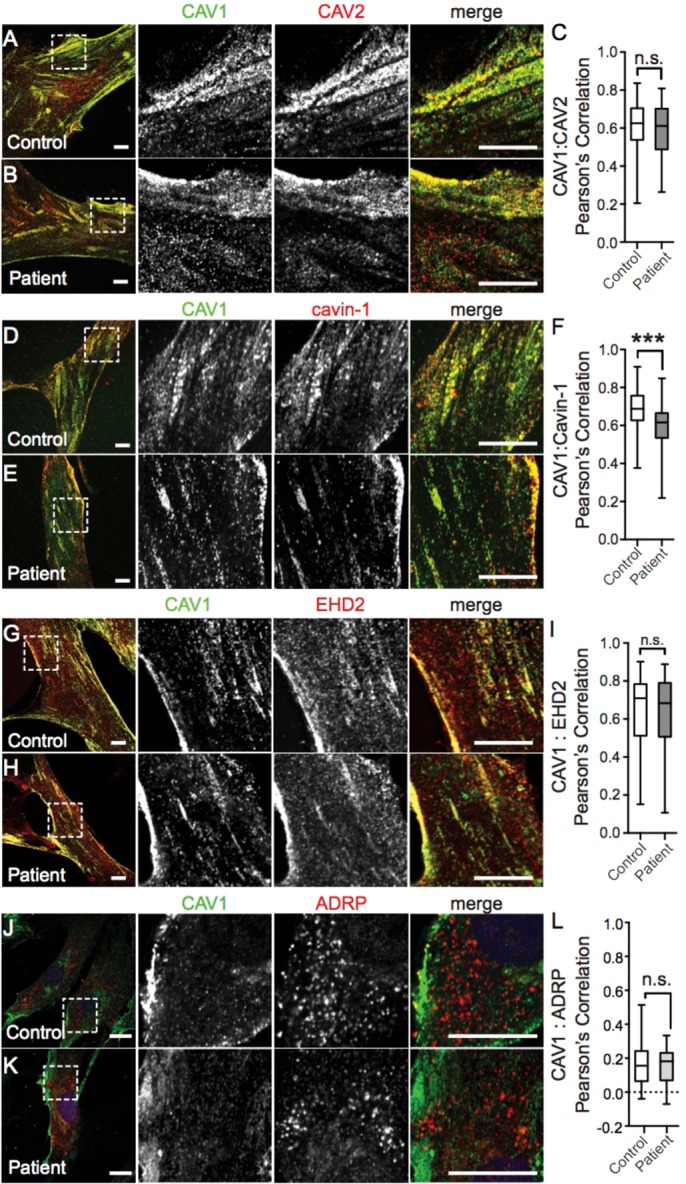
The subcellular distribution of CAV1 and caveolaer accessory proteins is normal in patient skin fibroblasts. Representative immunofluorescence images of control and patient skin fibroblasts costained for CAV1 and CAV2 (A, B), Cavin-1 (D, E), EHD2 (G, H), or ARDP (J, K). In the merged images, CAV1 fluorescence is shown in green. Scale bars, 10 μm. (C, F, I, L) Quantification of the extent of colocalization of CAV1 and the indicated proteins using Pearson’s correlation coefficient. p values were calculated with a nonparametric two-tailed Mann–Whitney U test. Data are representative of two to three independent experiments for three control and three patient cell lines. n.s., not significant; ***, p < 0.001. The numbers of cells analyzed are as follows: CAV1/CAV2, 46 control and 46 patient cells; CAV1/Cavin-1 54 control and 61 patient cells; CAV1/EHD2 96 control and 93 patient cells; CAV1/ADRP, 41 control and 64 patient cells.
To independently verify that caveolae are present in patient fibroblasts, we performed electron microscopy. Morphologically defined caveolae were readily detected in patient cells (Figure 7A). However, the abundance of caveolae was reduced in patient fibroblasts (Figure 7B), consistent with the results of a recent report (Marsboom et al., 2017). To determine whether this difference in caveolae abundance could be accounted for by decreased CAV1 protein levels, we performed Western blotting. Using an N-terminally directed antibody that detects both wild-type and mutant CAV1, we detected significantly lower levels of CAV1 in patient fibroblasts (Figure 7, C and D). To determine whether the decreased CAV1 protein levels were due primarily to poor expression of CAV1 P158 or also as the result of decreased levels of WT CAV1, we probed for CAV1 protein with a C-terminal CAV1 antibody that is unable to detect the novel C-terminus of the frameshift mutant. As reported by this antibody, CAV1 protein levels were less than 50% of control values, suggesting that levels of WT CAV1 are also decreased in the patient fibroblasts (Figure 7, C and D). Consistent with the decreased CAV1 levels, levels of CAV2, Cavin-1, and EHD2 were also decreased in patient cells compared with controls (Figure 7, E and F). Flotillin-1 and flotillin-2 levels were likewise diminished, while PACSIN-2 levels were unaffected (Figure 7, E and F).
FIGURE 7:

The density of caveolae and caveolar protein levels are reduced in patient cells expressing CAV1-P158. (A) Cropped electron micrographs of control (top panel) and patient (bottom panel) skin fibroblasts. Images were acquired at 30,000× magnification. For purposes of illustration, the density of caveolae in these images is higher than the average values quantified in B. Scale bar, 500 nm. (B) Quantification of number of caveolae per micrometer of plasma membrane in patient and control fibroblasts. Caveolae were counted in 25 images each from three patients and three control cell lines in two independent experimental replicates and one experiment for one control and one patient cell line. **, p < 0.007, nonparametric Mann–Whitney U test. (C, D) Representative Western blots and densitometry analysis of CAV1 in control and patient fibroblasts as detected using N-term and C-term specific antibodies. b-Tubulin was blotted as a loading control. Densitometry data were averaged over three control and patient cell lines and the mean ± SD are indicated. *, p < 0.05, **, p < 0.01, Student’s t test. Data are representative of three independent experiments. (E, F) Representative Western blots and densitometry analysis of caveolar accessory proteins in control and patient skin fibroblasts. β-tubulin was blotted as a loading control. Densitometry data were averaged of three control and patient cell lines. n.s., not significant; *, p < 0.05, **, p < 0.01, Student’s t test. Data are representative of three independent experiments. (G) Tandem mass spectrometry was used to determine if CAV1-P158 protein is expressed in patient fibroblasts. CAV1 protein was immunoprecipitated using CAV1 N-term from lysates of a patient fibroblast cell line, separated by SDS–PAGE, and Coomassie stained. Immunoprecipitated CAV1 was also detected in immunoblots with an anti-CAV1 mAb 2297. The region of the SDS–PAGE gel that corresponded to the position of the CAV1 band in the immunoblot was excised and analyzed by mass spectrometry. Unique peptides of the novel C-terminus of CAV1-P158 were detected in patient cells.
We also directly tested whether the mutant protein is expressed using mass spectrometry. The frameshift mutation of CAV1 is predicted to generate a novel protein sequence that is not normally present in the human genome by BLAST analysis (Figure 7G). We thus tested for the presence of the peptide that corresponds to the region encompassing the mutant C-terminus in patient cells using mass spectrometry. Three MS/MS spectra were identified that correspond to regions of the novel C-terminus of CAV1 P158 (Figure 7G and Supplemental Figure 2), confirming that CAV1 P158 protein is expressed in the patient fibroblasts.
CAV1 forms high-molecular-weight oligomers correctly in patient fibroblasts
The finding that mutant CAV1 can at least partially incorporate into caveolae in patient fibroblasts raises the possibility that some features of these caveolae may be defective. Because the C-terminus is important for stabilizing homo-oligomers, a building block of caveolae (Song et al., 1997), we first asked whether the stability of oligomeric complexes is maintained in patient cells.
One method to detect caveolin complexes is using blue native polyacrylamide gel electrophoresis (BN–PAGE). Endogenous caveolin-1 typically migrates as ∼600-kDa complexes using this approach (Ren et al., 2004; Han et al., 2015), likely corresponding to the core unit of 8S caveolin-1 oligomer. As a control for these experiments, we studied a caveolin-1 mutant that has known defects in oligomerization, P132L (Lee et al., 2002; Ren et al., 2004). We prepared a series of control cell lysates from untransfected HeLa cells or HeLa cells expressing enhanced green fluorescent protein (EGFP), a wild-type caveolin-1 EGFP fusion protein (CAV1-GFP), or the aggregate-prone P132L mutant caveolin-1 EGFP fusion protein (P132L-GFP). Consistent with our recent report (Han et al., 2015), we found that when cells are transiently transfected with CAV1-GFP, CAV1-GFP and endogenous CAV1/2 form two separate high-molecular-weight complexes of ∼800 kDa (green arrow) and ∼600 kDa (red arrow), respectively. This is because endogenous caveolin is already assembled into long-lived complexes and caveolae that exclude transiently expressed caveolins (Han et al., 2015). In contrast, P132L-GFP migrates as an ∼800-kDa band, an irregular smear, and a weak ∼300-kDa band (Figure 8A), indicating defective assembly or stability of complexes/aggregates formed by this mutant. In patient fibroblasts, endogenous CAV1 formed high-molecular-weight complexes similar to those observed in control fibroblasts and HeLa cells (Figure 8A). This strongly suggests that core units of CAV1 complexes are intact in patient cells. However, a small shift in mobility was reproducibly observed. This implies that the size and/or shape of the CAV1 complexes is altered as the result of incorporation of the CAV1 P158 mutant, perhaps as a result of the novel C-terminus of P158 disrupting the normal conformation of the Cav1 complexes. Furthermore, CAV1 and CAV2 comigrated in both control and patient cells (Figure 8B). Thus the presence of the mutant protein also does not appear to interfere with the formation of CAV1/CAV2 hetero-oligomers.
FIGURE 8:

CAV1 incorporates correctly into CAV1/CAV2 hetero-oligomers in patient fibroblasts as assessed by BN–PAGE. Control and patient skin fibroblasts were subjected to BN–PAGE and blotted for the indicated proteins. Untransfected HeLa cells and HeLa cells transiently expressing EGFP, CAV1-GFP, or P132L-GFP were used as controls. Equal amounts of protein were loaded in each lane. (A) Western blots from BN–PAGE were blotted using CAV1 N-term (red in merge) or an anti-GFP antibody (green in merge). The red arrow indicates the position of complexes containing endogenous CAV1, and the green arrow shows complexes containing CAV1-GFP or P132L-GFP. Data are representative of at least three independent experiments. (B) As in A except blots were probed using CAV1 N-term (red in merge) and an antibody against CAV2 (green in merge). Note the strong overlap between CAV1 and CAV2 signals in the merged image. Data are representative of at least two independent experiments.
To further assess the integrity and stability of CAV1 complexes in patient cells, we used rate-zone sucrose gradient centrifugation to test for the presence of 8S and 70S complexes known to form as the protein traverses the secretory pathway (Hayer et al., 2010a). CAV2, which normally forms complexes and cofractionates with CAV1, was also examined. Both CAV1 and CAV2 localized normally in fractions corresponding to 8S and 70S complexes in patient cells relative to control fibroblasts (Figure 9A). This overall distribution of CAV1 was similar to the behavior of cotransfected WT CAV1 and P158 CAV1 in cotransfected Cav1–/– MEFs (Figure 4). This result again suggests that the introduction of the novel C-terminus in the frameshift mutant does not significantly interfere with the ability of CAV1 to form the homo- and hetero-oligomers that ultimately are incorporated into caveolae. We did, however, observe slightly more protein in 8S complexes and a less uniform distribution of the larger complexes in corresponding fractions in patient fibroblast than in control cells (Figure 9, A and B). It is thus possible that in the patient cells there is a slight defect in the incorporation of 8S complexes into caveolae or the 70S complex may be more easily disassembled into 8S complexes under the extraction conditions of the procedure.
FIGURE 9:
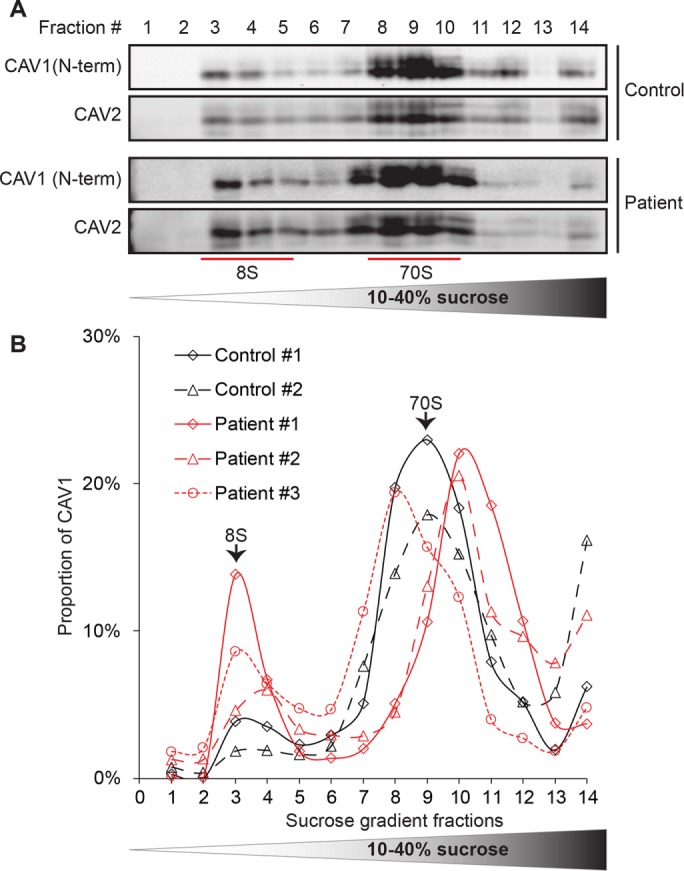
CAV1 associates normally with 8S and 70S complexes in patient fibroblasts. (A) Extracts prepared from control and patient fibroblasts were run through 10–40% sucrose velocity gradients and the resulting fractions analyzed by Western blot for CAV1 and CAV2. Both CAV1 and CAV2 associated with 8S and 70S oligomeric species in both control and patient cells. For each fraction, 10 μl of sample was loaded. Blots are shown for control cell line #1 and patient cell line #3. (B) Quantification of CAV1 levels for individual control (black lines) and patient (red lines) cell lines. Note that all three patient cells lines contained slightly larger amounts of 8S complexes than were seen in control cells. The broad range in sedimentation of the larger complex in patient cells is most likely due to technical variations between experiments rather than biological variability, as the fractions were collected by hand. Data are representative of one to two independent experiments per cell line.
CAV1 exhibits decreased detergent resistance in patient skin fibroblasts
CAV1 is a highly detergent-resistant protein and typically associates with buoyant detergent-resistant fractions (Parton and Simons 2007). The C-terminus is important for its association with DRMs (Song et al., 1997). Several additional proteins associated with caveolae are also known to at least partially cofractionate with DRMs, such as CAV2 (Davalos et al., 2010) and cavin-1 (Liu and Pilch, 2008; Davalos et al., 2010). We therefore next tested whether caveolae-associated proteins from patient cells maintain full detergent resistance (Figure 10). Flotillin-1, a protein whose detergent resistance does not depend on CAV1 expression (Rajendran et al., 2007), was used as a positive control for DRMs for these studies, and calnexin served as a marker for detergent-soluble fractions.
FIGURE 10:

Caveolae of patient fibroblasts display decreased detergent resistance. Detergent-resistant membranes (DRMs) were isolated from control and patient fibroblasts extracted using either 0.5% or 1% of cold TX-100. (A, B) Western blots of fractions from sucrose density gradients. Ten microliters of each fraction/sample was loaded, and fractions were probed with antibodies against the indicated proteins. Fraction 1 is the top of the gradient, and fraction 14 is the bottom. Fractions corresponding to DRM fractions are indicated with red lines. Note the marked decrease in levels of CAV1 in DRMs in patient cells extracted in 1.0% TX-100 relative to control cells. (C) Quantification of Western blots in A and B by densitometry. Patient cell lines are shown in red and control cells in black. Data in A and B are shown for a single control and single patient cell line and are representative of individual experiments carried out for each of the three patient and three control cell lines. All proteins were assessed for all six cell lines except for the following: for 0.5% TX-100 samples, Pacsin-2 data were collected for only one control and one patient cell line, and for 1.0% TX-100 data, CAV2 data were collected for only one control and one patient cell line.
In cells extracted with cold 0.5% TX-100, both CAV1 and CAV2 accumulated in detergent-resistant fractions to a similar extent in patient and control cells. CAV2 cofractionated extensively with CAV1 in both patient and control cells, and Cavin-1 partially cofractionated with CAV1. However, in patient cells, cavin-1 was completely lost from caveolar fractions (Figure 10A). These results suggest that the interaction of this accessory protein with caveolae/DRMs is weakened in patient cells.
We also assessed the effect of increasing detergent concentration to determine whether caveolae in patient cells were equally detergent resistant as in control cells. When cells were extracted with 1% TX-100 instead of 0.5% TX-100, differences between the patient and control cells become more pronounced (Figure 10B). Caveolae from control fibroblasts largely retained their resistance to the increased detergent, whereas a substantial shift of CAV1 from detergent resistant to more detergent-soluble fractions was observed for the patient cells (Figure 10C). These results suggest that the biochemical properties of patient caveolae are also altered.
Patient fibroblasts demonstrate decreased resistance to osmotic stress
One function of caveolae is to serve as a membrane reservoir (Sinha et al., 2011; Nassoy and Lamaze, 2012). Caveolae flatten in response to mechanical or osmotic stress, and cells that contain fewer caveolae show decreased viability following stress or in response to increased cardiac output (Sinha et al., 2011; Cheng et al., 2015). Because patient fibroblasts contain fewer caveolae than wild-type cells and also exhibit various defects, we hypothesized that they may exhibit lower buffering capacity. To test this, we subjected cells to osmotic stress by a 10-min incubation in hypotonic media (10-fold dilution) (Figure 11).
FIGURE 11:
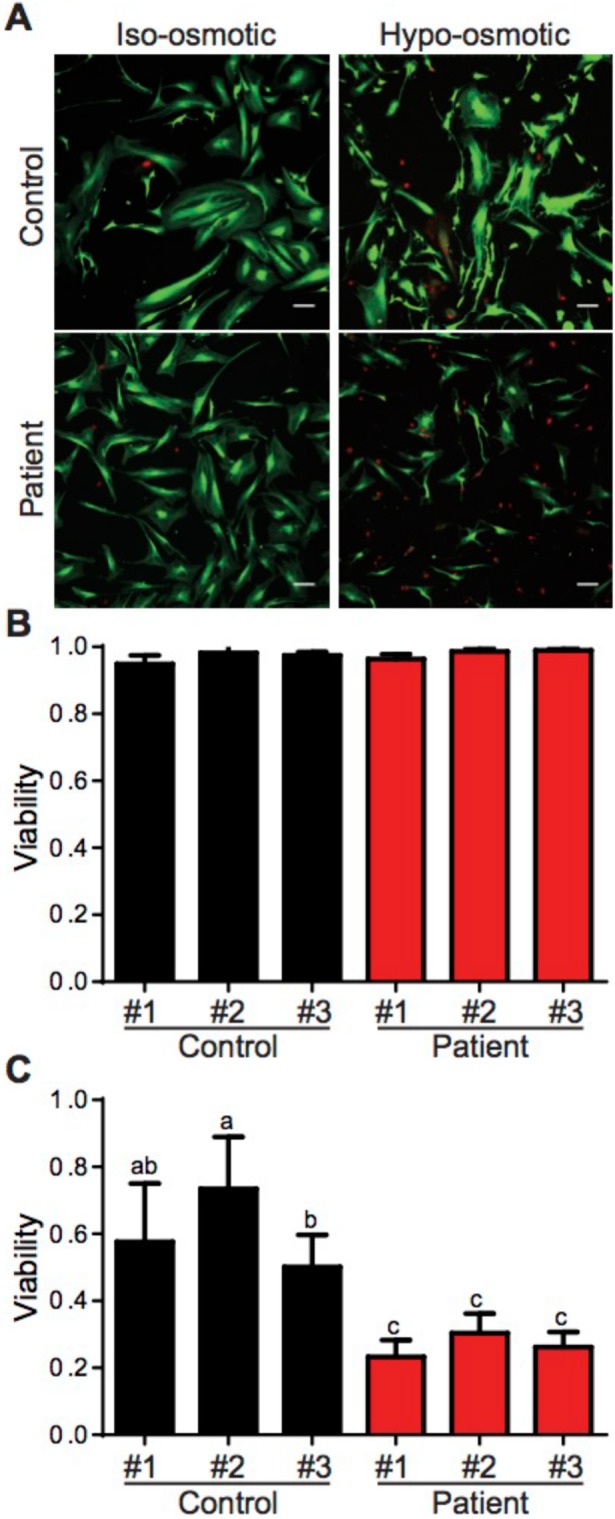
Patient fibroblasts demonstrate increased susceptibility to hypo-osmotic stress challenge. (A) Representative images of control and patient fibroblasts before and after 10 min of hypo-osmotic challenge. Live cells are shown in green and dead cells are red. Bar, 10 μm. (B, C) Quantification of cell viability under (B) control and (C) hypo-osmotic challenge conditions. Data represent the mean ± SD of at least four independent experiments in which at least nine fields of cells were analyzed for each cell line. Results for control cell lines are shown in black and in red for patient cell lines. One-way ANOVA was used for statistical analysis. Any two means that do not share the same letter were significantly different after running the ANOVA.
Cell viability was assessed using a live/dead assay in which the nuclei of dead cells are stained red and the cytoplasm of live cells are stained green. Before treatment, the vast majority of cells was viable and had a fibroblastic spindle shape (Figure 11, A and B). After 10 min treatment, over 50% of wild-type cells were still alive, compared with around 25% of patient cells (Figure 11, A and C). These results indicate that the expression of CAV1 P158 sensitizes the fibroblasts to osmotic stress.
DISCUSSION
Although CAV1 has been linked to a number of diseases, relatively few examples of disease-associated mutations of the protein have been identified. The recent discovery of heterozygous mutations in CAV1 associated with PAH (Austin et al., 2012; Garg et al., 2015; Schrauwen et al., 2015; Han, Copeland, et al., 2016) thus offers a unique opportunity to study the role of CAV1 and caveolae in both normal cellular function and disease. In the current study, we investigated the impact of the expression of a familial PAH-associated frameshift CAV1 mutant, CAV1 P158, on caveolae assembly in reconstituted Cav1–/– MEFs and patient skin fibroblasts.
Our results suggest a model in which the frameshift mutation introduces a gain of function by generating a de novo ER retention signal in the C-terminus of CAV1-P158 (Figure 12). The ER retention signal is functional when the protein is expressed in the absence of WT CAV1 and results in retention of the mutant protein in the ER and lipid droplets as well as enhanced turnover rates (Figure 12B). Despite its novel C-terminus, CAV1-P158 retains the ability to oligomerize with WT CAV1 when the two are coexpressed, forming mixed hetero-oligomers (Figure 12C). These complexes have several possible fates (Figure 12C). A subset of these mixed complexes is likely targeted for degradation, ultimately reducing CAV1 protein levels. This is accompanied by a concomitant decrease in accessory protein levels and caveolae numbers, as documented both here and in a recent report (Marsboom et al., 2017). We speculate that in patient cells these CAV1 complexes are cleared efficiently without overwhelming the degradation pathway, preventing the protein from being detectable in the ER. In contrast, under conditions of overexpression, the degradation machinery likely becomes saturated, resulting in the accumulation of CAV1 in the ER and lipid droplets. Our findings also suggest a subset of hetero-oligomeric complexes is capable of successfully exiting the ER and trafficking correctly to the plasma membrane. Once having reached the plasma membrane, these hetero-oligomers become incorporated into caveolae. What determines why some complexes containing a mixture of CAV1 P158 and WT CAV1 can assemble into caveolae while others fail to escape the ER is not entirely clear, but we speculate that these may depend on the relative expression levels of the two proteins (Marsboom et al., 2017). Furthermore, the determinants involved in targeting complexes containing the CAV1 mutant for degradation have yet to be elucidated. Future investigation is necessary to address these unanswered questions.
FIGURE 12:

Proposed model of CAV1-P158 trafficking in the absence or presence of WT CAV1. (A) In control cells, newly synthesized WT CAV1 forms 8S complexes and efficiently exits the ER. It is subsequently transported from the Golgi complex to the plasma membrane in the form of 70S complexes where it recruits cavin1 and forms caveolae. (For simplicity, other accessory proteins are not shown.) (B) Unlike WT CAV1, CAV1-P158 is incapable of supporting caveolae formation when expressed on its own due to the introduction of an ER retention signal by the frameshift mutation. Instead, it is retained in the ER and lipid droplets where it can also be targeted for degradation. (C) Coexpression of WT CAV1 and CAV1 P158 results in the formation of hybrid complexes of the two proteins. These hybrid complexes have at least two possible fates. A fraction of the hybrid complexes go on to form 8S and 70S complexes and are targeted to the cell surface where they form hybrid caveolae. These hybrid caveolae are apparently morphologically normal but are less detergent resistant than WT caveolae. A second fraction of hybrid complexes are unable to exit the ER and/or are rapidly degraded, thus decreasing overall CAV1 expression levels. What determines whether the hybrid WT/ P158 complexes ultimately reach the cell surface or are retained in the ER/degraded is unknown but is likely to reflect the relative ratio of WT and mutant CAV1 within a given complex.
Importantly, the hybrid caveolae containing a mixture of CAV1 P158 and WT CAV1 exhibit several defects, including decreased resistance to detergent extraction. While the mechanism underlying this shift in detergent resistance is not yet known, similar defects have been reported upon disruption of the C-terminus of CAV1 (Song et al., 1997) as well as for a different CAV1 mutant, F160X, that lacks the last 19 residues of the C-terminus (Han, Copeland, et al., 2016). The ability of caveolae to function as membrane reservoirs in patient fibroblasts carrying the CAV1 P158 mutant is also impaired in response to a hypo-osmotic challenge. We speculate that either the decreased numbers of caveolae in patient cells and/or biochemical changes in the properties of these hybrid caveolae may contribute to this phenotype.
Interestingly, a closely related CAV1 mutant identified in a patient with an idiopathic form of PAH, P158HfsX22, is predicted to be essentially identical in amino acid sequence to CAV1 P158 with the exception of residue 158 (Austin et al., 2012). Our results strongly predict that P158HfsX22, which also contains an ER retention signal, should behave in a similar manner as CAV1 P158. The behavior of CAV1 P158 is quite different, however, from that of another recently described PAH-associated mutation in CAV1, F160X (Garg et al., 2015; Schrauwen et al., 2015; Han, Copeland, et al., 2016). F160X was identified in a patient with both PAH and congenital generalized lipodystrophy and thus at first glance might be expected to exhibit more severe defects in caveolae formation. Despite this, the overall expression levels of CAV1 are similar in F160X patient and control skin fibroblasts, F160X is properly targeted to caveolae either when expressed either on its own or when coexpressed with WT CAV1, and membrane buffering activity in response to hypo-osmotic shock is similar in control and patient fibroblasts (Han, Copeland, et al., 2016). Thus the impact of F160X on caveolae assembly is more subtle than that of P158. Caveolae in the F160X patient cells are not entirely normal, however: they exhibit reduced colocalization of cavin-1 with CAV1, decreased stability of 8S and 70S complexes, and reduced DRM affinity (Han, Copeland, et al., 2016). It will be important in future studies to further compare the impact of the F160X and P158 CAV1 mutants on caveolae function to better discern how caveolar defects give rise to cellular dysfunction and disease progression, as well as to better understand what determines their tissue selectivity and why they do not exhibit a broader disease phenotype.
It is not yet clear how the defects in CAV1 oligomerization and caveolae assembly described here affect their functions or whether they directly or indirectly promote disease. The modulation of numerous cell signaling pathways is influenced by caveolae, and the stability of these domains may be important for these processes. The diminished abundance of caveolae that occurs in patient cells as a result of the expression of CAV1 P158 could also potentially contribute to development of PAH. Because the lung is composed of cells that are especially rich in CAV1, decreased numbers of caveolae in the pulmonary arteries may help explain why the CAV1 mutation preferentially causes defects in pulmonary-vascular function (Austin et al., 2012). Indeed, there is evidence that in both experimental and clinical forms of PAH, there is a loss of endothelial CAV1 (Mathew, 2011). In response to mechanical stress-induced membrane stretch, caveolae flatten and the caveolar accessory protein Cavin-1 is quickly released (Sinha et al., 2011; Gambin et al., 2014). Although the downstream consequences of the release of Cavin-1 and flattening are still not fully understood, it is likely that they are important for adaptive cellular responses to mechanical stimuli. In patient tissues primarily affected by disease, elevated vessel pressure/resistance associated with PAH may thus provide a persistent mechanical stimulus that triggers the pathological compensatory tissue remodeling observed in PAH. Further evaluation of the effects of mechanical stress on caveolae in endothelial and smooth muscle cells and mouse models should be able to test this hypothesis in the future.
CAV1 is also known to coordinate the activity of key modulators of vasoreactive pathways in endothelial cells to maintain vascular tone (Zhao et al., 2009). CAV1 deficiency causes pathological vascular remodeling and induces pulmonary hypertension in mice and humans (Zhao et al., 2009). In addition to down-regulation of CAV1, we also observed decreased levels of CAV2 and the caveolar accessory proteins Cavin-1 and EHD-2 in patient cells. Levels of noncaveolae raft proteins flotillin-1 and flotillin-2 were also decreased, despite their expression being considered independent of CAV1, at least in the case of Cav1–/– MEFs (Rajendran et al., 2007). This observation also raises the possibility that the P158 mutation may cause disease through a mechanism involving signaling from caveolae not directly coupled to the defects in trafficking and assembly reported here. Indeed, alterations in accessory protein levels could potentially contribute to disease pathology given that several of these proteins are themselves key regulators of human health and disease (Razani et al., 2002a, b; Nassar et al., 2013; Low and Nicholson 2015). Thus down-regulation of CAV1 and/or caveolae accessory proteins may have multiple consequences in signaling pathways relevant to PAH. Consistent with this, a number of signaling defects were recently reported in patient skin fibroblasts (Marsboom et al., 2017). Changes in CAV1 expression in patient cells may also differ between cell types. For example, although decreased levels of CAV1 were observed in endothelial cells in sections obtained from a lung biopsy from a PAH patient expressing a P158 frameshift mutant, CAV1 staining was still present in surrounding cell types (Austin et al., 2012). Further work will be required to determine how expression of the CAV1 mutant impacts caveolae formation and function on a cell and tissue-specific level to better understand the key factors that contribute to the development of PAH.
MATERIALS AND METHODS
Cell culture
Wild-type and Cav1–/– MEFs (purchased from the American Type Culture Collection) were maintained in DMEM supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin, and 1% l-glutamine and were tested two times over the course of these studies for mycoplasma contamination. Control and patient human dermal fibroblasts were as described previously (Austin et al., 2012). They were maintained in DMEM supplemented with 20% FBS, 1% penicillin/streptomycin, and 1% l-glutamine. All cells were cultured at 37°C in 5% CO2. Where indicated, transient transfections were performed according to the manufacture’s protocol with Lipofectamine 2000 (Thermo Scientific).
Plasmids
CAV1-mEmerald N-10 (Addgene No. 54026) and mEmerald-CAV1 C-10 (Addgene No. 54025) were gifts from Mike Davidson (National High Magnetic Field Laboratory, The Florida State University, Tallahassee) and were used to generate constructs for the current study as detailed below. Myc-CAV1 and HA-CAV1 were as recently described (Han, Copeland, et al., 2016). P132L-HA was subcloned from a previously described P132L-MycHis construct (Han, Copeland, et al., 2016) into a pcDNA3.1 backbone using the following primers: forward primer: 5′-CGGGATCCATGTCTGGGGGCAAATACGTAG-3′ and reverse primer: 5′-CGGAATTCTTAGCTAGCGTAGTCTGGGACGTCGTATGGGTATATTTCTTTCTGCAAGTTGATGCG-3′. pCMV-HA-N (No. 82017) empty vector plasmid DNA was used to generate HA-tagged constructs (ThermoFisher Scientific, Rockford, IL).
Standard PCR, site-directed mutagenesis (QuickChange Site-Directed mutagenesis kit, Agilent Technologies, Santa Clara, CA), and subcloning (all restriction enzymes were purchased from NEB) were used to generate novel constructs as follows:
mEmerald-tagged CAV1-P158 was generated by introducing the naturally occurring PAH-associated frameshift mutation (c.Δ474A/ p.P158PfsX22, single nucleotide deletion) into both mEmerald-Caveolin-1 C-10 and Caveolin-1-mEmerald N-10 constructs (5′ ACACCGTCTGTGACCCCTCTTTGAAGCTGTTG 3′) with site-directed mutagenesis.
HA-CAV1. Wild-type full-length CAV1 was excised from Caveolin-1-mEmerald N10 construct and subcloned into the pCMV-HA-N vector using BamHI and NheI restriction enzyme sites.
HA-CAV1-P158. CAV1-P158 was excised from CAV1-P158-mEmerald N10 construct and subcloned into the pCMV-HA-N vector using BamHI and NheI restriction enzyme sites.
HA-CAV1-P158-AAYK. HA-CAV1-P158 was mutagenized with the primers 5′ CATCAACTTGCAGAGCGAAATATAAATAGC3′ and 5′ CATCAACTTGCAGAGCGGCATATAAATAGC 3′ to introduce the K167A and K177A mutations, respectively to disrupt the ER-retention/retrieval signal.
HA-CAV1-P158-ΔKKYK. HA-CAV1-P158-ΔKKYK was generated by GenScript USA, using HA-CAV1 P158 as the template. Briefly, a nonsense mutation was introduced at the codon encoding K176 (AAG→TAG) using NheI and BamHI restriction enzyme sites to prematurely terminate reading before the KKYK ER-retention/retrieval.
HA-CAV1-KKYK. mEmerald-CAV1-KKYK was generated by GenScript USA, using mEmerald-Caveolin-1 C-10 plasmid DNA as a template. Briefly, BglII and SacII restriction enzyme sites were used to subclone the CAV1-P158 ER-retention/retrieval signal (KKYK) into the C-terminus of the WT CAV1 protein. CAV1-KKYK was then excised from mEmerald-CAV1-KKYK with BglII and EcoRI and subcloned into the linearized (BamHI/EcoRI) pCMV-HA-N empty vector.
Antibodies
Anti-Cav1 rabbit polyclonal (610060) (referred to here as N-term), anti-Cav1 clone 2234 monoclonal (610494), anti-Cav2 monoclonal (610684), anti-flotillin-1 mouse monoclonal (610820), anti-flotillin-2 mouse monoclonal (610383) and anti-calnexin mouse monoclonal (610523) were purchased from BD Transduction Laboratories. Anti-Caveolin-1 [E249] rabbit monoclonal (ab32577) (referred to here as C-term), anti-Cavin-1/PTRF rabbit polyclonal (ab48824), and anti-EHD2 goat polyclonal (ab23935) antibodies were purchased from Abcam. Anti-PACSIN2 rabbit polyconal (AP8088b) was obtained from ABGENT. Anti-Myc monoclonal [9B11] (#2276) and anti-HA-tag monoclonal [6E2] (#2367) antibodies were purchased from Cell Signaling Technology. Anti-ADRP guinea pig polyclonal [hNT] (GP46) antibody was purchased from PROGEN Biotechnik. Anti–β-tubulin monoclonal [E7] (AB_2315513) antibody was purchased from the University of Iowa Developmental Studies Hybridoma Bank. Anti-calreticulin rabbit polyclonal antibody (C4606) was purchased from Sigma Aldrich. Anti-GFP mouse monoclonal (JL-8; 632381) was obtained from Clontech. All Alexa-Fluor dye conjugated secondary antibodies were purchased from Thermo Scientific. The specificity of Cav1 antibodies was confirmed by comparing their labeling in Cav1+/+ and Cav1–/– MEFs as well as their ability to detect transfected CAV1.
Immunofluorescence microscopy
Immunofluorescence microscopy was performed as recently described (Han, Copeland, et al., 2016). Primary antibodies were used at the following concentrations: Cav1 N-term (1:300), Cav1 clone 2234 (1:50), calreticulin (1:100), ADRP (1:100), HA (1:100), Myc (1:100), Cavin-1 (1:100), Anti-Cav2 (1:50), and EHD2 (1:250). Secondary antibodies were used at 1:500–1:1000.
Colocalization analysis
Colocalization analysis was performed as recently described (Han, Copeland, et al., 2016). Statistical analysis of colocalization data was performed using Graphpad. Differences between two groups were assessed with an unpaired (two-tailed) Student’s u test (with Welch’s correction if SDs were not the same) or nonparametric Mann–Whitney U test. For colocalization experiments comparing three or more groups, a one-way analysis of variance (ANOVA) with post hoc Bonferroni’s multiple comparisons test were used in statistical analyses. For nonparametric data, a Kluskal–Wallis analysis was used with post hoc Dunn’s multiple comparison’s test. D’Agostino’s and Pearson’s tests were used to confirm normality. A minimum of two (majority three) experimental replicates were analyzed; p-values and sample sizes (n) are indicated in the figure legends.
SDS–PAGE and Western blotting
SDS–PAGE and Western blotting were performed as recently described (Han, Copeland, et al., 2016). Antibodies were used at the following concentrations for Western blotting: Myc (1:1000), HA (1:1000), CAV1 N-term (1:10,000), β-tubulin (1:3000), GFP (1:5000), CAV2 (1:1000), CAV1 C-term (1:1000), flotillin-1 (1:1000), flotillin-2 (1:1000), EHD-2 (1:1000), PACSIN-2 (1:1000), CAV1 2297 (1:1000), and calnexin (1:1000). Secondary antibodies were used at 1:10,000. Statistical analysis of densitometry results was performed using Graphpad.
Velocity gradient centrifugation isolation of 8S and 70S complexes
8S and 70S complexes were isolated as recently described (Han, Copeland, et al., 2016).
Preparation of detergent-resistant membranes
Detergent-resistant membranes were isolated as recently described (Han, Copeland, et al., 2016).
CAV1 half-life analysis
Cycloheximide (CHX) treatments were carried out 24 h after transfection. Transfected Cav1–/– MEFs were incubated at 37°C in cell culture media supplemented with 100 μg/ml CHX in dimethyl sulfoxide (DMSO) from 0 to 15 h. The cells were harvested at 0, 1.5, 3, 6, 9, or 15 h with CelLytic M buffer (Sigma-Aldrich) supplemented with a protein inhibitor cocktail and phosphatase inhibitors (Roche). Equal protein amounts were loaded on each gel as determined by BCA protein assay, and SDS–PAGE electrophoresis and Western blotting were conducted as previously described (Han, Copeland, et al., 2016). β-tubulin was blotted as an internal control. Densitometry analysis was conducted using ImageJ (http://fiji.sc/Fiji).
Electron microscopy
Electron microscopy was performed as recently described (Han, Copeland, et al., 2016). In brief, cells were grown to confluency in 10-cm culture plates, washed, and fixed in 2.5% glutaraldehyde diluted in 0.1 M cacodylate buffer, postfixed in osmium tetroxide, uranyl acetate stained en bloc, and epon embedded. Thin sections were counterstained with lead citrate viewed on a Philips/FEI T-12 transmission electron microscope operated at 80 kV. Twenty-five images of cell sections were captured from each sample, and caveolae were counted. Caveolae were defined as 50- to 80-nm-diameter flask-shaped or circular structures, either plasma membrane attached or internalized vesicles no more than 200 nm from plasma membrane. The number of caveolae was normalized to the total length of plasma membrane determined using the Trace tool in ImageJ. Data were collected from 25 images per experiment for two independent experiments for each cell line (three patient lines and three control lines) and an additional experiment including one patient and one control cell line. Data were collected and analyzed in a blinded manner.
Mass spectrometry identification of mutant CAV1 in patient fibroblasts
CAV1 was immunoprecipitated from patient fibroblasts using the Dynabeads Protein G Immunoprecipitation Kit (Life Technologies). Five micrograms of pAb h1-97 immunoglobulin G (20 μl) was cross-linked to 50 μl Dynabeads Protein G by BS3 (Sulfo-DSS; Thermo Scientific Pierce) according to the manufacturer’s suggested protocol. About 7 × 106 patient skin fibroblasts (PPH48MW98) were lysed at 4°C with 1000 μl CelLytic M buffer (Sigma-Aldrich). The lysed cells were centrifuged for 15 min at 12,000 × g at 4°C to pellet the cellular debris. The protein-containing supernatant was transferred into a new tube containing Dynabeads-antibody complex and incubated with rotation for 10 min at room temperature. The tube was placed on a magnet and the supernatant (“unbound”) transferred to a clean tube for further analysis. The Dynabeads-antibody-antigen complex was washed 4 times using 200 μl washing buffer for each wash. The final wash was conducted in a new clean tube. The wash solution (“wash”) was collected for further analysis. The Dynabeads-antibody-antigen complex was resuspended with 20 μl elution buffer, 10 μl premixed NuPAGE LDS Sample Buffer, and NuPAGE Sample Reducing Agent and heated for 10 min at 70°C. The supernatant was loaded onto the gel for Western blot and mass spectrometry identification.
Bands corresponding to caveolin-1 were excised and subjected to in-gel trypsin digestion. The resulting peptides were analyzed by a 70-min data-dependent LC-MS/MS analysis. Briefly, peptides were autosampled onto a 200 mm by 0.1 mm (Jupiter 3 micron, 300A), self-packed analytical column coupled directly to an LTQ (ThermoFisher) using a nanoelectrospray source and resolved using an aqueous to organic gradient. A series of a full scans followed by five data-dependent tandem mass spectra (MS/MS) was collected throughout the separation. Dynamic exclusion was enabled to minimize acquisition of redundant peptide spectra. MS/MS spectra were searched via SEQUEST against a human database (UniprotKB–reference proteome set) to which the mutant caveolin sequence had been appended and that also containing reversed version for each of the entries (http://www.ncbi.nlm.nih.gov/pubmed/7741214). Identifications were filtered and collated at the protein level using Scaffold (Proteome Software).
Blue native gel PAGE
BN–PAGE was performed as recently described (Han, Copeland, et al., 2016).
Osmotic swelling and cell viability experiments
Osmotic swelling and cell viability experiments were performed as recently described (Han, Copeland, et al., 2016). One-way ANOVAs were performed using Graphpad.
Supplementary Material
Acknowledgments
The assistance of the Proteomics Core of the Vanderbilt Mass Spectrometry Research Center is gratefully acknowledged. We also thank Kimberly Drake for expert technical assistance. We acknowledge support by R01 HL111259 and R01 HL111259-01S1 from the National Institutes of Health (NIH). Experiments were performed in part through the use of the VUMC Cell Imaging Shared Resource (supported by NIH grants CA-68485, DK-20593, DK-58404, HD-15052, DK-59637, and EY-008126). The funding sources had no role in the study design, collection, analysis or interpretation of data, writing the report, or the decision to submit the paper for publication. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Abbreviations used:
- ADRP
adipose differentiation related protein
- DMSO
dimethyl sulfoxide
- DRM
detergent-resistant membrane
- EGFP
enhanced green fluorescent protein
- PAH
pulmonary arterial hypertension
- ROIs
regions of interest
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E17-06-0421) on September 13, 2017.
REFERENCES
Boldface names denote co–first authors.
- Ariotti N, Parton RG. SnapShot: caveolae, caveolins, and cavins. Cell. 2013;154:704–704 e701. doi: 10.1016/j.cell.2013.07.009. [DOI] [PubMed] [Google Scholar]
- Austin ED, Loyd JE. The genetics of pulmonary arterial hypertension. Circ Res. 2014;115:189–202. doi: 10.1161/CIRCRESAHA.115.303404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin ED, Ma L, LeDuc C, Berman Rosenzweig E, Borczuk A, Phillips JA 3rd, Palomero T, Sumazin P, Kim HR, Talati MH, et al. Whole exome sequencing to identify a novel gene (caveolin-1) associated with human pulmonary arterial hypertension. Circ Cardiovasc Genet. 2012;5:336–343. doi: 10.1161/CIRCGENETICS.111.961888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhshi FR, Mao M, Shajahan AN, Piegeler T, Chen Z, Chernaya O, Sharma T, Elliott WM, Szulcek R, Bogaard HJ, et al. Nitrosation-dependent caveolin 1 phosphorylation, ubiquitination, and degradation and its association with idiopathic pulmonary arterial hypertension. Pulm Circ. 2013;3:816–830. doi: 10.1086/674753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busija AR, Patel HH, Insel PA. Caveolins and cavins in the trafficking, maturation, and degradation of caveolae: implications for cell physiology. Am J Physiol Cell Physiol. 2017;312:C459–C477. doi: 10.1152/ajpcell.00355.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhary N, Gomez GA, Howes MT, Lo HP, McMahon KA, Rae JA, Schieber NL, Hill MM, Gaus K, Yap AS, et al. Endocytic crosstalk: cavins, caveolins, and caveolae regulate clathrin-independent endocytosis. PLoS Biol. 2014;12:e1001832. doi: 10.1371/journal.pbio.1001832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng JP, Mendoza-Topaz C, Howard G, Chadwick J, Shvets E, Cowburn AS, Dunmore BJ, Crosby A, Morrell NW, Nichols BJ, et al. Caveolae protect endothelial cells from membrane rupture during increased cardiac output. J Cell Biol. 2015;211:53–61. doi: 10.1083/jcb.201504042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng JP, Nichols BJ. Caveolae: one function or many? Trends Cell Biol. 2016;26:177–189. doi: 10.1016/j.tcb.2015.10.010. [DOI] [PubMed] [Google Scholar]
- Cohen AW, Combs TP, Scherer PE, Lisanti MP. Role of caveolin and caveolae in insulin signaling and diabetes. Am J Physiol Endocrinol Metab. 2003a;285:E1151–E1160. doi: 10.1152/ajpendo.00324.2003. [DOI] [PubMed] [Google Scholar]
- Cohen AW, Razani B, Schubert W, Williams TM, Wang XB, Iyengar P, Brasaemle DL, Scherer PE, Lisanti MP. Role of caveolin-1 in the modulation of lipolysis and lipid droplet formation. Diabetes. 2004;53:1261–1270. doi: 10.2337/diabetes.53.5.1261. [DOI] [PubMed] [Google Scholar]
- Cohen AW, Razani B, Wang XB, Combs TP, Williams TM, Scherer PE, Lisanti MP. Caveolin-1-deficient mice show insulin resistance and defective insulin receptor protein expression in adipose tissue. Am J Physiol Cell Physiol. 2003b;285:C222–C235. doi: 10.1152/ajpcell.00006.2003. [DOI] [PubMed] [Google Scholar]
- Collins BM, Davis MJ, Hancock JF, Parton RG. Structure-based reassessment of the caveolin signaling model: do caveolae regulate signaling through caveolin-protein interactions. Dev Cell. 2012;23:11–20. doi: 10.1016/j.devcel.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosson P, Lefkir Y, Demolliere C, Letourneur F. New COP1-binding motifs involved in ER retrieval. EMBO J. 1998;17:6863–6870. doi: 10.1093/emboj/17.23.6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos A, Fernandez-Hernando C, Sowa G, Derakhshan B, Lin MI, Lee JY, Zhao H, Luo R, Colangelo C, Sessa WC. Quantitative proteomics of caveolin-1 regulated proteins: Characterization of PTRF/Cavin-1 in endothelial cells. Mol Cell Proteom. 2010;9:2109–2124. doi: 10.1074/mcp.M110.001289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drab M, Verkade P, Elger M, Kasper M, Lohn M, Lauterbach B, Menne J, Lindschau C, Mende F, Luft FC, et al. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science. 2001;293:2449–2452. doi: 10.1126/science.1062688. [DOI] [PubMed] [Google Scholar]
- Galbiati F, Engelman JA, Volonte D, Zhang XL, Minetti C, Li M, Hou H, Jr, Kneitz B, Edelmann W, Lisanti MP. Caveolin-3 null mice show a loss of caveolae, changes in the microdomain distribution of the dystrophin-glycoprotein complex, and t-tubule abnormalities. J Biol Chem. 2001;276:21425–21433. doi: 10.1074/jbc.M100828200. [DOI] [PubMed] [Google Scholar]
- Galbiati F, Volonte D, Minetti C, Bregman DB, Lisanti MP. Limb-girdle muscular dystrophy (LGMD-1C) mutants of caveolin-3 undergo ubiquitination and proteasomal degradation. Treatment with proteasomal inhibitors blocks the dominant negative effect of LGMD-1C mutants and rescues wild-type caveolin-3. J Biol Chem. 2000;275:37702–37711. doi: 10.1074/jbc.M006657200. [DOI] [PubMed] [Google Scholar]
- Galbiati F, Volonte D, Minetti C, Chu JB, Lisanti MP. Phenotypic behavior of caveolin-3 mutations that cause autosomal dominant limb girdle muscular dystrophy (LGMD-1C). Retention of LGMD-1C caveolin-3 mutants within the golgi complex. J Biol Chem. 1999;274:25632–25641. doi: 10.1074/jbc.274.36.25632. [DOI] [PubMed] [Google Scholar]
- Gambin Y, Ariotti N, McMahon KA, Bastiani M, Sierecki E, Kovtun O, Polinkovsky ME, Magenau A, Jung W, Okano S, et al. Single-molecule analysis reveals self assembly and nanoscale segregation of two distinct cavin subcomplexes on caveolae. Elife. 2014;3:e01434. doi: 10.7554/eLife.01434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg A, Kircher M, Del Campo M, Amato RS, Agarwal AK. Whole exome sequencing identifies de novo heterozygous CAV1 mutations associated with a novel neonatal onset lipodystrophy syndrome. Am J Med Genet A. 2015;167:1796–1806. doi: 10.1002/ajmg.a.37115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han B, Copeland CA, Kawano Y, Rosenzweig EB, Austin ED, Shahmirzadi L, Tang S, Raghunathan K, Chung WK, Kenworthy AK. Characterization of a caveolin-1 mutation associated with both pulmonary arterial hypertension and congenital generalized lipodystrophy. Traffic. 2016;17:1297–1312. doi: 10.1111/tra.12452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han B, Tiwari A, Kenworthy AK. Tagging strategies strongly affect the fate of overexpressed caveolin-1. Traffic. 2015;16:417–438. doi: 10.1111/tra.12254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen CG, Howard G, Nichols BJ. Pacsin 2 is recruited to caveolae and functions in caveolar biogenesis. J Cell Sci. 2011;124:2777–2785. doi: 10.1242/jcs.084319. [DOI] [PubMed] [Google Scholar]
- Hanson CA, Drake KR, Baird MA, Han B, Kraft LJ, Davidson MW, Kenworthy AK. Overexpression of caveolin-1 is sufficient to phenocopy the behavior of a disease-associated mutant. Traffic. 2013;14:663–677. doi: 10.1111/tra.12066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayer A, Stoeber M, Bissig C, Helenius A. Biogenesis of caveolae: stepwise assembly of large caveolin and cavin complexes. Traffic. 2010a;11:361–382. doi: 10.1111/j.1600-0854.2009.01023.x. [DOI] [PubMed] [Google Scholar]
- Hayer A, Stoeber M, Ritz D, Engel S, Meyer HH, Helenius A. Caveolin-1 is ubiquitinated and targeted to intralumenal vesicles in endolysosomes for degradation. J Cell Biol. 2010b;191:615–629. doi: 10.1083/jcb.201003086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MM, Bastiani M, Luetterforst R, Kirkham M, Kirkham A, Nixon SJ, Walser P, Abankwa D, Oorschot VM, Martin S, et al. PTRF-Cavin, a conserved cytoplasmic protein required for caveola formation and function. Cell. 2008;132:113–124. doi: 10.1016/j.cell.2007.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeper MM, Bogaard HJ, Condliffe R, Frantz R, Khanna D, Kurzyna M, Langleben D, Manes A, Satoh T, Torres F, et al. Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol. 2013;62:D42–D50. doi: 10.1016/j.jacc.2013.10.032. [DOI] [PubMed] [Google Scholar]
- Jackson MR, Nilsson T, Peterson PA. Retrieval of transmembrane proteins to the endoplasmic reticulum. J Cell Biol. 1993;121:317–333. doi: 10.1083/jcb.121.2.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasmin JF, Mercier I, Dupuis J, Tanowitz HB, Lisanti MP. Short-term administration of a cell-permeable caveolin-1 peptide prevents the development of monocrotaline-induced pulmonary hypertension and right ventricular hypertrophy. Circulation. 2006;114:912–920. doi: 10.1161/CIRCULATIONAHA.106.634709. [DOI] [PubMed] [Google Scholar]
- Kirkham M, Nixon SJ, Howes MT, Abi-Rached L, Wakeham DE, Hanzal-Bayer M, Ferguson C, Hill MM, Fernandez-Rojo M, Brown DA, et al. Evolutionary analysis and molecular dissection of caveola biogenesis. J Cell Sci. 2008;121:2075–2086. doi: 10.1242/jcs.024588. [DOI] [PubMed] [Google Scholar]
- Kovtun O, Tillu VA, Ariotti N, Parton RG, Collins BM. Cavin family proteins and the assembly of caveolae. J Cell Sci. 2015;128:1269–1278. doi: 10.1242/jcs.167866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Park DS, Razani B, Russell RG, Pestell RG, Lisanti MP. Caveolin-1 mutations (P132L and null) and the pathogenesis of breast cancer: caveolin-1 (P132L) behaves in a dominant-negative manner and caveolin-1 (-/-) null mice show mammary epithelial cell hyperplasia. Am J Pathol. 2002;161:1357–1369. doi: 10.1016/S0002-9440(10)64412-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Brown D, McKee M, Lebrasseur NK, Yang D, Albrecht KH, Ravid K, Pilch PF. Deletion of Cavin/PTRF causes global loss of caveolae, dyslipidemia, and glucose intolerance. Cell Metab. 2008;8:310–317. doi: 10.1016/j.cmet.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Pilch PF. A critical role of cavin (polymerase I and transcript release factor) in caveolae formation and organization. J Biol Chem. 2008;283:4314–4322. doi: 10.1074/jbc.M707890200. [DOI] [PubMed] [Google Scholar]
- Low JY, Nicholson HD. Epigenetic modifications of caveolae associated proteins in health and disease. BMC Genet. 2015;16:71. doi: 10.1186/s12863-015-0231-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig A, Howard G, Mendoza-Topaz C, Deerinck T, Mackey M, Sandin S, Ellisman MH, Nichols BJ. Molecular composition and ultrastructure of the caveolar coat complex. PLoS Biol. 2013;11:e1001640. doi: 10.1371/journal.pbio.1001640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luetterforst R, Stang E, Zorzi N, Carozzi A, Way M, Parton RG. Molecular characterization of caveolin association with the Golgi complex: identification of a cis-Golgi targeting domain in the caveolin molecule. J Cell Biol. 1999;145:1443–1459. doi: 10.1083/jcb.145.7.1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machleidt T, Li WP, Liu P, Anderson RG. Multiple domains in caveolin-1 control its intracellular traffic. J Cell Biol. 2000;148:17–28. doi: 10.1083/jcb.148.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsboom G, Chen Z, Yuan Y, Zhang Y, Tiruppathi C, Loyd JE, Austin ED, Machado RF, Minshall RD, Rehman J, et al. Aberrant caveolin-1-mediated Smad signaling and proliferation identified by analysis of adenine 474 deletion mutation (c.474delA) in patient fibroblasts: a new perspective on the mechanism of pulmonary hypertension. Mol Biol Cell. 2017;28:1177–1185. doi: 10.1091/mbc.E16-11-0790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew R. Cell-specific dual role of caveolin-1 in pulmonary hypertension. Pulm Med. 2011;2011:573432. doi: 10.1155/2011/573432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew R, Huang J, Gewitz MH. Pulmonary artery hypertension: caveolin-1 and eNOS interrelationship: a new perspective. Cardiol Rev. 2007;15:143–149. doi: 10.1097/01.crd.0000249381.49138.b9. [DOI] [PubMed] [Google Scholar]
- Mathew R, Huang J, Shah M, Patel K, Gewitz M, Sehgal PB. Disruption of endothelial-cell caveolin-1alpha/raft scaffolding during development of monocrotaline-induced pulmonary hypertension. Circulation. 2004;110:1499–1506. doi: 10.1161/01.CIR.0000141576.39579.23. [DOI] [PubMed] [Google Scholar]
- Monier S, Parton RG, Vogel F, Behlke J, Henske A, Kurzchalia TV. VIP21-caveolin, a membrane protein constituent of the caveolar coat, oligomerizes in vivo and in vitro. Mol Biol Cell. 1995;6:911–927. doi: 10.1091/mbc.6.7.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moren B, Shah C, Howes MT, Schieber NL, McMahon HT, Parton RG, Daumke O, Lundmark R. EHD2 regulates caveolar dynamics via ATP-driven targeting and oligomerization. Mol Biol Cell. 2012;23:1316–1329. doi: 10.1091/mbc.E11-09-0787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassar ZD, Moon H, Duong T, Neo L, Hill MM, Francois M, Parton RG, Parat MO. PTRF/Cavin-1 decreases prostate cancer angiogenesis and lymphangiogenesis. Oncotarget. 2013;4:1844–1855. doi: 10.18632/oncotarget.1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassoy P, Lamaze C. Stressing caveolae new role in cell mechanics. Trends Cell Biol. 2012;22:381–389. doi: 10.1016/j.tcb.2012.04.007. [DOI] [PubMed] [Google Scholar]
- Ostermeyer AG, Paci JM, Zeng Y, Lublin DM, Munro S, Brown DA. Accumulation of caveolin in the endoplasmic reticulum redirects the protein to lipid storage droplets. J Cell Biol. 2001;152:1071–1078. doi: 10.1083/jcb.152.5.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parton RG, del Pozo MA. Caveolae as plasma membrane sensors, protectors and organizers. Nat Rev Mol Cell Biol. 2013;14:98–112. doi: 10.1038/nrm3512. [DOI] [PubMed] [Google Scholar]
- Parton RG, Hanzal-Bayer M, Hancock JF. Biogenesis of caveolae: a structural model for caveolin-induced domain formation. J Cell Sci. 2006;119:787–796. doi: 10.1242/jcs.02853. [DOI] [PubMed] [Google Scholar]
- Parton RG, Simons K. The multiple faces of caveolae. Nat Rev Mol Cell Biol. 2007;8:185–194. doi: 10.1038/nrm2122. [DOI] [PubMed] [Google Scholar]
- Patel HH, Zhang S, Murray F, Suda RY, Head BP, Yokoyama U, Swaney JS, Niesman IR, Schermuly RT, Pullamsetti SS, et al. Increased smooth muscle cell expression of caveolin-1 and caveolae contribute to the pathophysiology of idiopathic pulmonary arterial hypertension. Faseb J. 2007;21:2970–2979. doi: 10.1096/fj.07-8424com. [DOI] [PubMed] [Google Scholar]
- Prewitt AR, Ghose S, Frump AL, Datta A, Austin ED, Kenworthy AK, de Caestecker MP. Heterozygous null bone morphogenetic protein receptor type 2 mutations promote SRC kinase-dependent caveolar trafficking defects and endothelial dysfunction in pulmonary arterial hypertension. J Biol Chem. 2015;290:960–971. doi: 10.1074/jbc.M114.591057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendran L, Le Lay S, Illges H. Raft association and lipid droplet targeting of flotillins are independent of caveolin. Biol Chem. 2007;388:307–314. doi: 10.1515/BC.2007.034. [DOI] [PubMed] [Google Scholar]
- Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, Marks CB, Macaluso F, Russell RG, Li M, Pestell RG, et al. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem. 2001;276:38121–38138. doi: 10.1074/jbc.M105408200. [DOI] [PubMed] [Google Scholar]
- Razani B, Lisanti MP. Caveolin-deficient mice: insights into caveolar function human disease. J Clin Invest. 2001;108:1553–1561. doi: 10.1172/JCI14611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razani B, Wang XB, Engelman JA, Battista M, Lagaud G, Zhang XL, Kneitz B, Hou H, Jr, Christ GJ, Edelmann W, et al. Caveolin-2-deficient mice show evidence of severe pulmonary dysfunction without disruption of caveolae. Mol Cell Biol. 2002a;22:2329–2344. doi: 10.1128/MCB.22.7.2329-2344.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razani B, Woodman SE, Lisanti MP. Caveolae: from cell biology to animal physiology. Pharmacol Rev. 2002b;54:431–467. doi: 10.1124/pr.54.3.431. [DOI] [PubMed] [Google Scholar]
- Ren X, Ostermeyer AG, Ramcharan LT, Zeng Y, Lublin DM, Brown DA. Conformational defects slow Golgi exit, block oligomerization, and reduce raft affinity of caveolin-1 mutant proteins. Mol Biol Cell. 2004;15:4556–4567. doi: 10.1091/mbc.E04-06-0480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Root KT, Plucinsky SM, Glover KJ. Recent progress in the topology, structure, and oligomerization of caveolin: a building block of caveolae. Curr Top Membr. 2015;75:305–336. doi: 10.1016/bs.ctm.2015.03.007. [DOI] [PubMed] [Google Scholar]
- Rothberg KG, Heuser JE, Donzell WC, Ying YS, Glenney JR, Anderson RG. Caveolin, a protein component of caveolae membrane coats. Cell. 1992;68:673–682. doi: 10.1016/0092-8674(92)90143-z. [DOI] [PubMed] [Google Scholar]
- Scheiffele P, Verkade P, Fra AM, Virta H, Simons K, Ikonen E. Caveolin-1 and -2 in the exocytic pathway of MDCK cells. J Cell Biol. 1998;140:795–806. doi: 10.1083/jcb.140.4.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlegel A, Lisanti MP. A molecular dissection of caveolin-1 membrane attachment and oligomerization. Two separate regions of the caveolin-1 C-terminal domain mediate membrane binding and oligomer/oligomer interactions in vivo. J Biol Chem. 2000;275:21605–21617. doi: 10.1074/jbc.M002558200. [DOI] [PubMed] [Google Scholar]
- Schrauwen I, Szelinger S, Siniard AL, Kurdoglu A, Corneveaux JJ, Malenica I, Richholt R, Van Camp G, De Both M, Swaminathan S, et al. A frame-shift mutation in CAV1 is associated with a severe neonatal progeroid and lipodystrophy syndrome. PLoS One. 2015;10:e0131797. doi: 10.1371/journal.pone.0131797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senju Y, Itoh Y, Takano K, Hamada S, Suetsugu S. Essential role of PACSIN2/syndapin-II in caveolae membrane sculpting. J Cell Sci. 2011;124:2032–2040. doi: 10.1242/jcs.086264. [DOI] [PubMed] [Google Scholar]
- Sinha B, Koster D, Ruez R, Gonnord P, Bastiani M, Abankwa D, Stan RV, Butler-Browne G, Vedie B, Johannes L, et al. Cells respond to mechanical stress by rapid disassembly of caveolae. Cell. 2011;144:402–413. doi: 10.1016/j.cell.2010.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song KS, Tang ZL, Li SW, Lisanti MP. Mutational analysis of the properties of caveolin-1. A novel role for the C-terminal domain in mediating homo-typic caveolin-caveolin interactions. J Biol Chem. 1997;272:4398–4403. doi: 10.1074/jbc.272.7.4398. [DOI] [PubMed] [Google Scholar]
- Stoeber M, Stoeck IK, Hanni C, Bleck CK, Balistreri G, Helenius A. Oligomers of the ATPase EHD2 confine caveolae to the plasma membrane through association with actin. EMBO J. 2012;31:2350–2364. doi: 10.1038/emboj.2012.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari A, Copeland CA, Han B, Hanson CA, Raghunathan K, Kenworthy AK. Caveolin-1 is an aggresome-inducing protein. Sci Rep. 2016;6:38681. doi: 10.1038/srep38681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinten J, Johnsen AH, Roepstorff P, Harpoth J, Tranum-Jensen J. Identification of a major protein on the cytosolic face of caveolae. Biochim Biophys Acta. 2005;1717:34–40. doi: 10.1016/j.bbamem.2005.09.013. [DOI] [PubMed] [Google Scholar]
- Zhao YY, Zhao YD, Mirza MK, Huang JH, Potula HH, Vogel SM, Brovkovych V, Yuan JX, Wharton J, Malik AB. Persistent eNOS activation secondary to caveolin-1 deficiency induces pulmonary hypertension in mice and humans through PKG nitration. J Clin Invest. 2009;119:2009–2018. doi: 10.1172/JCI33338. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.