

Abstract
Mitochondria play fundamental roles in the regulation of life and death of eukaryotic cells. They mediate aerobic energy conversion through the oxidative phosphorylation (OXPHOS) system, and harbor and control the intrinsic pathway of apoptosis. As a descendant of a bacterial endosymbiont, mitochondria retain a vestige of their original genome (mtDNA), and its corresponding full gene expression machinery. Proteins encoded in the mtDNA, all components of the multimeric OXPHOS enzymes, are synthesized in specialized mitochondrial ribosomes (mitoribosomes). Mitoribosomes are therefore essential in the regulation of cellular respiration. Additionally, an increasing body of literature has been reporting an alternative role for several mitochondrial ribosomal proteins as apoptosis-inducing factors. No surprisingly, the expression of genes encoding for mitoribosomal proteins, mitoribosome assembly factors and mitochondrial translation factors is modified in numerous cancers, a trait that has been linked to tumorigenesis and metastasis. In this article, we will review the current knowledge regarding the dual function of mitoribosome components in protein synthesis and apoptosis and their association with cancer susceptibility and development. We will also highlight recent developments in targeting mitochondrial ribosomes for the treatment of cancer.
Keywords: Mitochondrial ribosomes, Cancer, Apoptosis, Therapy
1. Introduction
Mitochondria play central roles in cancer cell physiology [1]. These eukaryotic organelles perform numerous bioenergetic and biosynthetic functions. Among them, mitochondria house pathways ranging from fatty acid oxidation to the TCA cycle, the oxidative phosphorylation (OXPHOS) system and respiration, and from the synthesis of amino acids, lipids and nucleotides to the biosynthesis of heme and iron-sulfur clusters [2]. Despite the fact that functional mitochondria are essential for the cancer cell, mitochondrial physiology is distinct in cancer and non-malignant cells [2], what initially brought to the hypothesis by Otto Warburg that a decline in mitochondrial energy metabolism might lead to the development of cancer [3]. In addition to providing fuel for life, mitochondria perform fundamental roles in the regulation of cell death pathways [4]. Considering that most tumor cells are by definition resistant to apoptosis, mitochondria may be regarded as the master regulators of both cellular life and death.
As an endosymbiotic organelle, the mitochondrion has retained a vestige of its bacterial ancestor genome, the mitochondrial DNA (mtDNA). In humans, where mitochondria contain more than 1100 proteins, the mtDNA encodes for only 13 hydrophobic subunits of the OXPHOS system, as well as for two rRNAs and a set of 22tRNAs. The vast majority of proteins are therefore encoded in the nuclear genome, synthesized by cytoplasmic ribosomes and imported into mitochondria. The expression of mtDNA-encoded genes, which depends on an organelle-specific machinery, ends with the synthesis of the 13 proteins in mitochondrial ribosomes or mitoribosomes [5]. Mitoribosomes are macrostructures of dual genetic origin, formed by 3 mitoribosomal RNA components encoded in the mtDNA and 89 specific protein components encoded in the nuclear DNA. Mitoribosomes differ from their bacterial and cytoplasmic counterparts in their composition, structure and mechanistic intricacies [6–9]. Their high degree of specialization has resulted in accelerated evolution so that significant differences exist even among mitoribosomes from different species [5–9]. However, as remnants of their eubacterial origin, mitoribosomes are as sensitive to similar antibiotics as bacteria and have many conserved proteins and RNA moieties [5, 10, 11].
The past decade has revealed new roles for the mitochondrial translation machinery and mitoribosome proteins in apoptotic signaling and the regulation of cell proliferation [12, 13]. Therefore, it is not surprising that gene expression studies have found alterations in the levels of mitoribosome proteins or assembly factors associated with the development of cancer [14, 15]. As a consequence, therapeutic approaches targeting mitochondrial protein synthesis have emerged as new interventions to combat specific types of malignancies [16–18].
Following this line of argumentation, the current review highlights the mitochondrial ribosome components and functions that have been associated with the development of cancer, and the underlying pathways. After an overview of mitoribosome composition and structure, we first describe the extraribosomal role of mitoribosome proteins in cell death regulation. Next, we address the association of mitoribosome proteins with cancer and examine their potential as biomarkers. Finally, we provide an overview of the recent evidence denoting the reliance of some cancer cells on OXPHOS, which has suggested mitochondrial translation as a target for cancer therapy.
2. Mitoribosomes: structure, biogenesis and function
2.1. Mitochondrial protein synthesis
The mitochondrial translation system evolved from that of the bacterial ancestor of mitochondria. This is evident from the fact that the proteins and RNA domains of mitochondria and bacteria that contribute to decoding and peptide bond formation, explained below, share a high degree of similarity. Therefore, mitoribosomes are sensitive to antibiotics such as chloramphenicol and tetracyclines, a property that is being exploited in the treatment of some types of cancer. Translation factors are conserved, and several mitochondrial factors can even functionally replace their homologs in bacteria [19]. However, multiple substantial differences exist, including (i) deviations in the genetic code [20–22], (ii) changes in the actual process of translation [23, 24] and (iii) the formation of mitochondrial ribosomes that differ in structure and composition, not only in comparison to their bacterial relatives but also among different species [6–9].
The mitochondrion has a simplified decoding system that allows a reduced set of mitochondrial tRNAs to recognize all the codons [25, 26]. The 22 tRNAs required in human mitochondria are encoded in the mtDNA. On the contrary, all translation factors are encoded in the nuclear genome [23, 24]. Mitochondrial mRNAs lack Shine-Dalgarno elements that could facilitate their interaction with ribosomes, and it is unclear how translation initiation is accomplished. Mitochondria have homologs of bacterial initiation factors IF2 (the functional equivalent of bacterial IF1 and IF2 [27]) and IF3 [28–32]. In mammals, IF2-mt promotes the binding of the initiator formylmethionine-tRNA (fMet-tRNA) to the AUG codon at the ribosomal peptidyl-site [33]. IF3-mt encourages the dissociation of fMet-tRNA that has bound to a small subunit in the absence of a correctly loaded mRNA [34]. Following translation initiation, it is assumed that elongation of the peptide chain occurs through mechanisms almost identical to those in bacteria [35–37] and involves homologs of the elongation factors EF-Tu, EF-G and EF-Ts [35]. However, termination of translation and recycling show stunning deviations from the bacterial pathway [38–41]. For example, mammalian mitochondria utilize only one release factor, mtRF1a [42], to decode all stop codons [43, 44].
2.2. Mammalian mitoribosomes
In all living organisms, mature ribosomes are composed of two distinct subunits, the large subunit (LSU) and the small subunit (SSU). Whereas the LSU catalyzes the peptidyl-transferase reaction, the SSU provides the platform for mRNA binding and decoding. Mammalian mitoribosomes sediment as 55S particles composed of a 28S mt-SSU and a 39S mt-LSU (Figure 1). The mt-SSU is formed by a 12S rRNA and 29 mitoribosomal proteins (MRPs). The mt-LSU is formed by a 16S rRNA, a structural tRNA (tRNAVal in human cells) and 50 MRPs [35, 45–49]. The two rRNA components are transcribed from mtDNA genes. The tRNA, also encoded in the mtDNA, occupies the region in the central protuberance where the cytoplasmic and bacterial ribosomes contain a 5S rRNA.
Figure 1. Structure of mitochondrial ribosomes.

Cryo-EM structures of human 28S mt-SSU and 37S mt-LSU (PDB 3J9M). Individual proteins relevant to apoptosis and cancer, which are mentioned in the text, are depicted in different colors and labeled.
Concerning the MRPs (listed in Table 1), 16 of the 30 mt-SSU proteins and 28 of the 50 mt-LSU proteins are homologs of proteins present in E. coli while the rest are mitochondrion-specific proteins [7, 47, 49–51]. Moreover, most conserved proteins contain mitochondrion-specific N- or C-terminal extensions, whereas some RNA helices present in the bacterial ribosome have been lost. As a consequence, human 55S mitoribosomes differ from bacterial (70S) and cytoplasmic ribosomes (80S) in their lower RNA:protein ratio. The recent high-resolution cryo-EM reconstruction of the mammalian mitoribosome revealed that the MRP extensions predominantly interact with mitochondrion-specific proteins whereas only a few participate in filling the space of deleted rRNA. Most mitochondrion-specific proteins are peripherally distributed over the solvent-accessible surface forming clusters at the central protuberance, the L7/L12 stalk, and contiguous to the polypeptide exit site. Similar to the protein extensions, these MRPs occupy novel positions rather than compensate for the missing rRNA [47, 49, 50, 52, 53].
Table 1. Mitoribosome proteins, assembly factors and their expression in cancer.
Proteins under and over-represented in the indicated cancers tissues relative to corresponding normal tissue, collected from the “Protein Atlas” database (http://www.proteinatlas.org). The percentages and color code reflects the proportion of analyzed cancer tissues with lowered or elevated protein levels. The old nomenclature for mitochondrial ribosome proteins has been substituted by an unifying nomenclature [165, 166] where proteins with a prefix “u” (for universal) are present in all kingdoms of life, proteins with a prefix “b” are bacterial in origin and do not have a eukaryotic (or archaeal) homolog, and proteins with a prefix “m” are mitochondrion-specific.

|

|

|

|

|

|

|
Some mitochondrion-specific proteins serve to establish intersubunit bridges [7], a feature that is different from cytoplasmic ribosomes that typically contain RNA-RNA intersubunit connections. The number of intersubunit bridges is lower in mammalian mitoribosomes than in the bacterial ribosome, possibly to allow the two subunits to have flexibility in the conformation enabling them to tilt freely [52, 53]. Another major remodeling from the bacterial ribosome occurs at the aminoacyl and peptidyl-tRNA binding sites, where some proteins present in bacterial ribosomes (e.g. uL5 or bL25) have been lost to accommodate human tRNAs, which contain highly variable loops at the elbow. A unique property of mitoribosomes is the acquisition of an intrinsic GTPase activity through mt-SSU subunit mS29, a GTP-binding protein [10, 46, 48]. The GTPase activity is probably relevant to subunit association, given the localization of mS29 at the subunit interface, its involvement in coordinating two mitochondrion-specific intersubunit bridges, and the fact that the affinity for GTP is higher for mt-SSU 28S subunits than for the 55S monosome [10, 46, 48].
Mitoribosomes reside in the mitochondrial matrix and associate with the inner membrane to facilitate co-translational insertion of the nascent polypeptides into the inner membrane. In fact, the polypeptide exit tunnel is adapted to the transit of hydrophobic nascent peptides [47, 49]. The tunnel is formed by several conserved proteins that create a ring around the exit site, namely bL23m, bL29m, bL22m, bL24m and bL17m. This conserved core is surrounded by another protein layer consisting of bL33 and mL45, which promote anchoring of the mt-LSU to the inner membrane [46–49]. Another significant structural remodeling has been observed at the mRNA entrance of the mt-SSU [51–53], to accommodate mammalian mitochondrial mRNAs, which miss or have very short 5′-untranslated regions [54]. Since the structural data has disclosed the presence of mS39, a pentatricopeptide repeat (PPR) protein in the proximity of the channel entrance, it has been suggested that it could be involved in recruiting the leaderless mRNA during translation initiation [51–53].
2.3. Ribosome assembly
The assembly of ribosomes requires coordinated processing and modification of rRNAs with the temporal association of ribosomal proteins. In all systems, the process is regulated by several classes of ribosome biogenetic factors, including nucleases, rRNA-modifying enzymes, DEAD-box helicases, GTPases and chaperones [55–57]. Although the mitoribosome assembly pathway is still poorly characterized, several assembly factors have been already identified. They include rRNA modification enzymes (the 16S rRNA methyltransferases MRM1-3) and their co-factors [58–60]. Also, several poorly characterized conserved GTPases participate in the assembly of the mt-SSU (C4orf14) or the mt-LSU (MTG1 and MTG2) [61–63]. Unexpectedly, two mitochondrial transcription factor family proteins (mTERF3 and mTERF4) play roles in mt-LSU assembly, perhaps coordinating transcription and ribosome biogenesis. The precise role of mTERF3 (or mTERFD1) is not understood [64], but mTERF4 binds to the rRNA methyltransferase NSUN4 to promote its recruitment to the mt-LSU [65], possibly to facilitate monosome assembly [66]. A role in monosome formation has also been proposed for MPV17L2, a mtDNA maintenance factor [67], although its function remains intriguing. Two DEAD-box helicases, DDX28 and DHX30, participate in mt-LSU assembly [68, 69]. DDX28 interacts with the 16S rRNA and its yeast counterpart, termed Mrh4, catalyzes a late step of mt-LSU assembly required for the incorporation of proteins uL16 and bL33 to a large on-pathway intermediate [70]. FASTKD2, a member of the Fas-activated serine-threonine kinase family of proteins also binds to the 16S rRNA and participates in mt-LSU assembly [68]. Furthermore, C7orf30, a member of the DUF143 family of ribosomal silencing factors, interacts with uL14 and promotes its incorporation into the mt-LSU [71]. Finally, GRSF1 (G-rich sequence factor 1) is an RNA-binding protein that interacts with several mt-RNAs, including the 12S rRNA, and participates in mt-SSU biogenesis [72].
Mitoribosome assembly has been proposed to occur in contact with the inner membrane [70, 73] in a compartment located near the mtDNA nucleoid termed RNA granule or mitochondriolus [68, 69, 74]. The mitochondrioli contain ribosomal proteins, ribosomal RNA modifying enzymes, all the mitoribosome assembly factors listed above and a host of proteins involved in diverse aspects of mt-RNA metabolism [59, 68, 69, 72, 75]. The dynamics of these compartments in health and disease remains to be fully understood.
In eukaryotic systems, several post-transcriptional regulation events are highly linked and provide a powerful mechanism to control the fate of a cell, and therefore are of central importance in cancer research [76, 77]. Emerging data, summarized in the following sections, highlight the additional relevance of mitochondrial ribosome components and mitochondrial translation in controlling cellular proliferation in OXPHOS-dependent and independent manners, thus providing novel mechanisms for understanding tumorigenesis and novel targets for therapeutics design.
3. Mitoribosomal proteins and apoptosis
Mitochondria play crucial roles in the induction of apoptosis or programmed cell death [78]. This paradigm is central to malignant cellular transformation because altered mitochondrial function and defective apoptosis are well-known hallmarks of cancer cells. In fact, tumor initiation and progression involve the development by cancer cells of mechanisms to inhibit apoptosis at multiple stages. Recent investigations have uncovered apoptosis-regulatory roles of several protein components of the mammalian mitoribosome, which has opened the question of whether the mitoribosome, mitochondrial translation or moonlighting functions of these mitoribosome proteins could be involved in apoptotic signaling [7, 10, 12].
The caspase proteases that execute apoptosis can be activated through one of two signaling cascades responding to different signals: the extrinsic pathway, involving cell surface death-receptors (e.g. FASL, TRAIL and TNFα), and the intrinsic or mitochondrial pathway. The mitochondrial pathway can be induced by a variety of stimuli such as chemotherapeutic agents, serum/growth factor starvation, irradiation, DNA damage, free radicals and viral infections [78, 79]. These stimuli result in changes in the permeability of inner mitochondrial membrane owing to the opening of the mitochondrial permeability transition pore (MPT), which induces loss of the mitochondrial transmembrane potential, the release of pro-apoptotic proteins to the intermembrane space and disruption of mitochondrial functions. However, an essential event in mitochondria-mediated apoptosis is mitochondrial outer membrane permeabilization (MOMP), achieved through the formation of pores by pro-apoptotic members of the BCL-2 (B-cell lymphoma 2) family of proteins (e.g. Bax and Bak), in competition with anti-apoptotic members of the family (e.g. Bcl2 and Bcl-XL). The crucial event following MOMP is the release of mitochondrial pro-apoptotic proteins (cyctochrome c, Smac and Omi) to the cytosol, where they form the caspase-activating apoptosome (cytochrome c) or bind to inhibitors of apoptosis to relieve their inhibitory effects on caspase activity (e.g. Smac and Omi), resulting in the death of the cell [78, 79].
Cancer cells often escape apoptosis by shifting the balance between pro- and anti-apoptotic BCL-2 family proteins by inducing their down- and up-regulation, respectively, thereby preventing MOMP. In fact, the basis of effective cancer treatments such as chemotherapy highly depends on the levels of pro-apoptotic and anti-apoptotic BCL-2 proteins, because that balance determines the sensitivity of the tumor cells toward apoptosis [80]. Among the multiple regulators of the intrinsic pathway of apoptosis, recent studies have reported the involvement of three mitochondrial ribosomal proteins (mS29, mL41 and mL65), whose roles are discussed below.
3.1. mS29 (death-associated protein 3, DAP3)
As discussed in the previous section, mS29 is a GTP binding protein from the mt-SSU [81]. In the context of the mitoribosome (Figure 1), mS29 mediates contacts with intersubunit bridges, and its GTPase activity might fuel monosome formation [46, 48]. Additionally, a number of phosphorylation sites have been identified on mS29 that are in proximity to proteins forming the intersubunit bridges [82, 83]. Whether the phosphorylation status of mS29 might regulate mitoribosomal subunit association remains to be discovered.
mS29 was first discovered as a member of the death-associated protein (DAP) family and termed DAP3 [81, 84], before being identified as a mitoribosome protein [85]. mS29/DAP3 is known as one of the major positive mediators of apoptosis [86]. Mutations in a highly conserved P-loop motif significantly reduces DAP3-induced cell death, and overexpression of DAP3 enhances apoptosis [87]. However, the precise mechanism/s by which DAP3 induces apoptosis remain to be fully understood. mS29/DAP3 has been found outside mitochondria to initiate the extrinsic apoptotic pathway through its interactions with apoptotic factors such as Fas ligand, tumor necrosis factor-alpha (TNF-α), and interferon-gamma (IFNγ) [86, 88–90]. Nevertheless, additional studies have shown that once activated mS29/DAP3 co-localizes and strongly associates with Fas-associated death receptor (FADD) (Figure 2) [91]. Additionally, DAP3 interacts with the factor IPS-1 (interferon-β promoter stimulator 1) to activate caspases 3, 8, and 9, resulting in a type of extracellular apoptosis known as anoikis [88, 91]. However, contrasting evidence has suggested a role for DAP3 in the mitochondrial apoptotic pathway and indicated DAP3-independent Fas-induced and TRAIL (tumor necrosis factor-related apoptosis inducing ligand)-induced apoptosis [92–94]. Moreover, mS29/DAP3 induces mitochondria-mediated apoptosis of a human laryngeal carcinoma cell line (Hep2 cells) through the activation of p38 MAPK and JNK signaling [95, 96]. The intrinsic pro-apoptotic role of DAP3 is further highlighted by the fact that overexpression of human, mouse or yeast DAP3 leads to cell death resulting in mitochondrial fragmentation, perhaps affecting mitochondrial fission [94, 97] in a manner dependent on the GTP-binding ability of DAP3 and its mitochondrial localization [94].
Figure 2. Involvement of mitoribosome subunit mS29/DAP3 in activating the extrinsic and intrinsic pathways of apoptosis.

The schematic portraits the components and major events of the extrinsic and mitochondrial apoptotic pathways. The extrinsic pathway or the death receptor pathway is activated by the binding of death receptors to their ligands such as FASL (FAS ligand), TNFa, (tumor necrosis factor) or TRAIL (TNF-related apoptosis-inducing ligand). This promotes the recruitment of adaptor proteins such as FAS-associated death domain protein (FADD) that bind to death effector domain containing caspase −8 or −10. Formation of this complex results in dimerization and activation of caspase −8 that can induce two different pathways to apoptosis; the first one is the direct cleavage and activation of executioner caspases −3, −6 or −7. The other one is the cleavage and activation of the BH3-interacting domain death agonist (BID), the truncated product of which is tBID. tBID leads to the interaction of BCL-2-associated X protein (BAX) and BCL-2 antagonist or killer (BAK) and induction mitochondrial outer membrane permeability (MOMP), thereby generating a crosstalk between the extrinsic and intrinsic apoptotic pathways.
The intrinsic or mitochondrial pathway is initiated by different apoptotic stimuli or environmental stresses such as DNA damage, ROS, endoplasmic reticulum (ER) stress, irradiation, which activate B-cell lymphoma 2 (BCL-2) homology 3 (BH3)-only proteins leading to translocation of BAX and BAK to mitochondria and induction of MOMP. Anti-apoptotic BCL-2 proteins prevent MOMP by binding to BH3-only proteins, tBID and activated BAX or BAK. MOMP induces the release of cytochrome c from the mitochondrial intermembrane space (IMS) to the cytosol that promotes caspase activation and apoptosis. Cytochrome c binds to the apoptotic protease-activating factor 1 (APAF1) and promotes the formation of the apoptosome that subsequently recruits and activates the initiator caspase, caspase 9. Caspase 9 further cleaves and activates executioner caspases −3, −6 or −7 leading to apoptotic cell death. Other proapoptotic proteins that are released following MOMP are second mitochondria-derived activator of caspase (SMAC; also known as DIABLO) and OMI (also known as HTRA2) that interacts with the X-linked inhibitor of apoptosis protein (XIAP) to prevent its inhibitory function on caspase −9 and −3.
mS29 (death-associated protein 3, DAP3) is depicted in the figure as an example of one of the three mitoribosomal subunits involved in apoptosis. mS29/DAP3 is predicted to regulate mitochondrial translation and apoptosis in various ways. mS29/DAP3 mediate contact with intersubunit bridges thereby assisting in monosomes formation and mito translation. mS29/DAP3 is also involved in mitochondrial dynamics by regulating the phosphorylation of DRP1 (dynamin-related GTPase involved in mitochondrial fission). In addition, mS29/DAP3 can initiate the extrinsic apoptotic pathway through its interactions with ligands of death receptors such as FASL, TRAIL and TNFα. mS29/DAP3 also co-localizes with FADD and participates in the formation of Death-Inducing Signaling Complex (DISC), which exists in the cell in an inactive form by the action of protein kinase B (AKT/PKB). DAP3 also interacts with FADD through the involvement of interferon-β promoter stimulator 1 (IPS1), which is a caspase activation and recruitment domain (CARD) bearing protein, present in the mitochondrial outer membrane. This complex further recruits procaspase-8, thereby triggering further downstream cleavage and activation of caspase resulting in cell death. mS29/DAP3 induces mitochondria-mediated apoptosis through the activation of p38 MAPK and the JNK signaling pathway.
Two other pro-apoptotic mitoribosome LSU proteins, mL41/BMRP that interacts with 16S rRNA and associates with mL65/PDCD9, have been also being depicted in the schematic. However, their pro-apoptotic mechanism/s of action remain to be unveiled.
DAP3 is essential for life, and its knockout in embryos is lethal, resulting in abnormal, shrunken mitochondria with swollen cristae [98]. This study showed DAP3 as a bifunctional protein involved in mitochondrial protein synthesis and respiration and in the extrinsic pathway of apoptosis [98]. It is important to notice that although mS29/DAP3 localizes to the mitochondrial matrix, it is not released to the cytoplasm during induction of apoptosis [94, 97], indicating the existence of an extramitochondrial pool of mS29 to induce the extrinsic apoptotic pathway [91]. Nonetheless, the apoptotic activity of DAP3 in this compartment can be inhibited by AKT (PKB) phosphorylation [88, 91]. These observations suggest different levels of regulation of DAP3 in apoptosis by gene expression, protein localization and phosphorylation. However, the mechanism underlying the dual extraribosomal functions of DAP3 in mitochondrial physiology and cell death regulation remains unclear. In agreement with the DAP3 overexpression experiments mentioned earlier, a recent study has suggested a role of DAP3 in maintaining balanced mitochondrial dynamics (fusion and fission) by regulating the phosphorylation of Drp1 (dynamin-related GTPase involved in mitochondrial fission) [99]. Additionally, it was shown that depleting DAP3 inhibits autophagy thereby sensitizing the cells to intrinsic death stimuli [99]. Despite these efforts, it remains unclear whether the role of mS29/DAP3 in the regulation of mitochondrial translation is coupled to its role in the intrinsic apoptosis pathway.
3.2. mL41 (BCL-2 interacting mitochondrial ribosomal protein BMRP)
In the structure of the mt-LSU subunit, mL41 assumes a mostly extended conformation (Figure 1), and interacts with the 16S rRNA and several surrounding mitoribosomal proteins, including another pro-apoptotic protein, mL65 [49].
Beyond its role in mitochondrial protein synthesis, mL41 has been found to suppress the growth of cancer cells in nude mice, possibly by induction of p53-induced mitochondrion-dependent apoptosis [100]. In short, when p53 translocates to the mitochondria in response to cell death signals, mL41 would stabilize p53 and enhance its local abundance - thereby contributing to p53-induced apoptosis in response to inhibition of cellular proliferation. In addition, in the absence of p53, mL41 stabilizes the p27Kip1 protein and p21WAF1/CIP1, a cyclin-dependent kinase inhibitor, and arrests the cell cycle at the G1 phase [100, 101], suggesting that mL41 is a negative regulator of cell cycle and plays a strong tumor-suppressive role associated with p53, p27Kip1 and p21WAF1/CIP1 [100]. However, the mechanism/by which mL41 increases the stability of p53, p27 and p21 remains unknown.
mL41 has also been named BMRP (BCL-2 interacting mitochondrial ribosomal protein) [102], based on the observation that when overexpressed in human cells, mL41 induces cell death which is counteracted by BCL-2. mL41/BMRP-induced cell death was also repressed by p35, a caspase inhibitor, further suggesting its involvement in the regulation of apoptosis via its interaction with anti-apoptotic members of the BCL-2 family of proteins. Most probably, this interaction/s do not occur with mL41 embedded within the mt-LSU structure [47, 49]. In fact, the BCL-2 binding sites have been found near the N-terminus of mL41 [103, 104]. They correspond to motifs that are either absent in the mature protein owing to the cleavage of the mitochondrial targeting sequence after its import into mitochondria, or might be buried within the 16S rRNA - thereby making it inaccessible for interaction with BCL-2. Therefore, the interaction of mL41 with BCL-2 and its apoptotic activity are likely to occur in the cytosol before its import into mitochondrial and incorporation in mitoribosomes [10, 104]. Further studies are needed to unravel the mechanisms by which the mL41 precursor induces apoptosis, and those that control mL41 processing in non-malignant and cancer cells.
3.3. mL65 (programmed cell death protein 9, PDCD9)
Initially known as MRPS30 and considered to be a mt-SSU protein, structural studies found mS30 to be present as a single copy in the mt-LSU and was renamed mL65. In the mt-LSU, mL65 has close contacts with its homologous protein mL37 [47, 49, 105] and also Cα-Cα crosslinks with pro-apoptotic mL41 [49], as mentioned earlier.
mL65 has homology to the chicken pro-apoptotic protein p52 which, when overexpressed in mouse fibroblasts, induces apoptosis, upregulation of transcription factor c-Jun and activation of c-Jun N-terminal kinase 1 (JNK1), through a pathway distinct from the extrinsic death receptor-induced apoptosis [13]. The human counterpart of p52 was termed PDCD9 (programmed cell death 9), and the corresponding gene mapped to chromosome 5q11 [106]. However, little is known about the molecular mechanisms underlying the pro-apoptotic roles of mL65. Future studies should focus on disclosing whether mL65 acts independently or in conjunction with the other pro-apoptotic mitoribosomal proteins, particularly its interactor mL41.
4. Mitoribosome components as biomarkers and their association with cancer
Advances in tumor and cancer cell genomics and proteomics are providing remarkable insights into the genetic and metabolic basis of most cancers. The identification of novel mutations, epigenetic dysregulation, and aberrant gene expression or protein abundance patterns has yielded deep insights into specific cancer pathogeneses [15, 107, 108]. Genetic, genomic and proteomic variations may serve as markers for diagnostics and in some cases, when a causal relationship is established, as therapeutic targets as well. These studies have uncovered the value of mitoribosome components, mitoribosome assembly factors and mitochondrial translation function as biomarkers that could reflect the molecular functional profile of specific tumors [15, 109, 110]. This is not that surprising, considering the involvement of mitoribosome proteins in the induction of apoptotic pathways, described earlier.
In this section, we will describe the combination of results obtained from candidate gene studies (on mS29, mL41 and mL65), genome-wide association studies (GWAS) and analysis of the cancer proteome (http://www.proteinatlas.org/humanproteome/cancer), which are shedding light onto the contribution of mitoribosome composition and function to the transformation of normal cells to tumorigenic. For example, a GWAS study on human breast cancer cells yielded significantly increased transcripts from more than 90 genes that are associated with mitochondrial biogenesis and/or mitochondrial translation, nearly 40 of them being MRPs [110]. Also, immunohistochemical analysis revealed that antibodies directed against 15 markers of mitochondrial biogenesis and/or mitochondrial translation selectively labeled epithelial breast cancer cells and that the signal was extremely lowered or excluded from adjacent tumor stromal cells. These were a regulator of mitochondrial function through cAMP signaling (AKAP1), mitochondrial lipid biosynthesis markers (GOLPH3 and GOLPH3L), a transporter that regulates the uptake of high-energy mitochondrial fuels (MCT1), mitoribosome proteins (mL40, uS7m, uS15m, and mS22), nuclear transcription factors that regulate mitochondrial biogenesis (NRF1, NRF2, PGC1-α), mtDNA metabolism (POLRMT, TFAM) and components of the mitochondrial import machinery (TIMM9 and TOMM70A). These results suggested that human breast cancers contain two distinct metabolic compartments, namely mitochondria-rich epithelial cancer cells and their surrounding glycolytic tumor stroma cells, which would establish a metabolic symbiosis.
Protein profiling using immunohistochemistry has allowed the Human Protein Atlas Project for the assessment of the distribution and relative abundance of proteins in tumor tissues. We have presented in Table 1 an evaluation of the differential expression of mitoribosome proteins and assembly factors between each of ten forms of cancer and their corresponding normal tissue counterparts. As an overview, our analysis shows that certain proteins have enhanced abundance in most tumors, including uL10m, mL38, mL52, uS2m, bS18m, mS27, NSUN4 and TFB1m, although the latter is specifically attenuated in breast cancer. On the contrary, another group of proteins has decreased abundance in most tumors, including bL17m, bL33m, bS6m (although elevated in liver cancer) and C7Orf30 (although elevated in thyroid cancer). From a tumor perspective, some display a general decrease in the levels of all proteins, such as colon carcinomas and particularly renal tumors, suggesting that mitochondrial OXPHOS might have limited relevance in these cancer cells. On the contrary, liver tumors display a general increase in mitoribosome-related proteins, as it occurs in melanomas, although in this case, a few proteins (uL18m, uL24m, bL27m, mL42, bS6m, mS31) have decreased abundance. Finally, limited changes were observed between lung tumors and normal lung tissue because only the levels of a few proteins (uL11m, bL19m, bL33m and uS11m) were found decreased. As a conclusion, our analysis shows differential mitoribosome protein abundance among different forms of cancer, but also highlights some potential tumor biomarkers. Below, we will discuss the published evidence on the potential of individual mitoribosomal proteins as cancer diagnostic markers based on GWAS and proteomics studies.
4.1. uL11m
In the mitoribosome structure, uL11m is located in the L7/L12 stalk and is involved in the binding and recruitment of translation factors to support mitochondrial translation [50]. uL11m was considered a potential cancer biomarker when tissues from patients suffering from head and neck squamous cell carcinoma (HSNCC) presented altered expression of OXPHOS complexes and defective mitochondrial translation over the progression of HSNCC, associated with a decrease in MRPL11 expression [14]. This observation was in agreement with microarray analyses of HNSCC primary tumor tissues and cell lines and HN cancer cell lines showing that transcription of mitoribosomal genes MRPL11 and MRPL21 were aberrantly expressed in squamous cell carcinoma tissues [111]. In addition, expression of MRPL11 was different between the node positive and negative tumors. The prognostic genes were clustered into two groups based on their metastatic phenotypes, and MRPL11 and MRPS23 were grouped based their association with rapid proliferation, oxidative phosphorylation, invasiveness, and tumor size [112]. In yeast, the cytoplasmic ribosomal homolog RP11 binds and suppresses the E3 ubiquitin-protein ligase function of HDM2 and leads to the stabilization and activation of the p53 tumor suppressor protein [113, 114] under conditions of growth inhibition, DNA damage and oncogenic insults [115]. It is currently unclear whether there is any functional interaction between cytoplasmic and mitoribosomal L11 in the context of cancer.
4.2. bL20m
The expression level of MRPL20 could have a potential role in androgen resistance since it was observed significantly downregulated in androgen-independent prostate cancers [116]. Prostate cancer accounts for approximately one-third of all cancer diagnosed in men in the United States, and the survival and growth of prostate cancer cells are initially dependent on the presence of androgens. In the first stages, prostate cancer treatment based on androgen ablation works for almost all patients. However, when prostate cancers become resistant to hormone blockade, they turn into highly aggressive and metastatic cancers, which are clinically defined as androgen-independent (AIPC). Best et al. [116] analyzed the differentially expressed genes in tumor biopsies between 10 androgen-independent tumors and 10 primary and untreated prostate tumors. The 239 genes whose expression levels were statistically different were divided into two distinct groups; genes associated with ribosomes and protein synthesis and genes related to cell adhesion and the extracellular matrix. The first group included MRPL20, with a 0.65-fold decreased expression in AIPC.
4.3. uL16m and uL21m
In partial agreement with our analysis presented in Table 1, the expression of several MRP genes was found to be a potential prognostic marker in colorectal tumorigenesis and tumor growth [117]. Analysis of gene expression profiles in sporadic colorectal cancer biopsies identified 7 new differentially expressed genes that included MRPL21 and MRPL16, which were particularly upregulated in synchronous adenoma. Tumor location was the dominant factor influencing differential gene expression caused by canonical molecular changes [117], suggesting that location-specific analysis could identify location-associated pathways (OXPHOS, in the location where MRPL21 and MRPL16 expression were augmented) and enhance the accuracy of tumor class prediction.
4.4. bL12m and bL28m
Mitochondrial functions in metabolism and apoptosis are strongly associated with the altered metabolic profile of pancreatic cancer cells through differential expression of genes such as MRPL28 [118]. Knockdown of MRPL28 in pancreatic tumor cells results in decreased mitochondrial activity and increased glycolysis, accompanied by reduced cellular proliferation. In an interesting experiment, these MRPL28-silenced cells were injected into a nude mouse, which developed tumors with approximately 2-3-fold increased growth compared to non-silenced cells. However, this effect was not specific for MRPL28, because injection of nude mice with pancreatic cancer cells silenced for MRPL12 or COX4 (cytochrome c oxidase subunit 4) reproduced these phenotypes [118], suggesting that alteration in mitochondrial metabolic profiles, mainly OXPHOS, is central to the progression of pancreatic tumors.
4.5. bL36m
The association of bL36m with cancer is indirect and poorly characterized. It is based on the physical interaction of this protein with the mitochondrial inner membrane protein LETM1 (leucine zipper/EF-hand-containing transmembrane-1). It has been suggested that LETM1 acts as an anchor protein for complex formation with the mitoribosome via bL36m to regulate mitochondrial biogenesis [119]. LETM1 was first identified in association with Wolf-Hirschhorn syndrome, being deleted in most patients with the syndrome [119]. LETM1 overexpression can induce necrotic cell death in HeLa cells, and LETM1 levels have been suggested to correlate with multiple human malignancies and tumor progression. However, the pathological relevance of different expression levels in LETM1 may be tumor cell-dependent. LETM1 expression was found elevated in breast cancers [120]. Yet, in a mouse model of human non-small cell lung cancer, adenovirus-mediated LETM1 overexpression in lung cancer cells induced mitochondrial degeneration, ATP depletion and cell cycle inhibition while facilitating apoptosis, ultimately resulting in the suppression of lung cancer cell growth in vitro and in vivo [121]. Furthermore, the involvement of bL36m remains intriguing, particularly considering the data presented in Table 1, showing that bL36m protein levels are elevated in liver tumors and melanomas, whereas they are decreased in breast, colon and pancreatic tumors and unaltered in lung cancers.
4.6. mL37
The possible involvement of mL37 in cell transformation and tumor cell proliferation is supported by two observations. Treatment of human lymphocytes with concanavalin A, a mitogenic activator of cytokines, which stimulates cell proliferation and can induce apoptosis, resulted in specific overexpression of MRPL37 [105]. MRPL37 mRNA levels were also highly elevated in different lymphoma human tissues and cell lines. Notably, mL37 shares 45% homology with the proapoptotic mitoribosomal protein mL65/PDCD9. The relevance of the homologous region in these to their potential properties in apoptosis and tumorigenesis deserves further investigation.
4.7. mL38
Gene expression profiling of mouse precursor T-cell lymphoblastic lymphoma/leukemia (pre-T LBL) identified MRPL38 gene expression as an oncogenic pathway indicator and a prognostic marker [122]. MRPL38 was one of the two genes found to be overexpressed more than four-fold in thymic tumors from clinically-sick transgenic mice generated by overexpression of SCL, LMO1 or NHD13, three known pre-T LBL genes [122]. The relevance of mL38 to cancer prognosis was also highlighted in another study aiming to explore the alterations of mitochondrial protein abundance in ovarian cancer, by comparing the mitochondrial proteomics profile of a pair of human ovarian carcinoma cell lines (SKOV3/SKOV3.ip1) with different metastatic potentials [123]. SKOV3.ip1 cells, with higher migration potential, were derived from ascitic tumor cells of nude mice bearing a tumor of SKOV3 ovarian cancer cells. mL38 was one of the four proteins found to be more abundant in SKOV3.ip1 cells, and therefore positively correlated with the aggressiveness of the tumor. Consistently with these investigations, mL38 abundance has been found highly increased in most tumor types (Table 1).
4.8. mL41
Consistent with its role in promoting p53-induced apoptosis, MRPL41 expression was found lowered or absent in most tumor tissues and cell lines [100]. This is particularly true in colon cancers and lymphomas (Table 1).
4.9. mL49
Oncocytic neoplasms or oncocytomas are tumors composed of cells characterized by an aberrant proliferation of mitochondria that is responsible for their ultrastructural alterations [124]. A microarray analysis carried out on thyroid oncocytomas revealed 126 genes that were found overexpressed in thyroid, parathyroid and renal oncocytic tumors compared with normal tissue samples. The gene cluster included MRPL49 and 13 genes coding for subunits of the OXPHOS complexes [125]. In the same line, a genome-wide transcriptional profiling analysis in human breast cancer cells also reported increased levels of the MRPL49 transcript [110]. These data are in agreement with the mitoribosomal protein expression profiles presented in Table 1, where mL49 abundance was found low in colon, renal and pancreatic tumors, and elevated in melanoma, thyroid and breast tumors.
4.10. mL64
Fist known as CR6-interacting factor 1 (CRIF1), this protein has been found in mitochondria and the nucleus. Consistent with a role as a tumor suppressor, CRIF1 interacts with and inhibits CDK2 thereby inducing cell cycle arrest in leukemia cells [126]. CRIF1-induced cell cycle arrest can also be mediated though its interaction with other nuclear proteins, including GADD45 family of proteins [127]. Similar to the tumor suppressor role of nuclear CRIF1, its depletion promotes leukemic T cell survival in the absence of growth factors [128]. CRIF1 also binds to the lymphocyte-specific protein tyrosine kinase (Lck), whose overexpression and hyperactivation have been associated with leukemia development. In a model, Lck would bind to CRIF1 and inhibit its function as a tumor suppressor [128]. Thus, the available data suggest that CRIF1 may play a role in negative regulation of cell cycle progression and cell growth. Expression of CRIF1 is hardly detectable in adrenal adenoma and papillary thyroid cancer and markedly lower than in adjacent normal tissue [127]. It remains to be determined how CDK2, Lck, GADD45 and other nuclear proteins compete in binding to CRIF1 and regulate its tumor suppressor activity. The role of CRIF1 in mitochondrial protein synthesis as mL64 may also be relevant to blood tumors that are highly dependent on OXPHOS, since, as explained in the next section, inhibition of mitochondrial translation has been identified as a potent therapeutic strategy for acute myeloid leukemia in human patients [129].
4.11. mL65
mL65/MRPS30 was first discovered as programmed cell death protein 9 or PDCD9. A study supports MRPS30 gene expression as a potential diagnostic marker to detect estrogen-responsive (ER) breast tumors. When searching for loci linked to gene expression in breast adenocarcinoma tissues by using a gene expression Quantitative Trait Locus (eQTL) approach, a single nucleotide polymorphism (SNP) on chromosome 5p12 (rs7716600), previously found to be specifically linked to ER-positive breast tumor susceptibility in several GWAS, was significantly associated with elevated expression of the nearby gene MRPS30 in ER-positive tumors [130]. The rs7716600 risk genotype was associated with decreased MRPS30 promoter methylation exclusively in ER-positive breast tumors. Based on their studies, the authors suggested that the 5p12 risk allele affects mS30 gene expression in ER tumor cells after tumor initiation by a mechanism affecting chromatin availability [130].
4.12. mS40 (formerly MRPS18B or S18-2)
When overexpressed, mS40/S18-2 binds to retinoblastoma protein (RB) and prevents the formation of the E2F1-R transcriptional complex [131]. The resulting elevated free E2F1 proteins in the nucleus subsequently promote S-phase entry and cell proliferation [131]. Also, overexpression of S18-2 in primary rat skin fibroblast induced malignant transformation and resulted in their immortalization. In human tumors, mS40 levels are not uniformly increased. They remain unchanged in most cancers, and are elevated only in liver tissues and lowered in renal and thyroid tumors (Table 1).
4.13. mS29/DAP3
Besides being the single ribosomal subunit with GTPase activity, mS29/DAP3 has been proposed to induce both the extrinsic and the intrinsic apoptotic pathways. As such, it is not surprising that mS29/DAP3 has been connected to numerous cancers. DAP3 expression was found to be from low to nonexistent in B-cell lymphoma cells, non-small cell lung cancers, as well as in head and neck, breast, colon and gastric cancers, possibly owing to hypermethylation of the DAP3 gene promoter [132, 133]. The role of DAP3 in cancer might be tissue-dependent. In some cases, DAP3 expression has been positively correlated with improved cancer prognosis, suggesting that DAP3 could combat cancer progression through its proapoptotic function [132, 133]. In these cases, DAP3 could serve as a potential biomarker to monitor the effectiveness of chemotherapy or other therapeutic treatments. However, in other cancers, such as thymoma and glioblastoma multiforme (GBM), DAP3 expression was found to be upregulated [134, 135]. Particularly interesting was the finding that DAP3 is overexpressed in the thyroid tumor cell line XTC-UC1, and that the expression of the DAP3 gene directly depends on the mtDNA level in these cells [136]. This observation has suggested that the variations of mtDNA levels, which are strongly involved in mtDNA-related disorders and various cancers, may dysregulate the balance between cellular life and death mediated by apoptotic processes.
4.14. Mitoribosome assembly factors
A recent large-scale analysis of gene expression profiles across nine distinct human tumors has indicated that RNA-binding protein-encoding genes, including ribosome assembly factors, are consistently differentially expressed in cancer tissues compared with other gene classes [137]. Among the mitochondrial proteins included, two mitoribosome assembly factors. The mt-SSU assembly factor GRSF1 presented a significant decrease in expression level while the mt-LSU assembly factor DDX28 showed a robustly elevated expression in cancer versus non-malignant counterpart tissues, particularly in colon and thyroid cancers [137], suggesting the existence of multiple mitoribosome assembly gene dysregulation patterns governing tumorigenesis. Indeed, DDX28 has been identified as a risk factor for colorectal cancer [15]. This study examined 18 of the 19 GWAS-identified colorectal cancer risk variants, most of which reside outside the coding regions of genes, for association with the expression of neighboring genes in 40 patients with colon cancer using paired adjacent normal and colon tumor samples [15]. The analysis indicated decreased expression of DDX28 in adjacent normal colon tissues of individuals carrying a particular variant associated with reduced risk of colorectal cancer, which has suggested that this gene may lower the risk of colorectal cancers by functioning to inhibit early events in colorectal carcinogenesis [15].
5. Targeting mitoribosome biogenesis and function for therapeutic interventions
5.1. Inhibitors of apoptosis
One of the hallmarks of tumorigenesis and tumor progression is the evasion of apoptosis by cancer cells. Dysregulated apoptosis can often translate into drug resistance of cancer because most of the conventional cancer treatments such as chemotherapy and radiation largely depend on their ability to induce apoptosis in cancer cells. Hence, over the past decade, a promising cancer therapeutic strategy has been targeting anti-apoptotic proteins that will restore the apoptotic pathway. Because none of the three mitoribosomal proteins shown to possess a role in apoptosis induction (mS29, mL41 and mL65) has been so far directly targeted for therapeutics, we refer the reader interested in the targeting of apoptosis pathways for cancer treatment to several extensive reviews published elsewhere [138–140].
5.2. Inhibitors of mitochondrial protein synthesis
Another hallmark of cancer is the extensive metabolic reprogramming that accompanies tumor progression and allows cancer cells to generate a large amount of ATP and biosynthetic precursors required for their growth. A well-described aspect of this metabolic remodeling is the Warburg effect or aerobic glycolysis. However, despite the increase in glycolytic flux, the mitochondrial function remains essential for cancer cell survival and proliferation, and it is emerging as a key target for therapeutic interventions [141]. Recent evidence has supported the “reverse Warburg effect” model, where some types of cancer cells utilize the energy-rich metabolites (e.g. lactate) provided by neighboring glycolytic and glutaminolytic stromal cells [142] or by cancer cells in more poorly oxygenated regions of the tumor [143] to drive ATP generation via OXPHOS [142]. Consistently, upregulation of mitochondrial translation has been reported in a subset of human tumors likely to meet the energy requirements of cancer cells [10]. Therefore, inhibition of mitochondrial translation can be viewed as a promising target for cancer therapy [144].
5.2.1. Targeting mitochondrial translation factors
Potentially, all components of the mitochondrial translational machinery, including tRNAs, rRNAs, mitoribosomal proteins and assembly factors, aminoacyl synthetases, tRNA modifying enzymes, as well as initiation, elongation and termination factors could be promising targets. As a case in point, shRNA-mediated transient silencing of mitochondrial translation elongation factor EF-Tu in acute myeloid leukemia cells reduced their growth rate and viability [129]. The suppression effect was associated with decreased mitochondrial membrane potential and oxygen consumption without changes in ROS generation, typical phenotypes of protein synthesis inhibition. The same study failed to observe a similar anti-leukemia effect by silencing of the mitochondrial initiation factor 3 (IF-3). However, the most plausible reason of the failure is that silencing in this case was not deep enough to affect mitochondrial translation given that the treatment did not disturb mitochondrial membrane potential or oxygen consumption capacity [129].
5.2.2. Targeting the mitoribosome
Another strategy has taken advantage of the susceptibility of mitochondrial ribosomes to antibiotics usually targeting bacterial protein synthesis [145]. The several classes of antibiotics that have been used in anti-cancer therapies include erythromycins, chloramphenicol, tetracyclines, glycylcyclines, actinonin, and aflatoxin B1 (Table 2).
Table 2. Mitochondrial protein synthesis inhibitory antibiotics tested for cancer treatment.
The chemical structures were obtained from Wikipedia (https://en.wikipedia.org).
| ANTIBIOTIC | MECHANISM OF ACTION | CHEMICAL STRUCTURE | TREATED CELL TYPES | REFS |
|---|---|---|---|---|
| Erythromycins | Binds to the LSU-rRNA complex in bacteria and mitochondria, and interferes with aminoacyl translocation, preventing the transfer of the tRNA bound at the A site of the rRNA complex to the P site of the rRNA complex. |
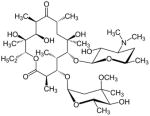
|
Breast cancer Neuroblastoma |
[1–4] |
| Chloramphenicol | Binds to the LSU-rRNA complex in bacteria and mitochondria, specifically binds to A2451 and A2452 residues in the 23S rRNA, and interferes with peptidyl transferase, preventing protein chain elongation. |
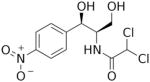
|
Breast cancer | [3] |
| Tetracyclines (Doxycycline) | Binds to the SSU-rRNA complex in bacteria and mitochondria, and prevents the interaction of aminoacyl-tRNA with the A site of the ribosome. The binding is reversible in nature. |
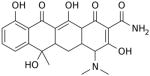
|
Mesothelioma Leukemia Renal & prostate carcinomas Nephroma Lymphoma* Leukemia |
[5–7] |
| Glycylcyclines (Tigecycline) | Mechanism of action like tetracyclines. |
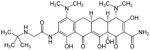
|
Breast cancer | [8–10] |
| Actinonin | A hydroxamate-containing compound and strong inhibitor of metalloenzymes, especially matrix metalloproteases (MMPs). The hydroxamate group of actinonin acts as the chelating group to bind the metal ion of the enzyme. |
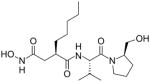
|
Leukemia Lymphoma |
[11–13] |
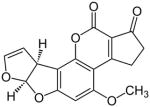
| Aflatoxin B1 | A strong genotoxic hepatocarcinogen that induces DNA adducts, leading to genetic changes in the target cells, which then cause DNA strand breakage, DNA base damage and oxidative, presumptively results in cancer. |
|
Hepatocarcinoma | [14–16] |
Denotes a Clinical trial of doxycycline monotherapy to ocular adnexal lymphoma patients.
References
C. van den Bogert, B.H. Dontje, A.M. Kroon, The antitumour effect of doxycycline on a T-cell leukaemia in the rat, Leukemia research 9(5) (1985) 617–23.
C. van den Bogert, B.H. Dontje, M. Holtrop, T.E. Melis, J.C. Romijn, J.W. van Dongen, A.M. Kroon, Arrest of the proliferation of renal and prostate carcinomas of human origin by inhibition of mitochondrial protein synthesis, Cancer research 46(7) (1986) 3283–9.
R. Lamb, B. Ozsvari, C.L. Lisanti, H.B. Tanowitz, A. Howell, U.E. Martinez-Outschoorn, F. Sotgia, M.P. Lisanti, Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: treating cancer like an infectious disease, Oncotarget 6(7) (2015) 4569–84.
J. Yongsheng, M. Xiaoyun, W. Xiaoli, L. Xin, Y. Haitao, L. Xiaoyan, Z. Jianquan, Antitumor activity of erythromycin on human neuroblastoma cell line (SH-SY5Y), Journal of Huazhong University of Science and Technology. Medical sciences = Hua zhong ke ji da xue xue bao. Yi xue Ying De wen ban = Huazhong keji daxue xuebao. Yixue Yingdewen ban 31(1) (2011) 33–8.
A.J. Ferreri, S. Govi, E. Pasini, S. Mappa, F. Bertoni, F. Zaja, C. Montalban, C. Stelitano, M.E. Cabrera, A. Giordano Resti, L.S. Politi, C. Doglioni, F. Cavalli, E. Zucca, M. Ponzoni, R. Dolcetti, Chlamydophila psittaci eradication with doxycycline as first-line targeted therapy for ocular adnexae lymphoma: final results of an international phase II trial, Journal of clinical oncology : official journal of the American Society of Clinical Oncology 30(24) (2012) 2988–94.
C. van den Bogert, B.H. Dontje, M. Holtrop, T.E. Melis, J.C. Romijn, J.W. van Dongen, A.M. Kroon, Arrest of the proliferation of renal and prostate carcinomas of human origin by inhibition of mitochondrial protein synthesis, Cancer Res. 46(7) (1986) 3283–9.
C. van den Bogert, B.H. Dontje, A.M. Kroon, The antitumour effect of doxycycline on a T-cell leukaemia in the rat, Leuk. Res. 9(5) (1985) 617–23.
B. Jhas, S. Sriskanthadevan, M. Skrtic, M.A. Sukhai, V. Voisin, Y. Jitkova, M. Gronda, R. Hurren, R.C. Laister, G.D. Bader, M.D. Minden, A.D. Schimmer, Metabolic adaptation to chronic inhibition of mitochondrial protein synthesis in acute myeloid leukemia cells, PLoS One 8(3) (2013) e58367.
M. Skrtic, S. Sriskanthadevan, B. Jhas, M. Gebbia, X. Wang, Z. Wang, R. Hurren, Y. Jitkova, M. Gronda, N. Maclean, C.K. Lai, Y. Eberhard, J. Bartoszko, P. Spagnuolo, A.C. Rutledge, A. Datti, T. Ketela, J. Moffat, B.H. Robinson, J.H. Cameron, J. Wrana, C.J. Eaves, M.D. Minden, J.C. Wang, J.E. Dick, K. Humphries, C. Nislow, G. Giaever, A.D. Schimmer, Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia, Cancer cell 20(5) (2011) 674–88.
G.A. Reed, G.J. Schiller, S. Kambhampati, M.S. Tallman, D. Douer, M.D. Minden, K.W. Yee, V. Gupta, J. Brandwein, Y. Jitkova, M. Gronda, R. Hurren, A. Shamas-Din, A.C. Schuh, A.D. Schimmer, A Phase 1 study of intravenous infusions of tigecycline in patients with acute myeloid leukemia, Cancer Med. 5(11) (2016) 3031–3040.
D.Z. Chen, D.V. Patel, C.J. Hackbarth, W. Wang, G. Dreyer, D.C. Young, P.S. Margolis, C. Wu, Z.J. Ni, J. Trias, R.J. White, Z. Yuan, Actinonin, a naturally occurring antibacterial agent, is a potent deformylase inhibitor, Biochemistry 39(6) (2000) 1256–62.
B. Peng, X. Zhang, F. Cao, Y. Wang, L. Xu, L. Cao, C. Yang, M. Li, G. Uzan, D. Zhang, Peptide deformylase inhibitor actinonin reduces celastrol’s HSP70 induction while synergizing proliferation inhibition in tumor cells, BMC Cancer 14 (2014) 146.
A. Sheth, S. Escobar-Alvarez, J. Gardner, L. Ran, M.L. Heaney, D.A. Scheinberg, Inhibition of human mitochondrial peptide deformylase causes apoptosis in c-myc-overexpressing hematopoietic cancers, Cell death & disease 5 (2014) e1152.
A.S. Hamid, I.G. Tesfamariam, Y. Zhang, Z.G. Zhang, Aflatoxin B1-induced hepatocellular carcinoma in developing countries: Geographical distribution, mechanism of action and prevention, Oncology letters 5(4) (2013) 1087–1092.
X. Peng, K. Chen, J. Chen, J. Fang, H. Cui, Z. Zuo, J. Deng, Z. Chen, Y. Geng, W. Lai, Aflatoxin B1 affects apoptosis and expression of Bax, Bcl-2, and Caspase-3 in thymus and bursa of fabricius in broiler chickens, Environmental toxicology 31(9) (2016) 1113–20.
N.K. Bhat, J.K. Emeh, B.G. Niranjan, N.G. Avadhani, Inhibition of mitochondrial protein synthesis during early stages of aflatoxin B1-induced hepatocarcinogenesis, Cancer research 42(5) (1982) 1876–80.
A recent study showed that antibiotics targeted to inhibit mitochondrial translation can be used to eradicate cancer stem cells across multiple cell types [16]. Lamb and coworkers tested several FDA-approved antibiotics to inhibit mitochondrial translation. They included erythromycins and chloramphenicol that selectively bind to the mitoribosomal large subunit and block the peptide exit channel or peptide bond formation, respectively. The study also included tetracyclines and their analogs glycylcyclines (tigecycline) that bind to the small subunits to block the codon-anticodon interaction. Twelve different cancer cell lines, representing 8 different tumor types (invasive/non-invasive breast, ovarian, prostate, pancreatic, lung, melanoma, and glioblastoma cancers), were treated with these antibiotics and screened for mammo-sphere and tumor-sphere formations.
Erythromycin and chloramphenicol
Treatment with 50 μM azithromycin, a potent erythromycin derivative, inhibited mammo-sphere formation in two different MCF7 and T47D breast cancer cell lines [16]. In addition, 250 μM treatment inhibited tumor-sphere formation in eight cancer cell lines, representing six different cancer types [16]. Consistently, treatment of human neuroblastoma cell line SH-SY5Y with erythromycin in a range from 62.5 μM to 500 μM inhibited cell proliferation in a concentration- and time-dependent manner [146]. In their report, Lamb et al. reported that chloramphenicol, which inhibited mammo-sphere generation of MCF7 cells at ~200 μM, was the weakest antibiotic tested in the study [16].
Tetracyclines
Regarding doxycycline, a tetracycline derivative with improved efficacy and stability, it has been reported to inhibit tumor growth through two potential mechanisms. It inhibits mitochondrial translation, and also inhibits matrix metalloproteinases, which appear to be essential for proliferation and dissemination of a variety of tumors [147]. Treatment with low doses of doxycycline induced cytostatic effects of variable extents in all eight mesothelioma cell lines tested but did not affect normal lung fibroblasts [148]. In addition, chronic doxycycline treatment inhibited induced T-cell type rat leukemia induced complete tumor eradication in a concentration-dependent manner [148]. Furthermore, treatment of epithelial-origin malignant cells, renal and prostate carcinomas with low tetracycline concentrations induced the inhibition of mitochondrial protein synthesis and arrest of proliferation [149, 150]. Similarly, tetracycline treatment strongly retarded the development of carcinogen-induced tumors and the development of a hypernephroma from human origin transplanted in the cheek pouch of the Syrian hamster [149, 150]. Moreover, treatment with 2 μM and 10 μM doxycycline prevented mammo-sphere formation in two breast cancer cell lines, MCF7 and T47D, and tumor-sphere formation in ten cancer cell lines, representing six different cancer types [16]. These data support the idea that tetracycline has a strong potential as a cytostatic agent in the mitochondrial translation system-targeting chemotherapy [151] and have encouraged some clinical trials. Clinical trials have proved the anti-lymphoma activity of doxycycline. Ocular adnexal lymphoma of the MALT type (OAL) is one of the most common forms of extragastric marginal zone B-cell lymphoma, openly associated with Chlamydia psittaci (Cp) infection. Ferreri and his group reported on a phase II trial testing the activity of first-line doxycycline therapy in a homogeneous cohort of patients with newly diagnosed stage-I OAMZL [152, 153]. Thirty-four patients who had measurable or parametrable disease were treated with 100 mg doxycycline administered orally twice daily for 3 weeks, and the response was assessed 3 and 12 months after treatment. Lymphoma regression was complete in 6 patients and partial in 16; stable in 11, and one had progressive disease. Moreover, 20 patients remained relapse-free at a median follow-up of 37 months, supporting the conclusion that lymphoma regression depends on Cp eradication upon doxycycline treatment.
Glycylcyclines
Among the glycylcyclines, which are tetracycline analogs, tigecycline is the only FDA-approved antibiotic. Tigecycline has been shown to have anti-leukemia properties in murine models [129]. Also, treatment of MCF7 and T47D breast cancer cell lines with 10 μM to 50 μM tigecycline suppressed mammo-sphere formation, and a single concentration of 50 μM significantly prevented tumor-sphere generation in eight different cancer cell lines [16]. Furthermore, treatment of 20 primary acute myeloid leukemia (AML) cell lines with tigecycline for 48 hrs selectively killed leukemia stem and progenitor cells with efficacy similar to EF-Tu silencing [129]. Preclinical efficacy and safety of tigecycline have been tested in patients with relapsed and refractory AML, but none of the patients had a clinical response in a phase I study of escalating tigecycline doses [154].
Actinonin
Actinonin is a naturally occurring antibacterial agent against gram-positive and gram-negative microorganisms [155], able to induce a progressive growth arrest in 16 different cancer cell lines [17]. Actinonin treatment induces a time-dependent loss of mitoribosomal proteins with different degrees of severity [156, 157]. This might be the effect of the peptidyl deformylase activity of Actinonin. Mitochondrial translation requires a formylated methionine-tRNA for the initiation of protein synthesis, and this formyl group is removed by the peptide deformylase as a part of N-terminal methionine excision (NME). Human mitochondrial peptide deformylase (HsPDF) is able to remove formyl groups from N-terminal methionines of several newly synthesized mitochondrial proteins to unmask the amino group of the first methionine, which is a prerequisite for the subsequent action of methionine aminopeptidase. HsPDF is overexpressed in many different cancer cell lines and primary myeloid leukemias [158]. Actinonin inhibition of HsPDF in cancer cells induced repression of mitochondrial translation [159], caused apoptotic death of Burkitt’s lymphoma [158], and rendered the side effects of the anti-tumor agent celastrol by controlling the heat shock response [160]. Treatment with actinonin stalled HsPDF on the mitoribosomal large subunit, near the exit channel, and triggered mitochondrial ribosome and RNA decay pathways [156]. This could explain why the inhibition of mitochondrial protein synthesis also affects cancer cells that depend on cytoplasmic glycolytic metabolism [156, 161]. Interestingly, the oncogene c-myc indirectly regulates the expression of HsPDF, and actinonin inhibition of HsPDF occurred exclusively in myc-positive cells.
Aflatoxin B1
Aflatoxins are poisonous and cancer-causing chemicals that are produced by the fungi Aspergillus. An early study had shown that treatment of hepatocytes with aflatoxin B1 (AFB1) progressively inhibited mitochondrial translation and transcription in the early stages of hepatocarcinogenesis, even 24 hr after carcinogen treatment [162]. However, the deleterious side effects induced by treatment with AFB1, which directly binds to the mtDNA altering transcription and mitochondrial polypeptide patterns [163, 164], should prevent its use in human patients.
6. Conclusion and Perspectives
The last decade has witnessed multiple advances in the understanding of the mitochondrial role in cancer physiology. Contrary to an old dogma, recent evidence has indicated that some types of cancer cells are highly dependent on mitochondrial oxidative phosphorylation. Because several mitoribosome proteins perform extra-ribosomal pro-apoptotic activities and many are differentially expressed in tumor tissues, they have the potential of becoming tumor-specific biomarkers. Further work is required to precisely decipher the mechanism underlying apoptosis induction by mS29, mL41 and mL65 proteins. In those tumor cells that are OXPHOS-dependent, inhibition of the mitochondrial translation machinery is emerging as a promising therapeutic approach, with the use of tetracycline derivatives leading the path to cancer treatment.
Acknowledgments
This research was supported by NIGMS-RO1 grants GM071775, GM105781 and GM112179 (to AB), NIGMS-MIRA grant R35GM118141 (to AB), Muscular Dystrophy Association Research Grant MDA-381828 (to AB), and an American Heart Association predoctoral fellowship (to HJK).
Abbreviations
- AIPC
Androgen-independent prostate cancer
- AKAP1
cAMP signaling
- AML
acute myeloid leukemia
- BAK
Bcl-2 homologous antagonist killer
- BAX
Bcl-2-associated X protein
- BCL-2
B-cell lymphoma 2
- BMRP
BCL-2 interacting mitochondrial ribosomal protein
- C7Orf30
chromosome 7 open reading frame 30
- CIFR1
CR6-interacting factor 1
- DAP3
Death Associated Protein 3
- DDX28
DEAD-Box Helicase 28
- DHX30
DEAH-Box Helicase 30
- DEAD box
helicase family of proteins characterized by the presence of an Asp-Glu-Ala-Asp (DEAD) motif
- DRP1
Dynamin-related protein 1
- EF-G
elongation factor G
- EF-Ts
elongation factor thermos-stable
- EF-Tu
elongation factor thermo-unstable
- FADD
Fas-Associated protein with Death Domain
- FASL
FAS ligand
- FASTKD2
Fas-activated serine/threonine kinase domain-containing protein 2
- fMet-tRNA
formylmethionine-tRNA
- GOLPH3/3L
Golgi phosphoprotein 3/3L
- GRSF1
G-Rich RNA Sequence Binding Factor 1
- GTP
guanosine triphosphate
- GWAS
genome-wide association studies
- HDM2
human double minute 2
- HSNCC
head and neck squamous cell carcinoma
- HsPDF
human mitochondrial peptide deformylase
- IF-3
initiation factor 3
- IF2-mt
mitochondrial translation initiation factor 2
- IFN-γ
Interferon-gamma
- IPS-1
interferon-γ promoter stimulator 1
- JNK
c-Jun N-terminal kinases
- Lck
lymphocyte-specific protein tyrosine kinase
- SNP
single nucleotide polymorphism
- LETM1
leucine zipper/EF hand-containing transmembrane-1
- LSU
large subunit
- MAPK
mitogen-activated protein kinases
- MCT1
monocarboxylate transporter 1
- MPV17L2
MPV17 Mitochondrial Inner Membrane Protein Like 2
- Mrh4
4th Mitochondrial DEAD-Box RNA Helicase
- MRM1-3
mitochondrial rRNA methyltransferases
- mRNA
messenger RNA
- MRPs
mitoribosomal proteins
- mtDNA
mitochondrial DNA
- MTERF
mitochondrial transcription termination factor
- MTG1-MTG2
mitochondrial ribosome associated GTPase1 and 2
- mtRF1a
mitochondrial translation release factor
- NRF1
nuclear respiratory factor 1
- NRF2
nuclear respiratory factor 2
- NSUN4
NOP2/Sun RNA Methyltransferase Family Member 4
- OAMZL
ocular adnexal marginal zone lymphoma
- OXPHOS
oxidative phosphorylation
- PDCD9
programmed cell death protein 9
- PGC1-α
PPARγ coactivator-1
- PKB
protein kinase B
- POLRMT
mitochondrial RNA polymerase
- PPR
pentatricopeptide repeat protein
- ROS
reactive oxygen species
- rRNA
ribosomal RNA
- SMAC
Second mitochondria-derived activator of caspase
- SSU
small subunit
- TCA
tricarboxylic acid cycle
- TFAM
transcription factor A, mitochondrial
- TFB1m
mitochondrial transcription factor B1
- TIMM9
mitochondrial import inner membrane translocase subunit Tim9
- TNFα
tumor necrosis factor alpha
- MOMP
mitochondrial outer membrane permeabilization
- TOMM70A
translocase of outer mitochondrial membrane 70A
- TRAIL
tumor necrosis factor-related apoptosis-inducing ligand
- tRNA
transfer RNA
Footnotes
Conflict of interest statement.
None declared.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gogvadze V, Orrenius S, Zhivotovsky B. Mitochondria in cancer cells: what is so special about them? Trends Cell Biol. 2008;18(4):165–73. doi: 10.1016/j.tcb.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 2.Wallace DC. Mitochondria and cancer. Nat Rev Cancer. 2012;12(10):685–98. doi: 10.1038/nrc3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–14. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 4.Lopez J, Tait SW. Mitochondrial apoptosis: killing cancer using the enemy within. Br J Cancer. 2015;112(6):957–62. doi: 10.1038/bjc.2015.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Silva D, Tu YT, Amunts A, Fontanesi F, Barrientos A. Mitochondrial ribosome assembly in health and disease. Cell Cycle. 2015;14(14):2226–50. doi: 10.1080/15384101.2015.1053672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sharma MR, Booth TM, Simpson L, Maslov DA, Agrawal RK. Structure of a mitochondrial ribosome with minimal RNA. Proc Natl Acad Sci U S A. 2009;106(24):9637–42. doi: 10.1073/pnas.0901631106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sharma MR, Koc EC, Datta PP, Booth TM, Spremulli LL, Agrawal RK. Structure of the mammalian mitochondrial ribosome reveals an expanded functional role for its component proteins. Cell. 2003;115(1):97–108. doi: 10.1016/s0092-8674(03)00762-1. [DOI] [PubMed] [Google Scholar]
- 8.Smits P, Smeitink JA, van den Heuvel LP, Huynen MA, Ettema TJ. Reconstructing the evolution of the mitochondrial ribosomal proteome. Nucleic Acids Res. 2007;35(14):4686–703. doi: 10.1093/nar/gkm441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mears JA, Sharma MR, Gutell RR, McCook AS, Richardson PE, Caulfield TR, Agrawal RK, Harvey SC. A structural model for the large subunit of the mammalian mitochondrial ribosome. J Mol Biol. 2006;358(1):193–212. doi: 10.1016/j.jmb.2006.01.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Greber BJ, Ban N. Structure and function of the mitochondrial ribosome. Annu Rev Biochem. 2016;85:103–32. doi: 10.1146/annurev-biochem-060815-014343. [DOI] [PubMed] [Google Scholar]
- 11.Ott M, Amunts A, Brown A. Organization and regulation of mitochondrial protein synthesis. Annu Rev Biochem. 2016;85:77–101. doi: 10.1146/annurev-biochem-060815-014334. [DOI] [PubMed] [Google Scholar]
- 12.Cavdar Koc E, Ranasinghe A, Burkhart W, Blackburn K, Koc H, Moseley A, Spremulli LL. A new face on apoptosis: death-associated protein 3 and PDCD9 are mitochondrial ribosomal proteins. FEBS Lett. 2001;492(1–2):166–70. doi: 10.1016/s0014-5793(01)02250-5. [DOI] [PubMed] [Google Scholar]
- 13.Sun L, Liu Y, Fremont M, Schwarz S, Siegmann M, Matthies R, Jost JP. A novel 52 kDa protein induces apoptosis and concurrently activates c-Jun N-terminal kinase 1 (JNK1) in mouse C3H10T1/2 fibroblasts. Gene. 1998;208(2):157–66. doi: 10.1016/s0378-1119(97)00626-4. [DOI] [PubMed] [Google Scholar]
- 14.Koc EC, Haciosmanoglu E, Claudio PP, Wolf A, Califano L, Friscia M, Cortese A, Koc H. Impaired mitochondrial protein synthesis in head and neck squamous cell carcinoma. Mitochondrion. 2015;24:113–21. doi: 10.1016/j.mito.2015.07.123. [DOI] [PubMed] [Google Scholar]
- 15.Loo LW, Cheng I, Tiirikainen M, Lum-Jones A, Seifried A, Dunklee LM, Church JM, Gryfe R, Weisenberger DJ, Haile RW, Gallinger S, Duggan DJ, Thibodeau SN, Casey G, Le Marchand L. cis-Expression QTL analysis of established colorectal cancer risk variants in colon tumors and adjacent normal tissue. PLoS One. 2012;7(2):e30477. doi: 10.1371/journal.pone.0030477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lamb R, Ozsvari B, Lisanti CL, Tanowitz HB, Howell A, Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: treating cancer like an infectious disease. Oncotarget. 2015;6(7):4569–84. doi: 10.18632/oncotarget.3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee MD, She Y, Soskis MJ, Borella CP, Gardner JR, Hayes PA, Dy BM, Heaney ML, Philips MR, Bornmann WG, Sirotnak FM, Scheinberg DA. Human mitochondrial peptide deformylase, a new anticancer target of actinonin-based antibiotics. J Clin Invest. 2004;114(8):1107–16. doi: 10.1172/JCI22269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jaras M, Ebert BL. Power cut: inhibiting mitochondrial translation to target leukemia. Cancer cell. 2011;20(5):555–6. doi: 10.1016/j.ccr.2011.10.028. [DOI] [PubMed] [Google Scholar]
- 19.Spremulli LL, Coursey A, Navratil T, Hunter SE. Initiation and elongation factors in mammalian mitochondrial protein biosynthesis. Prog Nucleic Acid Res Mol Biol. 2004;77:211–61. doi: 10.1016/S0079-6603(04)77006-3. [DOI] [PubMed] [Google Scholar]
- 20.Osawa S, Ohama T, Jukes TH, Watanabe K, Yokoyama S. Evolution of the mitochondrial genetic code. II. Reassignment of codon AUA from isoleucine to methionine. J Mol Evol. 1989;29(5):373–80. doi: 10.1007/BF02602907. [DOI] [PubMed] [Google Scholar]
- 21.Jukes TH, Osawa S. The genetic code in mitochondria and chloroplasts. Experientia. 1990;46(11–12):1117–26. doi: 10.1007/BF01936921. [DOI] [PubMed] [Google Scholar]
- 22.Bender A, Hajieva P, Moosmann B. Adaptive antioxidant methionine accumulation in respiratory chain complexes explains the use of a deviant genetic code in mitochondria. Proc Natl Acad Sci U S A. 2008;105(43):16496–501. doi: 10.1073/pnas.0802779105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perez-Martinez X, Funes S, Camacho-Villasana Y, Marjavaara S, Tavares-Carreon F, Shingu-Vazquez M. Protein synthesis and assembly in mitochondrial disorders. Curr Top Med Chem. 2008;8(15):1335–50. doi: 10.2174/156802608786141124. [DOI] [PubMed] [Google Scholar]
- 24.Towpik J. Regulation of mitochondrial translation in yeast. Cell Mol Biol Lett. 2005;10(4):571–94. [PubMed] [Google Scholar]
- 25.Lipinski KA, Kaniak-Golik A, Golik P. Maintenance and expression of the S. cerevisiae mitochondrial genome--from genetics to evolution and systems biology. Biochim Biophys Acta. 2010;1797(6–7):1086–98. doi: 10.1016/j.bbabio.2009.12.019. [DOI] [PubMed] [Google Scholar]
- 26.Bonitz SG, Berlani R, Coruzzi G, Li M, Macino G, Nobrega FG, Nobrega MP, Thalenfeld BE, Tzagoloff A. Codon recognition rules in yeast mitochondria. Proc Natl Acad Sci U S A. 1980;77(6):3167–70. doi: 10.1073/pnas.77.6.3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gaur R, Grasso D, Datta PP, Krishna PD, Das G, Spencer A, Agrawal RK, Spremulli L, Varshney U. A single mammalian mitochondrial translation initiation factor functionally replaces two bacterial factors. Mol Cell. 2008;29(2):180–90. doi: 10.1016/j.molcel.2007.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liao HX, Spremulli LL. Initiation of protein synthesis in animal mitochondria. Purification and characterization of translational initiation factor 2. J Biol Chem. 1991;266(31):20714–9. [PubMed] [Google Scholar]
- 29.de Cock E, Springer M, Dardel F. The interdomain linker of Escherichia coli initiation factor IF3: a possible trigger of translation initiation specificity. Mol Microbiol. 1999;32(1):193–202. doi: 10.1046/j.1365-2958.1999.01350.x. [DOI] [PubMed] [Google Scholar]
- 30.Anvret A, Ran C, Westerlund M, Thelander AC, Sydow O, Lind C, Hakansson A, Nissbrandt H, Galter D, Belin AC. Possible involvement of a mitochondrial translation initiation factor 3 variant causing decreased mRNA levels in Parkinson’s disease. Parkinsons Dis. 2010;2010:491751. doi: 10.4061/2010/491751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vambutas A, Ackerman SH, Tzagoloff A. Mitochondrial translational-initiation and elongation factors in Saccharomyces cerevisiae. Eur J Biochem. 1991;201(3):643–52. doi: 10.1111/j.1432-1033.1991.tb16325.x. [DOI] [PubMed] [Google Scholar]
- 32.Atkinson GC, Kuzmenko A, Kamenski P, Vysokikh MY, Lakunina V, Tankov S, Smirnova E, Soosaar A, Tenson T, Hauryliuk V. Evolutionary and genetic analyses of mitochondrial translation initiation factors identify the missing mitochondrial IF3 in S. cerevisiae. Nucleic Acids Res. 2012;40(13):6122–34. doi: 10.1093/nar/gks272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spencer AC, Spremulli LL. The interaction of mitochondrial translational initiation factor 2 with the small ribosomal subunit. Biochim Biophys Acta. 2005;1750(1):69–81. doi: 10.1016/j.bbapap.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 34.Christian BE, Spremulli LL. Evidence for an active role of IF3mt in the initiation of translation in mammalian mitochondria. Biochemistry. 2009;48(15):3269–78. doi: 10.1021/bi8023493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Christian BE, Spremulli LL. Mechanism of protein biosynthesis in mammalian mitochondria. Biochim Biophys Acta. 2012;1819(9–10):1035–54. doi: 10.1016/j.bbagrm.2011.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kehrein K, Ott M. Conserved and organelle-specific molecular mechanisms of translation in mitochondria. In: Bullerwell CE, editor. Organelle Genetics. Springer-Verlag; Berlin: 2012. pp. 401–429. [Google Scholar]
- 37.Schwartzbach CJ, Spremulli LL. Bovine mitochondrial protein synthesis elongation factors. Identification and initial characterization of an elongation factor Tu-elongation factor Ts complex. J Biol Chem. 1989;264(32):19125–31. [PubMed] [Google Scholar]
- 38.Chrzanowska-Lightowlers ZM, Pajak A, Lightowlers RN. Termination of protein synthesis in mammalian mitochondria. J Biol Chem. 2011;286(40):34479–85. doi: 10.1074/jbc.R111.290585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Teyssier E, Hirokawa G, Tretiakova A, Jameson B, Kaji A, Kaji H. Temperature-sensitive mutation in yeast mitochondrial ribosome recycling factor (RRF) Nucleic Acids Res. 2003;31(14):4218–26. doi: 10.1093/nar/gkg449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Callegari S, Gregory PA, Sykes MJ, Bellon J, Andrews S, McKinnon RA, de Barros Lopes MA. Polymorphisms in the mitochondrial ribosome recycling factor EF-G2mt/MEF2 compromise cell respiratory function and increase atorvastatin toxicity. PLoS Genet. 2012;8(6):e1002755. doi: 10.1371/journal.pgen.1002755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rorbach J, Richter R, Wessels HJ, Wydro M, Pekalski M, Farhoud M, Kuhl I, Gaisne M, Bonnefoy N, Smeitink JA, Lightowlers RN, Chrzanowska-Lightowlers ZM. The human mitochondrial ribosome recycling factor is essential for cell viability. Nucleic Acids Res. 2008;36(18):5787–99. doi: 10.1093/nar/gkn576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Towpik J, Chacinska A, Ciesla M, Ginalski K, Boguta M. Mutations in the yeast mrf1 gene encoding mitochondrial release factor inhibit translation on mitochondrial ribosomes. J Biol Chem. 2004;279(14):14096–103. doi: 10.1074/jbc.M312856200. [DOI] [PubMed] [Google Scholar]
- 43.Temperley R, Richter R, Dennerlein S, Lightowlers RN, Chrzanowska-Lightowlers ZM. Hungry codons promote frameshifting in human mitochondrial ribosomes. Science. 2010;327(5963):301. doi: 10.1126/science.1180674. [DOI] [PubMed] [Google Scholar]
- 44.Soleimanpour-Lichaei HR, Kuhl I, Gaisne M, Passos JF, Wydro M, Rorbach J, Temperley R, Bonnefoy N, Tate W, Lightowlers R, Chrzanowska-Lightowlers Z. mtRF1a is a human mitochondrial translation release factor decoding the major termination codons UAA and UAG. Mol Cell. 2007;27(5):745–57. doi: 10.1016/j.molcel.2007.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O’Brien TW, O’Brien BJ, Norman RA. Nuclear MRP genes and mitochondrial disease. Gene. 2005;354:147–51. doi: 10.1016/j.gene.2005.03.026. [DOI] [PubMed] [Google Scholar]
- 46.Amunts A, Brown A, Toots J, Scheres SHW, Ramakrishnan V. The structure of the human mitochondrial ribosome. Science. 2015;348(6230):95–8. doi: 10.1126/science.aaa1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brown A, Amunts A, Bai XC, Sugimoto Y, Edwards PC, Murshudov G, Scheres SH, Ramakrishnan V. Structure of the large ribosomal subunit from human mitochondria. Science. 2014;2:1258026. doi: 10.1126/science.1258026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Greber BJ, Bieri P, Leibundgut M, Leitner A, Aebersold R, Boehringer D, Ban N. Ribosome. The complete structure of the 55S mammalian mitochondrial ribosome. Science. 2015;348(6232):303–8. doi: 10.1126/science.aaa3872. [DOI] [PubMed] [Google Scholar]
- 49.Greber BJ, Boehringer D, Leibundgut M, Bieri P, Leitner A, Schmitz N, Aebersold R, Ban N. The complete structure of the large subunit of the mammalian mitochondrial ribosome. Nature. 2014;515(7526):283–6. doi: 10.1038/nature13895. [DOI] [PubMed] [Google Scholar]
- 50.Greber BJ, Boehringer D, Leitner A, Bieri P, Voigts-Hoffmann F, Erzberger JP, Leibundgut M, Aebersold R, Ban N. Architecture of the large subunit of the mammalian mitochondrial ribosome. Nature. 2014;505(7484):515–9. doi: 10.1038/nature12890. [DOI] [PubMed] [Google Scholar]
- 51.Kaushal PS, Sharma MR, Booth TM, Haque EM, Tung CS, Sanbonmatsu KY, Spremulli LL, Agrawal RK. Cryo-EM structure of the small subunit of the mammalian mitochondrial ribosome. Proc Natl Acad Sci U S A. 2014;111(20):7284–9. doi: 10.1073/pnas.1401657111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mailloux RJ, Jin X, Willmore WG. Redox regulation of mitochondrial function with emphasis on cysteine oxidation reactions. Redox Biol. 2014;2:123–39. doi: 10.1016/j.redox.2013.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Handy DE, Loscalzo J. Redox regulation of mitochondrial function. Antioxid Redox Signal. 2012;16(11):1323–67. doi: 10.1089/ars.2011.4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Temperley RJ, Wydro M, Lightowlers RN, Chrzanowska-Lightowlers ZM. Human mitochondrial mRNAs--like members of all families, similar but different. Biochim Biophys Acta. 2010;1797(6–7):1081–5. doi: 10.1016/j.bbabio.2010.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen SS, Williamson JR. Characterization of the ribosome biogenesis landscape in E. coli using quantitative mass spectrometry. J Mol Biol. 2013;425(4):767–79. doi: 10.1016/j.jmb.2012.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shajani Z, Sykes MT, Williamson JR. Assembly of bacterial ribosomes. Annu Rev Biochem. 2011;80:501–26. doi: 10.1146/annurev-biochem-062608-160432. [DOI] [PubMed] [Google Scholar]
- 57.Shoji S, Dambacher CM, Shajani Z, Williamson JR, Schultz PG. Systematic chromosomal deletion of bacterial ribosomal protein genes. 2011;413:751–761. doi: 10.1016/j.jmb.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee KW, Bogenhagen DF. Assignment of 2’-O-methyltransferases to modification sites on the mammalian mitochondrial large subunit 16S rRNA. J Biol Chem. 2014;289(36):24936–42. doi: 10.1074/jbc.C114.581868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee KW, Okot-Kotber C, Lacomb JF, Bogenhagen DF. Mitochondrial rRNA methyltransferase family members are positioned to modify nascent rrna in foci near the mtDNA nucleoid. J Biol Chem. 2013;288(43):31386–99. doi: 10.1074/jbc.M113.515692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rorbach J, Boesch P, Gammage PA, Nicholls TJ, Pearce SF, Patel D, Hauser A, Perocchi F, Minczuk M. MRM2 and MRM3 are involved in biogenesis of the large subunit of the mitochondrial ribosome. Mol Biol Cell. 2014;9:01–0014. doi: 10.1091/mbc.E14-01-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kotani T, Akabane S, Takeyasu K, Ueda T, Takeuchi N. Human G-proteins, ObgH1, Mtg1, associate with the large mitochondrial ribosome subunit and are involved in translation and assembly of respiratory complexes. Nucleic Acids Res. 2013;41:3713–22. doi: 10.1093/nar/gkt079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.He J, Cooper HM, Reyes A, Di Re M, Kazak L, Wood SR, Mao CC, Fearnley IM, Walker JE, Holt IJ. Human C4orf14 interacts with the mitochondrial nucleoid and is involved in the biogenesis of the small mitochondrial ribosomal subunit. Nucleic Acids Res. 2012;40:6097–108. doi: 10.1093/nar/gks257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Barrientos A, Korr D, Barwell KJ, Sjulsen C, Gajewski CD, Manfredi G, Ackerman S, Tzagoloff A. MTG1 codes for a conserved protein required for mitochondrial translation. Mol Biol Cell. 2003;14(6):2292–302. doi: 10.1091/mbc.E02-10-0636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wredenberg A, Lagouge M, Bratic A, Metodiev MD, Spahr H, Mourier A, Freyer C, Ruzzenente B, Tain L, Gronke S, Baggio F, Kukat C, Kremmer E, Wibom R, Polosa PL, Habermann B, Partridge L, Park CB, Larsson NG. MTERF3 regulates mitochondrial ribosome biogenesis in invertebrates and mammals. PLoS Genet. 2013;9(1):e1003178. doi: 10.1371/journal.pgen.1003178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Camara Y, Asin-Cayuela J, Park CB, Metodiev MD, Shi Y, Ruzzenente B, Kukat C, Habermann B, Wibom R, Hultenby K, Franz T, Erdjument-Bromage H, Tempst P, Hallberg BM, Gustafsson CM, Larsson NG. MTERF4 regulates translation by targeting the methyltransferase NSUN4 to the mammalian mitochondrial ribosome. Cell Metab. 2011;13(5):527–39. doi: 10.1016/j.cmet.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 66.Metodiev MD, Spahr H, Loguercio Polosa P, Meharg C, Becker C, Altmueller J, Habermann B, Larsson NG, Ruzzenente B. NSUN4 is a dual function mitochondrial protein required for both methylation of 12S rRNA and coordination of mitoribosomal assembly. PLoS Genet. 2014;10(2):e1004110. doi: 10.1371/journal.pgen.1004110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dalla Rosa I, Durigon R, Pearce SF, Rorbach J, Hirst EM, Vidoni S, Reyes A, Brea-Calvo G, Minczuk M, Woellhaf MW, Herrmann JM, Huynen MA, Holt IJ, Spinazzola A. MPV17L2 is required for ribosome assembly in mitochondria. Nucleic Acids Res. 2014;42(13):8500–15. doi: 10.1093/nar/gku513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Antonicka H, Shoubridge EA. Mitochondrial RNA granules are centers for posttranscriptional RNA processing and ribosome biogenesis. Cell Rep. 2015;10(6):920–932. doi: 10.1016/j.celrep.2015.01.030. [DOI] [PubMed] [Google Scholar]
- 69.Tu YT, Barrientos A. The human mitochondrial DEAD-box protein DDX28 resides in RNA granules and functions in mitoribosome assembly. Cell Rep. 2015;10(6):854–864. doi: 10.1016/j.celrep.2015.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.De Silva D, Fontanesi F, Barrientos A. The DEAD-Box protein Mrh4 functions in the assembly of the mitochondrial large ribosomal subunit. Cell Metab. 2013;18:712–725. doi: 10.1016/j.cmet.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fung S, Nishimura T, Sasarman F, Shoubridge EA. The conserved interaction of C7orf30 with MRPL14 promotes biogenesis of the mitochondrial large ribosomal subunit and mitochondrial translation. Mol Biol Cell. 2013;24(3):184–93. doi: 10.1091/mbc.E12-09-0651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Antonicka H, Sasarman F, Nishimura T, Paupe V, Shoubridge EA. The mitochondrial RNA-binding protein GRSF1 Localizes to RNA granules and Is required for posttranscriptional mitochondrial gene expression. Cell Metab. 2013;17(3):386–98. doi: 10.1016/j.cmet.2013.02.006. [DOI] [PubMed] [Google Scholar]
- 73.Kaur J, Stuart RA. Truncation of the Mrp20 protein reveals new ribosome-assembly subcomplex in mitochondria. EMBO Rep. 2011;12(9):950–5. doi: 10.1038/embor.2011.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Barrientos A. Mitochondriolus: assembling mitoribosomes. Oncotarget. 2015;6(19):16800–1. doi: 10.18632/oncotarget.4646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jourdain AA, Koppen M, Wydro M, Rodley CD, Lightowlers RN, Chrzanowska-Lightowlers ZM, Martinou JC. GRSF1 Regulates RNA Processing in Mitochondrial RNA Granules. Cell Metab. 2013;17(3):399–410. doi: 10.1016/j.cmet.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wurth L. Versatility of RNA-Binding Proteins in Cancer. Comp Funct Genomics. 2012;2012:178525. doi: 10.1155/2012/178525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kim MY, Hur J, Jeong S. Emerging roles of RNA and RNA-binding protein network in cancer cells. BMB Rep. 2009;42(3):125–30. doi: 10.5483/bmbrep.2009.42.3.125. [DOI] [PubMed] [Google Scholar]
- 78.Wang C, Youle RJ. The role of mitochondria in apoptosis. Annu Rev Genet. 2009;43:95–118. doi: 10.1146/annurev-genet-102108-134850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Indran IR, Tufo G, Pervaiz S, Brenner C. Recent advances in apoptosis, mitochondria and drug resistance in cancer cells. Biochim Biophys Acta. 2011;1807(6):735–45. doi: 10.1016/j.bbabio.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 80.Sarosiek KA, Chi X, Bachman JA, Sims JJ, Montero J, Patel L, Flanagan A, Andrews DW, Sorger P, Letai A. BID preferentially activates BAK while BIM preferentially activates BAX, affecting chemotherapy response. Mol Cell. 2013;51(6):751–65. doi: 10.1016/j.molcel.2013.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Denslow ND, Anders JC, O’Brien TW. Bovine mitochondrial ribosomes possess a high affinity binding site for guanine nucleotides. J Biol Chem. 1991;266(15):9586–90. [PubMed] [Google Scholar]
- 82.Miller JL, Koc H, Koc EC. Identification of phosphorylation sites in mammalian mitochondrial ribosomal protein DAP3. Protein Sci. 2008;17(2):251–60. doi: 10.1110/ps.073185608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Miller JL, Cimen H, Koc H, Koc EC. Phosphorylated proteins of the mammalian mitochondrial ribosome: implications in protein synthesis. J Proteome Res. 2009;8(10):4789–98. doi: 10.1021/pr9004844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Saveanu C, Fromont-Racine M, Harington A, Ricard F, Namane A, Jacquier A. Identification of 12 new yeast mitochondrial ribosomal proteins including 6 that have no prokaryotic homologues. J Biol Chem. 2001;276(19):15861–7. doi: 10.1074/jbc.M010864200. Epub 2001 Feb 14. [DOI] [PubMed] [Google Scholar]
- 85.Cavdar Koc E, Burkhart W, Blackburn K, Moseley A, Spremulli LL. The small subunit of the mammalian mitochondrial ribosome. Identification of the full complement of ribosomal proteins present. J Biol Chem. 2001;276(22):19363–74. doi: 10.1074/jbc.M100727200. [DOI] [PubMed] [Google Scholar]
- 86.Kissil JL, Deiss LP, Bayewitch M, Raveh T, Khaspekov G, Kimchi A. Isolation of DAP3, a novel mediator of interferon-gamma-induced cell death. J Biol Chem. 1995;270(46):27932–6. doi: 10.1074/jbc.270.46.27932. [DOI] [PubMed] [Google Scholar]
- 87.Morgan CJ, Jacques C, Savagner F, Tourmen Y, Mirebeau DP, Malthiery Y, Reynier P. A conserved N-terminal sequence targets human DAP3 to mitochondria. Biochem Biophys Res Commun. 2001;280(1):177–81. doi: 10.1006/bbrc.2000.4119. [DOI] [PubMed] [Google Scholar]
- 88.Wazir U, Orakzai MM, Khanzada ZS, Jiang WG, Sharma AK, Kasem A, Mokbel K. The role of death-associated protein 3 in apoptosis, anoikis and human cancer. Cancer Cell Int. 2015;15:39. doi: 10.1186/s12935-015-0187-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Cohen O, Inbal B, Kissil JL, Raveh T, Berissi H, Spivak-Kroizaman T, Feinstein E, Kimchi A. DAP-kinase participates in TNF-alpha- and Fas-induced apoptosis and its function requires the death domain. J Cell Biol. 1999;146(1):141–8. doi: 10.1083/jcb.146.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kissil JL, Cohen O, Raveh T, Kimchi A. Structure-function analysis of an evolutionary conserved protein, DAP3, which mediates TNF-alpha- and Fas-induced cell death. EMBO J. 1999;18(2):353–62. doi: 10.1093/emboj/18.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Miyazaki T, Shen M, Fujikura D, Tosa N, Kim HR, Kon S, Uede T, Reed JC. Functional role of death-associated protein 3 (DAP3) in anoikis. J Biol Chem. 2004;279(43):44667–72. doi: 10.1074/jbc.M408101200. [DOI] [PubMed] [Google Scholar]
- 92.Berger T, Kretzler M. TRAIL-induced apoptosis is independent of the mitochondrial apoptosis mediator DAP3. Biochem Biophys Res Commun. 2002;297(4):880–4. doi: 10.1016/s0006-291x(02)02310-0. [DOI] [PubMed] [Google Scholar]
- 93.Berger T, Kretzler M. Interaction of DAP3 and FADD only after cellular disruption. Nat Immunol. 2002;3(1):3–5. doi: 10.1038/ni0102-3b. [DOI] [PubMed] [Google Scholar]
- 94.Mukamel Z, Kimchi A. Death-associated protein 3 localizes to the mitochondria and is involved in the process of mitochondrial fragmentation during cell death. J Biol Chem. 2004;279(35):36732–8. doi: 10.1074/jbc.M400041200. [DOI] [PubMed] [Google Scholar]
- 95.SN, BJ, AYK, SN S29 ribosomal protein induces mitochondria mediated apoptosis of Hep2 cells via the activation of p38 MAPK and JNK signaling. Int J Integr Biol. 2009;5:49–57. [Google Scholar]
- 96.Maeda Y. A long-range foresight for the medical application of apoptosis specifically induced by Dd-MRP4, Dictyostelium mitochondrial ribosomal protein S4, to cancer therapy. Biomolecules. 2015;5(1):113–20. doi: 10.3390/biom5010113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Berger T, Brigl M, Herrmann JM, Vielhauer V, Luckow B, Schlondorff D, Kretzler M. The apoptosis mediator mDAP-3 is a novel member of a conserved family of mitochondrial proteins. J Cell Sci. 2000;113(Pt 20):3603–12. doi: 10.1242/jcs.113.20.3603. [DOI] [PubMed] [Google Scholar]
- 98.Kim HR, Chae HJ, Thomas M, Miyazaki T, Monosov A, Monosov E, Krajewska M, Krajewski S, Reed JC. Mammalian DAP3 is an essential gene required for mitochondrial homeostasis in vivo and contributing to the extrinsic pathway for apoptosis. FASEB J. 2007;21(1):188–96. doi: 10.1096/fj.06-6283com. [DOI] [PubMed] [Google Scholar]
- 99.Xiao L, Xian H, Lee KY, Xiao B, Wang H, Yu F, Shen HM, Liou YC. Death-associated protein 3 regulates mitochondrial-encoded protein synthesis and mitochondrial dynamics. J Biol Chem. 2015;290(41):24961–74. doi: 10.1074/jbc.M115.673343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yoo YA, Kim MJ, Park JK, Chung YM, Lee JH, Chi SG, Kim JS, Yoo YD. Mitochondrial ribosomal protein L41 suppresses cell growth in association with p53 and p27Kip1. Mol Cell Biol. 2005;25(15):6603–16. doi: 10.1128/MCB.25.15.6603-6616.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kim MJ, Yoo YA, Kim HJ, Kang S, Kim YG, Kim JS, Yoo YD. Mitochondrial ribosomal protein L41 mediates serum starvation-induced cell-cycle arrest through an increase of p21(WAF1/CIP1) Biochem Biophys Res Commun. 2005;338(2):1179–84. doi: 10.1016/j.bbrc.2005.10.064. [DOI] [PubMed] [Google Scholar]
- 102.Chintharlapalli SR, Jasti M, Malladi S, Parsa KV, Ballestero RP, Gonzalez-Garcia M. BMRP is a Bcl-2 binding protein that induces apoptosis. J Cell Biochem. 2005;94(3):611–26. doi: 10.1002/jcb.20292. [DOI] [PubMed] [Google Scholar]
- 103.Malladi S, Parsa KV, Bhupathi D, Rodriguez-Gonzalez MA, Conde JA, Anumula P, Romo HE, Claunch CJ, Ballestero RP, Gonzalez-Garcia M. Deletion mutational analysis of BMRP, a pro-apoptotic protein that binds to Bcl-2. Mol Cell Biochem. 2011;351(1–2):217–32. doi: 10.1007/s11010-011-0729-1. [DOI] [PubMed] [Google Scholar]
- 104.Conde JA, Claunch CJ, Romo HE, Benito-Martin A, Ballestero RP, Gonzalez-Garcia M. Identification of a motif in BMRP required for interaction with Bcl-2 by site-directed mutagenesis studies. J Cell Biochem. 2012;113(11):3498–508. doi: 10.1002/jcb.24226. [DOI] [PubMed] [Google Scholar]
- 105.Levshenkova EV, Ukraintsev KE, Orlova VV, Alibaeva RA, Kovriga IE, Zhugdernamzhilyn O, Frolova EI. The structure and specific features of the cDNA expression of the human gene MRPL37. Bioorg Khim. 2004;30(5):499–506. doi: 10.1023/b:rubi.0000043788.86955.ea. [DOI] [PubMed] [Google Scholar]
- 106.Carim L, Sumoy L, Nadal M, Estivill X, Escarceller M. Cloning, expression, and mapping of PDCD9, the human homolog of Gallus gallus pro-apoptotic protein p52. Cytogenet Cell Genet. 1999;87(1–2):85–8. doi: 10.1159/000015397. [DOI] [PubMed] [Google Scholar]
- 107.Lawrence RT, Perez EM, Hernandez D, Miller CP, Haas KM, Irie HY, Lee SI, Blau CA, Villen J. The proteomic landscape of triple-negative breast cancer. Cell Rep. 2015;11(4):630–44. doi: 10.1016/j.celrep.2015.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chang CQ, Yesupriya A, Rowell JL, Pimentel CB, Clyne M, Gwinn M, Khoury MJ, Wulf A, Schully SD. A systematic review of cancer GWAS and candidate gene meta-analyses reveals limited overlap but similar effect sizes. Eur J Hum Genet. 2014;22(3):402–8. doi: 10.1038/ejhg.2013.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mints M, Mushtaq M, Iurchenko N, Kovalevska L, Stip MC, Budnikova D, Andersson S, Polischuk L, Buchynska L, Kashuba E. Mitochondrial ribosomal protein S18–2 is highly expressed in endometrial cancers along with free E2F1. Oncotarget. 2016;7(16):22150–8. doi: 10.18632/oncotarget.7905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sotgia F, Whitaker-Menezes D, Martinez-Outschoorn UE, Salem AF, Tsirigos A, Lamb R, Sneddon S, Hulit J, Howell A, Lisanti MP. Mitochondria “fuel” breast cancer metabolism: fifteen markers of mitochondrial biogenesis label epithelial cancer cells, but are excluded from adjacent stromal cells. Cell Cycle. 2012;11(23):4390–401. doi: 10.4161/cc.22777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sugimoto T, Seki N, Shimizu S, Kikkawa N, Tsukada J, Shimada H, Sasaki K, Hanazawa T, Okamoto Y, Hata A. The galanin signaling cascade is a candidate pathway regulating oncogenesis in human squamous cell carcinoma. Genes, chromosomes & cancer. 2009;48(2):132–42. doi: 10.1002/gcc.20626. [DOI] [PubMed] [Google Scholar]
- 112.Lyng H, Brovig RS, Svendsrud DH, Holm R, Kaalhus O, Knutstad K, Oksefjell H, Sundfor K, Kristensen GB, Stokke T. Gene expressions and copy numbers associated with metastatic phenotypes of uterine cervical cancer. BMC genomics. 2006;7:268. doi: 10.1186/1471-2164-7-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lohrum MA, Ludwig RL, Kubbutat MH, Hanlon M, Vousden KH. Regulation of HDM2 activity by the ribosomal protein L11. Cancer cell. 2003;3(6):577–87. doi: 10.1016/s1535-6108(03)00134-x. [DOI] [PubMed] [Google Scholar]
- 114.Bhat KP, Itahana K, Jin A, Zhang Y. Essential role of ribosomal protein L11 in mediating growth inhibition-induced p53 activation. EMBO J. 2004;23(12):2402–12. doi: 10.1038/sj.emboj.7600247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhang Y, Wolf GW, Bhat K, Jin A, Allio T, Burkhart WA, Xiong Y. Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress checkpoint pathway. Mol Cell Biol. 2003;23(23):8902–12. doi: 10.1128/MCB.23.23.8902-8912.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Best CJ, Gillespie JW, Yi Y, Chandramouli GV, Perlmutter MA, Gathright Y, Erickson HS, Georgevich L, Tangrea MA, Duray PH, Gonzalez S, Velasco A, Linehan WM, Matusik RJ, Price DK, Figg WD, Emmert-Buck MR, Chuaqui RF. Molecular alterations in primary prostate cancer after androgen ablation therapy. Clinical cancer research : an official journal of the American Association for Cancer Research. 2005;11(19 Pt 1):6823–34. doi: 10.1158/1078-0432.CCR-05-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kim JC, Kim SY, Roh SA, Cho DH, Kim DD, Kim JH, Kim YS. Gene expression profiling: canonical molecular changes and clinicopathological features in sporadic colorectal cancers. World J Gastroenterol. 2008;14(43):6662–72. doi: 10.3748/wjg.14.6662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chen Y, Cairns R, Papandreou I, Koong A, Denko NC. Oxygen consumption can regulate the growth of tumors, a new perspective on the Warburg effect. PLoS One. 2009;4(9):e7033. doi: 10.1371/journal.pone.0007033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Piao L, Li Y, Kim SJ, Byun HS, Huang SM, Hwang SK, Yang KJ, Park KA, Won M, Hong J, Hur GM, Seok JH, Shong M, Cho MH, Brazil DP, Hemmings BA, Park J. Association of LETM1 and MRPL36 contributes to the regulation of mitochondrial ATP production and necrotic cell death. Cancer Res. 2009;69(8):3397–404. doi: 10.1158/0008-5472.CAN-08-3235. [DOI] [PubMed] [Google Scholar]
- 120.Li N, Zheng Y, Xuan C, Lin Z, Piao L, Liu S. LETM1 overexpression is correlated with the clinical features and survival outcome of breast cancer. International journal of clinical and experimental pathology. 2015;8(10):12893–900. [PMC free article] [PubMed] [Google Scholar]
- 121.Hwang SK, Piao L, Lim HT, Minai-Tehrani A, Yu KN, Ha YC, Chae CH, Lee KH, Beck GR, Park J, Cho MH. Suppression of lung tumorigenesis by leucine zipper/EF hand-containing transmembrane-1. PLoS One. 2010;5(9):e12535. doi: 10.1371/journal.pone.0012535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lin YW, Aplan PD. Gene expression profiling of precursor T-cell lymphoblastic leukemia/lymphoma identifies oncogenic pathways that are potential therapeutic targets. Leukemia. 2007;21(6):1276–84. doi: 10.1038/sj.leu.2404685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wang Y, Dong L, Cui H, Shen DH, Wang Y, Chang XH, Fu TY, Ye X, Yao YY. Up-regulation of mitochondrial antioxidation signals in ovarian cancer cells with aggressive biologic behavior. J Zhejiang Univ Sci B. 2011;12(5):346–56. doi: 10.1631/jzus.B1000192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gasparre G, Romeo G, Rugolo M, Porcelli AM. Learning from oncocytic tumors: Why choose inefficient mitochondria? Biochim Biophys Acta. 2011;1807(6):633–42. doi: 10.1016/j.bbabio.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 125.Baris O, Savagner F, Nasser V, Loriod B, Granjeaud S, Guyetant S, Franc B, Rodien P, Rohmer V, Bertucci F, Birnbaum D, Malthiery Y, Reynier P, Houlgatte R. Transcriptional profiling reveals coordinated up-regulation of oxidative metabolism genes in thyroid oncocytic tumors. The Journal of clinical endocrinology and metabolism. 2004;89(2):994–1005. doi: 10.1210/jc.2003-031238. [DOI] [PubMed] [Google Scholar]
- 126.Ran Q, Hao P, Xiao Y, Xiang L, Ye X, Deng X, Zhao J, Li Z. CRIF1 interacting with CDK2 regulates bone marrow microenvironment-induced G0/G1 arrest of leukemia cells. PLoS One. 2014;9(2):e85328. doi: 10.1371/journal.pone.0085328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chung HK, Yi YW, Jung NC, Kim D, Suh JM, Kim H, Park KC, Song JH, Kim DW, Hwang ES, Yoon SH, Bae YS, Kim JM, Bae I, Shong M. CR6-interacting factor 1 interacts with Gadd45 family proteins and modulates the cell cycle. J Biol Chem. 2003;278(30):28079–88. doi: 10.1074/jbc.M212835200. [DOI] [PubMed] [Google Scholar]
- 128.Vahedi S, Chueh FY, Dutta S, Chandran B, Yu CL. Nuclear lymphocyte-specific protein tyrosine kinase and its interaction with CR6-interacting factor 1 promote the survival of human leukemic T cells. Oncol Rep. 2015;34(1):43–50. doi: 10.3892/or.2015.3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Skrtic M, Sriskanthadevan S, Jhas B, Gebbia M, Wang X, Wang Z, Hurren R, Jitkova Y, Gronda M, Maclean N, Lai CK, Eberhard Y, Bartoszko J, Spagnuolo P, Rutledge AC, Datti A, Ketela T, Moffat J, Robinson BH, Cameron JH, Wrana J, Eaves CJ, Minden MD, Wang JC, Dick JE, Humphries K, Nislow C, Giaever G, Schimmer AD. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer cell. 2011;20(5):674–88. doi: 10.1016/j.ccr.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Quigley DA, Fiorito E, Nord S, Van Loo P, Alnaes GG, Fleischer T, Tost J, Moen Vollan HK, Tramm T, Overgaard J, Bukholm IR, Hurtado A, Balmain A, Borresen-Dale AL, Kristensen V. The 5p12 breast cancer susceptibility locus affects MRPS30 expression in estrogen-receptor positive tumors. Mol Oncol. 2014;8(2):273–84. doi: 10.1016/j.molonc.2013.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Darekar SD, Mushtaq M, Gurrapu S, Kovalevska L, Drummond C, Petruchek M, Tirinato L, Di Fabrizio E, Carbone E, Kashuba E. Mitochondrial ribosomal protein S18–2 evokes chromosomal instability and transforms primary rat skin fibroblasts. Oncotarget. 2015;6(25):21016–28. doi: 10.18632/oncotarget.4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Jia Y, Ye L, Ji K, Zhang L, Hargest R, Ji J, Jiang WG. Death-associated protein-3, DAP-3, correlates with preoperative chemotherapy effectiveness and prognosis of gastric cancer patients following perioperative chemotherapy and radical gastrectomy. Br J Cancer. 2014;110(2):421–9. doi: 10.1038/bjc.2013.712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wazir U, Jiang WG, Sharma AK, Mokbel K. The mRNA expression of DAP3 in human breast cancer: correlation with clinicopathological parameters. Anticancer Res. 2012;32(2):671–4. [PubMed] [Google Scholar]
- 134.Han MJ, Chiu DT, Koc EC. Regulation of mitochondrial ribosomal protein S29 (MRPS29) expression by a 5’-upstream open reading frame. Mitochondrion. 2010;10(3):274–83. doi: 10.1016/j.mito.2009.12.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Jacques C, Fontaine JF, Franc B, Mirebeau-Prunier D, Triau S, Savagner F, Malthiery Y. Death-associated protein 3 is overexpressed in human thyroid oncocytic tumours. Br J Cancer. 2009;101(1):132–8. doi: 10.1038/sj.bjc.6605111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Jacques C, Chevrollier A, Loiseau D, Lagoutte L, Savagner F, Malthiery Y, Reynier P. mtDNA controls expression of the Death Associated Protein 3. Exp Cell Res. 2006;312(6):737–45. doi: 10.1016/j.yexcr.2005.11.027. [DOI] [PubMed] [Google Scholar]
- 137.Kechavarzi B, Janga SC. Dissecting the expression landscape of RNA-binding proteins in human cancers. Genome Biol. 2014;15(1):R14. doi: 10.1186/gb-2014-15-1-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Baig S, Seevasant I, Mohamad J, Mukheem A, Huri HZ, Kamarul T. Potential of apoptotic pathway-targeted cancer therapeutic research: Where do we stand? Cell death & disease. 2016;7:e2058. doi: 10.1038/cddis.2015.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bai L, Wang S. Targeting apoptosis pathways for new cancer therapeutics. Annu Rev Med. 2014;65:139–55. doi: 10.1146/annurev-med-010713-141310. [DOI] [PubMed] [Google Scholar]
- 140.Hassan M, Watari H, AbuAlmaaty A, Ohba Y, Sakuragi N. Apoptosis and molecular targeting therapy in cancer. Biomed Res Int. 2014;2014:150845. doi: 10.1155/2014/150845. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 141.DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. 2016;2(5):e1600200. doi: 10.1126/sciadv.1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, Casimiro MC, Wang C, Fortina P, Addya S, Pestell RG, Martinez-Outschoorn UE, Sotgia F, Lisanti MP. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. 2009;8(23):3984–4001. doi: 10.4161/cc.8.23.10238. [DOI] [PubMed] [Google Scholar]
- 143.Feron O. Pyruvate into lactate and back: from the Warburg effect to symbiotic energy fuel exchange in cancer cells. Radiother Oncol. 2009;92(3):329–33. doi: 10.1016/j.radonc.2009.06.025. [DOI] [PubMed] [Google Scholar]
- 144.Pearce S, Nezich CL, Spinazzola A. Mitochondrial diseases: translation matters. Mol Cell Neurosci. 2013;55:1–12. doi: 10.1016/j.mcn.2012.08.013. [DOI] [PubMed] [Google Scholar]
- 145.McKee EE, Ferguson M, Bentley AT, Marks TA. Inhibition of mammalian mitochondrial protein synthesis by oxazolidinones. Antimicrob Agents Chemother. 2006;50(6):2042–9. doi: 10.1128/AAC.01411-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Yongsheng J, Xiaoyun M, Xiaoli W, Xin L, Haitao Y, Xiaoyan L, Jianquan Z. Antitumor activity of erythromycin on human neuroblastoma cell line (SH-SY5Y) J Huazhong Univ Sci Technolog Med Sci. 2011;31(1):33–8. doi: 10.1007/s11596-011-0146-4. [DOI] [PubMed] [Google Scholar]
- 147.Rubins JB, Charboneau D, Alter MD, Bitterman PB, Kratzke RA. Inhibition of mesothelioma cell growth in vitro by doxycycline. J Lab Clin Med. 2001;138(2):101–6. doi: 10.1067/mlc.2001.116591. [DOI] [PubMed] [Google Scholar]
- 148.van den Bogert C, Dontje BH, Kroon AM. The antitumour effect of doxycycline on a T-cell leukaemia in the rat. Leuk Res. 1985;9(5):617–23. doi: 10.1016/0145-2126(85)90142-0. [DOI] [PubMed] [Google Scholar]
- 149.van den Bogert C, van Kernebeek G, de Leij L, Kroon AM. Inhibition of mitochondrial protein synthesis leads to proliferation arrest in the G1-phase of the cell cycle. Cancer Lett. 1986;32(1):41–51. doi: 10.1016/0304-3835(86)90037-6. [DOI] [PubMed] [Google Scholar]
- 150.van den Bogert C, Dontje BH, Holtrop M, Melis TE, Romijn JC, van Dongen JW, Kroon AM. Arrest of the proliferation of renal and prostate carcinomas of human origin by inhibition of mitochondrial protein synthesis. Cancer Res. 1986;46(7):3283–9. [PubMed] [Google Scholar]
- 151.Kroon AM, Dontje BH, Holtrop M, Van den Bogert C. The mitochondrial genetic system as a target for chemotherapy: tetracyclines as cytostatics. Cancer Lett. 1984;25(1):33–40. doi: 10.1016/s0304-3835(84)80023-3. [DOI] [PubMed] [Google Scholar]
- 152.Ferreri AJ, Govi S, Pasini E, Mappa S, Bertoni F, Zaja F, Montalban C, Stelitano C, Cabrera ME, Giordano Resti A, Politi LS, Doglioni C, Cavalli F, Zucca E, Ponzoni M, Dolcetti R. Chlamydophila psittaci eradication with doxycycline as first-line targeted therapy for ocular adnexae lymphoma: final results of an international phase II trial. J Clin Oncol. 2012;30(24):2988–94. doi: 10.1200/JCO.2011.41.4466. [DOI] [PubMed] [Google Scholar]
- 153.Ferreri AJ, Ponzoni M, Guidoboni M, Resti AG, Politi LS, Cortelazzo S, Demeter J, Zallio F, Palmas A, Muti G, Dognini GP, Pasini E, Lettini AA, Sacchetti F, De Conciliis C, Doglioni C, Dolcetti R. Bacteria-eradicating therapy with doxycycline in ocular adnexal MALT lymphoma: a multicenter prospective trial. J Natl Cancer Inst. 2006;98(19):1375–82. doi: 10.1093/jnci/djj373. [DOI] [PubMed] [Google Scholar]
- 154.Reed GA, Schiller GJ, Kambhampati S, Tallman MS, Douer D, Minden MD, Yee KW, Gupta V, Brandwein J, Jitkova Y, Gronda M, Hurren R, Shamas-Din A, Schuh AC, Schimmer AD. A Phase 1 study of intravenous infusions of tigecycline in patients with acute myeloid leukemia. Cancer Med. 2016;5(11):3031–3040. doi: 10.1002/cam4.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Chen DZ, Patel DV, Hackbarth CJ, Wang W, Dreyer G, Young DC, Margolis PS, Wu C, Ni ZJ, Trias J, White RJ, Yuan Z. Actinonin, a naturally occurring antibacterial agent, is a potent deformylase inhibitor. Biochemistry. 2000;39(6):1256–62. doi: 10.1021/bi992245y. [DOI] [PubMed] [Google Scholar]
- 156.Richter U, Lahtinen T, Marttinen P, Myohanen M, Greco D, Cannino G, Jacobs HT, Lietzen N, Nyman TA, Battersby BJ. A mitochondrial ribosomal and RNA decay pathway blocks cell proliferation. Curr Biol. 2013;23(6):535–41. doi: 10.1016/j.cub.2013.02.019. [DOI] [PubMed] [Google Scholar]
- 157.Battersby BJ, Richter U. Why translation counts for mitochondria - retrograde signalling links mitochondrial protein synthesis to mitochondrial biogenesis and cell proliferation. J Cell Sci. 2013;126(Pt 19):4331–8. doi: 10.1242/jcs.131888. [DOI] [PubMed] [Google Scholar]
- 158.Sheth A, Escobar-Alvarez S, Gardner J, Ran L, Heaney ML, Scheinberg DA. Inhibition of human mitochondrial peptide deformylase causes apoptosis in c-myc-overexpressing hematopoietic cancers. Cell death & disease. 2014;5:e1152. doi: 10.1038/cddis.2014.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Escobar-Alvarez S, Gardner J, Sheth A, Manfredi G, Yang G, Ouerfelli O, Heaney ML, Scheinberg DA. Inhibition of human peptide deformylase disrupts mitochondrial function. Mol Cell Biol. 2010;30(21):5099–109. doi: 10.1128/MCB.00469-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Peng B, Zhang X, Cao F, Wang Y, Xu L, Cao L, Yang C, Li M, Uzan G, Zhang D. Peptide deformylase inhibitor actinonin reduces celastrol’s HSP70 induction while synergizing proliferation inhibition in tumor cells. BMC Cancer. 2014;14:146. doi: 10.1186/1471-2407-14-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 162.Bhat NK, Emeh JK, Niranjan BG, Avadhani NG. Inhibition of mitochondrial protein synthesis during early stages of aflatoxin B1-induced hepatocarcinogenesis. Cancer Res. 1982;42(5):1876–80. [PubMed] [Google Scholar]
- 163.Peng X, Chen K, Chen J, Fang J, Cui H, Zuo Z, Deng J, Chen Z, Geng Y, Lai W. Aflatoxin B1 affects apoptosis, expression of Bax, Bcl-2, and Caspase-3 in thymus and bursa of fabricius in broiler chickens. Environ Toxicol. 2016;31(9):1113–20. doi: 10.1002/tox.22120. [DOI] [PubMed] [Google Scholar]
- 164.Peng X, Yu Z, Liang N, Chi X, Li X, Jiang M, Fang J, Cui H, Lai W, Zhou Y, Zhou S. The mitochondrial and death receptor pathways involved in the thymocytes apoptosis induced by aflatoxin B1. Oncotarget. 2016;7(11):12222–34. doi: 10.18632/oncotarget.7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ban N, Beckmann R, Cate JH, Dinman JD, Dragon F, Ellis SR, Lafontaine DL, Lindahl L, Liljas A, Lipton JM, McAlear MA, Moore PB, Noller HF, Ortega J, Panse VG, Ramakrishnan V, Spahn CM, Steitz TA, Tchorzewski M, Tollervey D, Warren AJ, Williamson JR, Wilson D, Yonath A, Yusupov M. A new system for naming ribosomal proteins. Curr Opin Struct Biol. 2014;24:165–9. doi: 10.1016/j.sbi.2014.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Amunts A, Brown A, Bai X, Llácer JL, Hussain T, Emsley P, Long F, Murshudov G, Scheres SHW, Ramakrishnan V. Structure of the yeast mitochondrial large ribosomal subunit. Science. 2014;343:1485–89. doi: 10.1126/science.1249410. [DOI] [PMC free article] [PubMed] [Google Scholar]