

Abstract
Patients supported by left ventricular assist devices (LVADs) often present with the loss of large von Willebrand factor (VWF) multimers. This VWF deficiency is believed to contribute to the bleeding diathesis of patients on LVAD support and is caused by excessive VWF cleavage by the metalloprotease ADAMTS-13 under high shear stress. However, only a small percentage of patients who have suffered the loss of large VWF multimers bleed. The actual rates of VWF cleavage in these patients have not been reported, primarily due to the lack of reliable detection methods. We have developed and validated a Selected Reaction Monitoring mass spectrometry method to quantify VWF cleavage as the ratio of the ADAMTS-13-cleaved peptide MVTGNPASDEIK to the ILAGPAGDSNVVK peptide. The rate of VWF cleavage was found to be 1.26 ± 0.36% in normal plasma. It varied significantly in patient samples, ranging from 0.23% to 2.5% of total VWF antigen, even though all patients had the loss of large VWF multimers. VWF cleavage was greater in post-LVAD samples from patients who had developed bleeding, but was mostly reduced in patients who had developed thrombosis. This SRM method is reliable to quantify the rate of VWF cleavage in patients on LVAD support.
Keywords: left ventricular assist device, von Willebrand Factor, mass spectrometry
Introduction
Left ventricular assist devices (LVADs) provide cardiac support for patients with end-stage heart disease, but device-related hemostatic complications remain common and are associated with poor outcomes. Bleeding is most common and is primarily found in the gastrointestinal track at the site of an arteriovenous malformation,1,2 leading to the hypothesis that it is caused by the loss of large VWF multimers3-5 in a condition called acquired von Willebrand syndrome (AVWS).3,4 AWAS is widely considered to be caused by excessive VWF cleavage by the metalloprotease ADAMTS-13 (a disintegrin and metalloprotease with thrombospondin type-1 repeats 13).5,6 VWF cleavage can indeed be induced by elevated shear stress7-9 similar to that of an LVAD-driven blood flow.10 However, several lines of evidence suggest that excessive cleavage by ADAMTS-13 is not the sole cause of AVWS. First, there is no direct evidence that VWF multimers are excessively cleaved by ADAMTS-13 in these patients. Second, the loss of large VWF multimers is observed in nearly all patients, but only 11% to 30% of them bleed.1,11 Third, high shear stress induce not only VWF cleavage, but also VWF binding to platelets and VWF-mediated platelet aggregation.12-14 These shear stress activated VWF multimers could bind to platelets and, thus, be removed from the circulation, leading the consumptive loss of large VWF multimers. Using a newly synthesized antibody specific for cleaved VWF, we have recently demonstrated that the two pathways could cause the loss of large VWF multimers in patients on LVAD support: excessive cleavage (loss-of-function) and shear-induced VWF activation (gain-of-function).15 Although both processes can result in AVWS, they can lead to distinct hemostatic defects that require different treatments. A technical limitation of the antibody study is its inability to accurately quantify VWF cleavage. To address this limitation, we have developed a targeted mass spectrometry to quantify VWF cleavage by ADAMTS-13 in plasma samples from patients on LVAD support.
Materials and Methods
Blood samples were collected from 14 patients (age 40-69 yrs, 50% females) before and three months after LVAD implants. The recruitment was approved by the institutional review boards of the Texas Heart Institute and the Bloodworks Research Institute. All patients had New York Heart Association (NYHA) Class IV symptoms and received a HeartMate II LVAD (Thoratec, Pleasanton, CA). Patients with malignancy, autoimmune disease, or were at a hypercoagulable state were excluded.
Selected Reaction Monitoring (SRM) was used to measure VWF and its rate of cleavage in plasma samples. VWF levels were measured by four tryptic VWF peptides: EGGPSQIGDALGFAVR, VTVFPIGIGDR and ILAGPAGDSNVVK in the A3 domain, and SGFTYVLHEGECCGR in the C domain. These peptides were chosen for being proteotypic and for having a unique signature (m/z ratio) in the proteome for every Q1/Q3 pair from the peptides, as determined by the in-house human proteomics database SRMAtlas (www.srmatlas.org).16 In addition, the ADAMTS-13-cleaved peptide M1606VTGNPASDEIK was detected. Internal peptide standards for all five VWF peptides were synthesized with heavy isotopic lysine (13C615N2) or arginine (13C615N4) at the C-termini (Thermo Fisher Scientific, Rockford, IL). The total peak area and the ratio of plasma peptide and its synthesized heavy counterpart were normalized to quantify VWF and its cleavage.
VWF purified from human cryoprecipitate and a recombinant human VWF A1-3 domain peptide was cleaved by ADAMTS-13 in vitro6 and tested as a control. VWF cleavage was also measured by immunoblots for data validation. VWF antigen was measured by a commercial ELISA.15
Results and Discussion
The baseline demographic, diagnosis, and laboratory results of patients examined in this study are presented in Table 1. Clinical indexes reported here have been selected to estimate vital organ functions: hemopoietic cells by bemoglobin (Hb, also for hemolysis) and white blood cell counts (WBC), hemostasis by platelets (PLT) and prothrombin time (PT), the nutritional status by albumin, the renal function by creatinine, and bilirubin (total and indirect) as the marker of baseline liver function and index of pre-implant right sided dysfunction. Normal control plasma was pooled from 61 healthy control subjects, allowing us to cost effectively test a large number of donors in order to cover the full range of normal values. The four uncleaved VWF peptides profiled in supplemental sFigure 1 were detected at comparable levels in all plasma samples (Figure 1A) with a reproducibility of 96.8%. The levels of the four peptides in the baseline samples before LVAD implantation were significantly higher than those in healthy control (Figure 1A, black bars). They were reduced after LVAD implantation, but remained higher than normal controls (Figure 1A, white bars), consistent with our recent report.15 The mass spectrometry results were validated by their close association with VWF antigen levels measured by ELISA (Figure 1B, r2 = 0.697, p < 0.001). The detection of these peptides was also validated using a recombinant VWF A1-3 domain polypeptide that was digested by ADAMTS-13 (sFigure 3). In contrast to the readily detection of these four VWF peptides, the cleaved peptide MVTGNPASDEIK (Figure 1C) was detected only after VWF was concentrated 20-fold by immunoprecipitation with a polyclonal VWF antibody. Consistent with SRM data, cleaved VWF was also primarily detected by immunoblots in plasma spiked with cleaved VWF (Figure 1D). Furthermore, M1606VTGNPASDEIK had been detected only in the methionine-oxidized form (molecular mass: 1277.41 vs. 1261.41 for oxidized and non-oxidized peptides, sFigure 3) in samples from the patients and from healthy subjects. The finding suggests that the Met1606 is highly sensitive to oxidation during sample processing and that the synthesized heavy peptide should be oxidized in order to detect and quantify VWF cleavage. Together, these data demonstrate that SRM reliably and quantitatively detected VWF antigen and its cleavage by ADAMTS-13, simultaneously.
Table 1. Demographic, diagnosis, and laboratory findings of patients enrolled in the study.
| ID* | Sex | Etiology** | Hb (g/dl) | PLT (×103/μl) | PT | Albumin (g/dl) | Creatinine (mg/dl) | WBC (×103/μl) | Bilirubin total (mg/dl) | Bilirubin indirect (mg/dl) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | M | NICMP | 16 | 122 | 11.6 | 3.2 | 0.84 | 5.3 | 0.4 | 0.2 |
| 2 | F | NICMP | 10.4 | 228 | 20.9 | 3.8 | 1.54 | 6.5 | 0.8 | 0.4 |
| 4 | F | NICMP | 10.9 | 234 | 9.9 | 3.5 | 1.51 | 11.6 | 1.5 | 1 |
| 6 | M | ICMP | 13.5 | 264 | 12 | 4.1 | 1.13 | 5.1 | 0.5 | 0.2 |
| 7 | F | NICMP | 12.1 | 317 | 24 | 3.8 | 0.63 | 8.9 | 1.4 | 0.8 |
| 8 | F | NICMP | 11.6 | 208 | 11.5 | 4.2 | 1.08 | 9.8 | 1.9 | 0.7 |
| 9 | M | ICMP | 9.3 | 208 | 12 | 3.4 | 1.73 | 6.2 | 0.5 | 0.3 |
| 10 | M | ICMP | 13.6 | 92 | 11.2 | 4.3 | 1.23 | 4.8 | 0.6 | 0.2 |
| 11 | M | ICMP | 12.8 | 284 | 12.3 | 3.5 | 0.95 | 12.9 | 0.7 | 0.3 |
| 14 | M | NICMP | 8.8 | 219 | 11.3 | 3.8 | 1.44 | 5.9 | 0.8 | 0.4 |
| 17 | F | NICMP | 12.7 | 185 | 21 | 3.8 | 1.35 | 7.7 | 1.2 | 0.4 |
| 18 | M | ICMP | 12.6 | 623 | 10.7 | 3.9 | 1.17 | 14.6 | 0.4 | 0.1 |
| 19 | F | ICMP | 7.4 | 235 | 10.8 | 4.1 | 1.34 | 7.4 | 0.6 | 0.3 |
| 20 | F | NICMP | 3.7 | 144 | 11.9 | 2.9 | 0.56 | 3.7 | 2 | 1.1 |
Patient 2, 5,11 were Coumadin at the time of enrolment for pre-existing conditions (i.e. Atrial fibrillation)
NICMP = non ischemic cardiomyopathy, ICMP= ischemic cardiomyopathy and all are baseline values prior to implant.
Clinical indexes reported have been selected as estimate of several organ functions, namely hemopoietic for Hemoglobin and WBC, hemostasis for PLT and PT, nutritional status for albumin, renal function for Creatinine, Bilirubin (total and indirect) as marker of baseline liver function and index of pre implant right sided dysfunction.
Figure 1. SRM detection of VWF cleavage by ADAMTS-13.
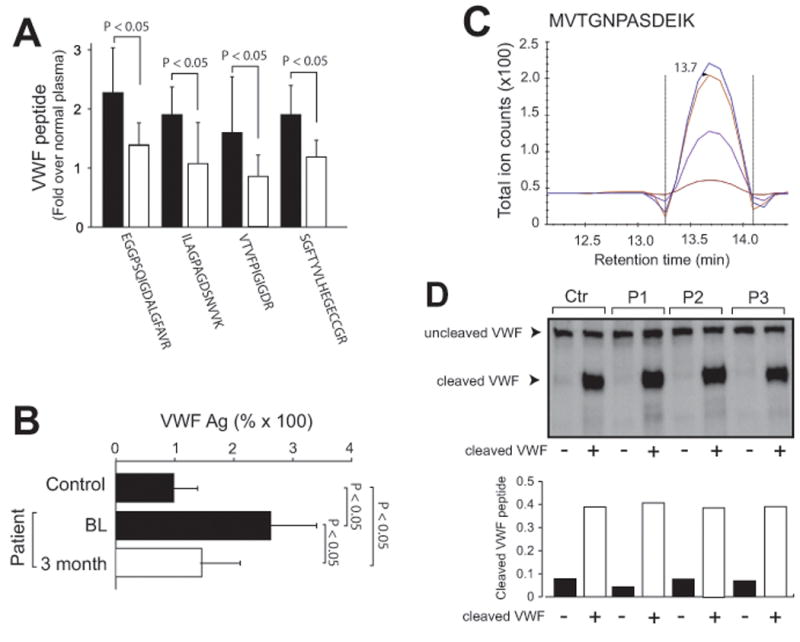
(A) Four VWF tryptic peptides detected by SRM in samples from patients at baseline before and 3 months after LVAD implantation (fold increase over normal plasma, paired t test, n = 15). (B) Plasma levels of VWF antigen in samples from healthy controls and from patients at the baseline before and 3 months after LVAD implantation (paired t test, n = 26). (C) A chromatogram of the oxidized M1606VTGNPASDEIK peptide detected in VWF immunoprecipitated from normal plasma (the four curves are explained in sFigure 1). (D) Top panel: immunoblots of VWF in plasma from three patients (P) and one control subject (Ctr) before and after 400 nM of cleaved VWF was added and bottom panel: the mass spectrometry detection of cleaved VWF in the same samples shown in the top panel.
Using this newly established SRM, we determined that the plasma levels of the cleaved M1606VTGNPASDEIK peptide in samples collected 3 months after LVAD implantation varied significantly, but were mostly below the level of normal plasma pooled from 61 healthy subjects (Figure 2A). To adjust for high VWF antigen in these patients (Figure 1B), we defined VWF cleavage as the ratio of M1606VTGNPASDEIK to ILAGPAGDSNVVK. This rate varied significantly in the post-LVAD samples, accounting for 0.23% to 2.5% of total VWF as compared to 1.26 ± 0.36% in normal plasma, even though the loss of large VWF multimers was found in 91.3% of patients as we have recently reported.15 The findings suggest that 1) circulating VWF was minimally cleaved in normal subjects and 2) there was no a drastic increase in VWF cleavage in post-LVAD samples. These findings raise a possibility that VWF was cleaved by ADAMTS-13 primarily to be released from its anchorage to endothelial cells as we previously shown.17 While further experiments are required to determine whether such small changes in VWF cleavage are sufficient to alter VWF reactivity, a subgroup analysis did find that VWF cleavage in post-LVAD samples was increased in patients who developed bleeding and was mostly reduced in patients who developed thrombosis (Figure 2B). Because this study was primarily focused on developing a quantitative method to measure VWF cleavage, it had a limited number of patients. This small patient cohort is not powered to determine the association of VWF cleavage with the development of hemostatic complications in patients on LVAD support.
Figure 2. Subgroup analyses of VWF cleavage in patients on LVAD support.
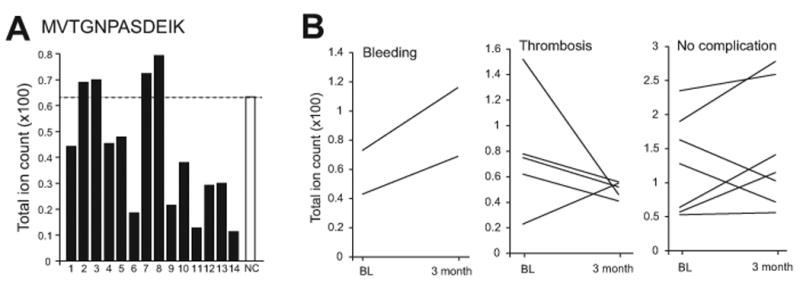
(A) Levels of the tryptic M1606VTGNPASDEIK peptide in plasma samples collected from 14 patients 3 months after LVAD implantation (NC: normal pooled plasma). (B) Changes in the rate of VWF cleavage before and after LVAD implants in patients with bleeding, thrombosis and no complications.
In summary, we have developed a reliable mass spectrometry-based method to quantify VWF cleavage by ADAMTS-13 in plasma. We made four novel observations. First, we provide first quantitative evidence that cleaved VWF accounts for less than 2% of total VWF in circulation. This finding suggests the multimer patterns of plasma VWF are not entirely resulted from ADAMTS-13 cleavage, but could also attribute to the redox regulation of the intra-multimer disulfide bounds as previously reported.18-20 Second, VWF cleavage was reduced in 71.4% of heart failure patients before LVAD implantation, consistent with reduced ADAMTS-13 activity in patients with myocardial infarction and inflammation.21 Third, VWF cleavage was not uniformly increased (as has been widely speculated), but rather varied significantly among patients on LVAD supports, even though AVWS is found in > 90% of patients. VWF cleavage patterns appear to correlate with adverse events in which increased VWF cleavage mirrors bleeding, whereas decreased cleavage is more common in patients with thrombotic complications. These findings, together with our recent report,15 suggest that the LVAD-associated AVWS can be caused not only by increasing cleavage (loss-of-function), but also by shear-induced VWF binding to platelets, and potentially to endothelial cells (gain-of-function).15 While both processes can result in the loss of large VWF multimers, they may require different treatments. The enhancement of VWF cleavage may be more effective for patients with the gain-of-function AWAS, whereas blocking VWF cleavage may be necessary for patients with the loss-of-function AWAS. This SRM method can therefore be used to define the relationship among VWF cleavage, AVWS and hemostatic complications in patients on LVAD support.
Supplementary Material
Table 2. VWF cleavage in plasma of patients on LVAD support.
| Patient | Outcome | Cleaved VWF (% of total) | |
|---|---|---|---|
|
| |||
| Baseline | 3 month | ||
| 1 | GI Bleeding | 0.43 | 0.69 |
| 2 | GI bleeding | 0.73 | 1.16 |
|
| |||
| 3 | No complication | 0.57 | 1.15 |
| 4 | No complication | 1.28 | 0.72 |
| 5 | No complication | 0.64 | 1.41 |
| 6 | No complication | 0.53 | 0.56 |
|
| |||
| 7 | Stroke | 0.23 | 0.35 |
| 8 | Stroke | 0.78 | 0.56 |
| 9 | Aortic thrombosis/NSTEMI** | 1.52 | 0.46 |
|
| |||
| 10 | No complication | 1.63 | 1.03 |
| 11 | No complication | 1.90 | 2.78 |
|
| |||
| 12 | Pump thrombosis | 2.20 | 1.61 |
|
| |||
| 13 | No complication | 2.35 | 2.59 |
|
| |||
| 14 | Stroke | 0.75 | 0.52 |
|
| |||
| Ctr* | 1.26 | ||
Pooled plasma from 61 healthy controls.
NSTEMI: non-ST segment elevation myocardial infarction
Footnotes
Conflict of Interests: The authors claim no conflict of interests related to this manuscript
Author contributions: Yong Zhou: performed mass spectrometry analyses of patient samples
Shizhen Qin: performed mass spectrometry analyses of patient samples
Tristan Hilton: performed experiments and analyzed data
Li Tang: performed mass spectrometry analyses of patient samples
Miguel Cruz: constructed and generated VWF A1-3 domain polypeptide
Joel L. Moake: edited the manuscript
O.H. Frazier: directed patient recruitments and wrote the manuscript
Qiang Tian: designed experiments
Jing-fei Dong: formulated hypothesis, designed the study, analyzed data, and wrote the manuscript
Angelo Nascimbene: design the study, recruited patients, performed clinical analyses, and wrote the manuscript
This work is supported by NIH grants HL71895, HL085769 and HL125957.
References
- 1.Letsou GV, Shah N, Gregoric ID, Myers TJ, Delgado R, Frazier OH. Gastrointestinal bleeding from arteriovenous malformations in patients supported by the Jarvik 2000 axial-flow left ventricular assist device. J Heart Lung Transplant. 2005;24(1):105–109. doi: 10.1016/j.healun.2003.10.018. [DOI] [PubMed] [Google Scholar]
- 2.Demirozu ZT, Radovancevic R, Hochman LF, et al. Arteriovenous malformation and gastrointestinal bleeding in patients with the HeartMate II left ventricular assist device. J Heart Lung Transplant. 2011;30(8):849–853. doi: 10.1016/j.healun.2011.03.008. [DOI] [PubMed] [Google Scholar]
- 3.Meyer AL, Malehsa D, Bara C, et al. Acquired von Willebrand syndrome in patients with an axial flow left ventricular assist device. Circ Heart Fail. 2010;3(6):675–681. doi: 10.1161/CIRCHEARTFAILURE.109.877597. [DOI] [PubMed] [Google Scholar]
- 4.Davis ME, Haglund NA, Tricarico NM, Matafonov A, Gailani D, Maltais S. Immediate recovery of acquired von Willebrand syndrome after left ventricular assist device explantation: implications for heart transplantation. ASAIO J. 2015;61(1):e1–4. doi: 10.1097/MAT.0000000000000157. [DOI] [PubMed] [Google Scholar]
- 5.Meyer AL, Malehsa D, Budde U, Bara C, Haverich A, Strueber M. Acquired von Willebrand syndrome in patients with a centrifugal or axial continuous flow left ventricular assist device. JACC Heart Fail. 2014;2(2):141–145. doi: 10.1016/j.jchf.2013.10.008. [DOI] [PubMed] [Google Scholar]
- 6.Crow S, Chen D, Milano C, et al. Acquired von Willebrand syndrome in continuous-flow ventricular assist device recipients. Ann Thorac Surg. 2010;90(4):1263–1269. doi: 10.1016/j.athoracsur.2010.04.099. discussion 1269. [DOI] [PubMed] [Google Scholar]
- 7.Wu T, Lin J, Cruz MA, Dong JF, Zhu C. Force-induced cleavage of single VWFA1A2A3 tridomains by ADAMTS-13. Blood. 2010;115(2):370–378. doi: 10.1182/blood-2009-03-210369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsai HM, Sussman II, Nagel RL. Shear stress enhances the proteolysis of von Willebrand factor in normal plasma. Blood. 1994;83(8):2171–2179. [PubMed] [Google Scholar]
- 9.Zhang X, Halvorsen K, Zhang CZ, Wong WP, Springer TA. Mechanoenzymatic cleavage of the ultralarge vascular protein von Willebrand factor. Science. 2009;324(5932):1330–1334. doi: 10.1126/science.1170905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nascimbene A, Neelamegham S, Frazier OH, Moake JL, Dong JF. Acquired von Willebrand syndrome associated with left ventricular assist device. Blood. 2016;127(25):3133–3141. doi: 10.1182/blood-2015-10-636480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goda M, Jacobs S, Rega F, et al. Time course of acquired von Willebrand disease associated with two types of continuous-flow left ventricular assist devices: HeartMate II and CircuLite Synergy Pocket Micro-pump. J Heart Lung Transplant. 2013;32(5):539–545. doi: 10.1016/j.healun.2013.02.006. [DOI] [PubMed] [Google Scholar]
- 12.Moake JL, Turner NA, Stathopoulos NA, Nolasco L, Hellums JD. Shear-induced platelet aggregation can be mediated by vWF released from platelets, as well as by exogenous large or unusually large vWF multimers, requires adenosine diphosphate, and is resistant to aspirin. Blood. 1988;71(5):1366–1374. [PubMed] [Google Scholar]
- 13.Auton M, Sowa KE, Smith SM, Sedlak E, Vijayan KV, Cruz MA. Destabilization of the A1 domain in von Willebrand factor dissociates the A1A2A3 tri-domain and provokes spontaneous binding to glycoprotein Ibalpha and platelet activation under shear stress. JBiolChem. 2010;285(30):22831–22839. doi: 10.1074/jbc.M110.103358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choi H, Aboulfatova K, Pownall HJ, Cook R, Dong JF. Shear-induced disulfide bond formation regulates adhesion activity of von willebrand factor. JBiolChem. 2007;282(49):35604–35611. doi: 10.1074/jbc.M704047200. [DOI] [PubMed] [Google Scholar]
- 15.Nascimbene Angelo, Konkle Barbara A, Moake Joel L, Frazier OH, Dong Jing-fei. von Willebrand factor proteolysis by ADAMTS-13 in patients on left ventricular assist device support. Journal of Heart and Lung Transplantation. 2016 doi: 10.1016/j.healun.2017.01.010. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kusebauch U, Campbell DS, Deutsch EW, et al. Human SRMAtlas: A Resource of Targeted Assays to Quantify the Complete Human Proteome. Cell. 2016;166(3):766–778. doi: 10.1016/j.cell.2016.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dong JF, Moake JL, Nolasco L, et al. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood. 2002;100(12):4033–4039. doi: 10.1182/blood-2002-05-1401. [DOI] [PubMed] [Google Scholar]
- 18.Yeh HC, Zhou Z, Choi H, et al. Disulfide bond reduction of von Willebrand factor by ADAMTS-13. JThrombHaemost. 2010;8(12):2778–2788. doi: 10.1111/j.1538-7836.2010.04094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nolasco L, Nolasco J, Feng S, Afshar-Kharghan V, Moake J. Human complement factor H is a reductase for large soluble von Willebrand factor multimers--brief report. Arterioscler Thromb Vasc Biol. 2013;33(11):2524–2528. doi: 10.1161/ATVBAHA.113.302280. [DOI] [PubMed] [Google Scholar]
- 20.C J, F X, W Y, et al. Oxidative modification of von Willebrand factor by neutrophil oxidants inhibits its cleavage by ADAMTS13. Blood. 2009 doi: 10.1182/blood-2009-03-213967. inpress. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maino A, Siegerink B, Lotta LA, et al. Plasma ADAMTS-13 levels and the risk of myocardial infarction: an individual patient data meta-analysis. J Thromb Haemost. 2015;13(8):1396–1404. doi: 10.1111/jth.13032. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.