

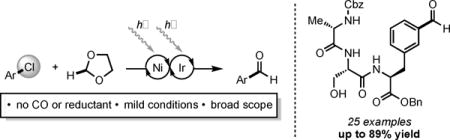
Abstract
We report a redox-neutral formylation of aryl chlorides that proceeds through selective 2-functionalization of 1,3-dioxolane via nickel and photoredox catalysis. This scalable, benchtop approach provides a distinct advantage over traditional reductive carbonylation in that no carbon monoxide, pressurized gas, or stoichiometric reductant is employed. Mild conditions enable unprecedented scope from abundant and complex aryl chloride starting materials.
Keywords: Formylation, nickel, C–H functionalization, radical photoelimination
TOC image
Aromatic aldehydes are generated from abundant aryl chlorides via nickel-photocatalyzed C–H functionalization of the inexpensive solvent 1,3-dioxolane. The absence of pressurized carbon monoxide and a stoichiometric reductant enables broad functional group tolerance and scope.
Aromatic aldehydes are among the most versatile intermediates in the synthesis of pharmaceuticals, fragrances, fine chemicals, and natural products.[1] Indeed, the functional group can be rapidly elaborated via an ever growing host of C–C and C–X bond-forming reactions. Despite the ubiquitous application of aryl aldehydes, synthetic methods for their preparation are limited. Classical approaches such as the Vilsmeier–Haack and Duff reactions proceed via electrophilic aromatic substitution; therefore their reactivity and regioselectivity are endogenous to the particular aryl substrate (Figure 1A).[2] As an alternative, traditional organometallic methods, such as the addition of Grignard reagents to DMF at cryogenic temperatures, impart regiocontrol at the expense of functional group compatibility.[3] To date, the most general method for the synthesis of aryl aldehydes is palladium catalyzed reductive carbonylation of aryl iodides and bromides, first reported by Heck in 1974 under 100 atm of syngas (1:1 H2:CO) at 150 °C.[4] Although this procedure has been performed on multi-ton scale,[5] the conditions are not ideal for benchtop synthesis due to the hazards associated with carbon monoxide and the specialized equipment required for handling high pressure syngas. To develop user-friendly protocols, several laboratories have reported alternative condensed phase reductants[6] and CO surrogates[7] such as the crystalline N‐formylsaccharin.[7b] Nevertheless, reductive formylations are still limited to simple arenes due to the general requirements of high reaction temperatures and stoichiometric reducing agents. Moreover, most methods are incapable of employing aryl chlorides,[8] by far the most abundant and diverse class of aryl halides.[9]
Figure 1.
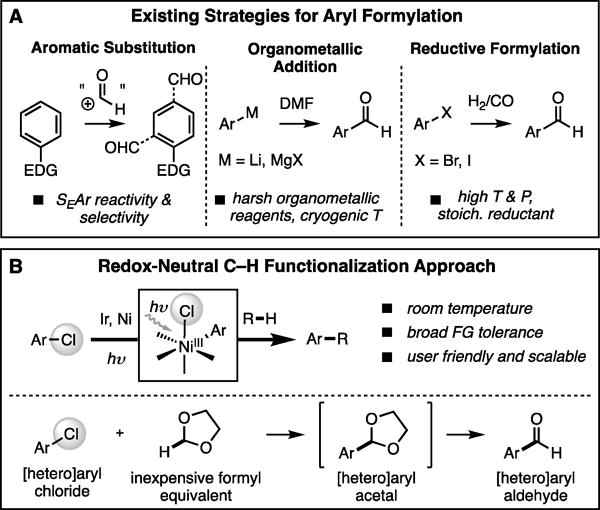
(a) Prior art in aryl formylation. (b) Proposed redox-neutral formylation of aryl chlorides via C–H activation of 1,3-dioxolane.
Recently, our lab[10] and others reported directing group free Csp3–H cross-coupling platforms capable of carrying out C–H arylations at room temperature. In contrast to systems reported by MacMillan[11] and Molander[12], which primarily employ aryl bromides and iodides, our method utilizes aryl chlorides by design. Here we demonstrate that this manifold can be leveraged to enable redox-neutral formylation by selective 2-arylation of the inexpensive and abundant solvent 1,3-dioxolane with aryl chlorides followed by a mild acidic workup (Figure 1B). Importantly this strategy overcomes the challenges of positional selectivity and functional group compatibility associated with classical formylation reactions and obviates the need for gaseous reagents, stoichiometric reductants and high reaction temperatures required for reductive carbonylation.
We envisioned that oxidative addition of Ni(0) (Figure 2, 1) into an aryl chloride would produce Ni(II) intermediate 2. Simultaneously, visible light irradiation of Ir(III) photocatalyst 3 would generate triplet excited *Ir(III) (τ0 = 2.3 μs, *E1/2 = 1.21 V vs SCE in MeCN) (4) which could engage 2 (EP = 0.85 V vs SCE in THF) in photoinduced electron transfer to give presumed Ni(III) intermediate 5.[10,13] According to our prior studies,[10] photolysis of 5 produces Ni(II) species 6 and a chlorine radical, capable of abstracting a hydrogen atom from 1,3-dioxolane. A concern in developing this reaction was that 1,3-dioxolane has two sets of chemically distinct α-oxy C–H bonds. Thermodynamic computations imply a moderate driving force for 2-functionalization (ΔBDFE = 1.4 kcal mol−1), which we expected could be influenced by the Ni catalyst that accepts the resultant carbon-centered radical. Reductive elimination from Ni(III) species 7 would then give the aryl-dioxolane acetal and hydrolytic workup would furnish the desired aryl aldehyde product. Finally, to close both cycles Ni(I) species 8 (E1/2[Ni(II)/(0)] = −1.2 V vs SCE in DMF) can be reduced by Ir(II) species 9 (E1/2 = −1.37 V vs SCE in MeCN).[13,14]
Figure 2.
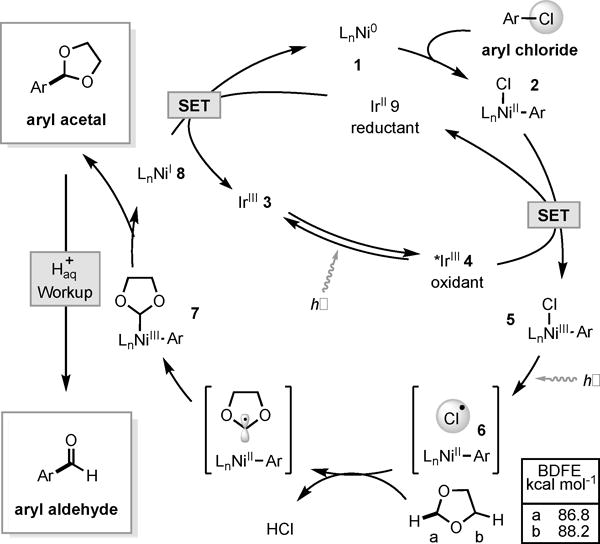
Proposed catalytic cycle.
We began studying the proposed formylation reaction with emphasis on the development of a user friendly and scalable protocol. We were pleased to find that the commercial, bench-stable precatalyst systems—Ir[dF(CF3)ppy]2(dtbbpy)PF6[15] and NiCl2·DME with 4,4′-di-t-butyl-2,2′-bipyridine ligand (dtbbpy)—combined with two equivalents of potassium phosphate under irradiation with blue LEDs enabled the selective 2‐functionalization of 1,3-dioxolane with numerous aryl chlorides. A subsequent mild acidic workup gave the desired aryl (Figure 3, 10–18), heteroaryl (19–22) and biologically relevant (23–34) aldehydes in good to excellent yields with minimal loss of material upon acetal hydrolysis (76% yield 10 vs. 74% yield 11). Interestingly, the average selectivity for the cross-coupling step favoured methylene over ethylene C–H functionalization by 9:1. This observation is in qualitative agreement with computed C–H BDFEs, which predict a selectivity of 5:1 purely based on thermodynamics and stoichiometry. However, the disparity suggests that other factors such as polar effects in the rebound of the radical to Ni(II) may also be important. Based on these observations, it is noteworthy that the reaction of chlormezanone afforded 23 in 89% yield—the maximum theoretical yield based on average selectivity. For most substrates, simple silica gel column chromatography was sufficient to obtain the desired product in high purity (see SI for details). However, in some cases, a mixture of aryl aldehyde and isomeric acetal resulting from 4-functionalization of dioxolane was isolated. Notably, regioisomerism can be avoided by employing 1,3,5-trioxane as a formyl source; for example, using 50 equivalents of 1,3,5-trioxane and benzene as a solvent, the trioxanyl acetal of 12 can be prepared in 57% yield under otherwise identical conditions. Control reactions demonstrated that in the absence of light, nickel or photoredox catalyst, no product was formed. Reactions carried out on the benchtop afforded comparable yields to those set up in a glovebox; for example, 23 was obtained in 81% yield (vs 89% yield) using Schlenk technique. In addition, the reaction is amenable to operationally simple batch scale-up; fenofibrate gave 1.47 g of 24 in 83% yield on 5 mmol scale. Moreover, benchtop scale-up reactions of 16 and 17 gave 64% yield and 70% yield respectively on gram scale.
Figure 3.
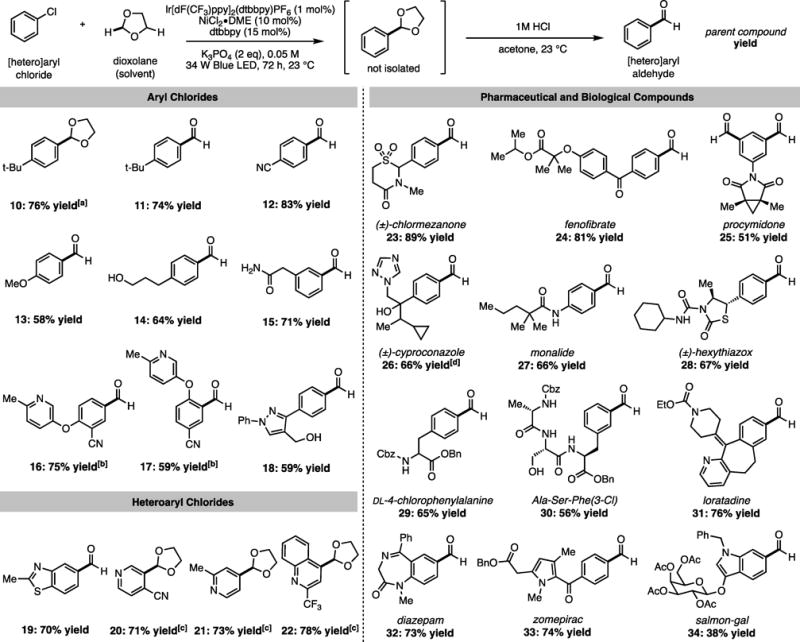
Formylation of aryl chlorides: substrate scope. Yields are an average of two runs on 0.25 mmol scale. For 23 – 34, the name of the parent compound is listed. [a]Acid hydrolysis omitted. [b]Benctop setup; single run. [c]Aldehyde does not form under hydrolysis conditions. [d]2.3:1 dr (conserved from starting material).
Reaction scope investigations indicate that the method is general across a broad range of electronically differentiated aryl chlorides. It was observed that electron-deficient aryl chlorides generally afforded higher yields than electron-rich substrates over the 72 hour reaction time. Time point experiments showed that while the reaction of 4-chlorobenzonitrile reached nearly full conversion within 48 hours, 4-chloroanisole continued to undergo cross coupling for the duration of the 96 hour monitoring period (Figure S5), indicating electron-rich substrates react at a reduced rate. Sterically encumbered chloroarenes bearing ortho substituents underwent coupling to give the corresponding formylated products (17, 21). Additionally, the dichloride fungicide procymidone underwent multiple functionalizations to form dialdehyde 25.
A broad range of reactive functional groups were well tolerated, highlighting the exceptionally mild reaction conditions. Aryl chlorides containing protic functionality underwent efficient formylation to yield primary alcohols 14 and 30, primary and tertiary benzylic alcohols 18 and 26, primary amide 15 and secondary amides 27–30. Functional groups susceptible to hydrogenation under typical reductive carbonylation conditions can be accommodated, such as alkene 31 and imine 32. Furthermore, formylation proceeds in the presence of diverse heterocycles, both distal and proximal, including pyridines 16, 17, 20, 21, and 31, pyrazole 18, benzothiazole 19, quinoline 22, triazole 26, thiazolidinone 28, benzodiazepine 32, pyrrole 33, and indole 34. We were pleased to find that numerous pharmaceuticals (23, 24, 31–33) and agrochemicals (25–28) were accommodated. Formylations that yielded amino ester 29, tripeptide 30, and glycoside 34 are particularly noteworthy, demonstrating a new strategy for synthesizing biomolecular aldehydes, a desirable functional handle in the field of bioconjugation.[16] More generally, the unparalleled scope of this transformation offers the possibility for late-stage formylation of typically inert aryl chlorides and thus the potential to disrupt current trends in synthesis in which aryl aldehydes are employed early in a synthetic sequence.[1a,1b]
We recognized that in some instances it may be advantageous to employ aryl bromides or iodides in this transformation. 4-Bromobenzonitrile and 4-bromobiphenyl performed modestly well in the coupling reaction, giving the corresponding aryl acetals (36-CN and 36-Ph) in 52% and 36% yield respectively (Figure 4). Notably, aryl bromides are significantly more selective for 2-functionalization, in both cases doubling the regioisomeric ratio. This observation is consistent with a halogen radical abstraction mechanism wherein selectivity for the initial C–H functionalization is governed by Hammond’s postulate—that is, the late transition state in bromine abstraction (BDEH–Br = 88 kcal∙mol−1 vs. BDEH–Cl = 103 kcal∙mol−1) results in higher selectivity for the thermodynamically favored alkyl radical product. Interestingly, the change in selectivity for each set of aryl chlorides and aryl bromides was moderately substituent dependent (Entry 1: Entry 4 = 1:1.3, Entry 2: Entry 5 = 1:1.1), in agreement with the proposed role of aryl Ni(II) (Figure 2, species 6) in the radical rebound step. Together these observations suggest the potential for both halide- and substrate-control over C–H functionalization selectivity. Although aryl iodides alone are incompetent (BDEH–I = 71 kcal∙mol−1), moderate yield can be attained through halide exchange with tetrabutylammonium chloride (TBACl).
Figure 4.
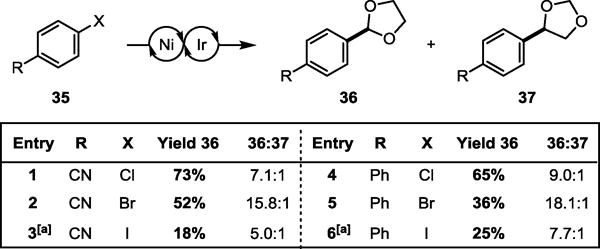
Aryl halide selectivity studies. Yields determined by GC-FID using 1-fluoronaphthalene as an external standard. See Tables S2 and S3 for additional experiments and reaction conditions. [a] With addition of 1 equiv TBACl.
In a preliminary evaluation of other substrate classes under unoptimized conditions, alkyl bromide (3-bromopropyl)benzene afforded 4-phenylbutanal in 6% yield, demonstrating the potential of this strategy to convert alkyl halides to homologated aldehydes. Moreover, the acyl chloride 4-methylbenzoyl chloride underwent coupling to give the corresponding protected glyoxal in 39% yield (See SI for details). By comparison, under reductive carbonylation conditions, acid halides are protodehalogenated to form aldehydes,[4] and the aryl glyoxals are typically only accessible through oxidative methods.
Although various oxidation states (ie. alcohol, carboxylic acid derivative) can be accessed from aldehydes, redox manipulations are not ideal for step economy and require stoichiometric oxidants or reductants. We recognized the generality of the redox-neutral Csp3–H functionalization strategy underlying our formylation reaction and hypothesized that direct access to other oxidation states could be afforded by judicious selection of C–H coupling partner. Gratifyingly, under unoptimized conditions the methyl ester and benzyl alcohol of 4-chlorobiphenyl could be accessed by direct arylation of trimethyl orthoformate, via a tertiary radical intermediate, and trimethyl orthoacetate to give 39 and 40 in 14% and 42% yield respectively (Figure 5). These preliminary examples provided promising results for further development.
Figure 5.
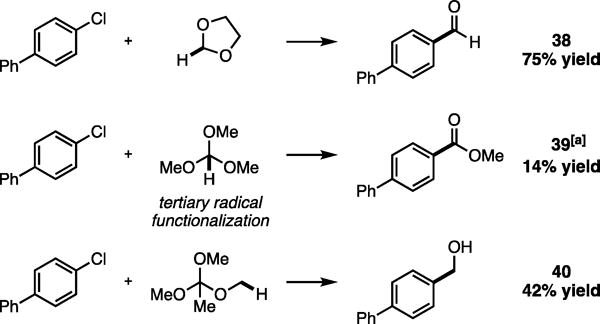
Direct access to formyl oxidation and reduction analogues via redox-neutral Csp3–H functionalization. Isolated yields. [a] Generates methoxy functionalization product in 15% yield. See SI for experimental details.
In conclusion, we have demonstrated a redox-neutral C–H functionalization approach to aryl formylation which proceeds under exceptionally mild conditions to give numerous aryl, heteroaryl, and biologically relevant aldehydes.
Supplementary Material
Acknowledgments
Financial support was provided by NIGMS (R01 GM100985) and Celgene Corporation.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.a) Baumann KL, Butler DE, Deering CF, Mennen KE, Millar A, Nanninga TN, Palmer CW, Roth BD. Tetrahedron Lett. 1992;33:2283–2284. [Google Scholar]; b) McNamara JM, Leazer JL, Bhupathy M, Amato JS, Reamer RA, Reider PJ, Grabowski EJJ. J Org Chem. 1989;54:3718–3721. [Google Scholar]; c) Fráter G, Bajgrowicz JA, Kraft P. Tetrahedron. 1998;54:7633–7703. [Google Scholar]; d) Woodward RB, Cava MP, Ollis WD, Hunger A, Daeniker HU, Schenker K. J Am Chem Soc. 1954;76:4749–4751. [Google Scholar]; e) Magnus P, Sane N, Fauber BP, Lynch V. J Am Chem Soc. 2009;131:16045–16047. doi: 10.1021/ja9085534. [DOI] [PubMed] [Google Scholar]
- 2.a) Reimer K, Tiemann F. Ber Dtsch Chem Ges. 1876;9:1268–1278. [Google Scholar]; b) Gattermann L, Koch JA. Chem Ber. 1897;30:1622–1624. [Google Scholar]; c) Attermann L, Berchelmann W. Ber Dtsch Chem Ges. 1898;31:1765–1769. [Google Scholar]; d) Vilsmeier A, Haack A. Chem Ber. 1927;60:119–122. [Google Scholar]; e) Duff JC, Bills EJ. J Chem Soc. 1932:1987–1988. [Google Scholar]
- 3.Bouveault L. Bull Soc Chim Fr. 1904;31:1306–1322. [Google Scholar]
- 4.Schoenberg A, Heck RF. J Am Chem Soc. 1974;96:7761–7764. [Google Scholar]
- 5.Klaus S, Neumann H, Zapf A, Strübing D, Hübner S, Almena J, Riermeier T, Groß P, Sarich M, Krahnert WR, Rossen K, Beller M. Angew Chem Int Ed. 2006;45:154–158. doi: 10.1002/anie.200502697. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2006;118:161–165. [Google Scholar]
- 6.a) Baillargeon VP, Stille JK. J Am Chem Soc. 1983;105:7175–7176. [Google Scholar]; b) Pri-Bar I, Buchman O. J Org Chem. 1984;49:4009–4011. [Google Scholar]
- 7.a) Okano T, Harada N, Kiji J. Bull Chem Soc Jpn. 1994;67:2329–2332. [Google Scholar]; b) Ueda T, Konishi H, Manabe K. Angew Chem Int Ed. 2013;52:8611–8615. doi: 10.1002/anie.201303926. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2013;125:8773–8777. [Google Scholar]; c) Yu B, Zhao Y, Zhang H, Xu J, Hao L, Gao X, Liu Z. Chem Commun. 2014;50:2330–2333. doi: 10.1039/c3cc49365b. [DOI] [PubMed] [Google Scholar]; d) Jiang X, Wang JM, Zhang Y, Chen Z, Zhu YM, Ji SJ. Org Lett. 2014;16:3492–3495. doi: 10.1021/ol5014262. [DOI] [PubMed] [Google Scholar]; e) Natte K, Dumrath A, Neumann H, Beller M. Angew Chem Int Ed. 2014;53:10090–10094. doi: 10.1002/anie.201404833. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2014;126:10254–10258. [Google Scholar]; f) Iranpoor N, Firouzabadi H, Etemadi-Davan E, Rostami A, Moghadam KR. Appl Organometal Chem. 2015;29:719–724. [Google Scholar]
- 8.Ben-David Y, Portnoy M, Milstein D. J Am Chem Soc. 1989;111:8742–8744. [Google Scholar]
- 9.A Ni/photoredox decarboxylative approach for formylating activated aryl chlorides was recently reported; Huang H, Li X, Yu C, Zhang Y, Mariano P, Wang W. Angew Chem Int Ed. 2017;56:1500–1505. doi: 10.1002/anie.201610108. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2017;129:1522–1527. [Google Scholar]
- 10.Shields BJ, Doyle AG. J Am Chem Soc. 2016;138:12719–12722. doi: 10.1021/jacs.6b08397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shaw MH, Shurtleff VW, Terrett JA, Cuthbertson JD, MacMillan DWC. Science. 2016;352:1304–1308. doi: 10.1126/science.aaf6635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heitz DR, Tellis JC, Molander GA. J Am Chem Soc. 2016;138:12715–12718. doi: 10.1021/jacs.6b04789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lowry MS, Goldsmith JI, Slinker JD, Rohl R, Pascal RA, Malliaras GG, Bernhard S. Chem Mater. 2005;17:5712–5719. [Google Scholar]
- 14.Durandetti M, Devaud M, Perichon J. New J Chem. 1996;20:659–667. [Google Scholar]
- 15.(4,4′-Di-t-butyl-2,2′-bipyridine)bis[3,5-difluoro-2-[5-trifluoromethyl-2-pyridinyl-κN)phenyl-κC]iridium(III) hexafluorophosphate [870987-63-6].
- 16.Carrico IS, Carlson BL, Bertozzi CR. Nat Chem Biol. 2007;3:321–322. doi: 10.1038/nchembio878. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.