

Graphical Abstract
This review highlights recent efforts in using photochemistry to exert dynamic control over the properties of hydrogel biomaterials.
1. Introduction
Methods to culture cells outside of their in vivo tissue microenvironments have engendered the study and understanding of many fundamental processes of life. For example, these simplified in vitro models have allowed researchers to uncover many of the molecular mechanisms behind stem cell maintenance and differentiation, as well as the reprogramming1 and transdifferentation2 of somatic cells. However, in many instances, traditional glass or plastic culture substrates are poor substitutes for the native extracellular matrix environment.3 These glassy materials are several orders of magnitude stiffer than most soft tissues, and two dimensional surfaces can lead to artificial polarization of adherent cells.
Only relatively recently have the effects of substrate mechanics and dimensionality on cell behavior been investigated. Bissell and coworkers provided some of the early clues related to the significant effect of dimensionality on cells in vitro. In their pioneering studies, results showed that, in contrast to monolayer culture, 3D culture of normal breast epithelial cells resulted in formation of spherical colonies with a central lumen and polarized epithelium, similar to the acini found in normal tissue.4 Furthermore, malignant breast cancer cells displayed a distinct phenotype in 3D that could be reversed by blocking integrin binding to the extracellular matrix (ECM),5 and overexpression of matrix-remodeling enzymes resulted in malignancy of normal epithelial cells.6 These studies highlighted the importance of cell-matrix interactions and the inclusion of such contextual cues in cell culture systems.
In addition, cultured cells have been shown to sense their mechanical surroundings through cell-generated traction forces. Multipotent human mesenchymal stem cells (hMSCs) preferentially commit to different lineages depending on the stiffness of their culture substrate. Soft matrices promote neurogenesis, those of intermediate stiffness support myogenesis, and rigid substrates enhance osteogenesis, corresponding to the respective relative stiffness of neural, muscle, and collagenous bone tissues.7 More recent efforts have revealed that cellular behavior can also be influenced by the viscoelastic8–11 and strain-stiffening12 properties of biomaterial matrices. The effects of matricellular interactions are now recognized as important in tissue morphogenesis13 and disease progression,14 as well as in guiding the self-renewal or differentiation of multipotent stem cells such as MSCs7,15–18 and pluripotent stem cells (PSCs).19–21 Insights into these interactions have allowed in vitro cultures to transition from dispersed single cells to vascularized tissues composed of multiple cell types,22,23 three-dimensional tumor models,24,25 functional human muscle,26 and more fanciful creations such as soft robotics powered by the optogenetic stimulation of mammalian muscle cells.27,28 Towards the goal of culturing more complex and realistic tissue models, many researchers are turning to self-organizing stem cell derived mini-tissues, known as organoids.29,30 Organoids of various tissues including intestine,31,32 optic cup,33 and brain34 have been developed, and the structural and functional similarity to their parent tissues is opening new avenues for drug discovery and testing35 and for tissue replacement therapies. Collectively, these technologies and methods are helping illuminate the underlying drivers of tissue morphogenesis and disease.
To accommodate this growing interest in advanced culture systems, complementary work is underway to create biomaterial scaffolds, so called synthetic ECM mimics, that capture the wide variety of properties found in both healthy and diseased tissues. Far from homogeneous, the ECM composition and mechanics vary spatially throughout a tissue, and both of these can change dramatically during a variety of important events, including development,13 cancer,14,36 fibrosis,37 aging,38 and wound healing.39 Beginning with embryonic development, changes in the matrix properties of tissues are widespread throughout our lives. The structure of fibrillar fibronectin ECM during development regulates cellular migration during gastrulation, the process of germ layer formation that marks the onset of differentiation.40 During subsequent morphogenesis, the progression of cellular organization into tissue structures is largely a mechanical phenomenon, driven by cell-generated traction forces and the folding, buckling, and branching of epithelia initially organized into simple sheets and tubes.13,41–43 These mechanical events are accompanied by precise signaling of morphogens, soluble or sequestered molecules whose gradients encode spatial cues for differentiating cells.44 The concerted action of these spatially and temporally regulated events gives rise to the vast diversity of tissues in adult animals. Similar shifts in matrix properties can occur in adult tissue. Tumor growth is accompanied by remodeling45 and stiffening of the ECM, driving elevated cytoskeletal tension through altered focal adhesions.46,47 This increase in cell tension can disrupt cell polarity and even lead to a malignant phenotype in a stiffness-dependent manner.46,47 The motility of cancer cells is further dependent on a changing chemical environment. For example, tumor associated macrophages secrete epidermal growth factor, a potent chemokine that promotes cancer cell invasion and metastasis.48
To mimic the complex and dynamic nature of the ECM for tissue engineering applications, it is increasingly evident that many targeted tissues require matrices that not only capture key aspects of the ECM, but that the cell-laden scaffolds can be modulated in real time with precise control. This review focuses on hydrogel biomaterials, as hydrogels are widely used as cell delivery systems and as matrices for regenerating soft tissues.49 Presently, most hydrogel scaffolds are fabricated with controlled initial properties that are often static and uniform, built and functionalized through spontaneously forming interactions, both covalent and associating. There are many strategies to control the mixing or printing of hydrogel precursors to create scaffolds with gradient properties,50–52 controlled porosities,53–55 or spatial loading of biological matter.56,57 These primarily focus on manufacturing and physical methods for spatiotemporal control of the hydrogel properties, and excellent reviews on these topics have been published.58–60 However, from a materials chemistry perspective, photochemistry provides a series of versatile reactions from which to capture and reinstate the ECM heterogeneity in hydrogel biomaterials. By controlling the light dose, location, and timing, users can achieve precise spatiotemporal control over a given chemical reaction, which can be further leveraged to alter the mechanical and chemical environment of biomaterials (Figure 1). Many of the reactions occur rapidly under ambient or physiological conditions, which allows for the direct encapsulation of cells, and in some instances, subsequent in situ modulation of biomaterial properties while observing and directing cellular responses to these changes. The insights gained from these types of experiments are being used to design new generations of biomaterials for use in regenerative medicine applications and beyond.
Figure 1.

Synthetic hydrogel matrices can provide a blank slate for encapsulated cells. Using photochemical reactions, researchers can modify these scaffolds to impart adhesivity and bioactivity and to change the mechanical properties, which can be used to direct cell behavior through cell-matrix interactions.
In the context of biomaterials, hydrogels constitute an ideal framework in which to perform photochemical reactions in the presence of cells. Hydrogels are crosslinked, hydrophilic polymer networks that imbibe large amounts of water. As a result, they mimic many of the physical properties of native soft tissues, and loosely crosslinked networks allow for diffusion of exogenously delivered nutrients, growth factors, and cytokines, and also permit facile transport of cell-secreted factors such as matrix molecules and paracrine signals. Hydrogel scaffolds have been synthesized from a wide range of macromolecular precursors, including poly(ethylene glycol), gelatin, hyaluronan, alginate, and poly(vinyl alcohol).49,61 Synthetic hydrogels are particularly tunable, both in terms of their chemical and mechanical properties, and have become an important biomaterial for 3D cell culture and tissue engineering. These matrices have been adapted to a variety of useful chemicals reactions that can be performed in the presence of biological material, and a subset of these have been adapted as photochemical reactions for this purpose (Table 1). Moreover, hydrogels are optically clear, facilitating photochemistry and imaging by light microscopy. This review will focus on recent advancements in photopatterning of hydrogel microenvironments to achieve spatial and temporal control over cell-material interactions and provide a perspective on current challenges and future opportunities for advancement in this field of biomaterial science.
Table 1.
Examples of photochemical reactions to form and functionalize hydrogel matrices for biomaterial applications
| Reaction | Example | Reference | light control | Section |
|---|---|---|---|---|
| Chain photopolymerization |
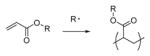
|
Many, see 62 | Photoinitation | 2 |
| Thiol-Michael |
|
63–65 | Photocaged thiol66 or photogenerated base67 | 6 |
| Thiol-ene |
|
63,68,69 | Photoinitation70,71 | 3 |
| Thiol-yne |
|
72,73 | Photoinitation72–74 | 3 |
| Enzymatic crosslinking |
|
75,76 | Photocaged peptide substrate77,78 | 6 |
| Diels alder: Normal |
|
79,80 | Photoenol reactions81 | 7 |
| Inverse electron demand |
|
82–84 | Oxidation of dihydrogen tetrazine85 | 7 |
| 1,3-dipolar cycloadditions |
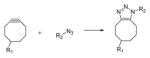
|
86–88 | Photodeprotection of cyclooctyne,89 photoreduction of Cu(II),90 tetrazole-alkene91 | 7 |
| Imine bond formation |
|
92,93 | Photocaged alkoxyamine,94–96 photogenerated aldehyde97–99 | 6 |
2. Photoinitiated chain polymerizations
Photoinitiated chain polymerizations represent one of the first and most widely used photochemical reactions to be applied in a biomaterials setting (for example dental restorative materials, contact lenses, and tissue sealants). From the perspective of 3D cell culture and regenerative biology applications, photopolymerization of water soluble macromolecular monomers are routinely used to embed cells in hydrogel environments. The usefulness of such photopolymerizations is due to several factors. First, as an established and easily implemented strategy, there is a plethora of well characterized platforms that have proven cytocompatible. Additionally, this strategy benefits from similarity to the photopolymer industry, and a diverse array of precursors (monomers, initiators) are commercially available and relatively inexpensive. These factors contribute to a low entry barrier for biomaterials researchers. Due to the chain reaction mechanism, radical photoinitiated polymerizations require lower light doses compared to other photochemistries, which makes experimentation more amenable for cell encapsulation and reduces the exposure of potentially fragile biological signals such as proteins and nucleic acids to light. While radical-induced damage to encapsulated cells is always a concern, care can be taken to operate in a regime where this is minimized.100–103 Further, there is evidence that the presence of polymerizable groups during radical generation can have a protective effect on biological activity when proteins or other fragile molecules may be present in the formulation.70,104–107
Early work with photopolymerized hydrogels in the biomaterials community primarily focused on synthesizing hydrogels with controlled initial properties (mechanics, swelling, transport) and the introduction of degradable linkers; however, the systems were generally spatially uniform. Photocrosslinkable macromers were developed based on a wide range of chemistries including poly(ethylene glycol) (PEG), poly(vinyl alcohol) (PVA), poly(2-hydroxyethyl methacrylate) (PHEMA), poly(acrylamide) (PAm), and poly(N-isopropyl acrylamide) (PNIPAAm).108 Natural polymers such as alginate,109 hyaluronic acid,110,111 and collagen (gelatin)112–114 are also easily adapted to photopolymerization. Major efforts concentrated on increasing the biological efficacy of such matrices by engineering degradability and bioactivity.108,115 Degradability has been introduced by synthesizing block copolymers with blocks that are susceptible to hydrolytic cleavage at physiological pH (e.g., PEG-b-poly(lactic acid))116 or by incorporating peptide epitopes that can be cleaved by cell secreted enyzmes, such as matrix metalloproteinases (MMPs).117,118 The latter approach localizes hydrogel degradation to pericellular regions, whereas the former occurs homogeneously throughout the bulk of highly swollen gels. For synthetic matrices, which lack intrinsic adhesion sites for cells, peptide sequences that recapitulate regions on adhesive proteins, for instance arginine-glycine-aspartatic acid (RGD),119 have been routinely incorporated into hydrogels to promote cell-matrix interactions through these integrin-binding ligands.120 Incorporation of full-length proteins into hydrogels is also readily achieved, for example by using succinimidyl esters to conjugate amine groups found in proteins to reactive end groups.121,122 However, care must be taken to ensure that the protein retains its biological activity through the functionalization and photopolymerization reactions.
Some of the early demonstrations that used photopolymerized hydrogels to encapsulate cells, included photoencapsulation of smooth muscle cells,121 fibroblasts,123,124 osteoblasts,125 and chrondrocytes,126–128 amongst others. Due to the on-demand polymerization, the hydrogels could be formed in situ at tissue interfaces109,129,130 and even polymerized through tissues, as demonstrated by subcutaneous gel formation after injection of monomer solutions followed by illumination of the dermis.131 By virtue of the photoinitiated mechanism, the materials can, by default, be adapted for biological applications that require spatiotemporal control of the gel evolution. In one approach, Hubbell and coworkers used a visible-light initiating system to graft poly(ethylene glycol) coronas to pancreatic islets of Langerhans.101 By addition of the photoactive compound Eosin Y, which adsorbs to the surface of islets, the photopolymerization reaction was localized to the pericellular region, and conformal PEG coronas, tens of microns thick, were grafted from the cell membranes. This approach provided an immunoprotective barrier that allowed for xenotransplantation of islets. A similar method was used to graft PEG layers to the inner wall of arteries, which was shown to reduce intimal thickening after injury.132 In these examples, spatial resolution of the hydrogel formulation was afforded by localizing the photoinitiator to a tissue or cell surface.
More commonly, spatial resolution is enabled by controlled light illumination, such as passing a collimated light source through a patterned photomask, using focused laser light, or digital light processing (DLP) maskless photolithography techniques (Figure 2).58,133 While each of these methods can provide high resolution in the xy plane (perpendicular to the light path), multiphoton techniques are necessary to achieve axial resolution at the micron scale.134,135 These light-directing techniques have been used to create a multitude of three-dimensionally patterned hydrogels for use in 3D cell culture and tissue engineering applications. Liu and Bhatia demonstrated the use of a photolithography technique to create patterned hydrogel constructs in the presence of encapsulated hepatic cells.136 In this example, light was passed through a patterned photomask to polymerize PEG-diacrylate (PEGDA) hydrogel constructs that were covalently bound to a glass slide. The feature height was controlled physically, by the dimension of silicon spacers, and multiple rounds of photopolymerization and washing allowed for the creation of interconnected cell-laden hydrogel structures. Feature resolution was tens of microns, a cellularly relevant size scale, and the authors demonstrated that complex tissue constructs could be fabricated using simple photolithography techniques. The work was further expanded to create microfabricated hepatic structures that mimicked the branched morphology of liver tissue,137 and was later combined with an electropatterning technique to spatially arrange cells prior to patterned photopolymerization.138 West and coworkers used similar techniques to photopattern acryloyl-RGD into PEGDA hydrogels and demonstrated the organization of adhered cells onto patterned adhesive regions in 2D.139 The authors demonstrated that multiple peptides could be incorporated in successive patterning steps and combined with photopatterned PEGDA to create structures with spatially patterned biochemical and topographical properties.139
Figure 2.

Strategies to control spatial illumination of hydrogels. a,b) Photomasks can be used to selectively block collimated light to create a) patterns or b) gradients in illumination that are uniform in the axial direction. c) A maskless photolithography technique is used to project patterns based on graphical inputs to a digital micromirror device (DMD). Reprinted from Ma et al.140 Copyright (2016) National Academy of Sciences. d) Alternatively, focused laser light can be used to illuminate regions with 3D resolution. The light illumination is illustrated by photobleaching uniformly bound fluorescein within a hydrogel at a single plane with light of 488 nm (single photon absorption) and 800 nm (two photon absorption). Two-photon absorption events are localized to the imaging plane. Photobleaching and confocal imaging performed with a 1.0 NA objective. Scale bar = 25 μm.
To achieve three-dimensional resolution on length scales similar to or smaller than most primary cells (~10 microns), multiphoton techniques have been used.134 In two-photon excitation, two photons of a longer wavelength are absorbed instead of one higher energy photon, and therefore, the excitation is dependent on the light intensity squared. The squared light intensity profile drops off rapidly outside of the focal volume, confining photochemical events to the focal plane.135 Because of the lack of absorption events outside the focal plane, these techniques are able to pattern deeper into samples without light attenuation. West and coworkers used two-photon photon photolithography to pattern RGD in precise, pre-defined regions to guide fibroblast migration in 3D.141 The extent of tethering can be controlled with the light dose, and objectives with increasing numerical aperture increase the axial resolution (Figure 3a).142 When two-photon photopolymerization was used to pattern RGD and vascular endothelial growth factor (VEGF) together in micron-scale patterns, cultured endothelial cells confined to these regions underwent enhanced tubulogenesis.143
Figure 3.

Three-dimensional patterning techniques using photoinitiated chain reactions. a) Two-photon patterning is used to immobilize acrylated peptides in 3D with high resolution (scale bar = 25 μm). The acrylated peptides are tagged with AlexaFluorÒ 488, 532 and 633. Reproduced from Hoffmann et al.142 with permission from the Royal Society of Chemistry. b) A stereolithographic approach using projection based methods to rapidly create three-dimensionally defined hydrogel structures. Cells in alternating layers are labelled with CellTracker® green and orange. Scale bar 50 μm. Reprinted from Gou et al.144 c) Stereolithography using focused laser light to achieve 3D printing of cell-laden scaffolds. Scale bar 1 mm. Reproduced from Chan et al.145 with permission from the Royal Society of Chemistry. d) Projection stereolithography used to photopolymerize methacrylated gelatin hydrogel structures. Images show the proliferation of human umbilical vein endothelial cells seeded onto the scaffolds over 4 days. Reprinted from Gauvin et al.146 copyright (2012) Elsevier.
Using techniques developed by the coatings industry, light absorbing dyes have also been used to control light penetration and patterning resolution in 3D hydrogel scaffolds.147 Strong absorbers of the initiating light limit photopolymerization reactions to the incident surface, and unreacted monomers can be readily washed away before a subsequent second layer is polymerized. Alternatively, oxygen flux through a permeable window can be used to maintain a liquid “dead zone” at the interface, and the photopolymerized object can be drawn out continuously without washing steps, greatly increasing production speed.148 In combination with digital light processing techniques, complex three-dimensional scaffolds can be created rapidly (Figure 3b–d).83,140,144,145 In one recent example, iPSC-derived hepatic cells were seeded into a 3D biomimetic liver construct, and endothelial and mesenchymal support cells were structurally patterned in a manner that more closely recapitulates the liver lobule in vivo.144 This micropatterned tri-culture platform was found to outperform the functions of hepatic cells cultured in monolayers or unstructured 3D hydrogels.
3. Thiol-ene and thiol-yne photoclick chemistry
The development of more sophisticated biomaterials platforms has benefited from advances in the chemistries that can be used to create and functionalize hydrogels. In particular, the field of “click” chemistry, and especially the growing number of bio-orthogonal reactions, has made a significant impact on the biomaterials field. First outlined in 2001 by Sharpless and colleagues,149 the term click chemistry was put forth to describe chemical reactions that create typically carbon-heteroatom linkages and proceed selectively to high yields under mild conditions. Prominent cases include the 1,3 dipolar cycloaddition, nucleophilic ring-opening reactions, oxime/hydrazone carbonyl chemistry, and additions to alkenes and alkynes. The rapid and mild conditions under which these reactions occur and the orthogonality towards other reactive groups make these reactions ideally suited for hydrogel formation in the presence of cells and biological material. Hydrogels formed in such a manner include the copper-free click reaction between cyclooctynes and azides,88,150 the formation of oxime and hydrazone crosslinked hydrogels,151–153 and thiol-ene reactions.70,71,154,155 Thiol-ene reactions – both radical mediated and base-catalyzed – have gained particular favor as crosslinking methods used by the biomaterials community due to the wide availability of starting materials (including cysteine residues) and ease of use. The two distinct reaction mechanisms should not be confused: the radical-mediated thiol-ene reaction will be our focus here, as it is one of the few photo-click reactions, whereas the based catalyzed Michael-type conjugate addition of thiols to electron deficient alkenes such as acrylates, vinyl sulfones, maleimides is reviewed elsewhere.63,65 However, it is worth mentioning that light-based control of thiol-Michael reaction has also been demonstrated using photocaged thiols and photogenerated bases, and will be discussed in Section 6. The thiol-yne reaction is conceptually similar to the thiol-ene reaction and bears the same advantages, with the major difference being that the alkyne is able to undergo two consecutive thiol additions to form a dithioether adduct.72 This effectively makes the alkyne difunctional, allowing the formation of highly functionalized substrates. However, compared to thiol-enes, the thiol-yne reaction has yet to gain real traction in a biomaterials setting, hence we will limit our discussion to the thiol-ene reaction.
The thiol-ene photoclick reaction can be initiated in a similar manner to other chain polymerizations, but the polymerization mechanism differs. Upon hydrogen abstraction from a thiol by an initiator radical species, a thiyl (sulfur centered) radical is generated. The thiyl radical then undergoes addition to the alkene species, creating a carbon-centered radical which then abstracts a proton from another thiol to regenerate the thiyl species, beginning the cycle again (Figure 4a). In this way, the thiol-ene cycle is able to rapidly proceed through a large number of addition reactions, and network growth proceeds in a stepwise manner. The choice of alkene species also affects the polymerization mechanism. In the ideal case, the C-S chain transfer rate is sufficiently higher than that of C-C propagation such that homopolymerization of the alkene species is negligible. This is generally achieved with electron-rich and/or strained alkenes such as vinyl ethers and norbornenes. In such systems, network formation proceeds in a purely stepwise manner. A mixed-mode reaction mechanism can also be achieved, for example using acrylates as the olefin species.156 A dependence of the overall rate on the initiation rate to the one-half power implies that termination occurs by radical recombination.68
Figure 4.

Thiol-ene photoclick chemistry is a mild and efficient reaction for hydrogel formation and functionalization. a) The thio-ene reaction mechanism, showing the cycle of propagation (thiyl radical addition to alkene) and chain transfer to generate a new thiyl radical. b) Under ambient conditions and identical photoinitiating schemes and functional group concentrations, hydrogels are formed by the thiol-ene reaction within 5 s; comparatively, hydrogels formed by homopolymerization of PEG-diacrylate are delayed. Reproduced from McCall et al.104 Copyright (2012) American Chemical Society.
The thiol-ene reaction possesses a number of benefits compared to chain polymerizations. Among these, the reaction rate under similar conditions is much faster. The crosslinking of PEGDA chain growth networks and the step growth thiol-ene polymerization between tetrafunctional PEG-norbornene and difunctional PEG-thiol have been directly compared.104 With identical functional group concentrations of 40 mM, initiator concentrations of 1 mM and light intensities of 10 mW/cm2, the PEG-DA network reaches 95% of its final modulus in 180 s, whereas the thiol-ene network achieves this in 5 s (Figure 4b). Thus, the thiol-ene network can be formed with a much lower total number of radicals generated, which was shown to decrease radical-associated damage to encapsulated lysozyme and transforming growth factor-β (TGFβ).104 The difference in rates is attributed, at least in part, to another advantage of the thiol-ene reaction: insensitivity to oxygen inhibition. The presence of molecular oxygen in radical polymerizations typically results in rate retardation by formation of inactive peroxy radical species.157 However, in the presence of thiols, the peroxy radical abstracts a proton from a thiol, forming a thiyl radical which continues the polymerization.69 In fact, the addition of a small amount of thiol to an acrylate polymerization can be used to achieve relative oxygen insensitivity.158 This insensitivity allows for the polymerization of thin films in the presence of air, which is difficult or impossible otherwise due to the constant flux of oxygen. Furthermore, the influx of oxygen into a polymerizing material can also cause lead to unintended spatial heterogeneities in the final network, for example, leading to a tacky surface that is at a lower conversion compared to the interior. As photopolymerizations in the presence of cells are performed in oxygenated conditions, this attribute shows a clear advantage of thiol-ene photopolymerizations for creating and functionalizing biomaterials.
Perhaps more importantly, thiol-ene photopolymerizations appear to be a milder method for cell encapsulation compared to (meth)acrylate photopolymerizations. In a direct comparison between thiol-ene and acrylate PEG hydrogels, thiol-ene polymerization resulted in lower measurable levels of damage to pancreatic β-cells in both gelling and non-gelling solutions, and allowed for encapsulation at lower cell densities than was possible using PEGDA.160 As one might expect, thiol-ene photopolymerizations can be initiated by numerous mechanisms, including directly by the type II photoinitiator Eosin Y, allowing for visible light photopolymerization. This visible light initiation strategy resulted in higher viability of hMSCs compared to the visible light photopolymerization of PEGDA, which requires addition of a co-initiator (triethanolamine) and comonomer (n-vinylpyrrolidone).161 In an effort to better understand the effect of photoencapsulation methods on cellular behavior, Roberts and Bryant investigated intracellular reactive oxygen species (ROS) produced during thiol-ene and chain growth hydrogel formation.162 Primary bovine chondrocytes displayed higher levels of intracellular ROS after encapsulation in PEGDA hydrogels compared to thiol-ene gels. The authors suggested that this may be because ROS formed during thiol-ene polymerizations can participate in the polymerization, whereas ROS in acrylate polymerizations cannot, leading to increased chain transfer to the chondrocytes. Furthermore, the quality of cartilage formed differed, with acrylate polymerization leading to hypertrophic cartilage and thiol-ene polymerization leading to hyaline cartilage and a greater total amount of sulfated glycosaminoglycans and collagen produced. Elevated intracellular oxidative stress was also observed in PEG-methacrylate (PEGMA) encapsulation of salivary gland cells, leading to a dramatic decrease in cell viability that could be circumvented by using a thiol-ene photopolymerization.163 By encapsulating multicellular spheroids in thiol-ene hydrogels and incorporating MMP-sensitive crosslinks, acinus formation and apicobasal polarization occurred, resembling the native salivary gland.164
The choice of thiol and alkene groups are important decisions for thiol-ene photopolymerizations, and the general reactivities of numerous functional groups have been catalogued.63,69 Norbornene is among the most widely employed alkenes for hydrogel formation. A low-cost and generally stable moiety, norbornene is a bicyclic compound, and benefits from significant ring strain relief when reacted. Multifunctional PEG-norbornene was introduced in 2009 as an efficient alkene for reaction with cysteine thiols by Fairbanks and coworkers (Figure 5a),71 and has since been widely used to create peptide functionalized PEG hydrogels. This approach was recently reviewed by Lin et al.165 In addition, norbornene functionalized hyaluronic acid (norHA) has been used to fabricate bulk166 and nanofibrous159 patterned hydrogels, and norbornene-modified gelatin has been introduced as well.167,168 When aligned nanofibrous norHA hydrogels were photopatterned with RGD, it was found that cells localized to adhesive regions but preferentially aligned along the fiber orientation (Figure 5b). Norbornene has the added benefit of also being able to undergo cycloadditions with nitrile oxides,169,170 and tetrazines83,84,171 in bioorthogonal click reactions. The latter was recently leveraged to form click based alginate hydrogels through an inverse electron demand Diels Alder (IEDDA) cycloaddition and then functionalized by reaction of a cysteine-containing peptide with excess norbornenes.172 Norbornene has also been genetically encoded,173 raising new possibilities for biomaterial formation and functionalization.
Figure 5.
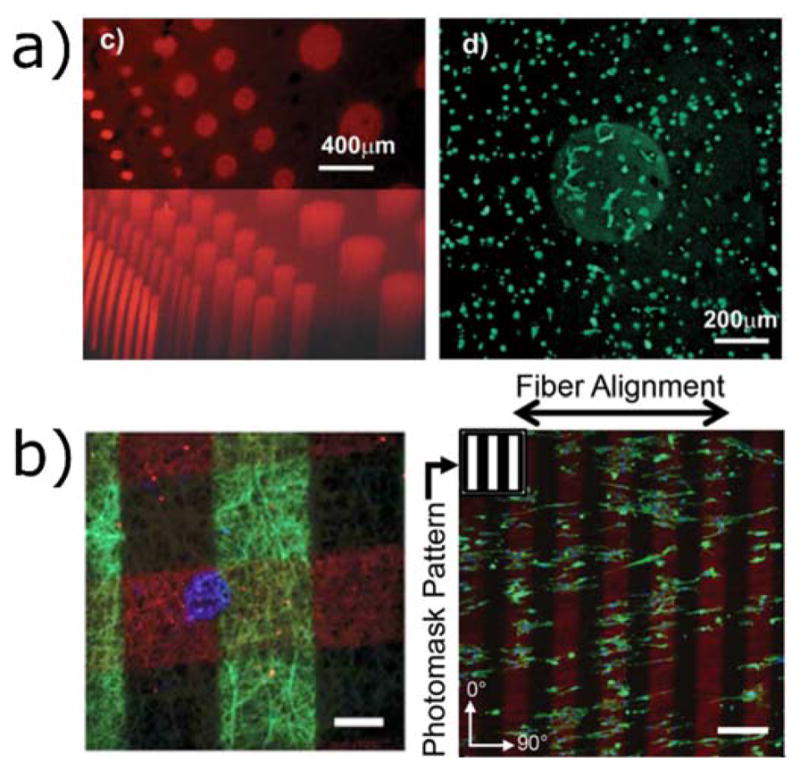
Thiol-ene photopolymerizations used to functionalize three-dimensional hydrogels. a) Norbornene-modified PEG can be crosslinked and functionalized with cysteine-containing peptides. hMSCs seeded in MMP-degradable hydrogels spread only in regions with photopatterned RGD. Reproduced from Fairbanks et al.71 copyright (2009) Wiley. b) Left: nanofibrous norbornene hyaluronic acid hydrogels patterned with three fluorescent peptides. Right: when RGD patterns are created perpendicular to the nanofiber alignment, cells localize to the RGD regions (indicated in red), but align with the fiber orientation. Scale bars 100 μm (left) and 200 μm (right). Reproduced from Wade et al.159 copyright (2015) Wiley.
One limitation of the thiol-ene reaction is that the thioether bond formed is generally irreversible, so when used for tethering pendant functional groups, the signal persists. If reversiblility is desired, other strategies, such as using orthogonal thiol-ene photoconjugation and photocleavage reactions, have been developed (Section 5). In this approach, however, the reversibility is limited to one cycle. Building from the reversible addition-fragmentation chain transfer (RAFT) literature, the allyl sulfide functionality has become an intriguing chemistry for the synthesis of networks integrating fully reversible thiol-ene reactions. When a thiyl radical adds to the alkene of the allyl sulfide, a fragmentation reaction occurs which releases a thiyl radical and regenerates an alkene (Figure 6a).174,175 In this way, it is possible to perform multiple successive rounds of photopatterning without loss of the reactive target. This materials chemistry was recently used to demonstrate the reversible incorporation of cysteine-containing RGD peptides in hydrogels, thereby controlling mesenchymcal stem cell interactions with the dynamically changing material interface.176 Successive rounds of two-photon photopatterning were used to sequentially tether three consecutive peptides in the same three-dimensional space, with each patterning step releasing the previous peptide (Figure 6b–d).
Figure 6.

Reversible thiol-ene photopatterning via allyl sulfide chemistry. a) Consecutive swapping of thiolated molecules can be performed, forming new thioether bonds and reactive alkenes. b) An RGD peptide containing a cysteine residue and AlexaFluor555 is tethered to the network by two-photon photolithography. c) A second thiolated peptide (with AlexaFluor488 dye) is photopatterned in the buffalo shape, simultaneously releasing the first peptide. d) A third peptide (unlabeled) is patterned in the shape of the CU logo, releasing the second peptide. Scale bar 100 μm. Adapted from Gandavarapu et al.176 copyright (2014) Wiley.
4. Sequential crosslinking strategies
One of the challenges in creating cell-laden hydrogels with tunable mechanical properties is that one often desires to modify the material properties of the network (e.g., by altering the network connectivity and crosslinking density) at a later point in time. This dynamic control is useful for investigating the role of mechanosensing of cells in stiffening or softening environments, for releasing cells that are embedded in 3D environments, or even coordinating proliferation, migration, or cell-cell interactions in 3D. In one example, Engler and colleagues used a Michael-type reaction to slowly stiffen hydrogel constructs, mimicking cardiac stiffening during embryonic development, and studied the effect of stiffening on cardiac myocytes.177 Dynamic stiffening of the substrate enhanced the differentiation of pre-cardiac cells, and motivated further exploration into photocrosslinking systems with their precise spatial and temporal control.
One versatile sequential crosslinking method developed by Burdick and coworkers takes advantage of the ability of acrylates and methacrylates to participate in both thiol-Michael additions and chain polymerizations. In a hydrogel with an excess of (meth)acrylates with respect to thiols, a two-step polymerization can be performed: a nucleophilic addition reaction of the thiol to olefin followed by a radical chain polymerization of the remaining (meth)acrylates. Using this strategy, Burdick and coworkers were able to spatially pattern cell-laden hydrogels and investigate the effects of degradability, adhesion sites, and crosslinking density on the ability of human mesenchymal stem cells (hMSCs) to remodel the network and spread in 3D.178 This work was then expanded to include the influence of spatial mechanical patterning on hMSC fate in 3D. Contrary to two-dimensional culture7 and 3D culture in ionically crosslinked networks,179 increased network stiffness led to decreased osteogenesis and increased adipogenesis, possibly due to the inability of the hMSCs to degrade the more tightly crosslinked, stiffened matrix, leading to a rounded morphology.180 A similar secondary crosslinking strategy employing hyaluronic acid functionalized with both maleimide and methacrylate functionalities (MeMaHA) was then leveraged to induce stiffening of hydrogels with embedded hMSCs after a week in culture (Figure 7a).17 Although the cells now were able to spread and assemble an actin cytoskeleton in the degradable gel prior to photopolymerization, the stiffened network again led to adipogenesis over osteogenesis (Figure 7b). However, this change was accompanied by a decrease in cellular traction forces, supporting the proposed hypothesis that hMSC fate decisions are dictated by their ability to interact with the matrix via degradation. Similar observations were reported by Mabry et al. studying heart valve fibroblasts, which become activated myofibroblasts on stiff 2D substrates compared to soft substrates, but the trend was reversed in 3D, irrespective of cell morphology.181 Collectively, secondary photocrosslinking strategies have allowed researchers to decouple cell morphology from matrix stiffness and answer questions related to mechanosensing in 3D.
Figure 7.

Secondary photocrosslinking strategies to change hydrogel mechanics in situ. a) Schematic representation of a sequential crosslinking strategy. Hyaluronic acid is functionalized with methacrylate and maleimide groups (MeMaHA). Maleimide reactive groups preferentially react with a dithiol MMP-degradable crosslinker, leaving methacrylates available for subsequent photopolymerization. hMSCs are encapsulated during the initial hydrogel network formation and are allowed to spread in the softer, degradable hydrogel. At a later point in time, methacrylates are photopolymerized (red dashed lines) to stiffen the network. b) Traction force microscopy images of encapsulated hMSCs after 7 days in growth medium and 14 days in mixed medium after stiffening. Although cells retain a spread morphology after secondary photocrosslinking, hMSCs undergo adipogenesis in mixed-media conditions, supporting the proposed hypothesis that hMSC cell fate decisions depend on the ability of the cells to interact with the matrix via remodeling. Scale bars 25 μm. Reprinted from Khetan et al.17 copyright (2013) Nature Publishing Group. c) Direct printing of a shear-thinning hydrogel into a self-healing network. Guest-host interactions between adamantane and cyclodextrin form a shear-thinning network that can be printed and then covalently crosslinked by photopolymerization of pendant methacrylates. The polymerized construct remains after the support has been washed away. Scale bar 500 μm. Reproduced from Highley et al.182 copyright (2015) Wiley.
To add to the diversity in material property control, other secondary crosslinking strategies have focused on stiffening or irreversibly crosslinking associating polymer networks, for example, those formed by the Diels Alder reaction between maleimide and furan. The oxanorbornene adduct contains a reactive alkene that can be targeted for radical thiol-ene coupling. This subsequent reaction has the interesting effect of “locking” the adduct in the product state and preventing the retro-Diels Alder reaction. Thus, the stiffening in the network coincides with a loss of reversibility, making it an intriguing platform to study potential combinatorial effects of reversibility and stiffness. This strategy was first applied to 3D photolithography in solid polymers,183 but has also been adapted to hydrogel photopatterning.184 A similar approach was adapted to hydrogels crosslinked by guest-host interactions,182,185 where hyaluronan was modified with both cyclodextrin and methacrylate (CDMeHA) or adamantane and methacrylate AdMeHA) (Figure 7c). Upon mixing of these macromolecular precursors, a reversible, shear-thinning network is created that can then be extruded through the needle of a 3D printing device. Once in place, the printed structure is irreversibly crosslinked upon exposure to light. Temperature responsive materials have also been developed with this strategy in mind. ABA triblock copolymers composed of partially methacrylated poly(N-(2-hydroxypropyl) methacrylamide lactate) A blocks and poly(ethylene glycol) B blocks stored at room temperature form associating networks when printed onto a plate heated to 37°C, and then can be subsequently photocrosslinked.186 Conversely, associating networks of methacrylamide-modified gelatin have been selectively crosslinked at room temperature, and then heated to 37°C to release the unexposed and unreacted gelatin regions187 or directly printed as a shear-thinning mixture and crosslinked in situ.188 Secondary hydrogel crosslinking strategies are becoming a valuable tool in the field of bioprinting and additive manufacturing, especially those reactions that are compatible with simultaneous cell printing and encapsulation.
5. Photocleavage reactions
In the context of spatiotemporal control over hydrogel crosslinking, photodegradation serves as the counterpart to photopolymerization or photoconjugation. Various strategies exist, but all achieve the same goal: to decrease the network crosslinking density – and therefore the mechanical integrity – in regions of the hydrogel that are exposed to light. Similarly, biomolecules tethered to the network through photolabile linkages can be released on demand. By controlling the light dose, either softening (decreased crosslinking density) or complete erosion of the matrix (cleavage to a conversion beyond the reverse gel point) can be achieved. Depending on which cell-material interactions one wishes to control or the end application of the resulting biomaterial, photodegradation has been used to create spatiotemporal differences in material stiffness or biochemical composition,189 varied topolographical features,190,191 or complete dissolution of a preformed gel.192
One of the most common methods to synthesize photodegradable hydrogels is to incorporate a photolabile functionality directly into the crosslinks of the polymer network. Upon irradiation with an appropriate wavelength of light, the crosslinking molecule absorbs light and undergoes cleavage, reducing the number of elastically effective chains. The resulting reaction rate depends on the intensity and wavelength of the light and the photophysical properties of the photodegradable linker. For molecules that undergo an alpha cleavage and for uniform illumination throughout the material, first-order kinetics describe the rate of cleavage:
Here, C is the concentration of absorbing species, and the kinetic rate constant k is a product of the quantum yield (ϕ) and absorptivity (ε) of the photoactive species at the wavelength used, along with the light intensity (I), light frequency (v), Planck’s constant (h), and Avogadro’s number (NA). The rate of cleavage of a photolabile crosslinker can be related to a hydrogel’s storage modulus (G’) and crosslink density (ρx) and used to predict changes in material properties and mass loss with time.189,194,195
The factor 2 is included for the case of linear photodegradable macromers containing two photodegradable groups, as cleavage on either side results in network degradation. Depending on the chemical structure and connectivity of the network (Figure 9a), statistical-kinetic models can be used to predict the mass loss profile and time to reverse gelation.194–196 For chain growth networks, the critical conversion to reach reverse gelation is given by
Figure 9.

Formation and degradation of photodegradable hydrogel networks. a) Idealized structures of chain and step polymerized networks and their photodegradation products. Networks of linear PEG-di(photodegradable acrylate) are formed by radical polymerization, resulting in acrylate kinetic chains (black) connected by PEG spacers (red). PEG-tetra(difluorocyclooctyne) undergoes a spontaneous crosslinking reaction when mixed with a photodegradable di-azide peptide. b) Irradiation of the bulk hydrogel with 365 nm light results in loss of crosslinks that can be tracked with photorheometry. Shuttering the light results in stabilization of the modulus readings. c) Two-photon photodegradation (λ=740 nm) was used to create defined three-dimensional topographies within the hydrogel volume. Scale bar 100 μm. Reprinted from Kloxin et al.189 copyright (2009) AAAS. d) Orthogonal photochemical strategies are used to pattern RGD (dotted line) and erode the hydrogel network. Fibroblasts migrate through the 3D void space only in the region containing the adhesive ligand. Scale bar 100 μm. Reproduced from DeForest et al.150 copyright (2011) Nature Publishing Group.
where N is the number of crosslinking molecules per chain. For step growth networks, the conversion at reverse gelation can be determined by Flory-Stockmayer theory197 and given by:
where Fa and Fb are the functionalities of the A and B terminated monomers, and r (≤1) is the stoichiometric ratio between them. In practice, chain growth networks have more crosslinks per kinetic chain, so they usually require a higher level of photodegradation to achieve reverse gelation, but this also allows a large dynamic range in the modulus during softening. In contrast, step growth networks, especially those formed from low functionality macromers, reach reverse gelation at low extents of degradation of the photolabile linkers, and this can be beneficial for release of encapsulated cells or to create topographical features in two or three-dimensions. Therefore, the end application should be kept in mind when selecting chain or step polymerized photodegradable hydrogels.
In the biomaterials community, one of the most prevalent photodegradable hydrogels are based on cleavage of ortho-nitrobenzyl (oNB) derivatives. This strategy was introduced by Kloxin and coworkers,189 using a PEG crosslinking molecule functionalized at each end with a nitrobenzyl ester functionality and an acrylate to allow for polymerization (Figure 9a). Initially, hydrogels were synthesized using a redox-initiated radical polymerization scheme to copolymerize mono- and di-acrylated PEGs. Gel degradation was induced via light exposure (365, 405, or 740 nm), and spreading of encapsulated hMSCs was found to depend on the local polymer density. Furthermore, an adhesion peptide sequence, RGD, was tethered to the gel through a photolabile linker and subsequently released on demand. Release of RGD after 10 days was shown to promote survival and chondrogenic differentiation of hMSCs compared to persistent presentation of the integrin-binding epitope.
Photodegradable hydrogels have also been used to create gradients in the crosslinking density of initially uniform materials. When optically thick hydrogels were irradiated, the resulting gradient in light intensity led to a gradient in mechanical properties as a function of depth into the gel. Studies with hMSC-laden photodegradable hydrogels revealed that cells near the surface (<20 microns) adopted a more spread morphology compared to those deeper (100 microns) into the network.198 Complementary to this work, gradients in the modulus at a hydrogel’s surface, in the xy plane, were created using a moving photomask to control the time of light exposure. The gradient hydrogel properties influenced heart valve fibroblasts interacting with the material surface, causing differences in myofibroblast activation depending on the extent of photodegradation.199 Surface gradients have also been generated using a maskless photolithography system that employed a digital micromirror device (DMD) to differentially expose regions of the hydrogel based on an input image (Figure 10b).200 Here, any arbitrary image can be projected onto the hydrogel surface without the need to create a specific photomask. Using this technique, the authors were able to investigate the response of hMSCs to a pattern of repeating gradients, and that cells preferentially adhered to the most degraded regions. This result was surprising, considering that cells typically migrate to regions of increased stiffness.51,201 Clearly, these collective studies expose the likely combinatorial effects of topography, network structure, adhesive site availability and elastic modulus on cell migration.
Figure 10.
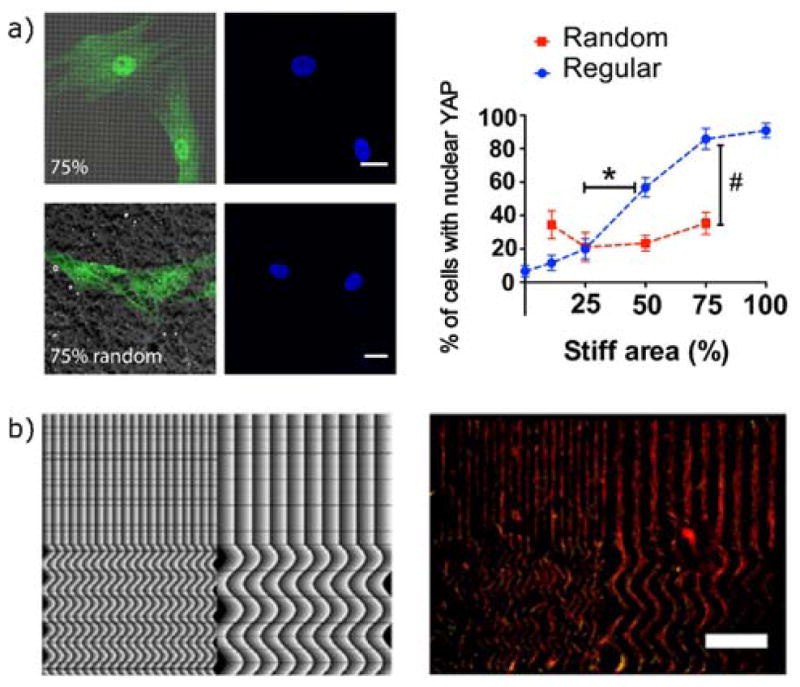
Cellular responses to mechanical surface patterning via photodegradation. a) Photomasks were used to create subcellular stiffness patterns with either regular or disordered spacing. Adherent hMSCs on regularly patterned substrates were more sensitive to changes in the overall fraction of stiff area, indicated by a greater change in the nuclear localization YAP. Scale bars 20 μm. Reproduced from Yang et al. copyright (2016) National Academy of Sciences. b) A maskless light projection technique was used to create repeating gradients in light exposure, leading to patterned photodegradation. hMSCs follow the patterns and localize to the more degraded regions. Scale bar 1 mm. Reprinted with permission from from Norris et al.200 copyright (2016) American Chemical Society.
Hydrogels with photolabile crosslinks are generally stable for long term culture (several days to weeks) when protected from initiating light wavelengths and intensities, and this allows for experiments on cell-laden hydrogels and subsequent temporal changes in crosslinking. In one example, hMSCs were allowed to spread on the surface of a hydrogel and 2-photon photodegradation was used to selectively degrade regions of the gel corresponding to focal adhesions.202 The subsequent cellular retraction was monitored and used to quantify aspects of the cytoskeletal response and dynamics. Photodegradable hydrogels and their temporally regulated mechanical properties have also been used to examine cell fate decisions. Namely, Yang et al. varied the culture time of hMSCs on stiff hydrogels before softening them in situ, and results showed that hMSCs retained a “mechanical memory” of the stiffness of past culture conditions and that there exists a threshold culture time upon which irreversible mechanical activation and differentiation occurs.18 Cell behavior and fate decisions also appear to depend on the spatial organization of mechanical surface patterning.203,204 When a photomask was used to create subcellular stiffness patterns, hMSCs preferentially formed focal adhesions on stiffer islands and on the interface between stiff and soft regions. Furthermore, cells seeded on surfaces with uniformly spaced patterns were more responsive to changes in the percentage of stiff regions, compared to cells in contact with disorganized patterns. This was evidenced by changes in mechanical activation and osteogenic differentiation (Figure 10a).203 Similarly, heart valve interstitial cells display larger focal adhesions, increased stress fiber formation, and an accelerated mechanical response to spatially organized matrix stiffness, which combine to increase myofibroblast activation.204 In total, these results highlight the effect of spatially varying substrate properties, and how one might design experiments to gain insight into the role that a disorganized ECM may play in fibrotic diseases or during injury or even how one might use biomaterials to manipulate cell function to regenerate healthy tissues in diseased microenvironments.
Beyond control of biomechanical and biochemical properties, photodegradable hydrogels also enable the generation of topographical features, either on the surface or within the bioscaffold networks. To create surface topographies, researchers leverage light attenuation, either existing or by the addition of competitive absorbers, to selectively degrade exposed regions. Depending on the light dose employed (i.e., the extent of degradation), either positive or negative features can be created on the surface. Light doses that lead to material degradation beyond the reverse gel point cause erosion of the gel from the top surface; conversely, lower light doses leave the gel intact, but reduce the local crosslink density and cause regional swelling and generation of positive topographical features.205 Kirschner and colleagues studied the effect of such surfaces on the phenotype of cultured hMSCs.190 By exposing hydrogel surfaces through photomasks of rectangular patterns of increasing aspect ratio, it was found that higher aspect ratio surface patterns led to higher alignment of hMSCs. In a separate study, heart valve interstitial cells (VICs) reversibly deactivated from their myofibroblast phenotype in response to in situ softening of the underlying hydrogel substrate. This deactivation, however, could be partially reversed by a subsequent patterning step to create surface topographies.206 Micropatterned hydrogel surfaces also promoted increased alignment and beating of neonatal rat cardiomyocytes cultured on a photodegradable gelatin substrate.191 While more challenging, three-dimensional topographies can be created within the bulk of hydrogel-cell scaffolds using focused laser light. This provides a precise means of guiding cellular interactions in 3D, and has been used to guide fibroblast migration (Figure 9d)150 and neurite extension207 in 3D environments.
Finally, photodegradation has been used in concert with other modes of degradation. For example, hydrogels formed through a thiol-Michael click reaction between an aromatic thiol and maleimide functional groups also undergo a “retro-Michael” reaction in glutathione-rich reducing environments.208,209 This strategy was combined with an ester-containing photodegradable hydrogel to create a network with three modes of degradation that varied across large time scales: photodegradation (minutes), thiol exchange (hours), and ester hydrolysis (days).210 In another study, photolysis was combined with cell-directed degradation by combining the oNB functionality into an MMP-sensitive peptide.211 The peptide was end-functionalized with azido groups for incorporation into a click-based network. Using controlled light illumination, the authors examined the role of 3D topographical cues on lung epithelial cells. Using a similar strategy, a photodegradable cell laden gel can be selectively eroded to produce microfluidic channels to produce vascularized hydrogel constructs.193 The adaptation of natural ECM components to photodegradable networks also integrates intrinsic degradability and cell adhesion sites, and photodegradable gelatin hydrogels have been introduced by reacting clickable212,213 or methacrylamide191 modified gelatin with photodegradable PEGs.
The absorbing wavelength and efficiency of photocleavage reactions are dictated by the structure of the photoactive species. For oNB photocages, the uncaging kinetics depend on the substituents of the aromatic ring and the benzylic position,214 and Kasko et al. have investigated the photodegradation kinetics of a number of oNB hydrogel networks.215 Hydrogels incorporating the coumarin functionality as a photolabile molecule have also been synthesized.216,217 Coumarin photocleavable units are advantageous because of their greatly increased 2-photon uncaging cross section compared to the oNB group.214 In one approach, researchers take advantage of the different spectral properties of some coumarin or nitrobenzyl species relative to each other. This opens the possibility of using orthogonal wavelengths of light to sequentially perform separate reactions,218,219 for example, to release two morphogens in a biologically relevant sequence from a single hydrogel depot.219 Orthogonal wavelengths of light also allow photopolymerization and photocleavage in hydrogels,150,220,221 by proper selection of the initiating strategy. In one example, Eosin Y has been used as the photoinitiator in a thiol-ene polymerization initiated by visible light (λ=490–650 nm), and subsequent exposure to UV light (365 nm) is used to cleave the nitrobenzyl linkages. This approach has been used to photopattern a cleavable RGD peptide and subsequently release it in a spatiotemporally controlled manner,220 pattern orthogonal chemical and mechanical environments (Figure 9d),150 and photopolymerize photodegradable hydrogels.221
In general, however, the photosensitivity of photodegradable hydrogels can be an obstacle for hydrogel formation, as it precludes the use of certain wavelengths for other photochemical reactions or even imaging of cell-laden hydrogels. And while photoinitiating and photocleaving strategies of different wavelengths have been combined, additional care must be taken to ensure that the photoinitiating scheme is truly independent of the photocleavage. In fact, photoactive species are routinely used to sensitize photocleavable linkers in both single- and two-photon applications.214 Thus, alternative strategies are often pursued in a complementary way to form three dimensional cell-laden photodegradable constructs. Bioorthogonal click chemistries that can be performed in the presence of living systems and biological materials are useful in this regard; these reactions proceed rapidly under cytocompatible conditions, and preserve photosensitive groups for later reactions.222–224 Of these, the strain-promoted azide alkyne cycloaddition (SPAAC) has, so far, proven most useful.88,96,150,192,207,212,213,220
Beyond the widely studied ortho-nitrobenzyl and coumarin containing photodegradable hydrogels, photodegradation strategies based on radical cleavage of hydrogel crosslinks have also been reported. In these reports, functional groups that are susceptible to radical cleavage, such as disulfides225 and allyl sulfides,192 are incorporated into the hydrogel backbone (Figure 11a). When a soluble photoinitiator is introduced and upon exposure to light, photogenerated radicals cause photocleavage reactions. Taking advantage of highly efficient radical photoinitiators such as lithium phenyl-2,4,6-trimethylbenzoyl phosphinate (LAP),103,226 this process occurs rapidly compared to oNB cleavage. By including a soluble monothiol species, the rate of photodegradation is further enhanced due to chain transfer to the soluble thiol, which can subsequently cause another degradation reaction, chain transfer event, and so on. The efficiency of the photocleavage reaction is enhanced through this mechanism, resulting in an amplification of the photon signal. For example, Brown et al. demonstrated the erosion of a 1 cm thick hydrogel sample in 1 minute of irradiation with 10 mW cm−2 of 365 nm light (Figure 11c).192 The radical-based photodegradation mechanism can also be used to allow network reorganization during irradiation, for example, the creation of topographical features by patterned stress relaxation during impression with a textured surface (Figure 11b).225 This photodegradation strategy is likely to find use in cases where thicker samples are used and/or when longer irradiation times are not practical. Further, these strategies are theoretically reversible, and hydrogels could be stiffened and softened repeatedly by changing the functionality of the soluble thiol species.
Figure 11.

Photodegradable and photoadaptable hydrogels based on radical processes. a) Reaction scheme for photodegradation of hydrogels containing disulfide or allyl sulfide linkages. b) A photoadaptable disulfide-crosslinked hydrogel is exposed to light while pressed against a textured surface, resulting in photoinitiated deformation. Scale bar 1.5 mm. Reproduced from Fairbanks et al.225 copyright (2011) American Chemical Society. c) Radical-mediated photodegradation requires lower concentration of photoactive species, allowing for the photodegradation of thicker monolithic samples. In this example, a 1 cm-thick hydrogel is degraded in approximately 1 min of irradiation (365 nm, 10 mW cm−2). Scale bar 5 mm. Reproduced from Brown et al.192 copyright (2017) Wiley.
6. Photocaging reactions
Although photoinitiated radical reactions remain a popular choice among biomaterials researchers, some concerns remain about the toxicity or oxidative stress that can be caused. Indeed, examples of the deleterious effects of radical exposure have been reported in the literature,227 and photodynamic therapies are routinely used to induce damage to cancer cells in a spatially controlled manner.228 While a broad range of photoinitiating reaction conditions have been identified that are biologically compatible, they do not meet the needs for all purposes. Alternative methods to create patterns in hydrogels exploit the ability to expose reactive functional groups by photocontrolled removal of a cage (i.e., photocaging). A range of functionalities have been revealed in this manner, increasing the breadth of photochemical and photo-click reactions available (Figure 12). Nitroaryl derivatives such as 2-nitrobenzyl (oNB) and nitrophenethyl (NPE) are popular choices. Upon irradiation, oNB undergoes isomerization to an aromatic nitrosocarbonyl and releases the protected group at the benzylic position. Conversely, NPE undergoes β-elimination to yield the uncaged molecule and a nitrostyrene or nitroso product.214,229 Derivatives of the (coumarin-4-yl)methyl protecting group have also found use in biomaterials applications. This photocage releases by heterolytic bond cleavage and reaction with the solvent, forming a new coumarylmethyl stable product and the released leaving group. By incorporating photocaged functional groups as a structural component of the network, photo-uncaging can degrade the network (as discussed in section 5).
Figure 12.
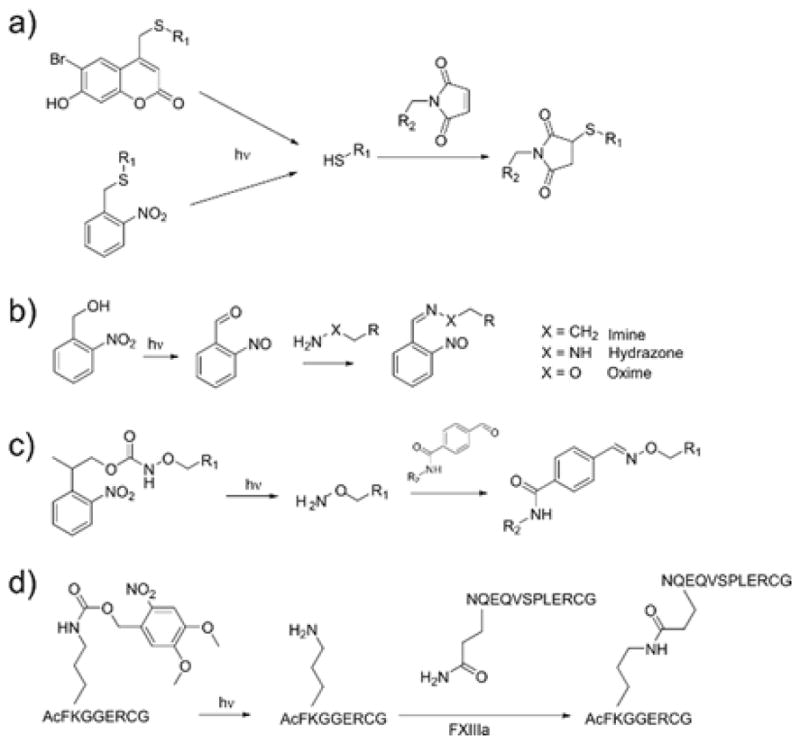
Photocaging strategies to reveal reactive groups upon irradiation.
Thiol functional groups (Figure 12a) are an attractive target for photocaging because, when released, they can undergo click reactions with a variety of substrates, including epoxides, isocyanates, and electron-deficient alkenes/alkynes.230 Shoichet and coworkers demonstrated such photochemical patterning by modifying hydrogels with o-NB and coumarin protected cysteines.66,231,232 Substituting a coumarin-based photocage due to its higher two-photon uncaging cross section, two-photon patterning (λ=740 nm) of multiple thiolated molecules was achieved with an axial resolution of around 20 μm.232 This strategy was then modified to achieve simultaneous immobilization of two full-length proteins: sonic hedgehog (SHH) and ciliary neurotrophic factor (CNTF).233 Sequential two-photon uncaging reactions was used to pattern maleimide-barnase and maleimide-streptavidin, and then barstar-SHH and biotin-CNTF were simultaneously diffused into the hydrogel and bound only to the regions containing their respective binding partners. Critically, the immobilized proteins retained their bioactivity, and the strategy provides a modular method for creating complex biochemical patterns that can theoretically be extended to virtually any protein. Thiol uncaging has also been used to fabricate hydrogels with light by employing a PEG-based hydrogel precursor containing coumarin-caged thiols.234 Upon exposure to light in the presence of a multifunctional PEG-maleimide, phototriggered hydrogel formation occurs. Single photon (405 nm) and two-photon (800 nm) patterning techniques were used to produce defined 3D hydrogel constructs, and the extent of crosslinking of the resulting gels could be tuned by controlling the light dose. This technique was used to encapsulate cells from human and mouse cell lines, and the viability compared favorably to photopolymerized PEG-diacrylate gels photoinitiated by Irgacure 2959. In an alternative strategy, spatiotemporal control of the thiol-Michael reaction can be achieved using photogenerated bases.67 However, this strategy may be difficult to implement in a biomaterials setting where pH must be maintained within a narrow range.
The precursors for oxime and hydrazone reactions have likewise been selected for uncaging reactions in biomaterials applications. Conveniently, the product of o-NB uncaging is a nitrosobenzaldehyde (Figure 12b), the generation of which can result in the formation of oxime or hydrazone bonds in the presence of alkoxyamines97,98 or hydrazines,99 respectively. Hydrazone-based hydrogel networks have been formed in such a manner, and used for cell encapsulation, while modulating the temporal network evolution with light dose and the spatial chemistry with controlled illumination.99 Alternative approaches involve caging the alkoxyamine substrate for subsequent reaction with a carbonyl species. 2-(2-nitrophenyl)propoxycarbonyl (NPPOC) is an appropriate photocage for this purpose because its uncaging does not form a carbonyl group that would undergo condensation with the liberated alkoxyamine (Figure 12c).94,95 DeForest and Tirrell adopted this strategy for the reversible photopatterning of full-length proteins.96 A hydrogel network containing pendant NPPOC-caged alkoxyamines was created, and proteins were modified with aldehydes through a photocleavable o-NB linkage (Figure 13). A first round of light exposure uncages reactive alkoxyamine groups for tethering the modified proteins. Ensuing light exposure cleaves o-NB linkages and releases the protein, and can simultaneously tether a second aldehyde-tagged protein. The authors demonstrated that tethered proteins (collagenase, primary antibodies, and Notch) retained their bioactivity when incorporated in this manner. Additionally, it was hypothesized that this reversible patterning approach could be used to influence hMSC cell fate. These cells are known to undergo osteogenic differentiation under conditions that promote a spread morphology in three dimensions.180 When the ECM protein vitronectin was photopatterned into these hydrogels, hMSCs underwent osteogenic, and this fate could be reverted by removal of the immobilized vitronectin after 4 days of culture.96 DeForest and coworkers have recently extended the use of NPPOC-caged alkoxyamine groups as a method to photopolymerize oxime hydrogels.235
Figure 13.

Reversible photopatterning of full length proteins using sequential photoconjugation and photocleavage reactions. a) Network alkoxyamines are uncaged and react with proteins conjugated to photocleavable aldehydes. A secondary round of light exposure releases the protein. b) A second protein can be incorporated during the release of the first, forming intricate patterns. Scale bar 50 μm. c) Sustained vitronectin immobilization leads to osteogenic differentiation of hMSCs, indicated by staining for osteocalcin (green). Reprinted from DeForest et al.96 copyright (2015) Nature Publishing Group.
In the preceding examples, proteins have been functionalized by non-specific modifications to allow for photopatterning, an approach that may compromise the bioactivity. Protein engineering strategies can be used to site-specifically incorporate reactivity, such as cysteine residues, non-natural amino acids, or specific amino acid sequences such as those recognized by the enzyme transglutaminase. Activated transglutaminase factor XIII (FXIIIa) acts by forming an isopeptide bond between lysine and glutamine residues.75,76 By caging the ε-amino group of lysine in the amino acid recognition sequence with a dimethoxy (nitroveratryl) derivative of an oNB cage, an inactive form of the substrate is obtained (Figure 12d). When exposed to 405 nm laser light in the presence of recombinant full length proteins containing the glutamine recognition sequence (NQEQVSPL) and FXIIIa, spatiotemporal enzyme-mediated ligation is achieved. This has been employed to immobilize peptides and proteins in three dimensional hydrogels.77,78 In particular, cellular adhesion or directed 3D migration could be achieved by patterning a peptide (RGD), a fibronectin fragment, or a full length protein (platelet-derived growth factor) (Figure 14).77 Such site specific-modifications may prove particularly useful for the immobilization of fragile biomolecules.
Figure 14.

Enzymatic photopatterning by the uncaging of the lysine residue in the transglutaminase recognition sequence. This strategy is used to induce migration out of a cluster of MSCs into the surrounding gel in regions patterned with a) RGD and b) fibronectin fragment. Scale bars 200 μm. Reproduced from Mosiewicz et al.77 copyright (2013) Nature Publishing Group
7. Other photoclick and photoswitchable reactions
Some photochemical reactions take advantage of innate photochemical activity of certain molecules, without the need for photoinitiating (Section 2, 3, & 4) or photocleavage (Sections 5 & 6) reactions. The intrinsic photoactivity of such molecules is advantageous because these reactions can often be performed without exogenous factors such as photoinitiators or releasing cleaved photocages into solution. Although yet to be fully explored in the biomaterials setting, these reactions are emerging to fill needs for fast and reversible conjugation strategies.
In addition to the photoinitiated thiol-ene reaction (Section 3), other photo-click reactions have recently been discovered or re-imagined.236 Notably among these are the tetrazole-alkene cycloaddition,91,237,238 the photo-Diels alder reaction,81 and the in situ conversion of cyclopropenone derivatives to cyclooctynes for copper-free cycloaddition with azides.89 In the photoactivated tetrazole-alkene cycloadditions, light absorbed by a 2,5 diaryl tetrazole causes the release of N2 and the generation of a nitrile imine dipole, which then undergoes a cycloaddition with an alkene dienophile. The photochemical step has a high quantum yield (0.5–0.9), and the cycloaddition proceeds rapidly, with rate constants up to 11.0 M−1 s−1.91 This reaction has the interesting side effect of generating a fluorescent product, which may aid in imaging of patterned regions, but could also hinder fluorescent imaging of cell structures. This cycloaddition reaction was recently demonstrated as a method to form hydrogels239 and as a photodegradation strategy to disrupt the structure of a supramolecular hydrogel.240 In the latter strategy, Zhang and coworkers designed a biaryl tetrazole capable of undergoing an intramolecular photoclick reaction that disrupts the pi-stacking of a self-assembled peptide hydrogel, resulting in relatively fast photodegradation and fluorescence turn-on.240
The Diels-Alder reaction is an attractive click chemistry in a biomaterials setting, and has been used to form hydrogels241 and in the controlled release of a model drug.242 The term “photoenol” is used to described dienols that are reversibly formed by the irradiation of o-methylphenylketones. These photoenols take part in efficient Diels-Alder reactions with dienophiles, imparting light-based control over this reaction. A different approach has recently been used to achieve photochemical control over the inverse electron demand Diels Alder reaction between tetrazine and norbornene using the photocatalytic oxidation of dihydrogen tetrazine with red light.85
In addition to rapid photochemical reactions, there is also a growing interest and demand for reversible reactions. From the perspective of spatiotemporal control, azobenzene and its photoisomerization have been exploited to synthesize hydrogels with reversible, photoswitchable properties. When irradiated with UV light (~365 nm), azobenzene isomerizes to the less stable cis conformation, and reverts to the trans conformation under irradiation by visible (420 nm) light. Perhaps more interestingly, the trans conformation participates in a host-guest complexation with cyclodextrin with a high affinity, whereas the cis isomer has a much lower affinity. This discrepancy has been used to achieve light-based control over the sol-gel transition in azobenzene hydrogels.243,244 In a system comprising multifunctional cyclodextrin- and azobenzene-functionalized polymers, an associating network is formed that can be reversibly disrupted by exposure to UV light. Exposing the resulting sol to visible light regenerates the hydrogel. This photochemical control of binding affinity has also been used to modulate the release of azobenzene drugs.245 The change in conformation can also induce a change in mechanics when azobenzene groups are incorporated directly into the network structure: isomerization to the cis state leads to a reversible decrease in elastic modulus, potentially due to disruption of hydrogen bonding.246 Similar strategies have been developed based on the reversible photodimerization of coumarin247 and anthracene248 functional groups. Strategies to create repeated, reversible changes in elastic properties of cell culture substrates are of particular interest for mechanobiology, for example to model the effects of repeated injury or fibrosis.
8. Summary and outlook
We have presented a perspective of the state of the art of hydrogel biomaterials with photochemical properties. The reactions presented herein are being used to form, functionalize, reshape, and degrade cell-laden hydrogels, and in doing so, are helping provide answers to some of the fundamental questions in mechanobiology, stem cell expansion and differentiation, tissue engineering, and cell-matrix signaling. However, this is far from a mature field. Research into click, biorthogonal, and photochemistries is continuously providing biomaterials researchers with new tools to create such scaffolds. As these scaffolds increase in sophistication, it will become possible to investigate the effects of multiple spatiotemporally controlled signals. Whereas current experiments often examine the effect of a single matrix property or signaling molecule in isolation, a myriad of likely interdependent changes may occur in vivo, and there is clearly a need for matrices to examine these combinatorial effects.
Structurally speaking, the amorphous, isotropic nature of many synthetic hydrogels is a poor match for the fibrillar, oriented, and topographically diverse ECM. This results in a mismatch between the two materials: natural ECM is highly viscoelastic and has non-linear properties, and while we are only beginning to understand how these may affect cell behavior, it is clear that mechanotransduction is dependent on more than the elastic modulus alone. In the biochemical landscape of tissues, Nature sequesters growth factors and signaling molecules with affinity binding interactions, which we have imitated with covalent immobilization. This approximation has allowed us to probe these interactions, but the utility of such designs is often limited by the permanence of covalent bonds. Therefore, look for reversible reactions and synthetic matrices that can selectively sequester molecules from the body to take on a bigger role.
Further, the advancements in spatiotemporal hydrogels will interface with advanced manufacturing techniques in a mutually beneficial manner. This is especially true in the context of tissue engineering. Our tissues are remarkable in their complexity, which spans several orders of magnitude, from nano-structured ECM, to the mesoscale organization of cell layers, to the macroscale form of the whole tissue. It is clear that improved techniques are needed to create such objects, and it is likely to come from a variety of sources. For example, stereolithographic techniques may be used in combination with bioprinting to bridge the gap between cell- and tissue-relevant size scales, and such a strategy could be complemented with self-assembling smart materials to impart nanoscale order. Moreover, these materials will be used in concert with tissue-specific stem cells that are capable of self organizing to varying degrees. Therefore, responsive and dynamic materials will also be used to coax these processes along the appropriate paths. On the other hand, as improvement in vascularization techniques allows us to create larger tissue constructs, we will no longer be able to rely on the relative clarity and low scattering of the hydrogels currently in use. Longer wavelengths of light including visible and near-infrared will be useful in this regard, and may be used with upconverting nanoparticles to perform many of the same reactions currently in place. Moving forward, we will also need improved tools to monitor not only how cells are behaving in these matrices, but also the effect they have on responsive or permissive scaffolds, and whether signals we have introduced remain in place and active. Dynamic matrices sit at a critical juncture between materials science and biology; as increased knowledge of 3D and 4D biological processes helps inform the design of spatiotemporal materials, these materials will in turn be used to further our understanding in the field of regenerative medicine.
Figure 8.

Photocleavage reactions of a) nitrobenzyl and b) coumarin photolabile linkages. When incorporated into the structure of the network, these cleavage reactions result in network degradation. c) Schematic showing the use of a photodegradable hydrogel to vascularize a cell-laden hydrogel construct in situ. Reprinted from Arakawa et al.193 copyright (2017) Wiley.
Acknowledgments
The authors are grateful to Alex Caldwell for helpful discussions and to E. Kyburz for assistance in figure illustration. Funding for this work was provided by the National Science Foundation (Division of Materials Research Grant 1408955), the National Institutes of Health (Grant DE016523), the NSF GRFP, and the NIH/CU Molecular Biophysics Training Program (T32 GM-065103).
Biographies

Kristi Anseth is the Tisone Professor of Chemical and Biological Engineering and Associate Director of the BioFrontiers Institute at the University of Colorado at Boulder. Dr. Anseth earned her B.S. degree from Purdue University, her Ph.D. degree from the University of Colorado, and completed post-doctoral research at MIT as an NIH fellow. Her research interests lie at the interface between biology and engineering where she designs new biomaterials for applications in drug delivery and regenerative medicine. She is an elected member of the National Academy of Engineering, National Academy of Medicine, National Academy of Sciences, and National Academy of Inventors.

Tobin Brown received his Bachelor’s degree in Chemical Engineering from McGill University in 2013. He is currently a PhD student at the University of Colorado Boulder in the research group of Professor Kristi Anseth, and his research interests lie in adaptable and dynamic hydrogel materials for use in regenerative medicine. He is a recipient of the NSF Graduate Research Fellowship and the Excellence in Graduate Polymer Research Award from the ACS Polymer Chemistry Division.
Footnotes
Conflicts of Interest
There are no conflicts of interest to declare.
References
- 1.Takahashi K, Yamanaka S. Cell. 2006;126:663–76. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 2.Graf T. Cell Stem Cell. 2011;9:504–516. doi: 10.1016/j.stem.2011.11.012. [DOI] [PubMed] [Google Scholar]
- 3.Cukierman E, Pankov R, Yamada KM. Curr Opin Cell Biol. 2002;14:633–639. doi: 10.1016/s0955-0674(02)00364-2. [DOI] [PubMed] [Google Scholar]
- 4.Petersen OW, Rønnov-Jessen L, Howlett AR, Bissell MJ. Proc Natl Acad Sci U S A. 1992;89:9064–9068. doi: 10.1073/pnas.89.19.9064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weaver VM, Petersen OW, Wang F, Larabell Ca, Briand P, Damsky C, Bissell MJ. J Cell Biol. 1997;137:231–245. doi: 10.1083/jcb.137.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sternlicht MD, Lochter A, Sympson CJ, Huey B, Rougier JP, Gray JW, Pinkel D, Bissell MJ, Werb Z. Cell. 1999;98:137–146. doi: 10.1016/s0092-8674(00)81009-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Engler AJ, Sen S, Sweeney HL, Discher DE. Cell. 2006;126:677–89. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 8.Murrell M, Kamm R, Matsudaira P. Biophys J. 2011;101:297–306. doi: 10.1016/j.bpj.2011.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cameron AR, Frith JE, Cooper-White JJ. Biomaterials. 2011;32:5979–5993. doi: 10.1016/j.biomaterials.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 10.Cameron AR, Frith JE, Gomez GA, Yap AS, Cooper-White JJ. Biomaterials. 2014;35:1857–1868. doi: 10.1016/j.biomaterials.2013.11.023. [DOI] [PubMed] [Google Scholar]
- 11.Chaudhuri O, Gu L, Klumpers D, Darnell M, Bencherif SA, Weaver JC, Huebsch N, Lee H, Lippens E, Duda GN, Mooney DJ. Nat Mater. 2015;15:326–334. doi: 10.1038/nmat4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Das RK, Gocheva V, Hammink R, Zouani OF, Rowan AE. Nat Mater. doi: 10.1038/nmat4483. [DOI] [PubMed] [Google Scholar]
- 13.Wozniak MA, Chen CS. Nat Rev Mol Cell Biol. 2009;10:34–43. doi: 10.1038/nrm2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jaalouk DE, Lammerding J. Nat Rev Mol Cell Biol. 2009;10:63–73. doi: 10.1038/nrm2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wen JH, Vincent LG, Fuhrmann A, Choi YS, Hribar KC, Taylor-Weiner H, Chen S, Engler AJ. Nat Mater. doi: 10.1038/nmat4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trappmann B, Gautrot JE, Connelly JT, Strange DGT, Li Y, Oyen ML, Cohen Stuart Ma, Boehm H, Li B, Vogel V, Spatz JP, Watt FM, Huck WTS. Nat Mater. 2012;11:642–9. doi: 10.1038/nmat3339. [DOI] [PubMed] [Google Scholar]
- 17.Khetan S, Guvendiren M, Legant WR, Cohen DM, Chen CS, Burdick Ja. Nat Mater. 2013;12:458–465. doi: 10.1038/nmat3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang C, Tibbitt MW, Basta L, Anseth KS. Nat Mater. 2014;13:645–52. doi: 10.1038/nmat3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Evans ND, Minelli C, Gentleman E, LaPointe V, Patankar SN, Kallivretaki M, Chen X, Roberts CJ, Stevens MM. Eur Cells Mater. 2009;18:1–13. doi: 10.22203/ecm.v018a01. [DOI] [PubMed] [Google Scholar]
- 20.Guilak F, Cohen DM, Estes BT, Gimble JM, Liedtke W, Chen CS. Cell Stem Cell. 2009;5:17–26. doi: 10.1016/j.stem.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caiazzo M, Okawa Y, Ranga A, Piersigilli A, Tabata Y, Lutolf MP. Nat Mater. doi: 10.1038/nmat4536. [DOI] [PubMed] [Google Scholar]
- 22.Kolesky DB, Truby RL, Gladman AS, Busbee TA, Homan KA, Lewis JA. Adv Mater. 2014;26:3124–3130. doi: 10.1002/adma.201305506. [DOI] [PubMed] [Google Scholar]
- 23.Miller JS, Stevens KR, Yang MT, Baker BM, Nguyen DHT, Cohen DM, Toro E, Chen AA, Galie PA, Yu X, Chaturvedi R, Bhatia SN, Chen CS. Nat Mater. 2012;11:768–774. doi: 10.1038/nmat3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang YS, Duchamp M, Oklu R, Ellisen LW, Langer R, Khademhosseini A. ACS Biomater Sci Eng. 2016 doi: 10.1021/acsbiomaterials.6b00246. acsbiomaterials.6b00246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fischbach C, Chen R, Matsumoto T, Schmelzle T, Brugge JS, Polverini PJ, Mooney DJ. Nat Methods. 2007;4:855–860. doi: 10.1038/nmeth1085. [DOI] [PubMed] [Google Scholar]
- 26.Madden L, Juhas M, Kraus WE, Truskey GA, Bursac N. Elife. doi: 10.7554/eLife.04885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park SJ, Gazzola M, Park KS, Park S, Di Santo V, Blevins EL, Lind JU, Campbell PH, Dauth S, Capulli AK, Pasqualini FS, Ahn S, Cho A, Yuan H, Maoz BM, Vijaykumar R, Choi JW, Deisseroth K, Lauder GV, Mahadevan L, Parker KK. Science (80- ) 2016;353:158–162. doi: 10.1126/science.aaf4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raman R, Cvetkovic C, Uzel SGM, Platt RJ, Sengupta P, Kamm RD, Bashir R. Proc Natl Acad Sci. 2016;113:3497–3502. doi: 10.1073/pnas.1516139113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yin X, Mead BE, Safaee H, Langer R, Karp JM, Levy O. Cell Stem Cell. 2016;18:25–38. doi: 10.1016/j.stem.2015.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gjorevski N, Ranga A, Lutolf MP. Development. 2014;141:1794–804. doi: 10.1242/dev.101048. [DOI] [PubMed] [Google Scholar]
- 31.Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, Clevers H. Nature. 2009;459:262–5. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 32.Gjorevski N, Sachs N, Manfrin A, Giger S, Bragina ME, Ordóñez-Morán P, Clevers H, Lutolf MP. Nature. doi: 10.1038/nature20168. [DOI] [PubMed] [Google Scholar]
- 33.Eiraku M, Takata N, Ishibashi H, Kawada M, Sakakura E, Okuda S, Sekiguchi K, Adachi T, Sasai Y. Nature. 2011;472:51–56. doi: 10.1038/nature09941. [DOI] [PubMed] [Google Scholar]
- 34.Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, Knoblich JA. Nature. 2013;501:373–379. doi: 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ranga A, Gjorevski N, Lutolf MP. Adv Drug Deliv Rev. 2014;69–70:19–28. doi: 10.1016/j.addr.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 36.Pathak A, Kumar S. Integr Biol. 2011;3:267. doi: 10.1039/c0ib00095g. [DOI] [PubMed] [Google Scholar]
- 37.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown Ra. Nat Rev Mol Cell Biol. 2002;3:349–63. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- 38.Frantz C, Stewart KM, Weaver VM. J Cell Sci. 2010;123:4195–4200. doi: 10.1242/jcs.023820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hinz B. J Biomech. 2010;43:146–155. doi: 10.1016/j.jbiomech.2009.09.020. [DOI] [PubMed] [Google Scholar]
- 40.Darribère T, Schwarzbauer JE. Mech Dev. 2000;92:239–250. doi: 10.1016/s0925-4773(00)00245-8. [DOI] [PubMed] [Google Scholar]
- 41.Mammoto T, Ingber DE. Development. 2010;137:1407–1420. doi: 10.1242/dev.024166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nelson CM, Gleghorn JP. Annu Rev Biomed Eng. 2012;14:129–154. doi: 10.1146/annurev-bioeng-071811-150043. [DOI] [PubMed] [Google Scholar]
- 43.Nelson CM. J Biomech Eng. 2016;138:21005. doi: 10.1115/1.4032128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.ichi Hironaka K, Morishita Y. Curr Opin Genet Dev. 2012;22:553–561. doi: 10.1016/j.gde.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 45.Conklin MW, Eickhoff JC, Riching KM, Pehlke CA, Eliceiri KW, Provenzano PP, Friedl A, Keely PJ. Am J Pathol. 2011;178:1221–1232. doi: 10.1016/j.ajpath.2010.11.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, Hammer DA, Weaver VM. Cancer Cell. 2005;8:241–254. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 47.Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SFT, Csiszar K, Giaccia A, Weninger W, Yamauchi M, Gasser DL, Weaver VM. Cell. 2009;139:891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goswami S, Sahai E, Wyckoff JB, Epidermal CF, Cammer M, Cox D, Pixley FJ, Stanley ER, Segall JE, Condeelis JS. Cancer Res. 2005;65:5278–5283. doi: 10.1158/0008-5472.CAN-04-1853. [DOI] [PubMed] [Google Scholar]
- 49.Slaughter BV, Khurshid SS, Fisher OZ, Khademhosseini A, Peppas Na. Adv Mater. 2009;21:3307–29. doi: 10.1002/adma.200802106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Burdick Ja, Khademhosseini A, Langer R. Langmuir. 2004;20:8–11. doi: 10.1021/la049298n. [DOI] [PubMed] [Google Scholar]
- 51.Lo CM, Wang HB, Dembo M, Wang Y. Biophys J. 2000;79:144–152. doi: 10.1016/S0006-3495(00)76279-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.DeLong SA, Moon JJ, West JL. Biomaterials. 2005;26:3227–3234. doi: 10.1016/j.biomaterials.2004.09.021. [DOI] [PubMed] [Google Scholar]
- 53.Mooney DJ, Baldwin DF, Suh NP, Vacanti JP, Langer R. Biomaterials. 1996;17:1417–1422. doi: 10.1016/0142-9612(96)87284-x. [DOI] [PubMed] [Google Scholar]
- 54.Griffin DR, Weaver WM, Scumpia PO, Di Carlo D, Segura T. Nat Mater. 2015;14:737–744. doi: 10.1038/nmat4294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huebsch N, Lippens E, Lee K, Mehta M, Koshy ST, Darnell MC, Desai RM, Madl CM, Xu M, Zhao X, Chaudhuri O, Verbeke C, Kim WS, Alim K, Mammoto A, Ingber DE, Duda GN, Mooney DJ. Nat Mater. doi: 10.1038/nmat4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alom Ruiz S, Chen CS. Soft Matter. 2007;3:168–177. doi: 10.1039/b613349e. [DOI] [PubMed] [Google Scholar]
- 57.Kang HW, Lee SJ, Ko IK, Kengla C, Yoo JJ, Atala A. Nat Biotechnol. 2016;34:312–319. doi: 10.1038/nbt.3413. [DOI] [PubMed] [Google Scholar]
- 58.Bajaj P, Schweller RM, Khademhosseini A, West JL, Bashir R. Annu Rev Biomed Eng. 2014;16:247–276. doi: 10.1146/annurev-bioeng-071813-105155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Uto K, Tsui JH, DeForest CA, Kim DH. Prog Polym Sci. 2016;65:53–82. doi: 10.1016/j.progpolymsci.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Murphy SV, Atala A. Nat Biotechnol. 2014;32:773–785. doi: 10.1038/nbt.2958. [DOI] [PubMed] [Google Scholar]
- 61.Zhang YS, Khademhosseini A. Science (80- ) 2017;356 doi: 10.1126/science.aaf3627. eaaf3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Odian G. Principles of Polymerization. 4. Wiley & Sons; New York: 2004. [Google Scholar]
- 63.Hoyle CE, Bowman CN. Angew Chem Int Ed Engl. 2010;49:1540–73. doi: 10.1002/anie.200903924. [DOI] [PubMed] [Google Scholar]
- 64.Lowe AB. Polym Chem. 2014;5:4820–4870. [Google Scholar]
- 65.Nair DP, Podgórski M, Chatani S, Gong T, Xi W, Fenoli CR, Bowman CN. Chem Mater. 2014;26:724–744. [Google Scholar]
- 66.Luo Y, Shoichet MS. Nat Mater. 2004;3:249–53. doi: 10.1038/nmat1092. [DOI] [PubMed] [Google Scholar]
- 67.Xi W, Krieger M, Kloxin CJ, Bowman CN. Chem Commun. 2013;49:4504–4506. doi: 10.1039/c3cc41123k. [DOI] [PubMed] [Google Scholar]
- 68.Morgan CR, Magnotta F, Ketley aD. J Polym Sci Polym Chem Ed. 1977;15:627–645. [Google Scholar]
- 69.Hoyle CE, Lee TY, Roper T. J Polym Sci Part A Polym Chem. 2004;42:5301–5338. [Google Scholar]
- 70.Aimetti AA, Machen AJ, Anseth KS. Biomaterials. 2009;30:6048–6054. doi: 10.1016/j.biomaterials.2009.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fairbanks BD, Schwartz MP, Halevi AE, Nuttelman CR, Bowman CN, Anseth KS. Adv Mater. 2009;21:5005–5010. doi: 10.1002/adma.200901808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fairbanks BD, Scott TF, Kloxin CJ, Anseth KS, Bowman CN. Macromolecules. 2009;42:211–217. doi: 10.1021/ma801903w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lowe AB, Hoyle CE, Bowman CN. J Mater Chem. 2010;20:4745. [Google Scholar]
- 74.Daniele MA, Adams AA, Naciri J, North SH, Ligler FS. Biomaterials. 2014;35:1845–1856. doi: 10.1016/j.biomaterials.2013.11.009. [DOI] [PubMed] [Google Scholar]
- 75.Sperinde JJ, Griffith LG. Macromolecules. 1997;30:5255–5264. [Google Scholar]
- 76.Hu BH, Messersmith PB. J Am Chem Soc. 2003;125:14298–14299. doi: 10.1021/ja038593b. [DOI] [PubMed] [Google Scholar]
- 77.Mosiewicz KA, Kolb L, van der Vlies AJ, Martino MM, Lienemann PS, Hubbell JA, Ehrbar M, Lutolf MP. Nat Mater. 2013;12:1072–8. doi: 10.1038/nmat3766. [DOI] [PubMed] [Google Scholar]
- 78.Griffin DR, Borrajo J, Soon A, Acosta-Vélez GF, Oshita V, Darling N, MacK J, Barker T, Iruela-Arispe ML, Segura T. ChemBioChem. 2014;15:233–242. doi: 10.1002/cbic.201300687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nicolaou KC, Snyder SA, Montagnon T, Vassilikogiannakis G. Angew Chemie Int Ed. 2002;41:1668–1698. doi: 10.1002/1521-3773(20020517)41:10<1668::aid-anie1668>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 80.Nimmo CM, Owen SC, Shoichet MS. Biomacromolecules. 2011;12:824–830. doi: 10.1021/bm101446k. [DOI] [PubMed] [Google Scholar]
- 81.Gruendling T, Oehlenschlaeger KK, Frick E, Glassner M, Schmid C, Barner-Kowollik C. Macromol Rapid Commun. 2011;32:807–812. doi: 10.1002/marc.201100159. [DOI] [PubMed] [Google Scholar]
- 82.Blackman ML, Royzen M, Fox JM. J Am Chem Soc. 2008;130:13518–13519. doi: 10.1021/ja8053805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Han HS, Devaraj NK, Lee J, Hilderbrand SA, Weissleder R, Bawendi MG. J Am Chem Soc. 2010;132:7838–7839. doi: 10.1021/ja101677r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Alge DL, Azagarsamy Ma, Donohue DF, Anseth KS. Biomacromolecules. 2013;14:949–53. doi: 10.1021/bm4000508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Truong VX, Tsang KM, Ercole F, Forsythe JS. Chem Mater. 2017;29:3678–3685. [Google Scholar]
- 86.Agard NJ, Prescher Ja, Bertozzi CR. J Am Chem Soc. 2004;126:15046–15047. doi: 10.1021/ja044996f. [DOI] [PubMed] [Google Scholar]
- 87.Baskin JM, Prescher Ja, Laughlin ST, Agard NJ, Chang PV, Miller Ia, Lo A, Codelli Ja, Bertozzi CR. Proc Natl Acad Sci U S A. 2007;104:16793–16797. doi: 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.DeForest Ca, Polizzotti BD, Anseth KS. Nat Mater. 2009;8:659–664. doi: 10.1038/nmat2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Poloukhtine AA, Mbua NE, Wolfert MA, Boons GJ, Popik VV. J Am Chem Soc. 2009;131:15769–15776. doi: 10.1021/ja9054096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Adzima BJ, Tao Y, Kloxin CJ, DeForest Ca, Anseth KS, Bowman CN. Nat Chem. 2011;3:256–259. doi: 10.1038/nchem.980. [DOI] [PubMed] [Google Scholar]
- 91.Lim RKV, Lin Q. Acc Chem Res. 2011;44:828–830. doi: 10.1021/ar200021p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kalia J, Raines RT. Angew Chemie - Int Ed. 2008;47:7523–7526. doi: 10.1002/anie.200802651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dirksen A, Hackeng TM, Dawson PE. Angew Chemie Int Ed. 2006;45:7581–7584. doi: 10.1002/anie.200602877. [DOI] [PubMed] [Google Scholar]
- 94.Dendane N, Hoang A, Guillard L, Defrancq E, Vinet F, Dumy P. Bioconjug Chem. 2007;18:671–676. doi: 10.1021/bc060254v. [DOI] [PubMed] [Google Scholar]
- 95.Mancini RJ, Li RC, Tolstyka ZP, Maynard HD. Org Biomol Chem. 2009;7:4954–9. doi: 10.1039/b904195h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.DeForest CA, Tirrell DA. Nat Mater. 2015;14:523–531. doi: 10.1038/nmat4219. [DOI] [PubMed] [Google Scholar]
- 97.Pauloehrl T, Delaittre G, Bruns M, Meißler M, Börner HG, Bastmeyer M, Barner-Kowollik C. Angew Chemie - Int Ed. 2012;51:9181–9184. doi: 10.1002/anie.201202684. [DOI] [PubMed] [Google Scholar]
- 98.Lee JH, Domaille DW, Noh H, Oh T, Choi C, Jin S, Cha JN. Langmuir. 2014;30:8452–8460. doi: 10.1021/la500744s. [DOI] [PubMed] [Google Scholar]
- 99.Azagarsamy MA, Marozas IA, Spaans S, Anseth KS. ACS Macro Lett. 2016;5:19–23. doi: 10.1021/acsmacrolett.5b00682. [DOI] [PubMed] [Google Scholar]
- 100.Bryant SJ, Nuttelman CR, Anseth KS. J Biomater Sci Polym Ed. 2000;11:439–457. doi: 10.1163/156856200743805. [DOI] [PubMed] [Google Scholar]
- 101.Cruise GM, Hegre OD, Scharp DS, Hubbell JA. Biotechnol Bioeng. 1998;57:655–665. doi: 10.1002/(sici)1097-0290(19980320)57:6<655::aid-bit3>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 102.Williams CG, Malik AN, Kim TK, Manson PN, Elisseeff JH. Biomaterials. 2005;26:1211–1218. doi: 10.1016/j.biomaterials.2004.04.024. [DOI] [PubMed] [Google Scholar]
- 103.Fairbanks BD, Schwartz MP, Bowman CN, Anseth KS. Biomaterials. 2009;30:6702–6707. doi: 10.1016/j.biomaterials.2009.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.McCall JD, Anseth KS. Biomacromolecules. 2012;13:2410–7. doi: 10.1021/bm300671s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.McCall JD, Lin CC, Anseth KS. Biomacromolecules. 2011;12:1051–1057. doi: 10.1021/bm101379v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lin CC, Sawicki SM, Metters AT. Biomacromolecules. 2008;9:75–83. doi: 10.1021/bm700782c. [DOI] [PubMed] [Google Scholar]
- 107.Fedorovich NE, Oudshoorn MH, van Geemen D, Hennink WE, Alblas J, Dhert WJA. Biomaterials. 2009;30:344–353. doi: 10.1016/j.biomaterials.2008.09.037. [DOI] [PubMed] [Google Scholar]
- 108.Ifkovits JL, Burdick Ja. Tissue Eng. 2007;13:2369–2385. doi: 10.1089/ten.2007.0093. [DOI] [PubMed] [Google Scholar]
- 109.Smeds KA, Grinstaff MW. J Biomed Mater Res. 2001;54:115–121. doi: 10.1002/1097-4636(200101)54:1<115::aid-jbm14>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 110.Leach JB, Bivens KA, Patrick CW, Schmidt CE. Biotechnol Bioeng. 2003;82:578–589. doi: 10.1002/bit.10605. [DOI] [PubMed] [Google Scholar]
- 111.Burdick JA, Chung C, Jia X, Randolph MA, Langer R. Biomacromolecules. 2005;6:386–391. doi: 10.1021/bm049508a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Van Den Bulcke AI, Bogdanov B, De Rooze N, Schacht EH, Cornelissen M, Berghmans H. Biomacromolecules. 2000;1:31–38. doi: 10.1021/bm990017d. [DOI] [PubMed] [Google Scholar]
- 113.Benton Ja, DeForest Ca, Vivekanandan V, Anseth KS. Tissue Eng Part A. 2009;15:3221–3230. doi: 10.1089/ten.tea.2008.0545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nichol JW, Koshy ST, Bae H, Hwang CM, Yamanlar S, Khademhosseini A. Biomaterials. 2010;31:5536–5544. doi: 10.1016/j.biomaterials.2010.03.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nguyen KT, West JL. Biomaterials. 2002;23:4307–4314. doi: 10.1016/s0142-9612(02)00175-8. [DOI] [PubMed] [Google Scholar]
- 116.Sawhney AS, Pathak CP, Hubbell JA. Macromolecules. 1993;26:581–587. [Google Scholar]
- 117.West JL, Hubbell JA. Macromolecules. 1999;32:241–244. [Google Scholar]
- 118.Lutolf MP, Lauer-Fields JL, Schmoekel HG, Metters AT, Weber FE, Fields GB, Hubbell JA. Proc Natl Acad Sci U S A. 2003;100:5413–8. doi: 10.1073/pnas.0737381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yamada Y, Kleinman HK. Curr Opin Cell Biol. 1992;4:819–23. doi: 10.1016/0955-0674(92)90105-l. [DOI] [PubMed] [Google Scholar]
- 120.Hern DL, Hubbell Ja. J Biomed Mater Res. 1998;39:266–276. doi: 10.1002/(sici)1097-4636(199802)39:2<266::aid-jbm14>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 121.Mann BK, Gobin AS, Tsai AT, Schmedlen RH, West JL. Biomaterials. 2001;22:3045–3051. doi: 10.1016/s0142-9612(01)00051-5. [DOI] [PubMed] [Google Scholar]
- 122.Gobin AS, West JL. Biotechnol Prog. 2003;19:1781–1785. doi: 10.1021/bp0341390. [DOI] [PubMed] [Google Scholar]
- 123.Gobin AS, West JL. FASEB J. 2002;16:1805–1807. [Google Scholar]
- 124.Schmedlen RH, Masters KS, West JL. Biomaterials. 2002;23:4325–4332. doi: 10.1016/s0142-9612(02)00177-1. [DOI] [PubMed] [Google Scholar]
- 125.Burdick Ja, Anseth KS. Biomaterials. 2002;23:4315–23. doi: 10.1016/s0142-9612(02)00176-x. [DOI] [PubMed] [Google Scholar]
- 126.Bryant SJ, Anseth KS. J Biomed Mater Res. 2002;59:63–72. doi: 10.1002/jbm.1217. [DOI] [PubMed] [Google Scholar]
- 127.Anseth KS, Metters AT, Bryant SJ, Martens PJ, Elisseeff JH, Bowman CN. J Control Release. 2002;78:199–209. doi: 10.1016/s0168-3659(01)00500-4. [DOI] [PubMed] [Google Scholar]
- 128.Elisseef J, McIntosh W, Anseth K, Riley S, Ragan P, Langer R. J Biomed Mater Res. 2000;51:164–171. doi: 10.1002/(sici)1097-4636(200008)51:2<164::aid-jbm4>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 129.Sawhney AS, Pathak CP, Hubbell JA. Biotechnol Bioeng. 1994;44:383–386. doi: 10.1002/bit.260440317. [DOI] [PubMed] [Google Scholar]
- 130.Piantino J, Burdick JA, Goldberg D, Langer R, Benowitz LI. Exp Neurol. 2006;201:359–367. doi: 10.1016/j.expneurol.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 131.Elisseeff J, Anseth K, Sims D, McIntosh W, Randolph M, Langer R. Proc Natl Acad Sci U S A. 1999;96:3104–3107. doi: 10.1073/pnas.96.6.3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Hill-West JL, Chowdhury SM, Slepian MJ, Hubbell JA. Proc Natl Acad Sci U S A. 1994;91:5967–71. doi: 10.1073/pnas.91.13.5967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Pereira RF, Bártolo PJ. Engineering. 2015;1:090–112. [Google Scholar]
- 134.Torgersen J, Qin XH, Li Z, Ovsianikov A, Liska R, Stampfl J. Adv Funct Mater. 2013;23:4542–4554. [Google Scholar]
- 135.Zipfel WR, Williams RM, Webb WW. Nat Biotechnol. 2003;21:1369–77. doi: 10.1038/nbt899. [DOI] [PubMed] [Google Scholar]
- 136.Liu VA, Bhatia SN. Biomed Microdevices. 2002;4:257–266. [Google Scholar]
- 137.Tsang VL, Chen AA, Cho LM, Jadin KD, Sah RL, DeLong S, West JL, Bhatia SN. FASEB J. 2007;21:790–801. doi: 10.1096/fj.06-7117com. [DOI] [PubMed] [Google Scholar]
- 138.Albrecht DR, Tsang VL, Sah RL, Bhatia SN. Lab Chip. 2005;5:111–8. doi: 10.1039/b406953f. [DOI] [PubMed] [Google Scholar]
- 139.Hahn MS, Taite LJ, Moon JJ, Rowland MC, Ruffino KA, West JL. Biomaterials. 2006;27:2519–2524. doi: 10.1016/j.biomaterials.2005.11.045. [DOI] [PubMed] [Google Scholar]
- 140.Ma X, Qu X, Zhu W, Li Y, Yuan S, Zhang H, Liu J, Wang P, Lai CSE, Zanella F, Feng G-S, Sheikh F, Chien S, Chen S. Proc Natl Acad Sci. 2016:201524510. doi: 10.1073/pnas.1524510113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lee SH, Moon JJ, West JL. Biomaterials. 2008;29:2962–2968. doi: 10.1016/j.biomaterials.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hoffmann JC, West JL. Soft Matter. 2010;6:5056. [Google Scholar]
- 143.Leslie-Barbick JE, Shen C, Chen C, West JL. Tissue Eng Part A. 2011;17:221–229. doi: 10.1089/ten.tea.2010.0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Gou M, Qu X, Zhu W, Xiang M, Yang J, Zhang K, Wei Y, Chen S. Nat Commun. 2014;5:3774. doi: 10.1038/ncomms4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Chan V, Zorlutuna P, Jeong JH, Kong H, Bashir R. Lab Chip. 2010;10:2062. doi: 10.1039/c004285d. [DOI] [PubMed] [Google Scholar]
- 146.Gauvin R, Chen YC, Lee JW, Soman P, Zorlutuna P, Nichol JW, Bae H, Chen S, Khademhosseini A. Biomaterials. 2012;33:3824–3834. doi: 10.1016/j.biomaterials.2012.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Han L, Mapili G, Chen S, Roy K. J Manuf Sci Eng. 2008;130:21005. [Google Scholar]
- 148.Tumbleston JR, Shirvanyants D, Ermoshkin N, Janusziewicz R, Johnson AR, Kelly D, Chen K, Pinschmidt R, Rolland JP, Ermoshkin A, Samulski ET, DeSimone JM. Science (80- ) 2015;347:1349–1352. doi: 10.1126/science.aaa2397. [DOI] [PubMed] [Google Scholar]
- 149.Kolb HC, Finn MG, Sharpless KB. Angew Chem Int Ed Engl. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 150.DeForest CA, Anseth KS. Nat Chem. 2011;3:925–931. doi: 10.1038/nchem.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Grover GN, Lam J, Nguyen TH, Segura T, Maynard HD. Biomacromolecules. 2012;13:3013–3017. doi: 10.1021/bm301346e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lin F, Yu J, Tang W, Zheng J, Defante A, Guo K, Wesdemiotis C, Becker ML. Biomacromolecules. 2013;14:3749–3758. doi: 10.1021/bm401133r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.McKinnon DD, Domaille DW, Cha JN, Anseth KS. Adv Mater. 2014;26:865–872. doi: 10.1002/adma.201303680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lutolf MP, Tirelli N, Cerritelli S, Cavalli L, Hubbell JA. Bioconjug Chem. 2001;12:1051–1056. doi: 10.1021/bc015519e. [DOI] [PubMed] [Google Scholar]
- 155.Phelps EA, Enemchukwu NO, Fiore VF, Sy JC, Murthy N, Sulchek TA, Barker TH, García AJ. Adv Mater. 2012;24:64–70. 2. doi: 10.1002/adma.201103574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Rydholm AE, Bowman CN, Anseth KS. Biomaterials. 2005;26:4495–4506. doi: 10.1016/j.biomaterials.2004.11.046. [DOI] [PubMed] [Google Scholar]
- 157.Ligon S, Husár B, Wutzel H, Holman R, Liska R. Chem Rev. 2014;114:557–589. doi: 10.1021/cr3005197. [DOI] [PubMed] [Google Scholar]
- 158.O’Brien AK, Cramer NB, Bowman CN. J Polym Sci Part A Polym Chem. 2006;44:2007–2014. [Google Scholar]
- 159.Wade RJ, Bassin EJ, Gramlich WM, Burdick JA. Adv Mater. 2015;27:1356–1362. doi: 10.1002/adma.201404993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Lin C-C, Raza A, Shih H. Biomaterials. 2011;32:9685–95. doi: 10.1016/j.biomaterials.2011.08.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Shih H, Lin CC. Macromol Rapid Commun. 2013;34:269–273. doi: 10.1002/marc.201200605. [DOI] [PubMed] [Google Scholar]
- 162.Roberts JJ, Bryant SJ. Biomaterials. 2013;34:9969–9979. doi: 10.1016/j.biomaterials.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Shubin AD, Felong TJ, Graunke D, Ovitt CE, Benoit DSW. Tissue Eng Part A. 2015;21:1733–1751. doi: 10.1089/ten.tea.2014.0674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Shubin AD, Felong TJ, Schutrum BE, Joe DSL, Ovitt CE, Benoit DSW. Acta Biomater. 2017;50:437–449. doi: 10.1016/j.actbio.2016.12.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Lin C-C, Ki CS, Shih H. J Appl Polym Sci. 2015;132 doi: 10.1002/app.41563. n/a-n/a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Gramlich WM, Kim IL, Burdick JA. Biomaterials. 2013;34:9803–9811. doi: 10.1016/j.biomaterials.2013.08.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Mũnoz Z, Shih H, Lin C-C. Biomater Sci. 2014;2:1063. doi: 10.1039/c4bm00070f. [DOI] [PubMed] [Google Scholar]
- 168.Greene T, Lin CC. ACS Biomater Sci Eng. 2015;1:1314–1323. doi: 10.1021/acsbiomaterials.5b00436. [DOI] [PubMed] [Google Scholar]
- 169.Gutsmiedl K, Wirges CT, Ehmke V, Carell T. Org Lett. 2009;11:2405–2408. doi: 10.1021/ol9005322. [DOI] [PubMed] [Google Scholar]
- 170.Truong VX, Zhou K, Simon GP, Forsythe JS. Macromol Rapid Commun. 2015;36:1729–1734. doi: 10.1002/marc.201500314. [DOI] [PubMed] [Google Scholar]
- 171.Truong VX, Ablett MP, Richardson SM, Hoyland JA, Dove AP. J Am Chem Soc. 2015;137:1618–1622. doi: 10.1021/ja511681s. [DOI] [PubMed] [Google Scholar]
- 172.Desai RM, Koshy ST, Hilderbrand SA, Mooney DJ, Joshi NS. Biomaterials. 2015;50:30–37. doi: 10.1016/j.biomaterials.2015.01.048. [DOI] [PubMed] [Google Scholar]
- 173.Lang K, Davis L, Torres-Kolbus J, Chou C, Deiters A, Chin JW. Nat Chem. 2012;4:298–304. doi: 10.1038/nchem.1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Evans RA, Rizzardo E. Macromolecules. 2000;33:6722–6731. [Google Scholar]
- 175.Scott TF, Schneider AD, Cook WD, Bowman CN. Science. 2005;308:1615–7. doi: 10.1126/science.1110505. [DOI] [PubMed] [Google Scholar]
- 176.Gandavarapu NR, Azagarsamy MA, Anseth KS. Adv Mater. 2014;26:2521–2526. doi: 10.1002/adma.201304847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Young JL, Engler AJ. Biomaterials. 2011;32:1002–1009. doi: 10.1016/j.biomaterials.2010.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Khetan S, Katz JS, Burdick Ja. Soft Matter. 2009;5:1601. [Google Scholar]
- 179.Huebsch N, Arany PR, Mao AS, Shvartsman D, Ali Oa, Bencherif Sa, Rivera-Feliciano J, Mooney DJ. Nat Mater. 2010;9:518–526. doi: 10.1038/nmat2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Khetan S, Burdick Ja. Biomaterials. 2010;31:8228–8234. doi: 10.1016/j.biomaterials.2010.07.035. [DOI] [PubMed] [Google Scholar]
- 181.Mabry KM, Lawrence RL, Anseth KS. Biomaterials. 2015;49:47–56. doi: 10.1016/j.biomaterials.2015.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Highley CB, Rodell CB, Burdick JA. Adv Mater. 2015;27:5075–5079. doi: 10.1002/adma.201501234. [DOI] [PubMed] [Google Scholar]
- 183.Adzima BJ, Kloxin CJ, DeForest CA, Anseth KS, Bowman CN. Macromol Rapid Commun. 2012;33:2092–2096. doi: 10.1002/marc.201200599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Yu F, Cao X, Li Y, Chen X. ACS Macro Lett. 2015;4:289–292. doi: 10.1021/mz5007427. [DOI] [PubMed] [Google Scholar]
- 185.Ouyang L, Highley CB, Rodell CB, Sun W, Burdick JA. ACS Biomater Sci Eng. 2016 doi: 10.1021/acsbiomaterials.6b00158. acsbiomaterials.6b00158. [DOI] [PubMed] [Google Scholar]
- 186.Censi R, Schuurman W, Malda J, Di Dato G, Burgisser PE, Dhert WJA, Van Nostrum CF, Di Martino P, Vermonden T, Hennink WE. Adv Funct Mater. 2011;21:1833–1842. [Google Scholar]
- 187.Ovsianikov A, Mühleder S, Torgersen J, Li Z, Qin XH, Van Vlierberghe S, Dubruel P, Holnthoner W, Redl H, Liska R, Stampfl J. Langmuir. 2014;30:3787–3794. doi: 10.1021/la402346z. [DOI] [PubMed] [Google Scholar]
- 188.Liu W, Heinrich MA, Zhou Y, Akpek A, Hu N, Liu X, Guan X, Zhong Z, Jin X, Khademhosseini A, Zhang YS. Adv Healthc Mater. 2017;1601451:1601451. doi: 10.1002/adhm.201601451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Kloxin AM, Kasko AM, Salinas CN, Anseth KS. Science. 2009;324:59–63. doi: 10.1126/science.1169494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kirschner CM, Anseth KS. Small. 2013;9:578–584. doi: 10.1002/smll.201201841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Tsang KMC, Annabi N, Ercole F, Zhou K, Karst DJ, Li F, Haynes JM, Evans Ra, Thissen H, Khademhosseini A, Forsythe JS. Adv Funct Mater. 2015;25:977–986. doi: 10.1002/adfm.201403124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Brown TE, Marozas IA, Anseth KS. Adv Mater. 2017:1605001. doi: 10.1002/adma.201605001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Arakawa CK, Badeau BA, Zheng Y, Deforest CA. doi: 10.1002/adma.201703156. [DOI] [Google Scholar]
- 194.Tibbitt MW, Kloxin AM, Anseth KS. J Polym Sci Part A Polym Chem. 2013;51:1899–1911. doi: 10.1002/pola.26574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Tibbitt MW, Kloxin AM, Sawicki L, Anseth KS. Macromolecules. 2013;46:2785–2792. doi: 10.1021/ma302522x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Reddy SK, Anseth KS, Bowman CN. Polymer (Guildf) 2005;46:4212–4222. [Google Scholar]
- 197.Stockmayer WH. J Chem Phys. 1943;11:45. [Google Scholar]
- 198.Kloxin AM, Tibbitt MW, Kasko AM, Fairbairn JA, Anseth KS. Adv Mater. 2010;22:61–6. doi: 10.1002/adma.200900917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Kloxin AM, Benton Ja, Anseth KS. Biomaterials. 2010;31:1–8. doi: 10.1016/j.biomaterials.2009.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Norris SCP, Tseng P, Kasko AM. ACS Biomater Sci Eng. 2016;2:1309–1318. doi: 10.1021/acsbiomaterials.6b00237. [DOI] [PubMed] [Google Scholar]
- 201.Tse JR, Engler AJ. PLoS One. doi: 10.1371/journal.pone.0015978. [DOI] [Google Scholar]
- 202.Tibbitt MW, Kloxin AM, Dyamenahalli KU, Anseth KS. Soft Matter. 2010;6:5100. doi: 10.1039/C0SM00174K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Yang C, DelRio FW, Ma H, Killaars AR, Basta LP, Kyburz KA, Anseth KS. Proc Natl Acad Sci. 2016:201609731. doi: 10.1073/pnas.1609731113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Ma H, Killaars AR, DelRio FW, Yang C, Anseth KS. Biomaterials. 2017;131:131–144. doi: 10.1016/j.biomaterials.2017.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Wong DY, Griffin DR, Reed J, Kasko AM. Macromolecules. 2010;43:2824–2831. [Google Scholar]
- 206.Kirschner CM, Alge DL, Gould ST, Anseth KS. Adv Healthc Mater. 2014;3:649–657. doi: 10.1002/adhm.201300288. [DOI] [PubMed] [Google Scholar]
- 207.McKinnon DD, Brown TE, Kyburz KA, Kiyotake E, Anseth KS. Biomacromolecules. 2014;15:2808–16. doi: 10.1021/bm500731b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Kharkar PM, Kloxin AM, Kiick KL. J Mater Chem B. 2014;2:5511. doi: 10.1039/c4tb00496e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Baldwin AD, Kiick KL. Polym Chem. 2013;4:133–143. doi: 10.1039/C2PY20576A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Kharkar PM, Kiick KL, Kloxin AM. Polym Chem. 2015;6:5565–5574. doi: 10.1039/C5PY00750J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Kloxin A, Lewis K, DeForest C, Seedorf G, Tibbitt MW, Balasubramaniam V, Anseth KS. Integr Biol. 2012:1540–1549. doi: 10.1039/c2ib20212c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Tamura M, Yanagawa F, Sugiura S, Takagi T, Sumaru K, Kanamori T. Sci Rep. 2015;5:15060. doi: 10.1038/srep15060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Truong VX, Tsang KM, Simon GP, Boyd RL, Evans RA, Thissen H, Forsythe JS. Biomacromolecules. 2015;16:2246–2253. doi: 10.1021/acs.biomac.5b00706. [DOI] [PubMed] [Google Scholar]
- 214.Klan P, Solomek T, Bochet CG, Givens R, Rubina M, Popik V, Kostikov A, Wirz J. Chem Rev. 2012;113:119–191. doi: 10.1021/cr300177k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Griffin DR, Kasko AM. J Am Chem Soc. 2012;134:13103–7. doi: 10.1021/ja305280w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Azagarsamy MA, McKinnon DD, Alge DL, Anseth KS. ACS Macro Lett. 2014;3:515–519. doi: 10.1021/mz500230p. [DOI] [PubMed] [Google Scholar]
- 217.de Gracia Lux C, Lux J, Collet G, He S, Chan M, Olejniczak J, Foucault-Collet A, Almutairi A. Biomacromolecules. 2015;16:3286–3296. doi: 10.1021/acs.biomac.5b00950. [DOI] [PubMed] [Google Scholar]
- 218.Kotzur N, Briand B, Beyermann M, Hagen V. J Am Chem Soc. 2009;131:16927–16931. doi: 10.1021/ja907287n. [DOI] [PubMed] [Google Scholar]
- 219.Azagarsamy MA, Anseth KS. Angew Chemie Int Ed. 2013;52:13803–13807. doi: 10.1002/anie.201308174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.DeForest CA, Anseth KS. Angew Chemie Int Ed. 2012;51:1816–1819. doi: 10.1002/anie.201106463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Ki CS, Shih H, Lin CC. Polym (United Kingdom) 2013;54:2115–2122. doi: 10.1016/j.polymer.2013.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Sletten EM, Bertozzi CR. Angew Chem Int Ed Engl. 2009;48:6974–98. doi: 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.McKay CS, Finn MG. Chem Biol. 2014;21:1075–1101. doi: 10.1016/j.chembiol.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Azagarsamy MA, Anseth KS. ACS Macro Lett. 2013;2:5–9. doi: 10.1021/mz300585q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Fairbanks BD, Singh SP, Bowman CN, Anseth KS. Macromolecules. 2011;44:2444–2450. doi: 10.1021/ma200202w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Majima T, Schnabel W, Weber W. Die Makromol Chemie. 1991;192:2307–2315. [Google Scholar]
- 227.Aruoma OI. J Am Oil Chem Soc. 1998;75:199–212. doi: 10.1007/s11746-998-0032-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Dolmans DEJGJ, Fukumura D, Jain RK. Nat Rev Cancer. 2003;3:380–387. doi: 10.1038/nrc1071. [DOI] [PubMed] [Google Scholar]
- 229.Zhao H, Sterner ES, Coughlin EB, Theato P. Macromolecules. 2012;45:1723–1736. [Google Scholar]
- 230.Hoyle CE, Lowe AB, Bowman CN. Chem Soc Rev. 2010;39:1355–1387. doi: 10.1039/b901979k. [DOI] [PubMed] [Google Scholar]
- 231.Musoke-Zawedde P, Shoichet MS. Biomed Mater. 2006;1:162–169. doi: 10.1088/1748-6041/1/3/011. [DOI] [PubMed] [Google Scholar]
- 232.Wosnick JH, Shoichet MS. Chem Mater. 2008;20:55–60. [Google Scholar]
- 233.Wylie RG, Ahsan S, Aizawa Y, Maxwell KL, Morshead CM, Shoichet MS. Nat Mater. 2011;10:799–806. doi: 10.1038/nmat3101. [DOI] [PubMed] [Google Scholar]
- 234.Liu Z, Lin Q, Sun Y, Liu T, Bao C, Li F, Zhu L. Adv Mater. 2014;26:3912–3917. doi: 10.1002/adma.201306061. [DOI] [PubMed] [Google Scholar]
- 235.Farahani PE, Adelmund SM, Shadish JA, DeForest CA. J Mater Chem B. doi: 10.1039/C6TB03400D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Herner A, Lin Q. Photo-Triggered Click Chemistry for Biological Applications. 2016;374 doi: 10.1007/s41061-015-0002-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Song W, Wang Y, Qu J, Madden MM, Lin Q. Angew Chemie - Int Ed. 2008;47:2832–2835. doi: 10.1002/anie.200705805. [DOI] [PubMed] [Google Scholar]
- 238.Wang Y, Song W, Hu WJ, Lin Q. Angew Chemie - Int Ed. 2009;48:5330–5333. doi: 10.1002/anie.200901220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Fan Y, Deng C, Cheng R, Meng F, Zhong Z. Biomacromolecules. 2013;14:2814–2821. doi: 10.1021/bm400637s. [DOI] [PubMed] [Google Scholar]
- 240.He M, Li J, Tan S, Wang R, Zhang Y. J Am Chem Soc. 2013;135:18718–21. doi: 10.1021/ja409000b. [DOI] [PubMed] [Google Scholar]
- 241.Nimmo CM, Owen SC, Shoichet MS. Biomacromolecules. 2011;12:824–830. doi: 10.1021/bm101446k. [DOI] [PubMed] [Google Scholar]
- 242.Koehler KC, Alge DL, Anseth KS, Bowman CN. Biomaterials. 2013;34:4150–8. doi: 10.1016/j.biomaterials.2013.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Zhao YL, Fraser Stoddart J. Langmuir. 2009;25:8442–8446. doi: 10.1021/la804316u. [DOI] [PubMed] [Google Scholar]
- 244.Tamesue S, Takashima Y, Yamaguchi H, Shinkai S, Harada A. Angew Chemie Int Ed. 2010;49:7461–7464. doi: 10.1002/anie.201003567. [DOI] [PubMed] [Google Scholar]
- 245.Nehls EM, Rosales A, Anseth KS. J Mater Chem B. 2016;4:1035–1039. doi: 10.1039/C5TB02004B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Rosales AM, Mabry KM, Nehls EM, Anseth KS. Biomacromolecules. 2015 doi: 10.1021/bm501710e. 150210101436008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Nagata M, Yamamoto Y. React Funct Polym. 2008;68:915–921. [Google Scholar]
- 248.Zheng Y, Micic M, Mello SV, Mabrouki M, Andreopoulos FM, Konka V, Pham SM, Leblanc RM. Macromolecules. 2002;35:5228–5234. [Google Scholar]