

Abstract
Background
The ST313 sequence type of Salmonella Typhimurium causes invasive non-typhoidal salmonellosis and was thought to be confined to sub-Saharan Africa. Two distinct phylogenetic lineages of African ST313 have been identified.
Methods
We analysed the whole genome sequences of S. Typhimurium isolates from UK patients that were generated following the introduction of routine whole-genome sequencing (WGS) of Salmonella enterica by Public Health England in 2014.
Results
We found that 2.7% (84/3147) of S. Typhimurium from patients in England and Wales were ST313 and were associated with gastrointestinal infection. Phylogenetic analysis revealed novel diversity of ST313 that distinguished UK-linked gastrointestinal isolates from African-associated extra-intestinal isolates. The majority of genome degradation of African ST313 lineage 2 was conserved in the UK-ST313, but the African lineages carried a characteristic prophage and antibiotic resistance gene repertoire. These findings suggest that a strong selection pressure exists for certain horizontally acquired genetic elements in the African setting. One UK-isolated lineage 2 strain that probably originated in Kenya carried a chromosomally located bla CTX-M-15, demonstrating the continual evolution of this sequence type in Africa in response to widespread antibiotic usage.
Conclusions
The discovery of ST313 isolates responsible for gastroenteritis in the UK reveals new diversity in this important sequence type. This study highlights the power of routine WGS by public health agencies to make epidemiologically significant deductions that would be missed by conventional microbiological methods. We speculate that the niche specialisation of sub-Saharan African ST313 lineages is driven in part by the acquisition of accessory genome elements.
Electronic supplementary material
The online version of this article (doi:10.1186/s13073-017-0480-7) contains supplementary material, which is available to authorized users.
Background
Serovars of Salmonella enterica cause infections in a diverse range of hosts. In humans, Salmonella are responsible for a broad range of clinical presentations, from gastroenteritis to invasion of normally sterile compartments such as the bloodstream or brain. Two serovars, Salmonella Typhi and Salmonella Paratyphi A, are particularly associated with both human-restricted and invasive disease. The clinical syndrome caused by these serovars is known as typhoid or enteric fever and this has led to the remaining 2600 serovars being loosely described as non-typhoidal. By inference, ‘non-typhoidal’ serovars have been considered to be non-invasive in immunocompetent individuals; this crude clinical distinction is misleading [1].
These ‘non-typhoidal’ Salmonella (NTS) serovars typically have a broad host-range and the majority of human cases are foodborne, often originating from zoonotic reservoirs [2]. While most infections are typically associated with self-limiting gastroenteritis, in a minority of cases (~5%) invasive disease occurs, frequently due to human host immunosuppression, for example advanced HIV infection [3]. NTS are a significant public health burden worldwide and S. Typhimurium and S. Enteritidis are the predominant serotypes observed in clinical cases in most countries. In England and Wales, 48.7% of the isolates referred to the Salmonella Reference Service were S. Typhimurium or S. Enteritidis in 2012 (Public Health England [PHE] data: https://www.gov.uk/government/publications/salmonella-by-serotype/salmonella-by-serotype-2000-to-2010).
The clinical distinction between typhoidal and non-typhoidal disease is particularly unhelpful in sub-Saharan Africa (sSA), where non-typhoidal serovars are among the most common cause of bloodstream infection, a clinical condition known as invasive non-typhoidal Salmonella (iNTS) disease [1]. While the high prevalence of immunosuppressive illness such as HIV and malaria in sSA are clear predisposing factors for the emergence of iNTS disease as a major public health problem, the huge burden of disease has led to further investigation into the serovars responsible for iNTS disease. S. Typhimurium is the serovar most commonly associated with this condition [4].
Multi-locus sequence typing (MLST) is a molecular approach for typing microorganisms and uses the allelic variation of seven highly conserved Salmonella housekeeping genes to infer bacterial phylogeny [5]. Whole-genome sequencing (WGS) studies of isolates collected from patients with iNTS disease in sSA initially identified a novel sequence type (ST), ST313, of S. Typhimurium in 2009 [6]. Subsequent genome-based studies confirmed two distinct phylogenetic lineages of ST313 associated with iNTS and spatio-temporal phylogenetic reconstruction suggested that lineage 1 emerged around 1960 in southwest Africa, whereas lineage 2 emerged around 1977 in Malawi [7]. Both lineages are associated with antimicrobial resistance (AMR) mediated by differing Tn21-like integrons on the virulence plasmid pSLT [6] and it was proposed that clonal replacement of lineage 1 by lineage 2 had occurred, driven by the acquisition of chloramphenicol resistance in lineage 2 [7]. S. Typhimurium ST313 has recently been detected in Brazil [8]. Unlike African ST313, the nine Brazilian ST313 isolates were predominantly associated with gastro-intestinal infection and were antibiotic-susceptible.
It has been suggested that the link between S. Typhimurium ST313 and iNTS disease in sSA is that, compared with the generalist S. Typhimurium ST19, ST313 has adapted to an extra-intestinal/invasive lifestyle via genome degradation [6, 9]. This would be consistent with the finding of an accumulation of pseudogenes in pathways associated with gastrointestinal colonisation, as observed in host-restricted Salmonella serovars such as S. Typhi and in Yersinia pestis, Shigella spp, Mycobacterium leprae and Bordetella pertussis [10–16]. Another observation from comparative genomic studies, which supports the hypothesised enhanced virulence of ST313 includes the detection of novel prophages BTP1 and BTP5 [17] including the reported BTP1 phage-encoded putative virulence gene, st313-td [18]. A number of phenotypic characteristics which distinguish ST313 from gastroenteritis-associated ST19 strains have been described, including a reduction in motility, flagellin expression, stationary-phase catalase activity and biofilm formation [19–21]. Despite these phenotypes, it remains to be proven whether ST313 are intrinsically capable of causing a higher level of systemic disease than ST19.
Since 1st April 2014, every presumptive Salmonella isolate received by the Salmonella Reference Service (SRS) of PHE has had WGS to allow identification, characterisation and typing in one laboratory process [22].
In this study, we investigated the prevalence of ST313 in cases of laboratory-confirmed S. Typhimurium infections reported in England and Wales, obtained clinical data regarding the origin of isolates (faeces or blood) and contacted patients to determine whether infection was associated with travel to high-incidence areas such as sSA. We used a phylogenetic approach to place the UK-isolated ST313 into the evolutionary context of ST313 lineages isolated in Africa (hereafter referred to as African ST313). We then investigated the accessory genome, multi-drug resistance (MDR) determinants and the presence of pseudogenes in the UK isolates to shed light on the population structure and evolution of ST313 S. Typhimurium. We also compared key phenotypes of the UK-isolated ST313 with the African ST313 lineage 2.
Methods
Strains and metadata
Genome sequences from a total of 333 S. Typhimurium isolates dating from 2012 (of which 314 were from human patients), and 3018 S. Typhimurium from 1st January 2014 to 31st May 2016 (of which 2833 were from human patients) were analysed for this study. Non-human isolates originated from animal, food or environmental sources. All Salmonella isolates sequenced by PHE are available from the NCBI SRA BioProject PRJNA248064; the accession numbers, along with isolation source, for all 3351 S. Typhimurium genomes sequenced by PHE within the timeframes stated above are listed in Additional file 1: Table S1.
Of the total S. Typhimurium genomes sequenced in 2012 and 2014–2016, 7/333 (2.1%) and 79/3018 (2.6%) were S. Typhimurium ST313, respectively. If only isolates from human patients are included, 7/314 (2.2%) and 77/2833 (2.7%) were S. Typhimurium ST313, respectively. Full strain metadata and genome accession numbers can be found in Additional file 2: Table S2. Sequence data (FASTQs) from 23 representative African ST313 isolates sequenced by Okoro et al. [7] were downloaded from the European Nucleotide Archive (accession numbers available in Additional file 2: Table S2) and analysed in the same way as sequence data generated from UK-isolated ST313 isolates.
Sequencing
DNA extraction for Illumina sequencing of Salmonella isolates was carried out using a modified protocol of the Qiasymphony DSP DNA midi kit (Qiagen). In brief, 0.7 mL of an overnight Salmonella culture in a 96-well plate was harvested. Bacterial cells were pre-lysed in 220 μL of ATL buffer (Qiagen) and 20 μL Proteinase K (Qiagen) and incubated with shaking for 30 min at 56 °C. A total of 4 μL of RNase (100 mg/mL; Qiagen) was added to the lysed cells and re-incubated for a further 15 min at 37 °C. This step increased the purity of the DNA for downstream sequencing. DNA from the treated cells was then extracted on the Qiasymphony SP platform (Qiagen) and eluted in 100 μL of sterile water. DNA concentration was derived using the GloMax system (Promega) and quality (optimal OD260/230 = 1.8–2.0) was determined using the LabChip DX system (Perkin Elmer). Extracted DNA was prepared using the NexteraXT sample preparation method and sequenced with a standard 2 × 100 bp protocol on a HiSeq 2500 instrument (Illumina, San Diego, CA, USA). Raw FASTQs were processed with Trimmomatic [23] and bases with a PHRED score < 30 removed from the trailing end.
Single molecule sequencing was performed on the PacBio RS II instrument at the Centre for Genomic Research, University of Liverpool. DNA was extracted from strain U2 using the Zymo Research Quick-DNA™ Universal Kit (cat. no. D4069) as per the Biological Fluids & Cells protocol. Extracted DNA was purified with Ampure beads (Agencourt) and the quantity and quality was assessed by Nanodrop and Qubit assays. In addition, the Fragment Analyser (VH Bio), was used to determine the average size of the DNA, using a high sensitivity genomic kit. DNA was sheared to approximately 10 kb using a Covaris g-tube and spinning at 5400 rpm in an Eppendorf centrifuge. The size range was checked on the Fragment Analyser. DNA was treated with exonuclease V11 at 37 °C for 15 min. The ends of the DNA were repaired as described by Pacific Biosciences. Samples were incubated for 20 min at 37 °C with damage repair mix supplied in the SMRTbell library kit (Pacific Biosciences). This was followed by a 5-min incubation at 25 °C with end-repair mix. DNA was cleaned using 1:1 volume ratio of Ampure beads and 70% ethanol washes. DNA was ligated to adapters overnight at 25 °C. Ligation was terminated by incubation at 65 °C for 10 min followed by exonuclease treatment for 1 h at 37 °C. The SMRTbell library was purified with 1:1 volume ratio of Ampure beads. The library was size-selected on the Blue Pippin (Sage) in the range 7–20 kb. The DNA was recovered and the quantity of library and therefore the recovery was determined by Qubit assay and the average fragment size determined by Fragment Analyser. SMRTbell libraries were annealed to sequencing primers at values predetermined by the ‘Binding Calculator’ software (Pacific Biosciences) and complexes made with the DNA polymerase (P6/C4 chemistry). The complexes were bound to Magbeads and loaded onto three SMRT cells. Sequencing was done using 360-min movie times. Sequence data from the three SMRT cells were assembled using the HGAP3/Quiver assembler. This resulted in two contigs representing the chromosome and the pSLT virulence plasmid. Terminal repeats were manually trimmed to represent circular molecules and the chromosome assembly was reordered so that the sequence started at the thrL locus in accordance with convention for Salmonella finished genomes. The closed sequences for the U2 chromosome and pSLT virulence plasmid were 4,811,399 bp and 93,862 bp, respectively. Prokka [24] was used to annotate the two sequences, using the –force flag to preferentially annotate CDS from reference databases FN424405 for the chromosome and AE006471 for the virulence plasmid. The finished U2 genome and annotation were submitted to Genbank and can be accessed using the Genbank accession number LT855376 (chromosome) and LT855377 (virulence plasmid).
Genomic analysis
The multi-locus ST was determined using a modified version of SRST [25]. For phylogenetic analysis, processed sequence reads were mapped to the S. Typhimurium LT2 reference genome (GenBank: AE006468) using BWA mem [26]. SNPs were called using GATK2 [27] in unified genotyper mode. Core genome positions that had a high-quality SNP (>90% consensus, minimum depth 10x, MQ ≥ 30) in at least one strain were extracted and IQ-TREE with parameters –m TVM + ASC –bb 1000 was used to construct a maximum likelihood phylogeny [28]. The TVM model was chosen after using the model test functionality built into IQ-TREE.
To examine the evolutionary history of ST313, four timed phylogenies were constructed using BEAST v1.8.0 [29], with varying clock rate models and tree priors. The resulting models were compared in terms of their tree likelihood and posterior and the strict exponential and strict constant models were found to be superior. A comparison using AICM calculated with Tracer v1.6.0 showed that the models had very similar values, tree topologies and branching support in terms of posterior probability were similar between the models. The 95% HPD for the exponential growth rate estimate was – 0.0026 to 0.0006; the strict, constant growth model was selected as the estimate of growth rate from the exponential model was around 0 (i.e. constant).
Accessory genome analysis was performed using de novo assemblies of quality processed FASTQs produced using SPAdes v2.5.1 using default parameters except –careful and –k 22, 33, 55, 77 [30]. Whole-genome assemblies were compared to the reference ST313 strain D23580 using BRIG [31].
Microbiology
Phenotypic antimicrobial susceptibility testing was carried out for all UK-isolated ST313 strains. The antimicrobial susceptibility testing was done using breakpoint concentrations. Briefly, an agar dilution method involving Iso-sensitest agar or Muller-Hinton agar was used to determine if isolates were sensitive or resistant to a set concentration of individual antimicrobials (Additional file 3: Table S3).
Swimming motility assays were performed based on methods previously described [32]. A 3-μL aliquot of bacteria grown overnight in LB (Lennox Broth; 10 g/L Bacto Tryptone, 5 g/L yeast extract and 5 g/L NaCl, pH7.0) was spotted onto LB (Lennox) plates containing 0.3% Bacto Agar (Difco). Plates were incubated at 37 °C. After exactly 5 h, the migration diameter was measured and plates were photographed.
Catalase activity and RDAR morphotypes were assayed based on methods used by Singletary et al. [21]. Briefly, for catalase activity, 20 μL of 20% aqueous H2O2 was added to 1 mL of bacteria grown overnight in LB (Lennox), in 1-cm diameter glass test tubes. Tubes were photographed after 5-min incubation at room temperature and the height of the bubble column measured. For RDAR morphology, 2 μL of bacteria grown overnight in LB (Lennox) were spotted onto LB plates without NaCl and supplemented with 40 μg/mL Congo red and 20 μg/mL Coomassie blue. Plates were incubated at 25 °C and 37 °C for seven days without inversion. All experiments were conducted in triplicate.
Epidemiology
Food poisoning is a notifiable disease in the UK and diagnostic laboratories are obliged to report the isolation of Salmonella from human clinical diagnostic samples. However, data are frequently incomplete and detailed exposure information for cases is not always available in retrospect. Therefore, targeted surveillance questionnaires were attempted to obtain enhanced information, focusing primarily on collection of information on clinical severity of disease; travel history and consumption of foods of African origin were utilised during telephone interviews for cases reported during 2014–2016 to collate relevant epidemiological data. Cases for which enhanced information were available are shown in Additional file 2: Table S2.
Collection of this epidemiological data was not attempted for the 2012 cases, but limited travel data had been recorded on the SRS Salmonella surveillance database for some isolates. It is important to emphasise that the travel information for the 2012 isolates is of low quality and the absence of reported travel does not mean that international travel had not occurred. Odds ratios (OR) were calculated using the medcalc.org website https://www.medcalc.org/calc/odds_ratio.php.
Results
Epidemiology of ST313 in the UK
Between January 2014 and May 2016, 3018 S. Typhimurium isolates from human, animal, food and environmental sources underwent WGS by PHE, of which 79 (2.6%) were of multi-locus ST ST313. WGS data were available for a further 333 S. Typhimurium isolates from 2012, of which seven (2.1%) were ST313. Of the total S. Typhimurium isolates derived from human patients in England and Wales in the two time periods, 2.7% (84/3147) were ST313.
The 86 UK-derived ST313 isolates (Additional file 2: Table S2) originated from 75 individual human patients (five patients had two isolates sequenced and one patient had five isolates sequenced); one isolate was from a dog and one was isolated from an unspecified raw food sample. The sample type was recorded for 72 of the 75 human patient isolates; 13 patients had one or more isolate from extra-intestinal sites (blood, pus or bronchial alveolar lavage) indicating iNTS disease and 59 isolates were from faeces alone (indicating gastrointestinal infection). Travel information was available for 51 of the 75 human patients, of whom eight reported travel to sSA during the estimated disease incubation period and one adult male reported consuming food of West African origin in London. Of the 51 patients with travel information, 48 had sample type recorded. Of the eight patients who reported travel to Africa, six had extra-intestinal infections. In contrast, just two of 40 patients for whom travel information was available and did not report travel to Africa had extra-intestinal infection, showing that travel to Africa is significantly associated with iNTS disease in the UK (OR = 57.0 [95% confidence interval {CI} = 6.7–484.8], p value = 0.0002) (Table 1A).
Table 1.
Summary of key epidemiological features of ST313 sampled by PHE. Full metadata for all isolates in this study is available in Additional file 2: Table S2
| A. Association between isolation source and travel to sSA. Total = 48a | ||
| Extra-intestinal infection | Gastro-intestinal infection | |
| Reported travel to sSA | 6 | 2 |
| No reported travel to sSA | 2 | 38 |
| OR = 57.0 (95% CI = 6.7–484.8), p value = 0.0002 | ||
| B. Association between lineage 2 infection and travel to sSA. Total = 51b | ||
| Lineage 2 | Non-lineage 2 | |
| Reported travel to sSA | 7 | 1 |
| No reported travel to sSA | 2 | 41 |
| OR = 143.5 (95% CI = 11.4–1802.9), p value = 0.0001 | ||
| C. Association between lineage 2 infection and extra-intestinal infection. Total = 72c | ||
| Lineage 2 | Non-lineage 2 | |
| Extra-intestinal infection | 10 | 3 |
| Gastro-intestinal infection | 1 | 57 |
| OR = 190.0 (95% CI = 17.9–2014.0), p value < 0.0001 | ||
a51 individual patient isolates with travel information, but three with source unknown
b51 individual patient isolates with travel information
c75 individual human patient isolates in total, but three with source unknown. One isolate not included in phylogeny due to poor quality sequence, so lineage unknown
Phylogenetic analysis reveals unprecedented diversity of ST313
All the UK-derived isolates in this study originated from diagnostic laboratories in England and Wales. To simplify the categorisation/differentiation of the lineages for discussion purposes, isolates belonging to lineage 1 and 2 (including those isolated in the UK) will be referred to as African lineages and the non-lineage 1 and 2 isolates will be referred to as UK-ST313.
Sequence data quality was sufficient to permit whole-genome single nucleotide polymorphism (SNP) phylogenetic analysis of the isolates from 76 of the 77 patient and non-human isolates. Within the wider phylogenetic context of S. Typhimurium, all ST313 isolates submitted to PHE formed a monophyletic group that clustered with previously described African ST313 isolates [7] (Fig. 1). A second maximum likelihood ST313 phylogeny was generated with a closely related ST19 strain also received by PHE (strain U21) as an outlier, to study phylogenetic relationships (Fig. 2a). Of the 76 isolates from distinct patients/sources, 12 belonged to the previously described lineage 2 and 64 did not fall within any ST313 lineage that has been reported to date. No lineage 1 isolates were identified. Where more than one isolate was derived from the same patient, there were no core genome SNP differences between the isolates. Furthermore, the African associated lineages 1 and 2 did not form a monophyletic group within the novel diversity observed. Both of the African lineages shared more recent common ancestors with UK-associated strains than with each other. Neither the food nor the animal isolate belonged to lineage 2.
Fig. 1.
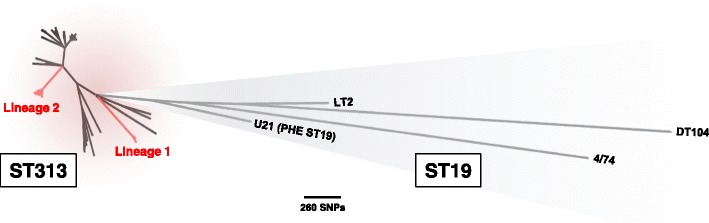
UK-ST313 isolates are phylogenetically distinct from African lineages. Unrooted maximum likelihood phylogeny of ST313 in the context of reference S. Typhimurium ST19 isolates. Isolate U21 was an ST19 isolate, closely related to ST313, that was used as an outgroup in further analyses. The African epidemic ST313 lineages 1 and 2 are labelled
Fig. 2.
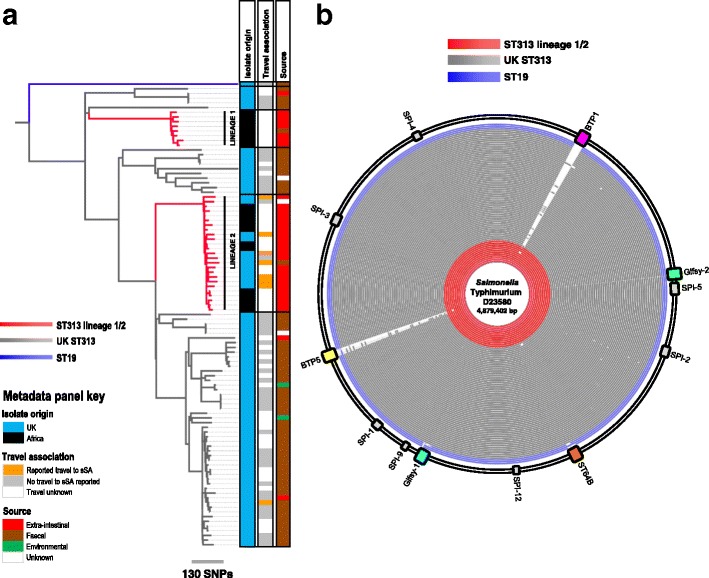
UK-ST313 are associated with gastrointestinal infection and do not harbour the African lineage associated prophages, BTP1 & BTP5. a Maximum likelihood phylogeny of 76 UK-isolated ST313 strains received by PHE in the context of 24 African ST313 sequenced by Okoro et al. [7]. Red branches indicate ST313 lineage 1 and 2. Adjacent metadata panel showing: 1. Country isolate was associated with; Africa - orange, not Africa - blue; 2. Source; extra-intestinal - red, faecal - brown, environmental - green, unknown - grey. b BLAST ring image showing blastn comparison of all UK-isolated ST313 genomes (red and grey rings) along with three reference ST19 strains (blue rings) against lineage 2 representative strain D23580. The position of the prophages (coloured blocks) and Salmonella pathogenicity islands (grey blocks) in lineage 2 strain D23580 are shown around the outside of the ring
The UK-ST313 isolates do not themselves form a coherent monophyletic cluster, revealing a previously unreported level of genetic diversity within ST313 (Fig. 1). To examine the evolutionary history of ST313, a maximum clade credibility tree was inferred using BEAST (Fig. 3) and the topology was largely congruent with respect to the Maximum Likelihood tree (Figs. 1 and 2). The most recent common ancestor (MRCA) of ST313 is estimated to have been in approximately 1787 (95% highest posterior distribution [HPD], 1735–1836). Lineage 1 diverged from other ST313 sampled in this study in approximately 1796 (95% HPD 1744–1842), while lineage 2 diverged from other ST313 sampled here in 1903 (95% HPD 1876–1927). The lineage 1 MRCA dated to around 1984 (95% HPD 1979–1987), while the lineage 2 MRCA dated to around 1991 (95% HPD 1986–1995). These confidence intervals overlap with the confidence intervals reported for the emergence of the two lineages by Okoro et al., 2012 [7]. The two African lineages do not form a monophyletic group and share an MRCA which is very close to that of ST313 as a whole. Both lineages 1 and 2 are separated from other sampled ST313 by long branches, indicating a distant MRCA with other isolates and suggesting that a tight population bottleneck has occurred relatively recently in evolutionary history.
Fig. 3.
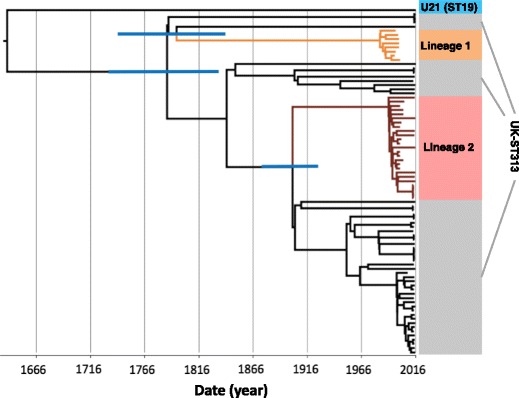
The timed phylogeny of all UK-isolated ST313 strains from this study and a representative sub-sample of ST313 genomes from Okoro et al. [7]. Figure shows the maximum clade credibility tree from BEAST. Branches 95% HPD are displayed in blue for key nodes defining lineage 1 and lineage 2 (for tree with all 95% HPD, see Additional file 3: Figure S4). Branches belonging to lineage 1 are coloured orange and branches belonging to lineage 2 are coloured brown
The association between phylogenetic context and travel to Africa
We investigated the association between reported travel to sSA and infection with the lineage 2 isolates. Of the eight UK patients reporting travel to sSA during the seven days before disease onset, seven were infected with a lineage 2 isolate. In contrast, of the 43 patients who reported no travel to sSA, two were infected with lineage 2 (Table 1B). This shows that travel to sSA was significantly associated with infection by ST313 lineage 2 (OR = 143.5 [95% CI = 11.4–1802], p value = 0.0001).
We investigated whether the ST313 lineage 2 isolates were more frequently associated with extra-intestinal or gastro-intestinal infection. Of the 11 patients infected with lineage 2 (and for which source of isolation data was available), ten patients had isolates that originated from extra-intestinal sites. In contrast, for the patients infected with UK-ST313 isolates, three of 60 isolates were from extra-intestinal sites (Table 1C). These data show that infection with lineage 2 is significantly associated with invasive disease (OR = 190.0 [95% CI = 17.9–2014.0], p value < 0.0001).
Salmonella isolates that are related by a common source exposure such as a shared contaminated food source are often within a single linkage cluster that varies by less than five core-genome SNP differences (termed a 5-SNP cluster). The UK-derived lineage 2 isolates did not belong to a single linkage cluster as they varied by at least 38 SNPs, with a median pairwise distance of 57 SNPs (Additional file 3: Figure S4A). In contrast, we identified groups of isolates that were very closely related within the UK-ST313 (Additional file 3: Figure S4B). There were three 5-SNP clusters which contained four, three and three isolates in each. This level of relatedness of these UK-ST313 is consistent with exposure to a common source.
Accessory genome signatures of ST313
MDR is a key phenotypic feature associated with both the African ST313 lineages and is encoded by Tn21-like integron insertions on the pSLT virulence plasmid. Analysis of the genome sequences indicated that all 76 ST313 isolates in this study carried the pSLT plasmid. However, the majority of the UK-ST313 isolates were antibiotic-sensitive (59/64 were sensitive to all antimicrobials tested) and no consistent AMR gene profile was identified. In contrast, 10/12 UK-isolated lineage 2 isolates contained the same pSLT-associated MDR locus as the African lineage 2 reference strain D23580 [6]. Four UK-isolated lineage 2 strains exhibited an atypical AMR gene profile; one isolate (U45) lacked the chloramphenicol resistance catA gene and another (U73) carried only the beta-lactamase gene bla TEM1. A third isolate, U1, had probably acquired resistance to fluoroquinolones via a mutation in the DNA gyrase subunit A gene, gyrA.
The fourth atypical UK-isolated lineage 2 isolate, U60, carried additional antibiotic resistance genes including extended-spectrum beta-lactamases (ESBL) bla CTX-M-15 and bla OXA-1, and genes conferring resistance to aminoglycosides, trimethoprim and tetracycline; aac(6′)-Ib-cr, dfrA-14, tet(A)-1 (Fig. 4a). Isolate U60 also encoded the tellurium heavy metal resistance operon (terBCDEF). Comparison to lineage 2 reference strain D23580 identified a 29-kb deletion in the pSLT-BT virulence plasmid [6], which corresponded to the conjugal transfer region. Additionally, sequence reads from strain U60 could be mapped to 97% of the IncHI2 pKST313 plasmid (Additional file 3: Figure S1A), a novel plasmid which has recently been reported in lineage 2 isolates from Kenya and is known to encode ESBL resistance loci [33].
Fig. 4.
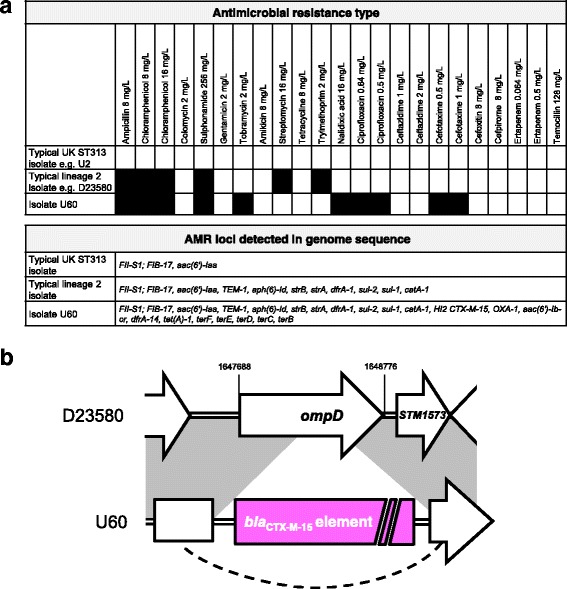
Isolate U60 contains additional resistance genes including a bla CTX-M-15 locus inserted into the chromosomal ompD locus. a Antimicrobial resistance typing data and resistance genes detected in genome sequences for isolate U60, compared to data for typical lineage 2 and UK-ST313 isolates. b Schematic illustrating the insertion of bla CTX-M-15 element into the chromosomal ompD locus in isolate U60. Further information is given in Additional file 3: Figure S1
A more detailed analysis of the genome of isolate U60 identified that the bla CTX-M-15 gene was inserted into the chromosome (location 1648104-1648109 on the D23580 reference genome), disrupting the ompD locus (Fig. 4b; Additional file 3: Figure S1). ESBL resistance genes have previously been reported in African ST313 isolates carrying plasmids such as pKST313 [33, 34], but this is the first report of a chromosomally encoded ESBL resistance gene in S. Typhimurium ST313.
The assembled genomes of the UK-ST313 isolates were compared to the African ST313 reference strain D23580 using BLAST (Fig. 2b). In agreement with published data [6, 9], the majority of the core genome, including the Salmonella Pathogenicity Island (SPI) repertoire was conserved in the ST313 isolates in this study and in three ST19 gastroenteritis isolates (Fig. 2b). The African ST313 lineages carry two prophages, BTP1 and BTP5, that are absent from ST19 strains [9, 17]. The entire BTP1 and BTP5 prophages were found in most ST313 isolates that belonged to African lineage 2 (12/13), but one UK-isolated lineage 2 strain, U68, lacked both prophages. The complete BTP1 and BTP5 prophages were not identified in any of the UK-ST313 isolates (Fig. 2b), though some isolates contained partial and fragmented identity to BTP1 and BTP5, indicating the presence of related prophages [35, 36] which may not occupy the same attachment site. As expected, the st313-td gene [18, 37] was carried by all 12 lineage 2 strains isolated from the UK that contained prophage BTP1. Only 1/51 UK-ST313 isolates contained the st313-td gene (isolate U76), where it was located on a prophage with 90% identity to BTP1.
To confirm the conservation of chromosomal organisation in the UK-ST313 isolates, a representative isolate U2 was re-sequenced by PacBio long-read sequencing. The assembly produced two closed circular contigs representing the chromosome and the pSLT virulence plasmid (Additional file 3: Figure S2). Comparison with the ST313 lineage 2 reference genome D23580 identified no large chromosomal re-arrangements, deletions or duplications, and confirmed that the BTP1 and BTP5 attachment sites were unoccupied and did not contain additional prophages. No additional plasmids larger than the detection limit of the PacBio sequencing (~10 kb) were detected in isolate U2. Overall, these data show that there is no particular prophage repertoire associated with the UK-ST313 strains.
Genome degradation and pseudogenes in UK and African ST313
The ST313 lineages 1 and 2 responsible for iNTS disease in Africa have undergone genome degradation [6, 9]. The pseudogenes identified in lineage 2 representative strain D23580 were put into the context of the high-quality finished genome of UK-ST313 isolate U2 (Fig. 5). The majority (34/44) of pseudogene mutations were conserved in U2. The only pseudogenes associated with characterised genes present in lineage 2 and intact in UK-ST313 strain U2 were macB, sseI and lpxO (Fig. 5). Of the 190 isolates (originating from 12 patients) of lineage 2, all had disruptions to the lpxO, ratB, allP, allB, pagO and pipD genes, showing genome degradation reported in African lineage 2 is conserved in lineage 2 strains isolated in the UK.
Fig. 5.

The majority of pseudogenes identified in lineage 2 strain D23580 are conserved in UK-ST313 representative strain U2. Heatmap adapted from Kingsley et al. [6] showing genome degradation in ST313 strain D23580 (first heatmap column) in the context of strain U2 (final column). Grey indicates pseudogenes conserved in both strains, while red indicates genes which are not degraded and therefore likely to be functional, in strain U2
UK and African ST313 strains share key phenotypes
Several studies have associated the ability of ST313 to cause iNTS disease with particular phenotypic characteristics, such as the lack of RDAR morphotype formation, reduced swimming motility and the inability to produce catalase at stationary phase [19–21, 38]. We investigated these phenotypic characteristics in the context of the UK-ST313 strains, using a subset of 16 UK-isolated ST313 that included 13 UK-ST313 isolates and three lineage 2 isolates. The phylogenetic context of these 16 isolates is shown in Additional file 3: Figure S3. Lineage 2 representative strain D23580 and ST19 representative strain 4/74 were included as positive and negative controls.
The swimming motility of UK-isolated ST313 was highly variable between isolates (Fig. 6a). One lineage 2 strain, U1, appeared to show low levels of motility but this strain was observed to have a growth defect (data not shown). The ST313 lineage 2 representative strain D23580 was less motile than ST19 strain 4/74, consistent with previous reports [19, 20]. However, there was no apparent association between motility (as measured by migration diameter) and phylogenetic context of the lineage 2 strains and the UK-ST313 strains.
Fig. 6.
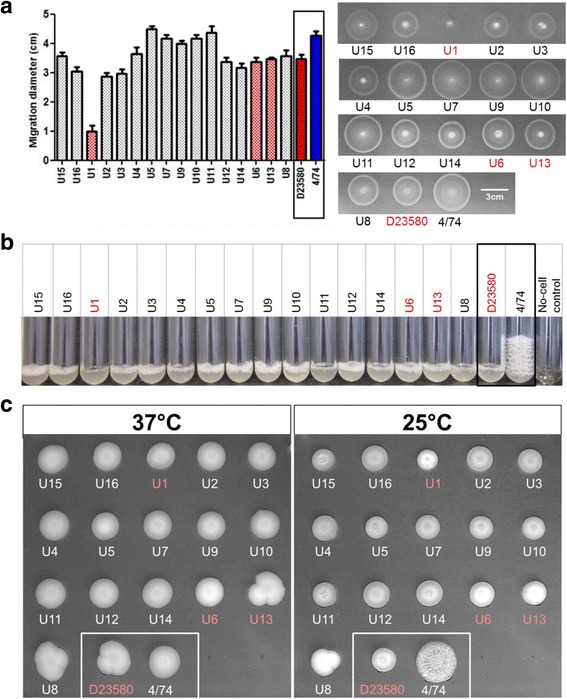
In vitro phenotypes of a subset of UK-isolated ST313 strains in the context of representative ST313 lineage 2 and ST19 strains D23580 and 4/74. UK-isolated strains that belong to African lineage 2 (U1, U6 and U13) are highlighted in red throughout. a Migration diameter after 5 h (average of three replicates is shown together with error bars representing standard deviation). A representative plate is shown, right. b Stationary phase catalase activity represented by bubble column height after 5 min exposure to 20 μL 20% H2O2. c RDAR morphology assay. RDAR phenotype forms after prolonged incubation at 25 °C but not at 37 °C
A non-synonymous SNP in the katE gene was reported to be responsible for the lack of catalase activity in ST313 lineage 2 [21]. All 16 UK-isolated strains were shown to be negative for stationary phase catalase activity, as was the lineage 2 representative strain D23580 (Fig. 6b). In contrast, ST19 strain 4/74 showed considerable stationary phase catalase activity, consistent with previous findings [21].
The RDAR morphotype of S. enterica is linked to resistance to desiccation and exogenous stresses [39]. African lineage 2 ST313 is reported to be defective in RDAR morphotype formation due to a truncated BcsG protein generated by a premature stop codon [21]. All the UK-isolated strains and the African lineage 2 reference strain D23580 did not exhibit the RDAR morphotype. In our experiments, only the ST19 strain 4/74 exhibited the RDAR morphotype (Fig. 6c).
These experiments did not identify any phenotypic differences that distinguished the UK-ST313 isolates from ST313 lineage 2 and future work is needed to identify African ST313-specific phenotypes.
Discussion
Recent reports of iNTS disease have been associated with S. Typhimurium ST313 in sSA [6, 7, 37] and it had been suggested that ST313 was geographically restricted to sSA. This prompted us to investigate the presence of ST313 among S. Typhimurium isolates from the UK.
We discovered that 2.7% of S. Typhimurium isolates referred to PHE from human patients are of MLST ST313 and that this sequence type is heterogeneous in terms of clinical presentation, genomic characteristics and epidemiology. The UK-isolated ST313 strains are predominantly fully antimicrobial-susceptible and cause gastroenteritis. This was also the case with the recently described ST313 strains from Brazil [8], raising the possibility that these isolates are more closely related to the UK-ST313 reported here than to African lineage 2. Further work is needed to show where Brazilian ST313 isolates fit into the population structure of S. Typhimurium ST313.
We identified a significant association between travel to Africa and infection with the previously described African-associated, ST313 lineage 2. The amount of diversity between the lineage 2 ST313 isolates from the UK (Additional file 3: Figure S4) was not consistent with an immediate, common source of exposure. Instead, we suggest that these isolates were associated with travel to Africa, a hypothesis that was supported by epidemiological data that linked the isolates to different African countries (Table 1). The UK-ST313 isolates, while having no large clusters that are typical of foodborne outbreaks, did contain three small clusters that were related at the 5-SNP level. This pattern of relatedness is consistent with UK-ST313 isolates being transmitted in a similar way to other gastrointestinal Salmonella in Europe.
Our study revealed novel diversity within ST313, which was previously restricted to two African lineages that had exhibited recent clonal expansion [7]. Here we place the African lineages into an evolutionary context by showing that lineages 1 and 2 do not form a monophyletic group within ST313, which is suggestive of two separate introductions of ST313 into sSA. African lineages 1 and 2 diverged from their inferred MRCA with UK lineages around 1796 and 1903, respectively. These findings reflect the limitations of classifying bacterial pathogens simply on the basis of sequence type and show that in the post-genomic era, the resolution offered by MLST may not be sufficient to describe epidemiologically relevant population structures.
It has been estimated that 9.2% of cases of Salmonellosis in the EU can be attributed to international travel and therefore sequencing Salmonella isolated in Europe can provide valuable information regarding the global diversity of Salmonella associated with human disease [40, 41]. The genome of one UK-isolated lineage 2 isolate associated with travel to Kenya, U60, contained sequences with high nucleotide similarity to a recently described IncHI2 plasmid, pKST313, that was carried by ceftriaxone-resistant ST313 isolates from Kenya [21]. Until now, the bla CTX-M-15 gene has only been found to be plasmid-associated in Salmonella. We discovered that the bla CTX-M-15 gene was chromosomally encoded in isolate U60, causing disruption of the ompD locus which has two implications. First, chromosomal integration ensures stability of ESBL-resistance even in the absence of the plasmid. Second, ompD encodes an outer membrane porin of S. Typhimurium that is highly immunogenic [42] and absent from S. Typhi. Accordingly, the disruption of ompD could enhance the reported ‘stealth’ phenotype of ST313 lineage 2 infection [19]. We note that OmpD is a potential vaccine target for iNTS [43] and the absence of OmpD from African ST313 populations could have implications for future iNTS vaccine development.
People infected with ST313 lineage 2 in the UK were significantly more likely to suffer from invasive disease than patients infected with UK-ST313 isolates. This observation provided an excellent opportunity to use comparative genomics to relate genetic findings that have been linked to the pathology of lineage 2 ST313 into the context of closely related, gastrointestinal-associated strains. We found that the only genetic characteristics common to both lineages 1 and 2 and absent from the UK-ST313 genomes were the BTP1 and BTP5 prophages and the plasmid-associated MDR loci. The two African lineages do not share a common ancestor that carried either prophage, suggesting independent acquisition of BTP1 and BTP5 by ST313 lineage 1 and 2. While the MDR loci of lineage 1 and 2 confer similar patterns of AMR, they are genetically distinct. The maintenance of the prophage and plasmid-encoded accessory genome in two distinct ST313 lineages in Africa implies a strong selection pressure that caused convergent evolution of the two African lineages. In contrast, we identified evidence for an assortment of distinct prophage repertoires in the UK-ST313 isolates, indicating an absence of selection for specific mobile elements.
Aside from the addition of mobile genetic elements and virulence factors, genome degradation by the accumulation of pseudogenes and deletion events accompanies adaption to a more invasive lifestyle [13, 44]. Initial analysis of the African ST313 representative strain D23580 genome reported 23 pseudogenes compared to the six present in ST19 strain SL1344 [6]. Here, we found that the majority of genome degradation found in lineage 2 strain D23580 was conserved in UK representative strain U2. The only pseudogenes associated with characterised genes that were found to be specific to African lineage 2 ST313 were the SPI2-secreted effector gene sseI, lipid A modification gene lpxO and macrolide efflux pump gene macB, each of which could play a role in infection dynamics [45–47].
A number of the in vitro phenotypes which have been reported for lineage 2 ST313, and could contribute to a host-adapted lifestyle [19–21, 38], were examined in the UK-ST313. Swimming motility was highly variable among the strains tested and UK-ST313 isolates behaved identically to African lineage 2 isolates in the catalase and RDAR morphotype assays. We detected no African-lineage-specific phenotypic characteristics and speculate that reduced motility, defective catalase activity and loss of RDAR formation are not be directly linked to iNTS disease.
A key contributing factor to iNTS disease is host immunosuppression and one limitation of this retrospective study was that the underlying health status of the patients was unknown. This study does highlight the extraordinary epidemiological insights that routine genomic surveillance of pathogens by public health agencies can offer and the ability to understand the pathogenesis of novel pathovars. In a clinical setting, the knowledge that an immune-compromised patient was infected with lineage 2 ST313 could impact clinical decision-making.
Conclusions
We have discovered previously unknown diversity in the ST313 sequence type that highlights the convergent evolution towards niche specialisation that has occurred in the African lineages. The routine genomic surveillance of pathogens is now being adopted internationally and will bring an unprecedented ability to monitor emerging threats, such as the appearance of extended-spectrum beta-lactamase resistance. WGS of clinical isolates represents a new window with which to view the epidemiology and microbiology of infectious diseases.
Additional files
Accession numbers, year of isolation and source details of all 3,351 S. Typhimurium genomes sequenced by PHE within the timeframe of this study. (XLSX 80 kb)
Complete metadata and accession numbers for all S. Typhimurium ST313 isolates included in this study. (XLSX 24 kb)
The antimicrobials used for susceptibility testing in this study. Figure S1. Isolate U60 contains additional resistance genes including a bla CTX-M-15 locus inserted into the chromosomal ompD locus. Figure S2. Circular representation of finished genome of UK-ST313 representative strain U2, showing the chromosome and the pSLT-U2 virulence plasmid. Figure S3. Fully labelled phylogenetic tree highlighting the context of subset of 16 UK-isolated ST313 strains included in phenotypic testing. Figure S4. The maximum clade credibility tree from BEAST showing the timed phylogeny of all ST313 isolated in this study and a representative sub-sample of African ST313 from Okoro et al. [7]. Figure S5. Distribution of pairwise SNP distances of lineage 2 ST313 and UK-ST313. (PDF 1193 kb)
Acknowledgements
We would like to acknowledge PHE Genomic Services and Development Unit, Infectious Disease Informatics and Salmonella Reference Service members, in particular Martin Day for antibiotic resistance typing. We are grateful to John Kenny, Margaret Hughes and Xuan Liu at the Centre for Genomic Research, University of Liverpool for assistance with PacBio sequencing. We also thank Rob Kingsley and members of the Hinton lab, in particular Rocío Canals, for helpful discussion and comments.
Funding
This work was supported by funding from the University of Liverpool and a Wellcome Trust Senior Investigator award to JCDH (Grant 106914/Z/15/Z).
Availability of data and materials
All data generated or analysed during this study are included in this published article (and its supplementary information files).
Abbreviations
- AMR
Antimicrobial resistance
- ESBL
Extended-spectrum beta-lactamase
- HPD
Highest posterior distribution
- iNTS
Invasive non-typhoidal Salmonella
- MDR
Multi-drug resistance
- MLST
Multi-locus sequence typing
- MRCA
Most recent common ancestor
- NTS
Non-typhoidal Salmonella
- PHE
Public Health England
- RDAR
Red dry and rough
- SRS
Salmonella Reference Service
- sSA
Sub-Saharan Africa
- ST
Sequence type
Authors’ contributions
PM, SO, CJ, EP, NF, JCDH and TD conceived experiments. PM, SO, LK, WR, CL, LL, SN contributed data or analysis. All authors contributed to, read and approved the final manuscript. JCDH and TD are joint senior authors. PM and SO contributed equally to this work.
Ethics approval and consent to participate
Ethical approval for the detection of gastrointestinal bacterial pathogens from faecal specimens, or the identification, characterisation and typing of cultures of gastrointestinal pathogens, submitted to the Gastrointestinal Bacteria Reference Unit is not required as it is covered by Public Health England’s surveillance mandate.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Jay C. D. Hinton and Timothy J. Dallman contributed equally as senior authors.
Electronic supplementary material
The online version of this article (doi:10.1186/s13073-017-0480-7) contains supplementary material, which is available to authorized users.
References
- 1.Feasey NA, Dougan G, Kingsley RA, Heyderman RS, Gordon MA. Invasive non-typhoidal salmonella disease: an emerging and neglected tropical disease in Africa. Lancet. 2012;379:2489–2499. doi: 10.1016/S0140-6736(11)61752-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mead PS, Slutsker L, Dietz V, McCaig LF, Bresee JS, Shapiro C, et al. Food-related illness and death in the United States. Emerg Infect Dis. 1999;5:607–625. doi: 10.3201/eid0505.990502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ao TT, Feasey NA, Gordon MA, Keddy KH, Angulo FJ, Crump JA. Global burden of invasive nontyphoidal Salmonella disease, 2010. Emerg Infect Dis. 2015;21:941–949. doi: 10.3201/eid2106.140999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reddy EA, Shaw AV, Crump JA. Community-acquired bloodstream infections in Africa: a systematic review and meta-analysis. Lancet Infect Dis. 2010;10:417–432. doi: 10.1016/S1473-3099(10)70072-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Achtman M, Wain J, Weill FX, Nair S, Zhou Z, Sangal V, et al. Multilocus sequence typing as a replacement for serotyping in Salmonella enterica. PLoS Pathog. 2012;8:e1002776. doi: 10.1371/journal.ppat.1002776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kingsley RA, Msefula CL, Thomson NR, Kariuki S, Holt KE, Gordon MA, et al. Epidemic multiple drug resistant Salmonella Typhimurium causing invasive disease in sub-Saharan Africa have a distinct genotype. Genome Res. 2009;19:2279–2287. doi: 10.1101/gr.091017.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Okoro CK, Kingsley RA, Connor TR, Harris SR, Parry CM, Al-Mashhadani MN, et al. Intracontinental spread of human invasive Salmonella Typhimurium pathovariants in sub-Saharan Africa. Nat Genet. 2012;44:1215–1221. doi: 10.1038/ng.2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Almeida F, Seribelli AA, da Silva P, Medeiros MIC, Dos Prazeres RD, Moreira CG, et al. Multilocus sequence typing of Salmonella Typhimurium reveals the presence of the highly invasive ST313 in Brazil. Infect Genet Evol J Mol Epidemiol Evol Genet Infect Dis. 2017;51:41–44. doi: 10.1016/j.meegid.2017.03.009. [DOI] [PubMed] [Google Scholar]
- 9.Okoro CK, Barquist L, Connor TR, Harris SR, Clare S, Stevens MP, et al. Signatures of adaptation in human invasive Salmonella Typhimurium ST313 populations from sub-Saharan Africa. PLoS Negl Trop Dis. 2015;9:e0003611. doi: 10.1371/journal.pntd.0003611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cole ST, Eiglmeier K, Parkhill J, James KD, Thomson NR, Wheeler PR, et al. Massive gene decay in the leprosy bacillus. Nature. 2001;409:1007–1011. doi: 10.1038/35059006. [DOI] [PubMed] [Google Scholar]
- 11.McClelland M, Sanderson KE, Clifton SW, Latreille P, Porwollik S, Sabo A, et al. Comparison of genome degradation in Paratyphi A and Typhi, human-restricted serovars of Salmonella enterica that cause typhoid. Nat Genet. 2004;36:1268–1274. doi: 10.1038/ng1470. [DOI] [PubMed] [Google Scholar]
- 12.McNally A, Thomson NR, Reuter S, Wren BW. “Add, stir and reduce”: Yersinia spp. as model bacteria for pathogen evolution. Nat Rev Microbiol. 2016;14:177–190. doi: 10.1038/nrmicro.2015.29. [DOI] [PubMed] [Google Scholar]
- 13.Nuccio S-P, Bäumler AJ. Comparative analysis of salmonella genomes identifies a metabolic network for escalating growth in the inflamed gut. MBio. 2014;5:e00929–14. doi: 10.1128/mBio.00929-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parkhill J, Wren BW, Thomson NR, Titball RW, Holden MTG, Prentice MB, et al. Genome sequence of Yersinia pestis, the causative agent of plague. Nature. 2001;413:523–527. doi: 10.1038/35097083. [DOI] [PubMed] [Google Scholar]
- 15.The HC. Thanh DP, Holt KE, Thomson NR, Baker S. The genomic signatures of Shigella evolution, adaptation and geographical spread. Nat Rev Micro. 2016;14:235–250. doi: 10.1038/nrmicro.2016.10. [DOI] [PubMed] [Google Scholar]
- 16.Yang F, Yang J, Zhang X, Chen L, Jiang Y, Yan Y, et al. Genome dynamics and diversity of Shigella species, the etiologic agents of bacillary dysentery. Nucleic Acids Res. 2005;33:6445–6458. doi: 10.1093/nar/gki954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Owen SV, Wenner N, Canals R, Makumi A, Hammarlöf DL, Gordon MA, et al. Characterization of the prophage repertoire of African Salmonella Typhimurium ST313 reveals high levels of spontaneous induction of novel phage BTP1. Front Microbiol. 2017;8:235. doi: 10.3389/fmicb.2017.00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Herrero-Fresno A, Wallrodt I, Leekitcharoenphon P, Olsen JE, Aarestrup FM, Hendriksen RS. The role of the st313-td gene in virulence of Salmonella Typhimurium ST313. PLoS ONE. 2014;9:e84566. doi: 10.1371/journal.pone.0084566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carden S, Okoro C, Dougan G, Monack D. Non-typhoidal Salmonella Typhimurium ST313 isolates that cause bacteremia in humans stimulate less inflammasome activation than ST19 isolates associated with gastroenteritis. Pathog Dis. 2015;73:ftu023. doi: 10.1093/femspd/ftu023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ramachandran G, Perkins DJ, Schmidlein PJ, Tulapurkar ME, Tennant SM. Invasive Salmonella Typhimurium ST313 with naturally attenuated flagellin elicits reduced inflammation and replicates within macrophages. PLoS Negl Trop Dis. 2015;9:e3394. doi: 10.1371/journal.pntd.0003394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singletary LA, Karlinsey JE, Libby SJ, Mooney JP, Lokken KL, Tsolis RM, et al. Loss of multicellular behavior in epidemic African nontyphoidal Salmonella enterica serovar Typhimurium ST313 strain D23580. MBio. 2016;7:e02265. doi: 10.1128/mBio.02265-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ashton PM, Nair S, Peters TM, Bale JA, Powell DG, Painset A, et al. Identification of Salmonella for public health surveillance using whole genome sequencing. PeerJ. 2016;4:e1752. doi: 10.7717/peerj.1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinforma Oxf Engl. 2014;30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 25.Inouye M, Conway TC, Zobel J, Holt KE. Short read sequence typing (SRST): multi-locus sequence types from short reads. BMC Genomics. 2012;13:338. doi: 10.1186/1471-2164-13-338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinforma Oxf Engl. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–274. doi: 10.1093/molbev/msu300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol J Comput Mol Cell Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alikhan N-F, Petty NK, Ben Zakour NL, Beatson SA. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics. 2011;12:402. doi: 10.1186/1471-2164-12-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bogomolnaya LM, Aldrich L, Ragoza Y, Talamantes M, Andrews KD, McClelland M, et al. Identification of novel factors involved in modulating motility of Salmonella enterica serotype typhimurium. PLoS ONE. 2014;9:e111513. doi: 10.1371/journal.pone.0111513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kariuki S, Okoro C, Kiiru J, Njoroge S, Omuse G, Langridge G, et al. Ceftriaxone-resistant Salmonella enterica serotype typhimurium sequence type 313 from Kenyan patients is associated with the blaCTX-M-15 gene on a novel IncHI2 plasmid. Antimicrob Agents Chemother. 2015;59:3133–3139. doi: 10.1128/AAC.00078-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Feasey NA, Cain AK, Msefula CL, Pickard D, Alaerts M, Aslett M, et al. Drug resistance in Salmonella enterica ser. Typhimurium bloodstream infection, Malawi. Emerg Infect Dis. 2014;20:1957–1959. doi: 10.3201/eid2011.141175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Casjens SR, Thuman-Commike PA. Evolution of mosaically related tailed bacteriophage genomes seen through the lens of phage P22 virion assembly. Virology. 2011;411:393–415. doi: 10.1016/j.virol.2010.12.046. [DOI] [PubMed] [Google Scholar]
- 36.Hatfull GF. Bacteriophage Genomics. Curr Opin Microbiol. 2008;11:447–453. doi: 10.1016/j.mib.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leekitcharoenphon P, Friis C, Zankari E, Svendsen CA, Price LB, Rahmani M, et al. Genomics of an emerging clone of Salmonella serovar Typhimurium ST313 from Nigeria and the Democratic Republic of Congo. J Infect Dev Ctries. 2013;7:696–706. doi: 10.3855/jidc.3328. [DOI] [PubMed] [Google Scholar]
- 38.Yang J, Barrila J, Roland KL, Kilbourne J, Ott CM, Forsyth RJ, et al. Characterization of the invasive, multidrug resistant non-typhoidal Salmonella strain D23580 in a murine model of infection. PLoS Negl Trop Dis. 2015;9:e0003839. doi: 10.1371/journal.pntd.0003839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.White AP, Gibson DL, Kim W, Kay WW, Surette MG. Thin aggregative fimbriae and cellulose enhance long-term survival and persistence of Salmonella. J Bacteriol. 2006;188:3219–3227. doi: 10.1128/JB.188.9.3219-3227.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guerin PJ, Grais RF, Rottingen JA, Valleron AJ. Using European travellers as an early alert to detect emerging pathogens in countries with limited laboratory resources. BMC Public Health. 2007;7:8. doi: 10.1186/1471-2458-7-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pires SM, Vieira AR, Hald T, Cole D. Source attribution of human salmonellosis: an overview of methods and estimates. Foodborne Pathog Dis. 2014;11:667–676. doi: 10.1089/fpd.2014.1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gil-Cruz C, Bobat S, Marshall JL, Kingsley RA, Ross EA, Henderson IR, et al. The porin OmpD from nontyphoidal Salmonella is a key target for a protective B1b cell antibody response. Proc Natl Acad Sci. 2009;106:9803–9808. doi: 10.1073/pnas.0812431106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ferreira RBR, Valdez Y, Coombes BK, Sad S, Gouw JW, Brown EM, et al. A highly effective component vaccine against nontyphoidal Salmonella enterica infections. MBio. 2015;6:e01421–01415. doi: 10.1128/mBio.01421-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Georgiades K, Raoult D. Genomes of the most dangerous epidemic bacteria have a virulence repertoire characterized by fewer genes but more toxin-antitoxin modules. PLoS ONE. 2011;6:e17962. doi: 10.1371/journal.pone.0017962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Andersen JL, He G-X, Kakarla P, KC R, Kumar S, Lakra WS, et al. Multidrug efflux pumps from Enterobacteriaceae, Vibrio cholerae and Staphylococcus aureus bacterial food pathogens. Int J Environ Res Public Health. 2015;12:1487–1547. doi: 10.3390/ijerph120201487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Carden SE, Walker GT, Honeycutt J, Lugo K, Pham T, Jacobson A, et al. Pseudogenization of the secreted effector gene sseI confers rapid systemic dissemination of S. Typhimurium ST313 within migratory dendritic cells. Cell Host Microbe. 2017;21:182–194. doi: 10.1016/j.chom.2017.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gibbons HS, Kalb SR, Cotter RJ, Raetz CRH. Role of Mg2+ and pH in the modification of Salmonella lipid A after endocytosis by macrophage tumour cells. Mol Microbiol. 2005;55:425–440. doi: 10.1111/j.1365-2958.2004.04409.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Accession numbers, year of isolation and source details of all 3,351 S. Typhimurium genomes sequenced by PHE within the timeframe of this study. (XLSX 80 kb)
Complete metadata and accession numbers for all S. Typhimurium ST313 isolates included in this study. (XLSX 24 kb)
The antimicrobials used for susceptibility testing in this study. Figure S1. Isolate U60 contains additional resistance genes including a bla CTX-M-15 locus inserted into the chromosomal ompD locus. Figure S2. Circular representation of finished genome of UK-ST313 representative strain U2, showing the chromosome and the pSLT-U2 virulence plasmid. Figure S3. Fully labelled phylogenetic tree highlighting the context of subset of 16 UK-isolated ST313 strains included in phenotypic testing. Figure S4. The maximum clade credibility tree from BEAST showing the timed phylogeny of all ST313 isolated in this study and a representative sub-sample of African ST313 from Okoro et al. [7]. Figure S5. Distribution of pairwise SNP distances of lineage 2 ST313 and UK-ST313. (PDF 1193 kb)
Data Availability Statement
All data generated or analysed during this study are included in this published article (and its supplementary information files).