

Abstract
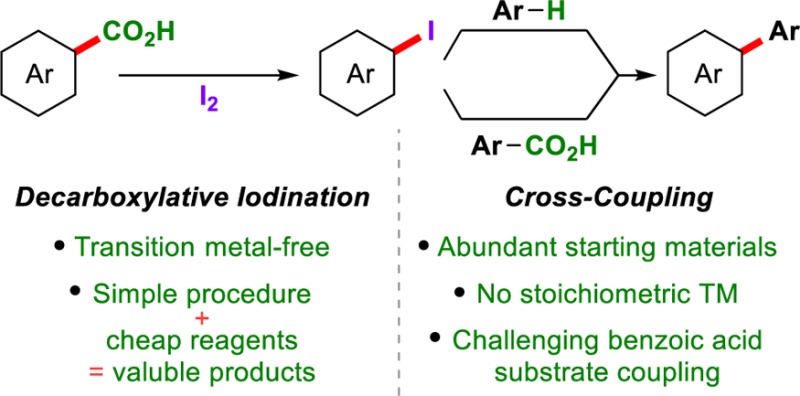
Constructing products of high synthetic value from inexpensive and abundant starting materials is of great importance. Aryl iodides are essential building blocks for the synthesis of functional molecules, and efficient methods for their synthesis from chemical feedstocks are highly sought after. Here we report a low-cost decarboxylative iodination that occurs simply from readily available benzoic acids and I2. The reaction is scalable and the scope and robustness of the reaction is thoroughly examined. Mechanistic studies suggest that this reaction does not proceed via a radical mechanism, which is in contrast to classical Hunsdiecker-type decarboxylative halogenations. In addition, DFT studies allow comparisons to be made between our procedure and current transition-metal-catalyzed decarboxylations. The utility of this procedure is demonstrated in its application to oxidative cross-couplings of aromatics via decarboxylative/C–H or double decarboxylative activations that use I2 as the terminal oxidant. This strategy allows the preparation of biaryls previously inaccessible via decarboxylative methods and holds other advantages over existing decarboxylative oxidative couplings, as stoichiometric transition metals are avoided.
1. Introduction
The advent of cross-coupling reactions revolutionized the thought process for C–C bond formation, particularly when constructing biaryls—common structures in many biologically active and functional molecules from blockbuster pharmaceuticals to day-to-day electronic devices.1,2 These methods generally consist of the coupling of an organometallic reagent with an aryl halide in the presence of a transition metal catalyst (Scheme 1, a). More recently, researchers in the areas of C–H and decarboxylative activation have looked to develop more efficient routes for cross-coupling.3,4 In particular, the coupling of benzoic acids with either an arene or a second benzoic acid has gained great interest, as both aryl donors are low-cost and readily available and, in an ideal scenario, H2O and CO2 are formed as the only waste products (Scheme 1, b).5,6 Progress in this area has already begun; however, current procedures generally yield poor reaction scope and require stoichiometric transition metals, thus limiting their applicability. Furthermore, the need for stoichiometric transition metals greatly reduces atom-economy and brings into question the true benefits of these procedures (Scheme 1, b) over traditional cross-couplings (Scheme 1, a).4i
Scheme 1. Comparison of Traditional Cross-Couplings, Decarboxylative Oxidative Couplings, and This Report.
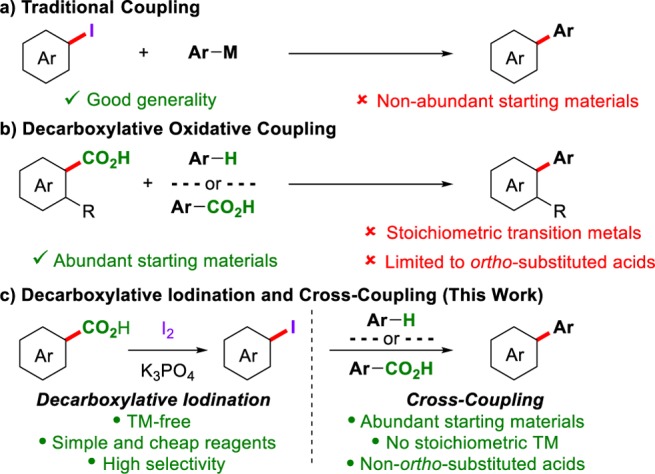
Aryl halides are prolific building blocks in organic synthesis.7 They have proved integral to the development of cross-coupling reactions, and they also undergo a variety of other transformations, such as nucleophilic substitution and metalation among many others.8 Due to their undeniable importance, efficient methods for aryl halide formation from chemical feedstocks are highly sought after.9 When developing a procedure for aryl halide formation, one should also consider the possibility of directly transforming the aryl halide in one-pot. This would allow for the streamlined synthesis of functional molecules from abundant starting materials via aryl halide intermediates (Scheme 1c). Whether the streamlined synthesis is successful or not will depend upon the compatibility of each step (iodination/functionalization) in the synthesis. By developing simple procedures for aryl halide formation, the chances of success are greatly improved, as the number of potentially inhibitory side products is reduced.
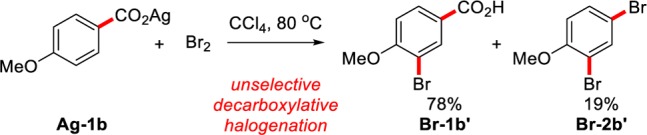
Using the carboxyl group as a site for selective transformations is an attractive prospect in synthesis. Benzoic acids are inexpensive and readily available, and their conversion through decarboxylative pathways holds potential in atom-economical processes. With regard to aryl halide formation, the Hunsdiecker reaction is a well-known process for the decarboxylative halogenation of anhydrous silver carboxylates with elemental halogens.10 This reaction affords good reactivity for aliphatic carboxylic acids; however, aromatic carboxylic acids have traditionally been poor substrates for this process. In particular, while some electron-deficient aromatics react with variable yields, electron-rich aromatics undergo electrophilic halogenation instead (Scheme 2).11 More recent efforts toward a decarboxylative halogenation of aromatic acids are of limited utility, as they (a) require stoichiometric transition metal additives, (b) show poor substrate scope, and/or (c) show poor selectivity.12,13 For these reasons, the conversion of aromatic benzoic acids into the corresponding aryl halides is generally carried out via a multi-step process.14 Therefore, the development of a method for the direct conversion of benzoic acids to aryl halides via decarboxylation remains an unsolved challenge.
Scheme 2. Current Status of the Aromatic Hunsdiecker Reaction.
We report here a general and cost-effective method for the decarboxylative iodination of (hetero)aromatic acids simply using readily available I2 (Scheme 1c). We detail the first examples of a transition-metal-free decarboxylative iodination of heteroaromatic acids and the first mechanistic study (through radical clock, DFT, and Hammett plot analyses) of an aromatic decarboxylative halogenation. The simplicity of the procedure also allows it to be applied to a streamlined synthesis of biaryls. In this process, the formed aryl halide can be subsequently cross-coupled with either an arene or a second benzoic acid using a copper catalyst in a one-pot process. This streamlined synthesis holds several advantages over current decarboxylative oxidative strategies, as stoichiometric transition metals are avoided and normally poorly reactive benzoic acids (e.g., electron-rich and non-ortho-substituted acids) are efficiently coupled.
2. Results and Discussion
2.1. Development of a Transition-Metal-Free Decarboxylative Iodination
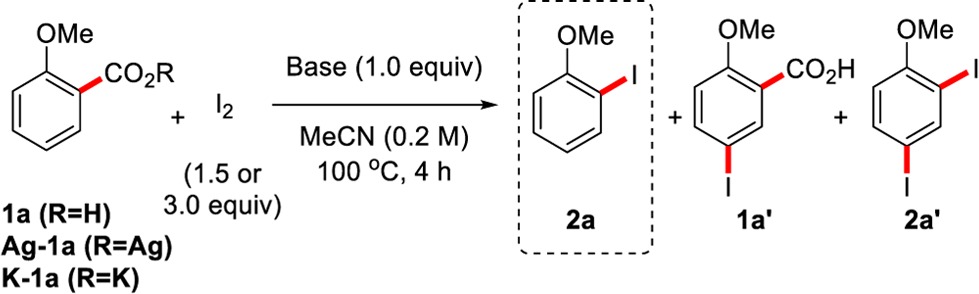
To begin our study, we investigated a classical aromatic Hunsdiecker-type decarboxylation—the reaction of the elemental halogens with the silver salts of benzoic acids—considering also that silver on its own can promote the decarboxylation of aromatic acids (Table 1).15 However, similarly to previous reports using Br2 (Scheme 2),11 the mixing of Ag(I)-2-methoxybenzoate Ag-1a with I2 gave a mixture of the unwanted iodinated acid 1a′ (74%) and the diiodinated product 2a′ (10%), but none of desired aryl iodide 2a (Table 1, entry 1). Thus, an undesired iodination process appears to be prevalent under Hunsdiecker-type conditions, preventing the desired decarboxylative iodination process. To our surprise, when the reaction was carried out in the absence of silver (using K-1a), the desired product 2a was formed in excellent yield and high selectivity (entry 2), revealing a previously unknown and remarkably simple transition-metal-free procedure for the decarboxylative iodination of aromatic acids. The reasoning for this switch in chemoselectivity is currently unclear, but our results suggest that a strong C–H iodinating agent is formed in the presence of Ag(I), and this is minimized when using the potassium benzoate K-1a.16 To avoid the preparation of benzoate salts, a screening of carbonate bases revealed that benzoic acids are suitable reagents and that, although all carbonate bases show some reactivity, the more soluble carbonate bases show improved reactivity (Table 1, entries 3–6). The screening of many inorganic bases revealed K3PO4 as the base of choice for this reaction, allowing the product 2a to be isolated in an excellent yield of 90% without the need for column chromatography (entry 7). A more atom-economic procedure can be had by decreasing the loading of I2 to 1.5 equiv and increasing the temperature of the reaction to 170 °C to provide the product in 73% yield (entry 8). Finally, control reactions revealed that (a) the reaction is sensitive to water (entry 9);17 (b) no loss of reactivity is observed when the experiment is conducted in the dark (entry 10); and (c) in the absence of a base, the reaction does not proceed (entry 11). Replacing I2 with Br2 led to the formation of the brominated acid Br-1a′ and dibrominated product Br-2a′, possibly due to the higher electrophilicity of Br2 (entry 12). Efforts toward a selective decarboxylative bromination are ongoing in our laboratory.
Table 1. Optimization of the Transition-Metal-Free Decarboxylative Iodinationa.
| entry | R | base | 1a | 2a | 1a′ | 2a′ |
|---|---|---|---|---|---|---|
| 1 | Ag | – | 14 | 0 | 74 | 10 |
| 2 | K | – | 9 | 90 | 2 | trace |
| 3 | H | Li2CO3 | 89 | 11 | trace | 0 |
| 4 | H | Na2CO3 | 76 | 23 | trace | 0 |
| 5 | H | K2CO3 | 64 | 31 | trace | 0 |
| 6 | H | Cs2CO3 | 57 | 38 | trace | trace |
| 7 | H | K3PO4 | 4 | 93 (90)b | 1 | trace |
| 8c | H | K3PO4 | 7 | 73 | 2 | 0 |
| 9d | H | K3PO4 | 62 | 34 | 0 | 0 |
| 10e | H | K3PO4 | 3 | 94 | 1 | trace |
| 11 | H | – | 94 | 0 | 2 | 0 |
| 12f | H | K3PO4 | trace | 0 | 23 | 72 |
Reaction conditions: benzoic acid/benzoate (0.2 mmol), I2 (0.6 mmol, 3.0 equiv), base (0.2 mmol, 1.0 equiv), MeCN (1.0 mL), 100 °C, 4 h.
Yield in parentheses is of isolated material. Isolated as a mixture with 2a′ (2a:2a′ > 100:1).
I2 (0.3 mmol, 1.5 equiv,), 170 °C, 16 h, 1,4-dioxane used as solvent.
1.0 equiv of H2O added.
Reaction performed in the dark.
I2 was replaced with Br2 to form the corresponding bromides Br-1a′ and Br-2a′.
2.2. Scope of the Transition-Metal-Free Decarboxylative Iodination
The development of such a simple route for the formation of aryl iodides was highly appealing, and we were keen to examine its applicability to other substrates (Scheme 3). Our first observation was that the system is not limited to ortho-substituted benzoic acids, in contrast to many transition-metal-catalyzed decarboxylations (2b, 2c).4,12 Although both ortho- and para-anisic acid (2a, 2b) are reactive, meta-anisic acid (2d) was unreactive, suggesting that the position of decarboxylation must be sufficiently nucleophilic.18 Increasing the electron density on the aromatic acid greatly improves the reactivity of the substrate and allows the temperature to be reduced, even to room temperature in some cases (2e–2h). These low-temperature decarboxylations are remarkable, as analogous transition-metal-catalyzed procedures require highly activated di-ortho-substituted benzoic acids and temperatures of 160 °C, and we are unaware of any other transition-metal-free decarboxylative transformations of aromatic acids occurring at these low temperatures.12f,19 Polymethylated benzoic acids have previously proved poorly reactive in decarboxylative processes;20 however, we observed good to excellent reactivity with these substrates (2i–2k), and even ortho-toluic acid (2l) showed some reactivity when both the temperature and loadings of I2 were increased. Likewise, unsubstituted 1-napthoic acid (1m) was reactive under more forcing conditions; however, a small amount of diiodination (2m′) was also observed. The position of diiodination in this and other products is highlighted by the position of the asterisks in each scheme. The reactivity and selectivity of napthoic acid can be improved by the introduction of a methyl group to this substrate (2n). Attempts to decarboxylate salicylic acids were unsuccessful; however, moderate to excellent yields were obtained upon protection of the hydroxyl group (2o, 2p). The system is tolerant of bearing amide and amino functionality, and, in some cases, the temperature could again be lowered to room temperature (2q–2t).21 Examining the reactivity of halo-substituted benzoic acids also clearly revealed that the presence of more electron-withdrawing substituents, while still producing good yields, reduces the reactivity of the system (2u–2ab).22 In light of this, we were surprised to observe that polyfluorinated benzoic acids showed excellent reactivity under these conditions (2ac–2ae); we are currently studying the reason for this unexpected reactivity.23 The procedure was also applied to the methylestrone-2-carboxylic acid 1af to provide the corresponding iodide in high yield (2af).24 In accord with the general trend of reactivity, substrates that do not bear electron-donating substituents were unreactive under these conditions (2ag–2aj).25 Finally, if a more atom-economical procedure is desired, the equivalents of I2 can be reduced to between 1.0 and 2.0 equiv by increasing the temperature of the reaction to 170 °C. The results for these atom-efficient couplings are provided in square brackets in both Schemes 3 and 4.26
Scheme 3. Scope of the Decarboxylative Iodination of Benzoic Acids.
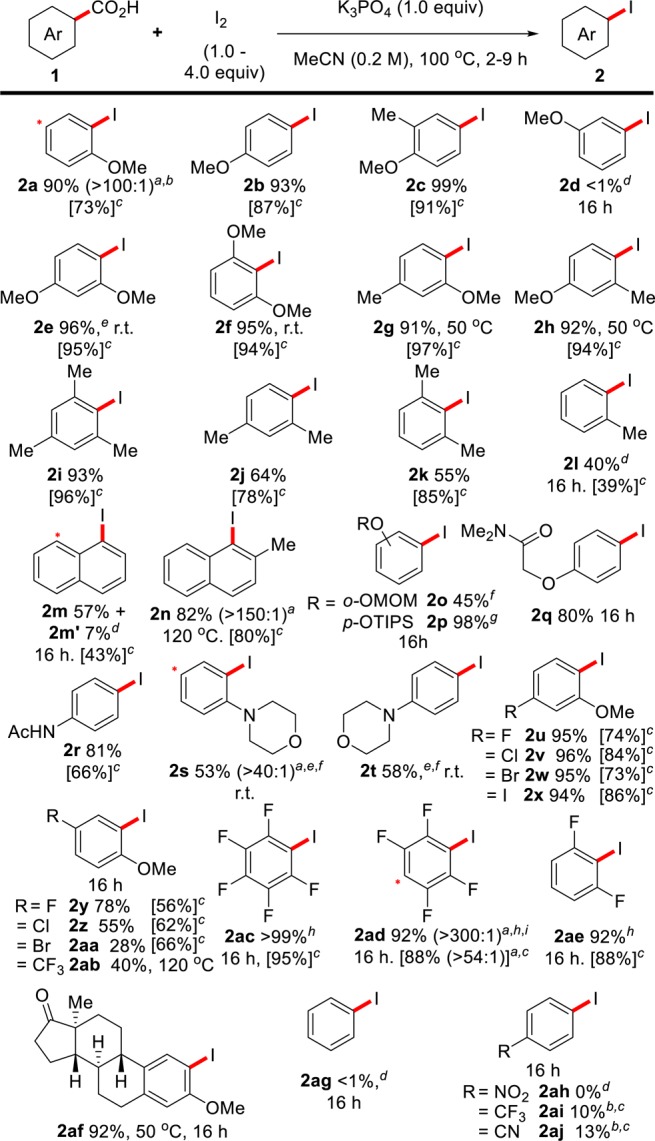
Reactions carried out at a 0.5 mmol scale of 1.
Ratios in brackets indicate mono-:diiodinated material by crude GC-FID analysis. Asterisk indicates position of diiodination.
I2 (3.0 equiv).
NMR yield for reactions employing I2 (1.0–2.0 equiv), 170 °C and 1,4-dioxane or o-DCB as solvent.
I2 (6.0 equiv), 140 °C.
I2 (2.0 equiv).
MeCN (5.0 mL).
I2 (3.0 equiv), 1,4-dioxane (1.0 M), 170 °C.
Yields determined by quantitative 19F NMR.
I2 (2.5 equiv).
Scheme 4. Scope of the Decarboxylative Iodination of Heterobenzoic Acids.
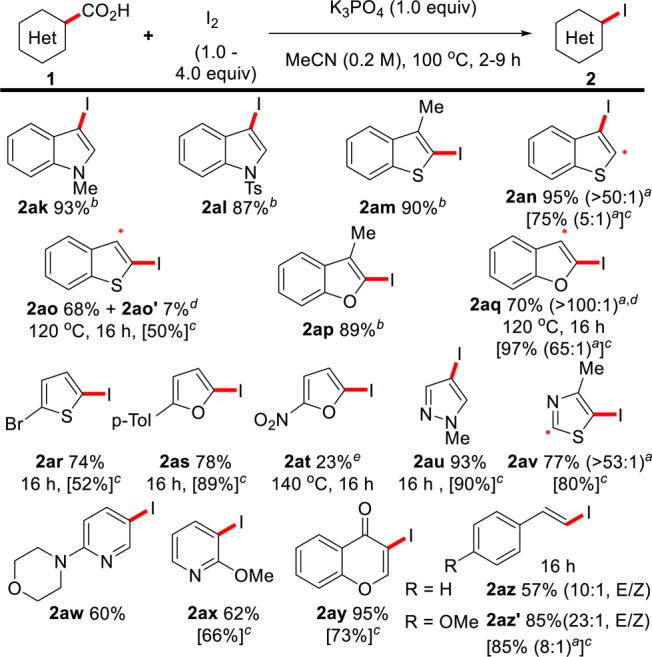
Reactions carried out at a 0.5 mmol scale of 1.
Ratios in brackets indicate mono-:diiodinated material by crude GC-FID analysis. Asterisk indicates position of diiodination.
I2 (2.0 equiv).
NMR yield for reactions employing I2 (1.0–2.0 equiv), 170 °C and 1,4-dioxane or o-DCB as solvent.
MeCN (5.0 mL).
I2 (6.0 equiv).
The compatibility of the reaction with various functional groups was investigated by means of a robustness screen (see Supporting Information (SI), Table S2a,b).27 We screened 30 additives in order to gain an informed view of the robustness of this reaction. This study revealed that many important functional groups, such as cyano, trifluoromethyl, nitro, tosylate, triflate, mesylate, and halo functionalities, were fully compatible with the reaction. On the other hand, hydroxyl and amino functionalities along with other nucleophilic (hetero)arenes were poorly tolerated. Likewise, alkene and alkyne additives were also detrimental to the reaction, although an internal alkyne additive was moderately tolerated. Most interesting was that aldehyde, ketone, and ester functionalities were all compatible with the reaction conditions.
Methods for the decarboxylative iodination of heteroaromatic acids are scarce and currently require stoichiometric transition metals and temperatures >160 °C;12,13 thus, we were keen to apply our conditions to these substrates (Scheme 4). Indoles bearing either methyl or tosyl protecting groups (2ak, 2al) showed good reactivity under our conditions, as did benzothiophenes and benzofurans (2am–2aq). Conveniently, the iodination can also be directed to the less nucleophilic C2 position of these substrates by simply using the C2-carboxyl-substituted substrates (2ao, 2aq). Similarly, thiophenes (2ar), furans (2as, 2at), pyrazoles (2au), and thiazoles (2av) were all compatible under these conditions. Once again, the yields provided in square brackets in Scheme 4 represent the outcome of the reaction when lower equivalents of I2 (1.0–2.0 equiv) and higher temperatures (170 °C) are used.26 In agreement with benzoic acids, less electron-rich heteroaromatic acids require more forcing conditions (2at). Pyridines bearing morpholino or methoxy substituents and chromone-3-carboxylic acid also display good reactivity (2aw–2ay). Finally, cinnamic acids are also reactive substrates; however, some isomerization of the C–C double bond was observed (2az–2az′).28
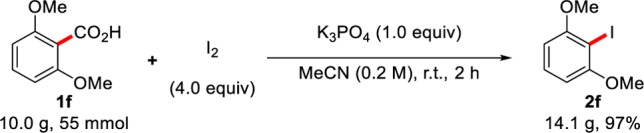
In order to further demonstrate the utility of our procedure, we looked to achieve a multi-gram synthesis of 1-iodo-2,6-dimethoxybenzene 2f (Scheme 5). Thus, when 10.0 g of benzoic acid 1f was subjected to the standard conditions at room temperature, the desired product 2f could be isolated with no detriment to the final yield (97%, 14.1 g)29 and without the requirement for silica gel chromatography. We believe that the broad availability of benzoic acid substrates along with the low-cost and scalability of this method has potential in large-scale synthesis.
Scheme 5. Multi-Gram-Scale Synthesis of 1-Iodo-2,6-dimethoxybenzene 2f.
2.3. Mechanistic Studies on the Transition-Metal-Free Decarboxylative Iodination
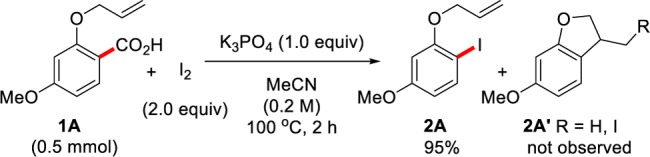
Our attention then turned to investigating the mechanism of this reaction. It is largely considered that the classical Hunsdiecker reaction—the decarboxylative halogenation of aliphatic silver carboxylates—proceeds via a radical pathway; therefore, we were interested to determine whether our system reacts in a similar manner.10 A possible mechanism via this route is given in Scheme 6; thus, upon formation of the benzoyl hypoiodite I(A)(30) from the corresponding benzoic acid (1a), base, and I2, homolytic bond breaking of the O–I bond would provide the benzoyl radical II and an iodine radical (Scheme 6, pathway A). A radical decarboxylation event would give the aryl radical III, and subsequent radical recombination would provide aryl iodide (2a). An alternative pathway that does not involve radical intermediates was also considered (Scheme 6, pathway B). This pathway may proceed via transition state IV(A) to give the aryl iodide 2a directly from the benzoyl hypoiodite I(A). This form of concerted decarboxylation–iodination mechanism is reminiscent of recently proposed pathways for transition-metal-catalyzed decarboxylations.31 We set out to delineate between these two pathways by initially conducting a radical clock experiment with substrate 1A (Scheme 7). Upon formation of the corresponding aryl radical from 1A, the rate constant for intramolecular cyclization to provide products of type 2A′ is of the order k = 8 × 109 s–1.32 Upon exposure of substrate 1A to our conditions, we observed only the iodinated product 2A and none of the cyclized product 2A′. This experiment strongly suggests that a radical mechanism is not in operation, in contrast to the classical Hunsdiecker reaction of aliphatic carboxylic acids and previous mechanistic proposals for transition-metal-free decarboxylative iodinations.10,13h,13i In order to assess the feasibility of our proposed non-radical pathway, we carried out a preliminary density functional theory (DFT) study (Scheme 6, pathway B, see values in parentheses).33 Decarboxylation via transition state IV(A) afforded a barrier of 27.6 kcal mol–1, which is reasonable for a reaction that can occur at 100 °C. Although further investigations are required, our current data support the pathway for decarboxylation via transition state IV(A).
Scheme 6. Possible Pathways for the Decarboxylative Iodination of Aromatic Acids.

Pathway A: radical decarboxylation–radical recombination pathway. Pathway B: concerted decarboxylation–iodination pathway.
Structures and energies calculated by DFT (B97D3/LanL2DZ for I, 6-31G(d) for other atoms); Gibbs free energies (G) are in kcal mol–1.
Total energy for 2a + CO2.
Scheme 7. Radical Clock Experiment with Benzoic Acid 1A.
Previous DFT studies by our group and others have proved highly useful when investigating the mechanism of transition-metal-catalyzed decarboxylations.31 In particular, they have proved essential for delineating why ortho-substituted benzoic acids are more reactive than non-ortho-substituted benzoic acids—a phenomenon commonly termed the ortho effect. Previous calculations have shown that the energy of the ortho-substituted substrate is higher than that of its meta and para isomers due to steric clash with the carboxyl group; however, the energies for the transition states of each isomer are close to equal. Overall, this causes the decarboxylation barrier to be lowered for ortho-substituted benzoic acids in transition-metal-catalyzed procedures. For example, the 2-methoxy substrate (I(C)) is destabilized with respect to the 4-methoxy-isomer (I(D)) by 5.8 kcal mol–1 in palladium-mediated decarboxylations; however, the transition states (IV(C/D)) only differ by 1.6 kcal mol–1 (Scheme 8, ii). Thus, the overall energy barrier is 4.2 kcal mol–1 lower for the ortho-substituted benzoic acid in palladium-mediated decarboxylations.31e Similarly, the difference in the decarboxylation barrier for silver-mediated decarboxylations is 6.0 kcal mol–1 due to the destabilization of the ortho-substituted substrate by 6.3 kcal mol–1 (I(E) vs I(F)) (Scheme 8, iii).31d We were keen to establish whether a similar ortho effect could be applied to our transition-metal-free system; therefore, we compared the barriers of decarboxylation for 2-methoxybenzoic acid and 4-methoxybenzoic acid (Scheme 8, i). From our experimental results (Scheme 3), we had observed that the corresponding aryl iodides (2a and 2b) were formed in high yield at 100 °C; however, whereas only 3.0 equiv of I2 was necessary for the reaction with 2-methoxybenzoic acid, 4.0 equiv was needed with 4-methoxybenzoic acid. This shows that, although the reactivity is similar, 2-methoxybenzoic acid is slightly more susceptible to decarboxylation under our conditions. Our DFT study supported this result, as we found that the barrier for decarboxylation with 2-methoxybenzoic acid was slightly lower than that for 4-methoxybenzoic acid by 0.9 kcal mol–1 (Scheme 8, ΔGA⧧ = 27.6 kcal mol–1 and ΔGB⧧ = 28.5 kcal mol–1 for 2-methoxy- and 4-methoxybenzoic acid, respectively). Further inspection of these data revealed that the 2-methoxy-substituted hypoiodite (I(A)) is 4.5 kcal mol–1 higher in energy than its 4-methoxy analogue (I(B)). This difference in energy is likely due to a steric effect when a group is present ortho to the carboxyl group and is consistent with the ortho effect that is witnessed in transition-metal-catalyzed procedures. However, whereas the difference in energy between ortho- and para-substituted transition states (IV) is small (<2.0 kcal mol–1) for transition-metal-catalyzed decarboxylation, we observe a significant energy difference of 3.6 kcal mol–1 for the decarboxylative iodination. In light of this, we suggest that an ortho effect resulting from steric destabilization is observed in our system, which causes ortho-substituted benzoic acids (e.g., 2-methoxybenzoic acid 1a) to be more susceptible to decarboxylation than non-ortho-substituted substrates (e.g., 4-methoxybenzoic acid 1b). However, this effect is small, especially when compared to transition-metal-catalyzed procedures; therefore, the reactivity difference between ortho- and non-ortho-substituted benzoic acids is minimized in our system. These results somewhat explain why, in our system, non-ortho-substituted benzoic acids are suitable substrates and further display the advantages of our procedure over those that require transition metals.
Scheme 8. Comparing the Ortho Effect of Transition-Metal-Mediated Decarboxylations and Transition-Metal-Free Decarboxylative Iodination.

(i) Investigating the ortho effect in the transition-metal-free decarboxylative iodination. Energies measured in kcal mol–1 for DFT modeling using an acetonitrile solvent correction. Structures and energies calculated by DFT (LanL2DZ for I, 6-31G(d) for other atoms); Gibbs free energies (G) are in kcal mol–1. (ii) The ortho effect in palladium-catalyzed decarboxylations as reported by Su et al.31e (iii) The ortho effect in silver-catalyzed decarboxylations as reported by Su et al.31d
Hammett plot analysis of the initial rates of decarboxylative iodination of a series of meta- and para-substituted 2-methoxybenzoic acids provided a rho (ρ) value of −4.6, consistent with a substantial buildup of positive charge in the transition state IV (Figure 1).22 This value is comparable to those previously reported for electrophilic aromatic substitution-type reactions and supports our observation that electron-rich (hetero)aromatic acids are preferred substrates in this system.22b,34 Our computational study (Scheme 6, pathway B) does not suggest a distinct Wheland-type intermediate is formed. However, the calculated natural bond orbital (NBO) charges indicate a buildup of positive charge in the transition state. For example, the carbon atom ipso to the methoxy group in the pre-transition state VI(A) (see SI) has a value of +0.345, which rises to +0.416 in the transition state TS-IV(A). Therefore, the proposed concerted decarboxylation–iodination process (Scheme 6, pathway B) is consistent with the experimental Hammett plot. Further studies are necessary to better understand the mechanism of this reaction.35
Figure 1.

Hammett plot of the decarboxylative iodination. Equation of fit: y = −4.59x + 0.09. R2= 0.92. Position (meta/para) of substituent is with respect to the carboxyl group.
2.4. Applying the Decarboxylative Iodination toward Decarboxylative Oxidative Couplings
The coupling of a benzoic acid with an arene or a second benzoic acid via a C–C/C–H or C–C/C–C double activation, respectively, is a highly appealing transformation. Currently, most procedures to carry out this transformation require stoichiometric transition metal additives with only a few exceptions.4i However, these methods have their own limitations, for example: (1) the copper-catalyzed coupling of benzoic acids with arenes is restricted to the coupling of ortho-nitrobenzoic acids with heteroarenes and requires high loadings of the catalyst;5p (2) although the coupling of ortho- and non-ortho-substituted benzoic acids with arenes is possible under silver or photoredox catalysis, solvent quantities of the arene are required.5o,5q Therefore, more economical and general methods for oxidative decarboxylative couplings are of high importance.
Our simple procedure for decarboxylative iodination does not require transition metal additives and is applicable to non-ortho-substituted benzoic acids. Thus, we envisaged that this new decarboxylative protocol could be used as the cornerstone to address some of the current limitations in C–C/C–C and C–C/C–H oxidative couplings. Our approach to these oxidative couplings would consist of generating the aryl iodide from the benzoic acid and then cross-coupling the aryl iodide with an arene or a second benzoic acid in a one-pot protocol. This strategy is reminiscent to that used by Daugulis and co-workers36 for the copper-catalyzed cross-coupling of two arenes via double C–H activations, which proceeds through an aryl iodide intermediate. In light of this precedent, we were confident that a decarboxylative oxidative cross-coupling should be possible.
We initially investigated the coupling of a benzoic acid with an arene (Scheme 9). In this procedure, the benzoic acid first undergoes decarboxylative iodination, followed by copper-catalyzed cross-coupling using conditions based on those reported by Daugulis and co-workers.36 Daugulis reported that the presence of iodine inhibits the copper-catalyzed coupling step,36b leading to very long reaction times of up to 9 days, although dimethylaniline could be added to quench excess iodine in a multi-step process. In our investigation, we found that excess iodine could be more efficiently eliminated by the addition of Et3N after the decarboxylative iodination has concluded and before the coupling step is carried out.37
Scheme 9. Scope of the Decarboxylative Oxidative Cross-Coupling between Benzoic Acids and Arenes.
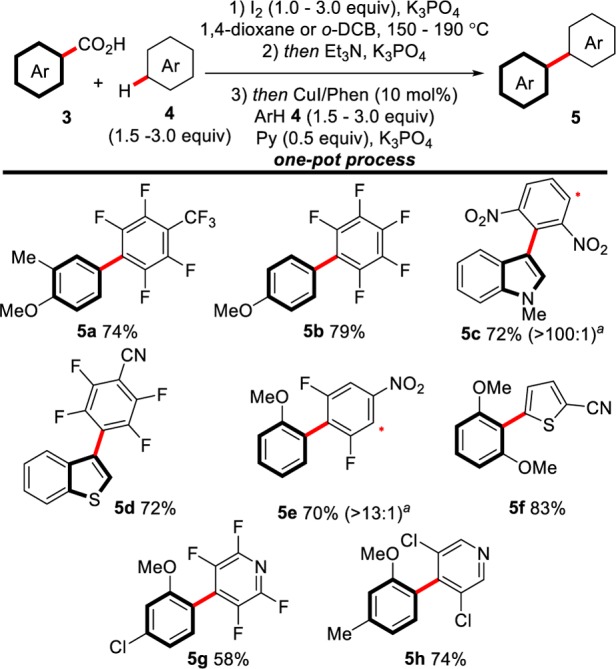
Reactions carried out at a 0.5 mmol scale of 4. All three steps are conducted between 150 and 190 °C. K3PO4 (6.5–8.0 equiv) total across three steps.
Ratios in brackets indicate ratio of regioisomeric products by crude GC-FID analysis. Asterisk indicates position of minor regioisomer.
Overall, the biaryl product is formed in a one-pot, three-step process consisting of (1) decarboxylative iodination, (2) iodine quench, and (3) cross-coupling, with the necessary reagents being added at each step.38 Importantly, the entire process is conducted in one-pot, and no workup or isolation is required between steps. Our initial results show that many combinations of benzoic acids and arenes can be coupled together. The reaction tolerates a variety of different functionalities, such as nitro (5c, 5e), chloro (5g, 5h), trifluoromethyl (5a), and cyano groups (5d, 5f). A range of aromatic acids are applicable in the cross-coupling, including methoxy- (e.g., 5b, 5f) and chloro-substituted (5g) benzoic acids and indole- (5c) and benzothiophene-carboxylic acids (5d). Most notable are the couplings of non-ortho-substituted benzoic acids 5a–5d, as these substrates are generally unreactive in current decarboxylative coupling procedures.
Our focus then turned to applying this procedure to the double decarboxylative cross-coupling of two aromatic acids (Scheme 10). Our strategy for the cross-coupling of benzoic acids with arenes could easily be applied to this process to provide a variety of biaryl products. Whereas current procedures provide high levels of homocoupled product (>9%), our procedure shows high selectivity for the cross-coupled product, with no homocoupled products, or only traces, being observed. A range of benzoic acids, including methoxy-substituted benzoic acids (e.g., 8a and 8b), benzothiophene-carboxylic acids (8c), and polyfluorinated benzoic acids (8d), could be coupled with polyfluorobenzoic acids in different combinations. These represent the first examples of double decarboxylative cross-couplings that do not require stoichiometric transition metal additives. Furthermore, products 8a and 8c represent the first examples of the coupling of a non-ortho-substituted benzoic acid in a double decarboxylative coupling. The power of this procedure can further be seen in the coupling of two electronically and sterically similar benzoic acids (8d–8f). Although the formation of products 8e and 8f requires stoichiometric transition metals and proceeds to moderate yields, we believe this represents the most efficient route for the coupling of near-identical benzoic acids to date.
Scheme 10. Scope of the Double Decarboxylative Oxidative Cross-Coupling between Two Benzoic Acids.
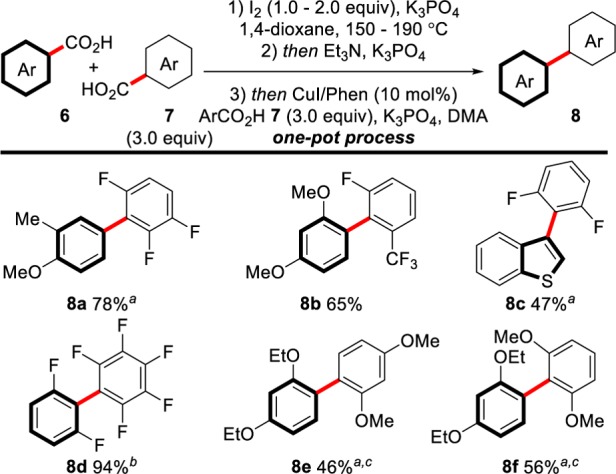
Reactions carried out at a 0.5 mmol scale of 6. K3PO4 (5.5–6.5 equiv) total across three steps.
The potassium salt of 7 was used in this case.
Yield determined by quantitative 19F NMR.
Step 3: no CuI/Phen added, PdCl2/BINAP (9 mol%), Ag2CO3 (3.20 equiv). The potassium salt of 6 was used in this case.
3. Conclusions
The field of decarboxylative activation is highly appealing, as it holds potential in efficient and atom-economic synthesis. We have reported a simple but effective transition-metal-free preparation of aryl iodides from readily available benzoic acids and I2. The procedure can be applied to a large range of electron-rich benzoic acids and to polyfluorinated aromatic acids. Importantly, this method overcomes some long-standing problems (e.g., poor selectivity and stoichiometric silver salts) of the classical aromatic Hunsdiecker decarboxylation. A combined theoretical and experimental study has shed light on the mechanism of this reaction, which we currently suggest proceeds via a concerted decarboxylation–halogenation-type process. To further demonstrate the potential applications of this procedure, we have developed a one-pot decarboxylative oxidative coupling. This process has several advantages over current procedures, namely the coupling of non-ortho-substituted benzoic acids, the removal of stoichiometric transition metal additives, and the coupling of nearly identical benzoic acids. We believe the scalability of the decarboxylative iodination holds potential for its application in preparative synthesis. Furthermore, the simplicity of the decarboxylative protocol, taken together with the breadth of methods for catalytic transformation of the resulting aryl iodides, should allow for the development of a variety of novel decarboxylative transformations, in addition to the oxidative cross-couplings reported in this article.
Acknowledgments
We gratefully acknowledge the Engineering and Physical Sciences Research Council (EPSRC, EP/I038578/1 and EP/L014017/2) for funding and the European Research Council for a Starting Grant (to I.L.). A.P. is funded by a scholarship from the Commonwealth Scholarship Commission.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b05155.
Experimental procedures and characterization data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Hassan J.; Sévignon M.; Gozzi C.; Schulz E.; Lemaire M. Chem. Rev. 2002, 102, 1359–1470. 10.1021/cr000664r. [DOI] [PubMed] [Google Scholar]; b Johansson Seechurn C. C. C.; Kitching M. O.; Colacot T. J.; Snieckus V. Angew. Chem., Int. Ed. 2012, 51, 5062–5085. 10.1002/anie.201107017. [DOI] [PubMed] [Google Scholar]; c Bolm C.; Hildebrand J. P.; Muniz K.; Hermanns N. Angew. Chem., Int. Ed. 2001, 40, 3284–3308. . [DOI] [PubMed] [Google Scholar]
- a Bringmann G.; Günther C.; Ochse M.; Schupp O.; Tasler S.. Biaryls in Nature: A Multi-Facetted Class of Stereochemically, Biosynthetically, and Pharmacologically Intriguing Secondary Metabolites. In Progress in the Chemistry of Organic Natural Products; Herz W., Falk H., Kirby G. W., Moore R. E., Eds.; Springer: Vienna, 2001; Vol. 82, pp 1–249. [DOI] [PubMed] [Google Scholar]; b Horton D. A.; Bourne G. T.; Smythe M. L. Chem. Rev. 2003, 103, 893–930. 10.1021/cr020033s. [DOI] [PubMed] [Google Scholar]; c Grimsdale A. C.; Chan K. L.; Martin R. E.; Jokisz P. G.; Holmes A. B. Chem. Rev. 2009, 109, 897–1091. 10.1021/cr000013v. [DOI] [PubMed] [Google Scholar]
- For selected reviews on C–H activation see:; a Alberico D.; Scott M. E.; Lautens M. Chem. Rev. 2007, 107, 174–238. 10.1021/cr0509760. [DOI] [PubMed] [Google Scholar]; b Ackermann L.; Vicente R.; Kapdi A. R. Angew. Chem., Int. Ed. 2009, 48, 9792–9826. 10.1002/anie.200902996. [DOI] [PubMed] [Google Scholar]; c Boorman T. C.; Larrosa I. Chem. Soc. Rev. 2011, 40, 1910–1925. 10.1039/C0CS00098A. [DOI] [PubMed] [Google Scholar]; d Wencel-Delord J.; Dröge T.; Liu F.; Glorius F. Chem. Soc. Rev. 2011, 40, 4740–4761. 10.1039/c1cs15083a. [DOI] [PubMed] [Google Scholar]; e Kuhl N.; Hopkinson M. N.; Wencel-Delord J.; Glorius F. Angew. Chem., Int. Ed. 2012, 51, 10236–10254. 10.1002/anie.201203269. [DOI] [PubMed] [Google Scholar]; f Kakiuchi F.; Kochi T.; Murai S. Synlett 2014, 25, 2390–2414. 10.1055/s-0034-1379210. [DOI] [Google Scholar]; g Ahlsten N.; Cambeiro X. C.; Perry G. J. P.; Larrosa I. In Topics in Heterocyclic Chemistry; Bandini M., Ed.; Springer International Publishing: Berlin, 2016; Vol. 46, pp 175–226. [Google Scholar]; h Font M.; Quibell J. M.; Perry G. J. P.; Larrosa I. Chem. Commun. 2017, 53, 5584–5597. 10.1039/C7CC01755C. [DOI] [PubMed] [Google Scholar]; i Simonetti M.; Cannas D. M.; Larrosa I. In Advances in Organometallic Chemistry; Pérez P. J., Ed.; Elsevier: Amsterdam,2017; Vol. 67, pp 299–399. [Google Scholar]; j Murakami K.; Perry G. J. P.; Itami K. Org. Biomol. Chem. 2017, 15, 6071–6075. 10.1039/C7OB00985B. [DOI] [PubMed] [Google Scholar]; k Yi H.; Zhang G.; Wang H.; Huang Z.; Wang J.; Singh A. K.; Lei A. Chem. Rev. 2017, 117, 9016–9085. 10.1021/acs.chemrev.6b00620. [DOI] [PubMed] [Google Scholar]
- For selected reviews on decarboxylative activation see:; a Gooßen L. J.; Gooßen K.; Rodríguez N.; Blanchot M.; Linder C.; Zimmermann B. Pure Appl. Chem. 2008, 80, 1725–1733. 10.1351/pac200880081725. [DOI] [Google Scholar]; b Gooßen L. J.; Rodríguez N.; Gooßen K. Angew. Chem., Int. Ed. 2008, 47, 3100–3120. 10.1002/anie.200704782. [DOI] [PubMed] [Google Scholar]; c Gooßen L. J.; Collet F.; Gooßen K. Isr. J. Chem. 2010, 50, 617–629. 10.1002/ijch.201000039. [DOI] [Google Scholar]; d Shang R.; Liu L. Sci. China: Chem. 2011, 54, 1670–1687. 10.1007/s11426-011-4381-0. [DOI] [Google Scholar]; e Rodríguez N.; Gooßen L. J. Chem. Soc. Rev. 2011, 40, 5030–5048. 10.1039/c1cs15093f. [DOI] [PubMed] [Google Scholar]; f Dzik W. I.; Lange P. P.; Gooßen L. J. Chem. Sci. 2012, 3, 2671–2678. 10.1039/c2sc20312j. [DOI] [Google Scholar]; g Cornella J.; Larrosa I. Synthesis 2012, 44, 653–676. 10.1055/s-0031-1289686. [DOI] [Google Scholar]; h Gooßen L. J.; Gooßen K.. Decarboxylative Coupling Reactions. In Topics in Organometallic Chemistry; Gooßen L. J., Ed.; Springer-Verlag: Berlin/Heidelberg, 2013; Vol. 44, pp 121–142. [Google Scholar]; i Perry G. J. P.; Larrosa I. Eur. J. Org. Chem. 2017, 2017, 3517–3527. 10.1002/ejoc.201700121. [DOI] [PMC free article] [PubMed] [Google Scholar]; For a review on de-amidative cross-coupling see:; j Liu C.; Szostak M. Chem. - Eur. J. 2017, 23, 7157–7173. 10.1002/chem.201605012. [DOI] [PubMed] [Google Scholar]
- For the coupling of benzoic acids with arenes see:; a Voutchkova A.; Coplin A.; Leadbeater N. E.; Crabtree R. H. Chem. Commun. 2008, 6312–6314. 10.1039/b813998a. [DOI] [PubMed] [Google Scholar]; b Wang C.; Piel I.; Glorius F. J. Am. Chem. Soc. 2009, 131, 4194–4195. 10.1021/ja8100598. [DOI] [PubMed] [Google Scholar]; c Cornella J.; Lu P.; Larrosa I. Org. Lett. 2009, 11, 5506–5509. 10.1021/ol902304n. [DOI] [PubMed] [Google Scholar]; d Zhou J.; Hu P.; Zhang M.; Huang S.; Wang M.; Su W. Chem. - Eur. J. 2010, 16, 5876–5881. 10.1002/chem.201000529. [DOI] [PubMed] [Google Scholar]; e Xie K.; Yang Z.; Zhou X.; Li X.; Wang S.; Tan Z.; An X.; Guo C.-C. Org. Lett. 2010, 12, 1564–1567. 10.1021/ol100296b. [DOI] [PubMed] [Google Scholar]; f Zhang F.; Greaney M. F. Angew. Chem., Int. Ed. 2010, 49, 2768–2771. 10.1002/anie.200906921. [DOI] [PubMed] [Google Scholar]; g Zhao H.; Wei Y.; Xu J.; Kan J.; Su W.; Hong M. J. Org. Chem. 2011, 76, 882–893. 10.1021/jo102175f. [DOI] [PubMed] [Google Scholar]; h Hu P.; Zhang M.; Jie X.; Su W. Angew. Chem., Int. Ed. 2012, 51, 227–231. 10.1002/anie.201106451. [DOI] [PubMed] [Google Scholar]; i Luo H.-Q.; Dong W.; Loh T.-P. Tetrahedron Lett. 2013, 54, 2833–2836. 10.1016/j.tetlet.2013.03.086. [DOI] [Google Scholar]; j Pei K.; Jie X.; Zhao H.; Su W. Eur. J. Org. Chem. 2014, 2014, 4230–4233. 10.1002/ejoc.201402278. [DOI] [Google Scholar]; k Suresh R.; Muthusubramanian S.; Kumaran R. S.; Manickam G. Asian J. Org. Chem. 2014, 3, 604–608. 10.1002/ajoc.201400013. [DOI] [Google Scholar]; l Yang K.; Wang P.; Zhang C.; Kadi A. A.; Fun H.-K.; Zhang Y.; Lu H. Eur. J. Org. Chem. 2014, 2014, 7586–7589. 10.1002/ejoc.201403234. [DOI] [Google Scholar]; m Chen L.; Ju L.; Bustin K. A.; Hoover J. M. Chem. Commun. 2015, 51, 15059–15062. 10.1039/C5CC06645J. [DOI] [PubMed] [Google Scholar]; n Zhao S.; Liu Y.-J.; Yan S.-Y.; Chen F.-J.; Zhang Z.-Z.; Shi B.-F. Org. Lett. 2015, 17, 3338–3341. 10.1021/acs.orglett.5b01560. [DOI] [PubMed] [Google Scholar]; o Kan J.; Huang S.; Lin J.; Zhang M.; Su W. Angew. Chem., Int. Ed. 2015, 54, 2199–2203. 10.1002/anie.201408630. [DOI] [PubMed] [Google Scholar]; p Patra T.; Nandi S.; Sahoo S. K.; Maiti D. Chem. Commun. 2016, 52, 1432–1435. 10.1039/C5CC08367B. [DOI] [PubMed] [Google Scholar]; q Candish L.; Freitag M.; Gensch T.; Glorius F. Chem. Sci. 2017, 8, 3618–3622. 10.1039/C6SC05533H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For the coupling of two benzoic acids see:; a Cornella J.; Lahlali H.; Larrosa I. Chem. Commun. 2009, 46, 8276–8278. 10.1039/c0cc01943g. [DOI] [PubMed] [Google Scholar]; b Xie K.; Wang S.; Yang Z.; Liu J.; Wang A.; Li X.; Tan Z.; Guo C.-C.; Deng W. Eur. J. Org. Chem. 2011, 2011, 5787–5790. 10.1002/ejoc.201100913. [DOI] [Google Scholar]; c Hu P.; Shang Y.; Su W. Angew. Chem., Int. Ed. 2012, 51, 5945–5949. 10.1002/anie.201200153. [DOI] [PubMed] [Google Scholar]; d Fu Z.; Li Z.; Xiong Q.; Cai H. RSC Adv. 2015, 5, 52101–52104. 10.1039/C5RA07771K. [DOI] [Google Scholar]
- For a comprehensive review on this subject see:Iodine chemistry and Applications; Kaiho T., Ed.; Wiley-VCH: Weinheim, 2015. [Google Scholar]
- For the use of aryl iodides in cross-couplings see ref (1). In nucleophilic substitution see:; a Bunnett J. F.; Zahler R. E. Chem. Rev. 1951, 49, 273–412. 10.1021/cr60153a002. [DOI] [Google Scholar]; b Hartwig J. F. Synlett 2006, 2006, 1283–1294. 10.1055/s-2006-939728. [DOI] [Google Scholar]; In metalation see:; c Knochel P.; Dohle W.; Gommermann N.; Kneisel F. F.; Kopp F.; Korn T.; Sapountzis I.; Vu V. A. Angew. Chem., Int. Ed. 2003, 42, 4302–4320. 10.1002/anie.200300579. [DOI] [PubMed] [Google Scholar]; d Tilly D.; Chevallier F.; Mongin F.; Gros P. C. Chem. Rev. 2014, 114, 1207–1257. 10.1021/cr400367p. [DOI] [PubMed] [Google Scholar]; e Handbook of Grignard Reagents; Silverman G. S., Rakita P. E., Eds.; Dekker: New York, 1996. [Google Scholar]; f Sotomayor N.; Lete E. Curr. Org. Chem. 2003, 7, 275–300. 10.2174/1385272033372987. [DOI] [Google Scholar]; In aryl radical formation see:; g Neumann W. P. Synthesis 1987, 1987, 665–683. 10.1055/s-1987-28044. [DOI] [Google Scholar]; h Kim H.; Lee C. Angew. Chem., Int. Ed. 2012, 51, 12303–12306. 10.1002/anie.201203599. [DOI] [PubMed] [Google Scholar]; i Nguyen J. D.; D’Amato E. M.; Narayanam J. M. R.; Stephenson C. R. J. Nat. Chem. 2012, 4, 854–859. 10.1038/nchem.1452. [DOI] [PubMed] [Google Scholar]; j Ghosh I.; Ghosh T.; Bardagi J. I.; Konig B. Science 2014, 346, 725–728. 10.1126/science.1258232. [DOI] [PubMed] [Google Scholar]; k Liu Y. X.; Xue D.; Wang J.-D.; Zhao C.-J.; Zou Q.-Z.; Wang C.; Xiao J. Synlett 2013, 24, 507–513. 10.1055/s-0032-1318155. [DOI] [Google Scholar]; In aryne generation see:; l Pellissier H.; Santelli M. Tetrahedron 2003, 59, 701–730. 10.1016/S0040-4020(02)01563-6. [DOI] [Google Scholar]; m Moss R. A.; Platz M. S.; Jones M. Jr. In Reactive Intermediate Chemistry; Winkler M., Wenk H. H., Sander W., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, 2005; pp 741–794. [Google Scholar]; As hypervalent iodine species see:; n Zhdankin V. V.; Stang P. J. Chem. Rev. 2008, 108, 5299–5358. 10.1021/cr800332c. [DOI] [PMC free article] [PubMed] [Google Scholar]; o Merritt E. A.; Olofsson B. Angew. Chem., Int. Ed. 2009, 48, 9052–9070. 10.1002/anie.200904689. [DOI] [PubMed] [Google Scholar]; p Deprez N. R.; Sanford M. S. Inorg. Chem. 2007, 46, 1924–1935. 10.1021/ic0620337. [DOI] [PubMed] [Google Scholar]; q Cambeiro X. C.; Boorman T. C.; Lu P.; Larrosa I. Angew. Chem., Int. Ed. 2013, 52, 1781–1784. 10.1002/anie.201209007. [DOI] [PubMed] [Google Scholar]; r Cambeiro X. C.; Ahlsten N.; Larrosa I. J. Am. Chem. Soc. 2015, 137, 15636–15639. 10.1021/jacs.5b10593. [DOI] [PubMed] [Google Scholar]; In natural products see:; s Wang L.; Zhou X.; Fredimoses M.; Liao S.; Liu Y. RSC Adv. 2014, 4, 57350–57376. 10.1039/C4RA09833A. [DOI] [Google Scholar]; t Markou K.; Georgopoulos N.; Kyriazopoulou V.; Vagenakis A. G. Thyroid 2001, 11, 501–510. 10.1089/105072501300176462. [DOI] [PubMed] [Google Scholar]; u Hair P. I.; McCormack P. L.; Curran M. P. Drugs 2008, 68, 1415–1434. 10.2165/00003495-200868100-00005. [DOI] [PubMed] [Google Scholar]; v Quinn M. J.; Fitzgerald D. J. Circulation 1999, 100, 1667–1672. 10.1161/01.CIR.100.15.1667. [DOI] [PubMed] [Google Scholar]; w Boger D. L. Med. Res. Rev. 2001, 21, 356–381. 10.1002/med.1014. [DOI] [PubMed] [Google Scholar]; x DeLeon A.; Patel N. C.; Crismon M. L. Clin. Ther. 2004, 26, 649–666. 10.1016/S0149-2918(04)90066-5. [DOI] [PubMed] [Google Scholar]; As medical imaging agents see:; y Seevers R. H.; Counsell R. E. Chem. Rev. 1982, 82, 575–590. 10.1021/cr00052a002. [DOI] [Google Scholar]; z Schlyer D. J. Ann. Acad. Med. Singapore 2004, 33, 146–154. [PubMed] [Google Scholar]; As organic materials see:; aa Tang M. L.; Bao Z. Chem. Mater. 2011, 23, 446–455. 10.1021/cm102182x. [DOI] [Google Scholar]
- For the preparation of aryl iodides via electrophilic aromatic substitution (SEAr) see:; a Taylor R.Electrophilic Aromatic Substitution; John Wiley: New York, 1990. [Google Scholar]; b Barluenga J.; Gonzalez J. M.; Garcia-Martin M. A.; Campos P. J.; Asensio G. J. Org. Chem. 1993, 58, 2058–2060. 10.1021/jo00060a020. [DOI] [Google Scholar]; c Rodriguez R. A.; Pan C.-M.; Yabe Y.; Kawamata Y.; Eastgate M. D.; Baran P. S. J. Am. Chem. Soc. 2014, 136, 6908–6911. 10.1021/ja5031744. [DOI] [PMC free article] [PubMed] [Google Scholar]; Via directed ortho metalation (DoM) see:; d Snieckus V. Chem. Rev. 1990, 90, 879–933. 10.1021/cr00104a001. [DOI] [Google Scholar]; Using directing groups see:; e Fahey D. R. J. Organomet. Chem. 1971, 27, 283–292. 10.1016/S0022-328X(00)80577-X. [DOI] [Google Scholar]; f Dick A. R.; Hull K. L.; Sanford M. S. J. Am. Chem. Soc. 2004, 126, 2300–2301. 10.1021/ja031543m. [DOI] [PubMed] [Google Scholar]; g Giri R.; Chen X.; Yu J.-Q. Angew. Chem., Int. Ed. 2005, 44, 2112–2115. 10.1002/anie.200462884. [DOI] [PubMed] [Google Scholar]; h Chen X.; Hao X.-S.; Goodhue C. E.; Yu J.-Q. J. Am. Chem. Soc. 2006, 128, 6790–6791. 10.1021/ja061715q. [DOI] [PubMed] [Google Scholar]; i Kalyani D.; Dick A. R.; Anani W. Q.; Sanford M. S. Tetrahedron 2006, 62, 11483–11498. 10.1016/j.tet.2006.06.075. [DOI] [Google Scholar]; j Kalyani D.; Dick A. R.; Anani W. Q.; Sanford M. S. Org. Lett. 2006, 8, 2523–2526. 10.1021/ol060747f. [DOI] [PubMed] [Google Scholar]; k Wan X.; Ma Z.; Li B.; Zhang K.; Cao S.; Zhang S.; Shi Z. J. Am. Chem. Soc. 2006, 128, 7416–7417. 10.1021/ja060232j. [DOI] [PubMed] [Google Scholar]; l Whitfield S. R.; Sanford M. S. J. Am. Chem. Soc. 2007, 129, 15142–15143. 10.1021/ja077866q. [DOI] [PubMed] [Google Scholar]; m Mei T.-S.; Giri R.; Maugel N.; Yu J.-Q. Angew. Chem., Int. Ed. 2008, 47, 5215–5219. 10.1002/anie.200705613. [DOI] [PubMed] [Google Scholar]; n Li J.-J.; Mei T.-S.; Yu J.-Q. Angew. Chem., Int. Ed. 2008, 47, 6452–6455. 10.1002/anie.200802187. [DOI] [PubMed] [Google Scholar]; o Whitfield S. R.; Sanford M. S. Organometallics 2008, 27, 1683–1689. 10.1021/om070095e. [DOI] [Google Scholar]; p Kakiuchi F.; Kochi T.; Mutsutani H.; Kobayashi N.; Urano S.; Sato M.; Nishiyama S.; Tanabe T. J. Am. Chem. Soc. 2009, 131, 11310–11311. 10.1021/ja9049228. [DOI] [PubMed] [Google Scholar]; q Zhao X.; Dimitrijević E.; Dong V. M. J. Am. Chem. Soc. 2009, 131, 3466–3467. 10.1021/ja900200g. [DOI] [PubMed] [Google Scholar]; r Song B.; Zheng X.; Mo J.; Xu B. Adv. Synth. Catal. 2010, 352, 329–335. 10.1002/adsc.200900778. [DOI] [Google Scholar]; s Bedford R. B.; Engelhart J. U.; Haddow M. F.; Mitchell C. J.; Webster R. L. Dalton Trans. 2010, 39, 10464–10472. 10.1039/c0dt00385a. [DOI] [PubMed] [Google Scholar]; t Mei T.-S.; Wang D.-H.; Yu J.-Q. Org. Lett. 2010, 12, 3140–3143. 10.1021/ol1010483. [DOI] [PubMed] [Google Scholar]; u Dudnik A. S.; Chernyak N.; Huang C.; Gevorgyan V. Angew. Chem., Int. Ed. 2010, 49, 8729–8732. 10.1002/anie.201004426. [DOI] [PMC free article] [PubMed] [Google Scholar]; v Powers D. C.; Xiao D. Y.; Geibel M. A. L.; Ritter T. J. Am. Chem. Soc. 2010, 132, 14530–14536. 10.1021/ja1054274. [DOI] [PMC free article] [PubMed] [Google Scholar]; w Wang W.; Pan C.; Chen F.; Cheng J. Chem. Commun. 2011, 47, 3978–3980. 10.1039/c0cc05557c. [DOI] [PubMed] [Google Scholar]; x Dubost E.; Fossey C.; Cailly T.; Rault S.; Fabis F. J. Org. Chem. 2011, 76, 6414–6420. 10.1021/jo200853j. [DOI] [PubMed] [Google Scholar]; y Bedford R. B.; Haddow M. F.; Mitchell C. J.; Webster R. L. Angew. Chem., Int. Ed. 2011, 50, 5524–5527. 10.1002/anie.201101606. [DOI] [PubMed] [Google Scholar]; z Schröder N.; Wencel-Delord J.; Glorius F. J. Am. Chem. Soc. 2012, 134, 8298–8301. 10.1021/ja302631j. [DOI] [PubMed] [Google Scholar]; aa John A.; Nicholas K. M. J. Org. Chem. 2012, 77, 5600–5605. 10.1021/jo300713h. [DOI] [PubMed] [Google Scholar]; bb Wang X.-C.; Hu Y.; Bonacorsi S.; Hong Y.; Burrell R.; Yu J.-Q. J. Am. Chem. Soc. 2013, 135, 10326–10329. 10.1021/ja4055492. [DOI] [PMC free article] [PubMed] [Google Scholar]; cc Chu L.; Xiao K.-J.; Yu J.-Q. Science 2014, 346, 451–455. 10.1126/science.1258538. [DOI] [PMC free article] [PubMed] [Google Scholar]; dd Haines B. E.; Xu H.; Verma P.; Wang X.-C.; Yu J.-Q.; Musaev D. G. J. Am. Chem. Soc. 2015, 137, 9022–9031. 10.1021/jacs.5b03410. [DOI] [PubMed] [Google Scholar]; ee Li B.; Liu B.; Shi B.-F. Chem. Commun. 2015, 51, 5093–5096. 10.1039/C5CC00531K. [DOI] [PubMed] [Google Scholar]; ff Teskey C. J.; Lui A. Y. W.; Greaney M. F. Angew. Chem., Int. Ed. 2015, 54, 11677–11680. 10.1002/anie.201504390. [DOI] [PMC free article] [PubMed] [Google Scholar]; From aryl diazonium salts see:; gg Sandmeyer T. Ber. Dtsch. Chem. Ges. 1884, 17, 1633–1635. 10.1002/cber.18840170219. [DOI] [Google Scholar]; hh Hodgson H. H. Chem. Rev. 1947, 40, 251–277. 10.1021/cr60126a003. [DOI] [PubMed] [Google Scholar]; ii Roglans A.; Pla-Quintana A.; Moreno-Mañas M. Chem. Rev. 2006, 106, 4622–4643. 10.1021/cr0509861. [DOI] [PubMed] [Google Scholar]; Via halogen exchange (HalEx) see:; jj Lindley J. Tetrahedron 1984, 40, 1433–1456. 10.1016/S0040-4020(01)91791-0. [DOI] [Google Scholar]; kk Klapars A.; Buchwald S. L. J. Am. Chem. Soc. 2002, 124, 14844–14845. 10.1021/ja028865v. [DOI] [PubMed] [Google Scholar]; ll Sheppard T. D. Org. Biomol. Chem. 2009, 7, 1043–1052. 10.1039/b818155a. [DOI] [PubMed] [Google Scholar]; mm Casitas A.; Canta M.; Solà M.; Costas M.; Ribas X. J. Am. Chem. Soc. 2011, 133, 19386–19392. 10.1021/ja2058567. [DOI] [PubMed] [Google Scholar]; nn Cant A. A.; Bhalla R.; Pimlott S. L.; Sutherland A. Chem. Commun. 2012, 48, 3993–3995. 10.1039/c2cc30956d. [DOI] [PubMed] [Google Scholar]; oo Font M.; Acuña-Parés F.; Parella T.; Serra J.; Luis J. M.; Lloret-Fillol J.; Costas M.; Ribas X. Nat. Commun. 2014, 5, 4373. 10.1038/ncomms5373. [DOI] [PubMed] [Google Scholar]; pp Serra J.; Whiteoak C. J.; Acuña-Parés F.; Font M.; Luis J. M.; Lloret-Fillol J.; Ribas X. J. Am. Chem. Soc. 2015, 137, 13389–13397. 10.1021/jacs.5b08756. [DOI] [PubMed] [Google Scholar]; qq Chen M.; Ichikawa S.; Buchwald S. L. Angew. Chem., Int. Ed. 2015, 54, 263–266. 10.1002/anie.201409595. [DOI] [PubMed] [Google Scholar]; rr Li L.; Liu W.; Zeng H.; Mu X.; Cosa G.; Mi Z.; Li C.-J. J. Am. Chem. Soc. 2015, 137, 8328–8331. 10.1021/jacs.5b03220. [DOI] [PubMed] [Google Scholar]
- a Borodine A. Justus Liebigs Ann. Chem. 1861, 119, 121–123. 10.1002/jlac.18611190113. [DOI] [Google Scholar]; b Hunsdiecker H.; Hunsdiecker C. Ber. Dtsch. Chem. Ges. B 1942, 75, 291–297. 10.1002/cber.19420750309. [DOI] [Google Scholar]; c Wilson C.Organic Reactions; John Wiley & Sons, Inc.: Hoboken, NJ, 2011; pp 332–387. [Google Scholar]; d Crich D. In Comprehensive Organic Synthesis; Trost B. M., Fleming I., Eds.; Elsevier: Amsterdam, 1991; Vol. 7, pp 717–734. [Google Scholar]; e Crich D.; Sasaki K. In Comprehensive Organic Synthesis II; Knochel P., Molander G. A., Eds.; Elsevier: Amsterdam, 2014; pp 818–836. [Google Scholar]
- a Dauben W. G.; Tilles H. J. Am. Chem. Soc. 1950, 72, 3185–3187. 10.1021/ja01163a105. [DOI] [Google Scholar]; b Barnes R. A.; Prochaska R. J. J. Am. Chem. Soc. 1950, 72, 3188–3191. 10.1021/ja01163a106. [DOI] [Google Scholar]; c Oldham J. W. H. J. Chem. Soc. 1950, 100–108. 10.1039/jr9500000100. [DOI] [Google Scholar]
- For reports that require stoichiometric transition metals see ref (11) and the following:; a Kiely J. S.; Nelson L. L.; Boudjouk P. J. Org. Chem. 1977, 42, 1480. 10.1021/jo00428a055. [DOI] [Google Scholar]; b Uemura S.; Tanaka S.; Okano M.; Hamana M. J. Org. Chem. 1983, 48, 3297–3301. 10.1021/jo00167a027. [DOI] [Google Scholar]; c Luo Y.; Pan X.; Wu J. Tetrahedron Lett. 2010, 51, 6646–6648. 10.1016/j.tetlet.2010.10.054. [DOI] [Google Scholar]; d Cornella J.; Rosillo-Lopez M.; Larrosa I. Adv. Synth. Catal. 2011, 353, 1359–1366. 10.1002/adsc.201100109. [DOI] [Google Scholar]; e Peng X.; Shao X.-F.; Liu Z.-Q. Tetrahedron Lett. 2013, 54, 3079–3081. 10.1016/j.tetlet.2013.03.135. [DOI] [Google Scholar]; f Fu Z.; Li Z.; Song Y.; Yang R.; Liu Y.; Cai H. J. Org. Chem. 2016, 81, 2794–2803. 10.1021/acs.joc.5b02873. [DOI] [PubMed] [Google Scholar]
- For reports that show limited scope and/or poor selectivity see:; a Barton D. H. R.; Lacher B.; Zard S. Z. Tetrahedron Lett. 1985, 26, 5939–5942. 10.1016/S0040-4039(00)98266-2. [DOI] [Google Scholar]; b Barton D. H. R.; Lacher B.; Zard S. Z. Tetrahedron 1987, 43, 4321–4328. 10.1016/S0040-4020(01)90307-2. [DOI] [Google Scholar]; c Singh R.; Just G. Synth. Commun. 1988, 18, 1327–1330. 10.1080/00397918808078799. [DOI] [Google Scholar]; d Camps P.; Lukach A. E.; Pujol X.; Vázquez S. Tetrahedron 2000, 56, 2703–2707. 10.1016/S0040-4020(00)00169-1. [DOI] [Google Scholar]; e Koo B.-S.; Kim E.-H.; Lee K.-J. Synth. Commun. 2002, 32, 2275–2286. 10.1081/SCC-120005997. [DOI] [Google Scholar]; f Putey A.; Popowycz F.; Joseph B. Synlett 2007, 2007, 419–422. 10.1055/s-2007-967953. [DOI] [Google Scholar]; g Hamamoto H.; Hattori S.; Takemaru K.; Miki Y. Synlett 2011, 2011, 1563–1566. 10.1055/s-0030-1260791. [DOI] [Google Scholar]; h Kulbitski K.; Nisnevich G.; Gandelman M. Adv. Synth. Catal. 2011, 353, 1438–1442. 10.1002/adsc.201100145. [DOI] [Google Scholar]; i Li Z.; Wang K.; Liu Z.-Q. Synlett 2014, 25, 2508–2512. 10.1055/s-0034-1378583. [DOI] [Google Scholar]
- A common route in the literature is (1) conversion of the carboxyl group to an acyl chloride, (2) conversion of the acyl chloride to the acyl azide and subsequent conversion to the aromatic amine (Curtius rearrangement), and (3) conversion of the aromatic amine to the diazonium salt and subsequent iodination (Sandmeyer reaction). For examples see:; a Moriconi A.; Cesta M. C.; Cervellera M. N.; Aramini A.; Coniglio S.; Colagioia S.; Beccari A. R.; Bizzarri C.; Cavicchia M. R.; Locati M.; Galliera E.; Di Benedetto P.; Vigilante P.; Bertini R.; Allegretti M. J. Med. Chem. 2007, 50, 3984–4002. 10.1021/jm061469t. [DOI] [PubMed] [Google Scholar]; b Zhang Y. J.; Wei H.; Zhang W. Tetrahedron 2009, 65, 1281–1286. 10.1016/j.tet.2008.12.056. [DOI] [Google Scholar]; c Yang X.; Sun G.; Yang C.; Wang B. ChemMedChem 2011, 6, 2294–2301. 10.1002/cmdc.201100384. [DOI] [PubMed] [Google Scholar]; d Zhang A. S.; Ho J. Z.; Braun M. P. J. Labelled Compd. Radiopharm. 2011, 54, 163–167. 10.1002/jlcr.1842. [DOI] [Google Scholar]; e Gluyas J. B. G.; Burschka C.; Dörrich S.; Vallet J.; Gronemeyer H.; Tacke R. Org. Biomol. Chem. 2012, 10, 6914–6929. 10.1039/c2ob25989c. [DOI] [PubMed] [Google Scholar]
- a Cornella J.; Sanchez C.; Banawa D.; Larrosa I. Chem. Commun. 2009, 7176–7178. 10.1039/b916646g. [DOI] [PubMed] [Google Scholar]; b Gooßen L. J.; Linder C.; Rodríguez N.; Lange P. P.; Fromm A. Chem. Commun. 2009, 7173–7175. 10.1039/b912509d. [DOI] [PubMed] [Google Scholar]; Lu P.; Sanchez C.; Cornella J.; Larrosa I. Org. Lett. 2009, 11, 5710–5713. 10.1021/ol902482p. [DOI] [PubMed] [Google Scholar]; c Seo S.; Taylor J. B.; Greaney M. F. Chem. Commun. 2012, 48, 8270–8272. 10.1039/c2cc33306f. [DOI] [PubMed] [Google Scholar]; d Grainger R.; Nikmal A.; Cornella J.; Larrosa I. Org. Biomol. Chem. 2012, 10, 3172–3174. 10.1039/c2ob25157d. [DOI] [PubMed] [Google Scholar]
- For example, Yu and co-workers have previously observed that mixing 2-methoxybenzoic acid 1a with I2 and AgOAc causes iodination para to the methoxy group to give 1a′, but on switching to CsOAc no para iodination is observed; see refs (9m) and (9b1).
- The reason for the sensitivity to water has not been thoroughly investigated. Currently we speculate that the benzoyl hypoiodite (ArCO2I) intermediate may react with water in an unproductive pathway. The addition of molecular sieves did not aid this reaction. Overall, the reaction system must be anhydrous; however, the reaction can be carried out on the bench if desired (see SI).
- The methoxy group on meta-anisic acid (2d) will donate electron density to the positions ortho/para to itself but will withdraw electron density at the position meta to itself. As the carboxyl group is meta to the methoxy group, it is deactivated in this substrate. Likewise, 3,5-dimethoxybenzoic acid was not successful in this reaction.
- For a room-temperature, transition-metal-catalyzed decarboxylative Heck reaction of highly activated, di-ortho-substituted benzoic acids see:Hossian A.; Bhunia S. K.; Jana R. J. Org. Chem. 2016, 81, 2521–2533. 10.1021/acs.joc.6b00100. [DOI] [PubMed] [Google Scholar]
- a Dickstein J. S.; Mulrooney C. A.; O’Brien E. M.; Morgan B. J.; Kozlowski M. C. Org. Lett. 2007, 9, 2441. 10.1021/ol070749f. [DOI] [PubMed] [Google Scholar]; b Dickstein J. S.; Curto J. M.; Gutierrez O.; Mulrooney C. A.; Kozlowski M. C. J. Org. Chem. 2013, 78, 4744. 10.1021/jo400222c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unfortunately, acyclic amine (e.g., NMe2) substituents were not tolerated under the reaction conditions and led to decomposition. The reason for decomposition has not been investigated.
- a Hammett L. P. J. Am. Chem. Soc. 1937, 59, 96–103. 10.1021/ja01280a022. [DOI] [Google Scholar]; b Hansch C.; Leo A.; Taft R. W. Chem. Rev. 1991, 91, 165–195. 10.1021/cr00002a004. [DOI] [Google Scholar]
- The reaction with 2-fluorobenzoic acid gave a poor yield (4% NMR yield). Further studies are necessary to describe the reactivity of polyfluorinated benzoic acids; however, previous transition-metal-promoted decarboxylative functionalizations have also shown unique reactivity for polyfluorinated benzoic acids. See ref (19) and the following:; a Shang R.; Xu Q.; Jiang Y.-Y.; Wang Y.; Liu L. Org. Lett. 2010, 12, 1000–1003. 10.1021/ol100008q. [DOI] [PubMed] [Google Scholar]; b Sardzinski L. W.; Wertjes W. C.; Schnaith A. M.; Kalyani D. Org. Lett. 2015, 17, 1256–1259. 10.1021/acs.orglett.5b00241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J.; Preciado S.; Xie P.; Larrosa I. Chem. - Eur. J. 2016, 22, 6798–6802. 10.1002/chem.201601114. [DOI] [PubMed] [Google Scholar]
- Although 4-substituted benzoic acids (NO2, CF3, and CN) are unreactive, these groups are tolerated in the reaction, as demonstrated in the robustness screen (see SI, Table S2a,b).
- See the SI for more detailed information on the conditions for these procedures.
- a Collins K. D.; Glorius F. Nat. Chem. 2013, 5, 597–601. 10.1038/nchem.1669. [DOI] [PubMed] [Google Scholar]; b Collins K. D.; Glorius F. Tetrahedron 2013, 69, 7817–7825. 10.1016/j.tet.2013.05.068. [DOI] [Google Scholar]; c Collins K. D.; Rühling A.; Lied F.; Glorius F. Chem. - Eur. J. 2014, 20, 3800–3805. 10.1002/chem.201304508. [DOI] [PubMed] [Google Scholar]; d Collins K. D.; Rühling A.; Glorius F. Nat. Protoc. 2014, 9, 1348–1353. 10.1038/nprot.2014.076. [DOI] [PubMed] [Google Scholar]
- For previous decarboxylative iodinations of cinnamic acids using other iodinating agents see:; a Naskar D.; Chowdhury S.; Roy S. Tetrahedron Lett. 1998, 39, 699–702. 10.1016/S0040-4039(97)10639-6. [DOI] [Google Scholar]; b Homsi F.; Rousseau G. Tetrahedron Lett. 1999, 40, 1495–1498. 10.1016/S0040-4039(99)00021-0. [DOI] [Google Scholar]; c Das J. P.; Roy S. J. Org. Chem. 2002, 67, 7861–7864. 10.1021/jo025868h. [DOI] [PubMed] [Google Scholar]
- The cost of the reagents (1f, I2, K3PO4, and MeCN) is approximately 10 times less than that using aryl iodide 2f (£300/mol vs £3080/mol, respectively). Prices are taken from Sigma Aldrich from April 2017.
- Attempts to establish benzoyl hypoiodite I as a definite intermediate were unsuccessful. However, the formation of acyl hypohalites is reported in the literature, and we believe it is reasonable to suggest its presence in this system. See:; a Kleinberg J. J. Chem. Educ. 1946, 23, 559–562. 10.1021/ed023p559. [DOI] [Google Scholar]; b Zingaro R. A.; Goodrich J. E.; Kleinberg J.; Vanderwerf A. J. Am. Chem. Soc. 1949, 71, 575–576. 10.1021/ja01170a054. [DOI] [Google Scholar]; c Tanner D. D.; Bunce N. J. In Acyl Halides; Patai S., Eds; John Wiley & Sons, Ltd.: Chichester, UK, 1972; pp 455–500. [Google Scholar]; d Barton D. H. R.; Faro H. P.; Serebryakov E. P.; Woolsey N. F. J. Chem. Soc. 1965, 2438–2444. 10.1039/jr9650002438. [DOI] [Google Scholar]; e Srivastava P. C.; Singh P.; Tangiri M.; Sinha A.; Bajpai S. J. Indian Chem. Soc. 1997, 74, 443–445. [Google Scholar]
- See ref (20a) and the following:; a Tanaka D.; Romeril S. P.; Myers A. G. J. Am. Chem. Soc. 2005, 127, 10323–10333. 10.1021/ja052099l. [DOI] [PubMed] [Google Scholar]; b Gooßen L. J.; Thiel W. R.; Rodríguez N.; Linder C.; Melzer B. Adv. Synth. Catal. 2007, 349, 2241–2246. 10.1002/adsc.200700223. [DOI] [Google Scholar]; c Gooßen L. J.; Rodríguez N.; Linder C.; Lange P. P.; Fromm A. ChemCatChem 2010, 2, 430–442. 10.1002/cctc.200900277. [DOI] [Google Scholar]; d Xue L.; Su W.; Lin Z. Dalton Trans. 2011, 40, 11926–11936. 10.1039/c1dt10771b. [DOI] [PubMed] [Google Scholar]; e Xue L.; Su W.; Lin Z. Dalton Trans. 2010, 39, 9815–9822. 10.1039/c0dt00491j. [DOI] [PubMed] [Google Scholar]; f Zhang S.-L.; Fu Y.; Shang R.; Guo Q.-X.; Liu L. J. Am. Chem. Soc. 2010, 132, 638–646. 10.1021/ja907448t. [DOI] [PubMed] [Google Scholar]; g Xie H.; Lin F.; Lei Q.; Fang W. Organometallics 2013, 32, 6957–6968. 10.1021/om400503x. [DOI] [Google Scholar]; h Grainger R.; Cornella J.; Blakemore D. C.; Larrosa I.; Campanera J. M. Chem. - Eur. J. 2014, 20, 16680–16687. 10.1002/chem.201402931. [DOI] [PubMed] [Google Scholar]; i Fromm A.; van Wüllen C.; Hackenberger D.; Gooßen L. J. J. Am. Chem. Soc. 2014, 136, 10007–10023. 10.1021/ja503295x. [DOI] [PubMed] [Google Scholar]
- a Johnston L. J.; Lusztyk J.; Wayner D. D. M.; Abeywickreyma A. N.; Beckwith A. L. J.; Scaiano J. C.; Ingold K. U. J. Am. Chem. Soc. 1985, 107, 4594–4596. 10.1021/ja00301a062. [DOI] [Google Scholar]; b Newcomb M.Encyclopedia of Radicals in Chemistry, Biology and Materials; John Wiley & Sons, Ltd.: Chichester, UK, 2012; pp 1–19. [Google Scholar]
- Ground state and transition state for the decarboxylation step were calculated by DFT using Gaussian 09. B97D3 and 6-31G(d) were used for C, H, O atoms, and LanL2DZ was used for I. See the SI for details.
- Okamoto Y.; Brown H. C. J. Org. Chem. 1957, 22, 485–494. 10.1021/jo01356a003. [DOI] [Google Scholar]
- Although our studies provide insight into the mechanism for the decarboxylative iodination of electron-rich benzoic acids, we do not believe this mechanism holds true for polyfluorobenzoic acids. Further studies are necessary to explain the unexpected reactivity of polyfluorobenzoic acids. These studies are ongoing in our laboratory.
- a Do H.-Q.; Daugulis O. Chem. Commun. 2009, 6433–6435. 10.1039/b912890e. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Do H.-Q.; Daugulis O. J. Am. Chem. Soc. 2011, 133, 13577–13586. 10.1021/ja2047717. [DOI] [PMC free article] [PubMed] [Google Scholar]; For related studies see:; c Do H.-Q; Daugulis O.. 2008, 129, 12404–12405. [DOI] [PMC free article] [PubMed]; d Do H.-Q.; Daugulis O. J. Am. Chem. Soc. 2008, 130, 1128–1129. 10.1021/ja077862l. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Do H.-Q.; Kashif Khan R. M.; Daugulis O. J. Am. Chem. Soc. 2008, 130, 15185–15192. 10.1021/ja805688p. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Daugulis O.; Do H.-Q.; Shabashov D. Acc. Chem. Res. 2009, 42, 1074–1086. 10.1021/ar9000058. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Do H.-Q.; Daugulis O. Org. Lett. 2009, 11, 421–423. 10.1021/ol802411f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The exact process by which the amine quenches iodine has not been determined, though the reaction between amines and iodine has been previously investigated. See:; a Bist H. D.; Person W. B. J. Phys. Chem. 1969, 73, 482–489. 10.1021/j100723a003. [DOI] [Google Scholar]; b Schug J. C.; Kogan M. J. J. Magn. Reson. 1973, 11, 406–415. 10.1016/0022-2364(73)90067-X. [DOI] [Google Scholar]; c Koepler O.; Laschat S.; Miehlich B.; Baro A.; Fischer P. Helv. Chim. Acta 2004, 87, 1927–1934. 10.1002/hlca.200490173. [DOI] [Google Scholar]
- In this report we demonstrate the use of this one-pot, three-step method for the cross-coupling of benzoic acids with arenes or a second benzoic acid; however, we believe this method may also be applicable to a range of cross-couplings with, for example, amines (Buchwald–Hartwig coupling), arylboronic acids (Suzuki coupling), and many others.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.