

Abstract
Clear cell carcinoma and endometrioid adenocarcinoma are histologic subtypes of ovarian and uterine cancer that demonstrate unique clinical behavior but share common underlying genomic aberrations and oncogenic pathways. ARID1A mutations are more frequently identified in these tumors, in comparison to other gynecologic histologies, and loss of ARID1A tumor suppressor function is thought to be an essential component of carcinogenic transformation. Several therapeutic targets in ARID1A mutated cancers are in development, including EZH2 inhibitors. EZH2 facilitates epigenetic methylation to modulate gene expression, and both uterine and ovarian cancers show evidence of EZH2 over expression. EZH2 inhibition in ARID1A mutated tumors acts in a synthetically lethal manner to suppress cell growth and promote apoptosis, revealing a unique new therapeutic opportunity. Several phase 1 and 2 clinical trials of EZH2 inhibitors are ongoing currently and there is considerable promise in translational trials for utilization of this new targeted therapy, both to capitalize on ARID1A loss of function and to increase sensitivity to platinum-based adjuvant chemotherapies. This review will synthesize the molecular carcinogenesis of these malignancies and their unique clinical behavior, as a foundation for an emerging frontier of targeted therapeutics – the synergistic inhibition of EZH2 in ARID1A mutated cancers.
Keywords: ARID1A, EZH2, Targeted therapy, Molecular carcinogenesis, Synthetic lethality
Introduction
Ovarian and uterine cancer represent gynecologic malignancies with significant morbidity and mortality in the advanced and recurrent settings. Clear cell carcinoma (CCC) and endometrioid adenocarcinoma are histologic subtypes of both ovarian and uterine cancer that demonstrate unique clinical behavior, although emerging literature suggests that they may share underlying genomic aberrations and oncogenic pathways. This review explores the evolving paradigm of targeted therapeutics, as the understanding of the pathogenesis of these cancers becomes paramount to exploring new therapeutic frontiers and achieving improved oncologic outcomes.
Low grade endometrioid and clear cell uterine carcinoma
Endometrial cancer is the most common gynecologic malignancy, with 61,380 estimated new cases and 10,920 estimated deaths in 2017 [1]. Approximately 75% of women are diagnosed with stage I disease, confined to the uterus, and have excellent 5-year survival [2]. While the majority of patients are diagnosed with well differentiated, International Federation of Gynecology and Obstetrics (FIGO) grade 1 and 2 endometrioid adenocarcinoma, a subset of patients have estrogen-independent histologies, including grade 3 endometrioid adenocarcinoma, serous carcinoma, clear cell carcinoma, carcinosarcoma, uterine sarcomas and undifferentiated cell types.
Clear cell endometrial carcinoma is rare, accounting for 1–6% of all endometrial cancers. Endometrial CCC is more likely to present with extra-uterine spread compared to low grade endometrioid histologies and is an independent predictor of poor prognosis. The proportion of patients with FIGO stage III-IV disease at the time of diagnosis is 36% with a 5-year disease specific survival of 68% [2]. Given its rarity, optimal management strategies are not well defined and treatment is extrapolated from large studies primarily comprised of the more common endometrioid histology. Management of endometrial CCC includes comprehensive surgical staging and typically adjuvant platinum-based chemotherapy and/or radiotherapy.
Endometrioid and clear cell ovarian carcinoma
Ovarian cancer, while less common than uterine cancer, is generally diagnosed at a more advanced stage, resulting in compromised long term outcomes, with an estimated 5-year survival of 46%. In 2017 there were an estimated 22, 440 new cases and 14, 080 deaths [1]. Ovarian cancers are broadly categorized by origin, into epithelial and non-epithelial neoplasms. The most common epithelial histology, high grade serous carcinoma, accounts for nearly 70% of ovarian cancers, and is disproportionately represented in clinical trials exploring novel therapeutic paradigms. In addition to high grade serous histology, epithelial ovarian cancers include endometrioid, clear cell, mucinous, carcinosarcoma, mixed epithelial and undifferentiated.
Ovarian endometrioid carcinoma accounts for approximately 10% of epithelial ovarian cancers. Clinically, these often present with disease confined to the pelvis, may be bilateral in up to 28% of cases, and are generally low grade and early stage, portending a more favorable prognosis when compared to high grade serous carcinoma [3]. Approximately 20% of patients with ovarian endometrioid carcinomas will also have simultaneous endometrial adenocarcinomas, and both histologies are thought to share molecular aberrations [4, 5]. Furthermore, endometriosis is highly prevalent in patients with endometrioid ovarian cancer, reported in up to 35.9% of cases [6]. Management paradigms are analogous to the more common high grade serous histology, and include surgical cytoreduction followed by adjuvant platinum based combination chemotherapy [7].
Ovarian CCC constitutes approximately 5–10% of all ovarian malignancies. Although commonly identified at an early stage, with ovarian confined disease, patients may be diagnosed with advanced stage disease, or suffer recurrence, both of which are difficult to treat given the relative resistance of this histology to standard chemotherapy [8]. When compared to high grade serous carcinoma, stage-for-stage survival is lower in patients with ovarian CCC [8]. As discussed above, treatment is dependent on surgical cytoreduction followed by combination platinum-based cytotoxic chemotherapy for patients with stage 1C or greater disease.
Given the rarity of these histologies as well as their unique characteristics, additional therapeutic strategies are urgently needed to improve survival in patients suffering from advanced stage or recurrent endometrioid and clear cell ovarian cancer.
Review
What is ARID1A?
Gene expression relies on the complex interplay of transcription factors, cofactors and chromatin regulators. The role of transcription dysregulation as a mechanism for cancer is well established. With the emergence of genome-wide analyses, the significant role of chromatin remodeling complexes on gene expression levels through modified transcription, replication, repair, recombination and methylation of DNA has been identified [9]. One such complex is the Switch/Sucrose Non-Fermentable (SWI/SNF) complex which is involved in activation and inhibition of transcription. The SWI/SNF complex plays a unique role in carcinogenesis and may be mutated in over 20% of human cancers [10]. ARID1A, an acronym for the gene AT-rich interacting domain-containing protein 1A, encodes a component of the SWI/SNF complex and has high mutation rates across multiple malignancies [11]. Mutations are typically frameshift or nonsense mutations, often occurring at either the nuclear-export signal sequence or at the ARID1A interaction site with the SWI/SNF complex resulting in protein complex instability [12]. This complex modifies expression of multiple genes, including p53, through direct interaction, SMAD3, CDKN1A (p21), MLH1 and PIK3IP1 through transcription regulation of downstream effectors, and transformation of cells through the PI3K/AKT pathway [13].
ARID1A as a tumor suppressor gene
ARID1A mutations typically results in loss of protein function with implications for cell proliferation, differentiation and apoptosis – essential roles of a tumor suppressor gene. Initial efforts into understanding its role in tumorigenesis began approximately a decade ago. The knockout of ARID1A in embryonic stem cells resulted in loss of self-renewal properties and substantially modified cellular differentiation [14]. In ARID1A knockout leukemia cell populations, Fas-mediated cell death is inhibited, supporting altered apoptosis [11, 15]. In 2011, Guan et al. restored wild-type ARID1A expression in ovarian cancer cells harboring deleterious mutations and noted reactivation of protein function. Additional work by Guan et al. in 2014 utilizing xenograft models confirmed that silencing ARID1A expression in nontransformed cells promoted cellular proliferation [13, 16]. These cumulative efforts were essential in establishing the role of ARID1A as a tumor suppressor gene.
ARID1A in ovarian cancers
As the molecular characterization of solid malignancies expanded, it became evident that ARID1A mutations were most pronounced in gynecologic cancers. Within ovarian cancer cohorts, mutation rates of 46–57% were identified in clear cell adenocarcinoma and 30% in endometrioid adenocarcinoma [17, 18]. This is contrasted with the absence of identifiable ARID1A mutations in high-grade serous carcinomas.
Endometrioid and clear cell ovarian carcinomas are uniquely associated with endometriosis, and have been recently referred to as endometriosis-associated ovarian cancer (EAOC). Between 14 and 42% of endometrioid and 20–36% of clear cell carcinomas are associated with endometriotic lesions or patient reported symptoms of endometriosis [6, 19]. Common genetic alterations, particularly in ARID1A, have been recognized within atypical endometriosis and adenocarcinoma, suggesting a significant carcinogenic role [20, 21]. Loss of ARID1A is more frequent in endometriosis associated neoplasms, with loss of ARID1A immunohistochemical expression in 61% of endometriosis associated clear cell carcinomas [22]. While it is unclear if ARID1A mutations alone are sufficient to induce cancer progression, concurrent mutations in alternate pathways, including the PI3K/AKT pathway are frequent and appear to occur simultaneously to facilitate tumorigenesis [22, 23].
ARID1A in endometrial cancers
ARID1A mutations are also found frequently among women with endometrial cancers, with mutation frequency of 40% in uterine endometrioid adenocarcinoma [24]. Low grade endometrioid carcinoma is the most predominant histology, with 29% of grade 1 and 2 tumors showing loss of expression, in contrast to 39% with grade 3 tumors, 26% of uterine clear cell carcinomas, and 18% of uterine serous carcinomas [25]. The Cancer Genome Atlas further confirmed that ARID1A is mutated in a high proportion of type I uterine cancers, particularly in the POLE hypermutated, microsatellite unstable (MSI) hypermutated, and copy-number low subgroups. These often occurred concurrently with inactivating PTEN mutations [26].
ARID1A as a biomarker
Given the heterogeneity of gynecologic cancers, a consistent relationship between the presence of an ARID1A mutation and prognosis has been elusive. In a 2015 meta-analysis of 5651 patients with a variety of tumor types, ARID1A was evaluated via genetic analysis and immunohistochemistry with findings that ARID1A deficient tumors had significantly increased cancer-specific mortality (HR = 2.55) and cancer recurrence (HR = 1.93) when compared to a matched ARID1A positive population [27]. Conversely, in patient cohorts with high grade endometrioid and clear cell endometrial cancer, ARID1A mutations were not associated with clinical stage, depth of myometrial invasion, lymph node metastasis, or overall survival [28–30]. In a separate study, Yokoyama et al. found that ARID1A expression levels using immunohistochemistry correlated with prognosis and chemoresistance in stage III and IV epithelial ovarian cancers. Women with low ARID1A expression more frequently had incomplete response to chemotherapy (p = 0.026) and were more likely to experience relapse after achieving a complete response (p = 0.07). ARID1A-negative tumors had significantly worse progression free survival than ARID1A-positive tumors [31]. Additionally, Itamochi et al. confirmed an association between ARID1A immunohistochemical expression and FIGO stage as well as prognosis, with stage I and II patients having 91% 5-year survival with normal ARID1A expression and 74% 5-year survival with negative ARID1A expression [32] (Fig. 1).
Fig. 1.
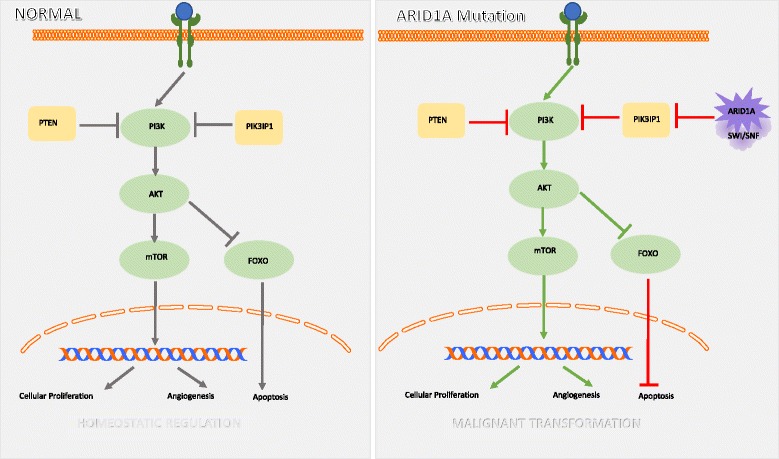
Normal and ARID1A cellular pathways. ARID1A mutation disrupts the homeostatic mechanisms of the PI3K/AKT/mTOR pathway, resulting in cellular proliferation, angiogenesis and inhibited apoptosis (Fig. 1)
Potential molecular therapeutic targets in ARID1A mutated cancers
Loss of ARID1A function facilitates tumorigenesis through its chromatin-mediated dysregulation of gene expression and loss of tumor suppressor gene function. As ARID1A plays an integral role in cell-cycle control and DNA damage repair pathways, the loss of ARID1A function with concurrent dysfunction in p53, p21, MLH1, or the PI3K/AKT/mTOR pathway allow malignant progression, and have revealed several potential therapeutic targets in ARID1A mutated cancers. These are detailed in Table 1 and Fig. 2 and several are being actively investigated for the treatment of epithelial ovarian cancer.
Table 1.
Therapeutic targets being explored in ARID1A mutated cancers
| EZH-2 | Epigenetic synthetic lethality, promotion of apoptosis | GSK126 | [33, 34] |
| PIK3IP1 mediated inhibition of PI3K/AKT pathway | |||
| mTOR | Inhibition of downstream regulator of PI3K/AKT pathway | Temsirolimus | [35, 36] |
| Ridaforolimuus | |||
| Everolimus | |||
| AP23573 | |||
| TP53 | Stabilization of wild-type p53 to overcome ARID1A loss, resume tumor suppressor function | Nutlin 3 | [13, 37] |
| PI3K/AKT | Inhibit upregulated AKT phosphorylation caused by concurrent mutations | Sorafenib | [38, 39] |
| Copanlisib | [36] | ||
| BKM120 | |||
| XL147 | |||
| BRCA | Enhance DNA-damaging effects of platinum chemotherapies EZH2 modulated function of BRCA1 |
[40, 41] | |
| ARID1B | Inhibition of residual AWI/SNF complex to suppress cell growth | [42, 43] | |
| Anti-IL6 | Inhibit inflammatory microenvironment and escape from anti-tumor immune response | [44] |
Fig. 2.
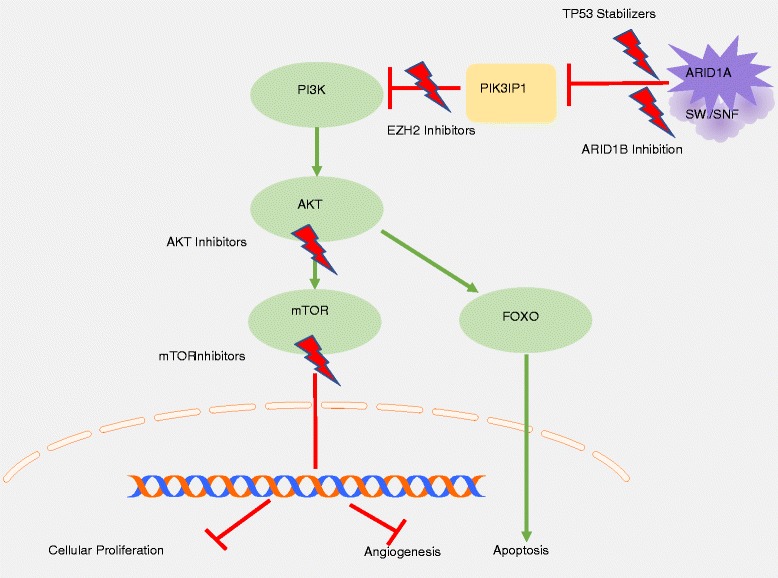
Therapeutic targets in ARID1A mutated cancers. Novel targeting of several key regulators in the PI3K/AKT/mTOR pathway and SWI/SNF complex modulate downstream effectors to inhibit cellular proliferation and promote apoptosis (Fig. 2)
The role of EZH2 in cancer
Functional studies of ovarian cancer cell lines reveal numerous gene targets of the SWI/SNF complex which may be impacted by ARID1A mutation, including cyclins, c-myc, and the Polycomb complexes [33, 34]. The enhancer of zeste homolog 2 (EZH2) protein is the enzymatically active core of the polycomb repressive complex 2/3 (PRC2), responsible for trimethylation of lysine 27 of histone H3 (H3K27) and can induce gene suppression through promoter binding [35, 36]. EZH2 plays a role in cancer progression through several mechanisms, including gain-of-function and loss-of-function mutations, overexpression of EZH2, mutations in the H3K27 demethylase gene, and through antagonistic mutations in the SWI/SNF chromatin remodeling complex. Within the gynecologic cancer arena, the predominant mechanisms are EZH2 overexpression and SWI/SNF antagonism of the Polycomb complex. EZH2 overexpression manifests through histone hypermethylation, resulting in tumor proliferation, cell cycle dysregulation, metastatic spread, and angiogenesis [37–39].
EZH2 in ovarian cancers
EZH2 overexpression is found in 50–85% of ovarian carcinomas, with high expression correlating with high grade, more advanced stage disease and poor survival [40, 41]. EZH2 plays an integral role in cellular proliferation, apoptosis and invasion in human epithelial ovarian cancer cell lines ([39, 42]. EZH2 is often overexpressed in ovarian clear cell carcinomas [42]. Additionally, these tumors are characterized by genomic instability and thus, epigenetic modification of gene expression may play a critical role in tumorigenesis.
EZH2 in uterine cancers
EZH2 targets genes which may exhibit modified function in endometrial cancer including: p16, E-cadherin, SFRP1, DKK3, and β-catenin [43–45]. EZH2 overexpression was identified in complex hyperplasia, atypical hyperplasia, and endometrial carcinoma but not in simple hyperplasia or normal endometrial tissue [45]. Overexpression has been associated with high proliferation rates and aggressive tumor subgroups of endometrial cancers ([43, 44]). EZH2 expression correlated with high tumor grade, deep myometrial invasion, lymphovascular space invasion and enhanced cellular proliferation, as well as decreased overall survival, suggesting a role as both a prognostic and therapeutic marker in endometrial cancer [44–46].
Synthetic lethality
Given the reversible epigenetic modifications which drive tumorigenesis, EZH2 methyltransferase activity is an ideal target for cancer therapeutics. Several new selective small molecules targeting EZH2 have been developed, including GSK126 [47], EPZ005687 [48] and EI1 [49], all of which inhibit EZH2 without otherwise effecting the PCR2 complex.
Homeostasis requires balanced action of ARID1A and EZH2 through chromatin-mediated gene expression. Loss of ARID1A expression results in imbalanced EZH2 activity, and is hypothesized to drive tumorigenesis (Figs. 3 and 4). Mechanistically, both ARID1A and EZH2 target PI3K-interacting protein 1 gene (PIK3IP1), with resultant silencing causing cell proliferation and promotion of anti-apoptotic effects through the PI3K-AKT pathway. In a pivotal study by Bitler et al., it was demonstrated that targeted EZH2 inhibition triggers apoptosis in ARID1A mutated cells and upregulates PIK3IP1 expression, thereby suppressing cell growth [50]. This cooperation of ARID1A mutation and EZH2 targeted inhibition represents a synthetically lethal interaction. This is particularly exciting in that drug-gene synthetic lethality often allows utilization of low concentrations of drugs, minimal toxicity, and limited treatment resistance (Table 2).
Fig. 3.
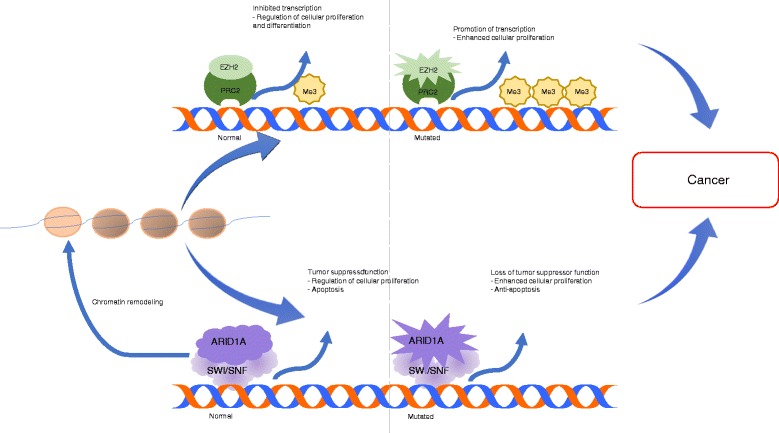
Mechanisms of both normal and mutated ARID1A and EZH2. Homeostasis requires balanced ARID1A and EZH2 function, while malignant transformation arises with dysregulation of either process (Fig. 3)
Fig. 4.
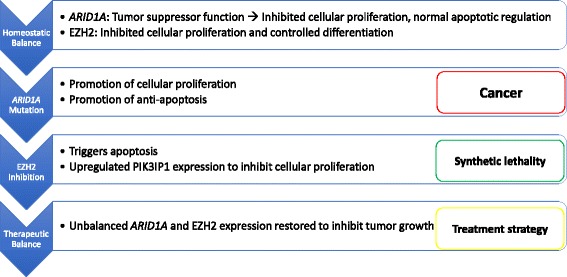
Mechanisms of EZH2 inhibitor synthetic lethality in ARID1A mutated tumors. Balanced action of EZH2 and ARID1A are necessary for normal cellular function
Table 2.
Ongoing EZH2 targeted clinical trials
| Targeted Agent | Trade Name | Route | Trial | Tumor | Status/Adverse Events (Grade 3–4) | References |
|---|---|---|---|---|---|---|
| EPZ-6438 | Tazemetostat | PO | Phase 1 | NHL | Thrombocytopenia Neutropenia Hypertension Anorexia Transaminitis |
[58] |
| PO | Phase 1/2 | B-cell lymphoma or advanced solid tumor | Recruiting actively | (Clinicaltrials.Gov. (2.13.2017)), (Epizyme pharmaceuticals website. (2.13.2017)) | ||
| NCT01897571 | ||||||
| Nausea | ||||||
| Asthenia | ||||||
| Thrombocytopenia | ||||||
| Neutropenia | ||||||
| Fatigue | ||||||
| PO | Phase 1 | Pediatric INI1 negative tumors or synovial sarcoma | Recruiting actively | (Clinicaltrials.Gov. (2.13.2017)), (Epizyme pharmaceuticals website. (2.13.2017)) | ||
| NCT02601937 | ||||||
| PO | Phase 2 | Adult INI1 negative tumors or synovial sarcoma | Recruiting actively | (Clinicaltrials.Gov. (2.13.2017)), (Epizyme pharmaceuticals website. (2.13.2017)) | ||
| NCT02601950 | ||||||
| PO | Phase 2 | Malignant mesothelioma | Recruiting actively | (Clinicaltrials.Gov. (2.13.2017)), (Epizyme pharmaceuticals website. (2.13.2017)) | ||
| NCT02860286 | ||||||
| GSK 2,816,126 | GSK126 | IV | Phase 1/2 | Diffuse large B-cell lymphoma | Fatigue | [59] |
| Nausea | ||||||
| Vomiting | ||||||
| Anemia | ||||||
| EPZ-5676 | Pinometostat | IV | Phase 1 | Leukemia | Hypophosphatemia | [60] |
| Neutropenia | ||||||
| Reduced ejection fraction | ||||||
| Transaminitis | ||||||
| CPI-1205 | IV | Phase 1 | B cell lymphoma | Recruiting actively | (Clinicaltrials.Gov. (2.13.2017)), | |
| NCT02395601 |
EZH2 targeted therapeutics
GSK126 was shown to be well tolerated in mouse xenograft models of ovarian CCC [47]. Of numerous available targeted small molecules, GSK126 exhibited the highest sensitivity against ARID1A deficient ovarian CCC cells [50]. Bitler et al. showed that GSK126 significantly decreased tumor burden and reduced the number of metastatic peritoneal tumor implants in a mouse clear cell ovarian xenograft model when compared to controls. On a mechanistic level, immunohistochemical analysis showed decreased H3K27Me3 levels, increased PIK3IP1 activity, as well as cleaved caspase 3 in the GSK126 treated mice. This further confirmed the PIK3PI1 shared gene target, allowing synthetic lethality in ARID1A-deficient tumors via EZH2 inhibition [50].
Resensitization to platinum based chemotherapies
In advanced clear cell ovarian carcinoma, approximately half of patients progress while on platinum-based chemotherapy, in comparison to 29% of those with high grade serous histology. Additionally, patients with advanced stage clear cell carcinoma have compromised survival outcomes, with a median overall survival overall of approximately 12 months, significantly less than those with high grade serous histology [51]. This compromised outcome in patients with clear cell carcinoma is thought to result from relative platinum resistance, and thus mechanisms to increase platinum sensitivity may significantly impact morbidity and mortality in this subset of patients. EZH2 downregulation in ovarian cancer has been shown to sensitize tumor cells to cisplatin and EZH2 overexpression is associated with resistance to cisplatin through H3K27 tri-methylation of drug-resistance genes [52, 53].
Translational research suggests that combination therapies utilizing platinum-based cytotoxic chemotherapy and EZH2 inhibitors, may be particularly potent and synergistic in ARID1A mutated tumors [54]. Within non-Hodgkin’s lymphoma tumors with EZH2 mutations, a combination of EPZ-6438 and traditional targeted chemotherapy prevented tumor growth [55]. Prostate cancer cell lines had increased tumor death when treated with combined etoposide and GSK126, again suggesting synergistic therapeutic effect [56]. Interestingly, EZH2 inhibition had varying effects in a preclinical study of non-small cell lung cancers, with increased sensitization to topoisomerase II inhibitors in the tumor subset demonstrating BRG1/SMARCA4 loss-of-function mutations or EGFR gain-of-function mutations [57]. These findings in non-gynecologic tumors provide a foundation for exploring the cooperative effects of chemotherapy and EZH2 inhibition within ovarian and uterine malignancies.
Future directions
The potential therapeutic role of EZH2 inhibition in ARID1A mutated gynecologic cancers may represent a novel and exciting treatment paradigm in a subset of patients with limited treatment options. Capitalizing on the concept of synthetic lethality induced in this population using EZH2 inhibitors, as well as the potential synergistic effects with platinum-based chemotherapies, may help translate into improved oncologic outcomes.
Given the frequency of ARID1A mutations in patients with clear cell and endometrioid ovarian and uterine cancer, and the prognostic implications of loss of ARID1A expression, it is natural to explore the impact of EZH2 inhibition in these patient subsets. Given the above, clinical trialists are currently designing a basket trial concept of the EZH2 inhibitor, Tazemetostat, in patients with recurrent, measurable, clear cell and endometrioid ovarian cancer, as well as recurrent endometrioid endometrial cancer. In this study, subjects with recurrent, measurable disease, will be enrolled and treated with single agent Tazemetostat at the recommended phase 2 dose of 800 mg orally twice daily until disease progression or unacceptable toxicity. ARID1A mutation status will be an integrated biomarker, and will be assessed via BAF250a IHC, as well as next generation sequencing, allowing investigators to determine the correlation between ARID1A mutation status and IHC expression. Furthermore, sequencing studies will facilitate assessment of the mutation status of all other SWI/SNF members.
Ultimately, if single agent regimens using EZH2 inhibitors are shown to be effective in this subset of patients, then novel combinatorial approaches utilizing platinum-based chemotherapy, TP53 stabilizers, PI3K/AKT inhibitors, or mTOR inhibitors may be warranted.
Conclusions
In an effort to expand the therapeutic portfolio for patients with advanced stage and recurrent ovarian and endometrial cancer, molecular characterization of theses lesions has emerged as a clinical priority. Using both institutional genomic data, and data from the NCI sponsored cancer genome atlas, ARID1A mutations were frequently identified as mutated tumor suppressor genes, in clear cell and endometrioid ovarian cancer as well as low grade endometrioid endometrial cancer. Ultimately, clinicians will look to capitalize on this molecular aberration via novel targeted therapies, such as tazemetostat, as single agents or in combination regimens. As our understanding of the molecular mechanisms underlying malignant transformation evolves, the discovery of novel targeted therapies that lead to meaningful survival gains may become a reality.
Acknowledgements
Not applicable.
Funding
This manuscript was supported by the Ruth L Kirschstein NRSA Institutional Training Research Grant, T32 CA06039611.
Availability of data and materials
Data sharing is not applicable to this article as not datasets were generated or analysed during the current study.
Abbreviations
- ARID1A
AT-rich interacting domain-containing protein 1A
- CCC
Clear cell carcinoma
- EZH2
Enhancer of zeste homolog 2
- FIGO
International Federation of Gynecology and Obstetrics
- PRC2
Polycomb repressive complex 2/3
- SWI/SNF
Switch/Sucrose Non-Fermentable
Authors’ contributions
Both JA and RE contributed to the literature review and manuscript preparation and editing. Both contributed to creation of novel figures and tables. Both authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests. RNE has served as a speaker and received honoraria from Genetech Roche, Clovis Oncology and AZ Oncology.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Jill K. Alldredge, Email: jalldred@uci.edu
Ramez N. Eskander, Phone: 858-822-6199, Email: reskander@ucsd.edu
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 2.Institute, N.C. Surveillance, epidemiology and end results program. https://seer.cancer.gov/statfacts/
- 3.Bouchard-Fortier G, Panzarella T, Rosen B, Chapman W, Gien LT. Endometrioid carcinoma of the ovary: outcomes compared to serous carcinoma after 10 years of follow-up. J Obstet Gynaecol Can. 2017;39:34–41. doi: 10.1016/j.jogc.2016.10.006. [DOI] [PubMed] [Google Scholar]
- 4.Zaino R, Whitney C, Brady MF, DeGeest K, Burger RA, Buller RE. Simultaneously detected endometrial and ovarian carcinomas--a prospective clinicopathologic study of 74 cases: a gynecologic oncology group study. Gynecol Oncol. 2001;83:355–362. doi: 10.1006/gyno.2001.6400. [DOI] [PubMed] [Google Scholar]
- 5.Eifel P, Hendrickson M, Ross J, Ballon S, Martinez A, Kempson R. Simultaneous presentation of carcinoma involving the ovary and the uterine corpus. Cancer. 1982;50:163–170. doi: 10.1002/1097-0142(19820701)50:1<163::AID-CNCR2820500131>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 6.Van Gorp T, Amant F, Neven P, Vergote I, Moerman P. Endometriosis and the development of malignant tumours of the pelvis. A review of literature. Best Pract Res Clin Obstet Gynaecol. 2004;18:349–371. doi: 10.1016/j.bpobgyn.2003.03.001. [DOI] [PubMed] [Google Scholar]
- 7.Chen S, Leitao MM, Tornos C, Soslow RA. Invasion patterns in stage i endometrioid and mucinous ovarian carcinomas: a clinicopathologic analysis emphasizing favorable outcomes in carcinomas without destructive stromal invasion and the occasional malignant course of carcinomas with limited destructive stromal invasion. Mod Pathol. 2005;18:903–911. doi: 10.1038/modpathol.3800366. [DOI] [PubMed] [Google Scholar]
- 8.Sugiyama T, Kamura T, Kigawa J, Terakawa N, Kikuchi Y, Kita T, Suzuki M, Sato I, Taguchi K. Clinical characteristics of clear cell carcinoma of the ovary: a distinct histologic type with poor prognosis and resistance to platinum-based chemotherapy. Cancer. 2000;88:2584–2589. doi: 10.1002/1097-0142(20000601)88:11<2584::AID-CNCR22>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 9.Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem. 2009;78:273–304. doi: 10.1146/annurev.biochem.77.062706.153223. [DOI] [PubMed] [Google Scholar]
- 10.Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, Crabtree GR. Proteomic and bioinformatic analysis of mammalian swi/snf complexes identifies extensive roles in human malignancy. Nat Genet. 2013;45:592–601. doi: 10.1038/ng.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guan B, Gao M, Wu C-H, Wang T-L, Shih I-M. Functional analysis of in-frame indel arid1a mutations reveals new regulatory mechanisms of its tumor suppressor functions. Neoplasia. 2012;14:986–993. doi: 10.1593/neo.121218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guan B, Wang TL, Shih Ie M. Arid1a, a factor that promotes formation of swi/snf-mediated chromatin remodeling, is a tumor suppressor in gynecologic cancers. Cancer Res. 2011;71:6718–6727. doi: 10.1158/0008-5472.CAN-11-1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao X, Tate P, Hu P et al. Es cell pluripotency and germ-layer formation require the swi/snf chromatin remodeling componenet baf250a. Proc Natl Acad Sci U S A. 2008;105(18):6656-61. [DOI] [PMC free article] [PubMed]
- 15.Luo B, W.C. Cheung, Subramanian A et al. Highly parallel identification of essential genes in cancer cells. Proc Natl Acad Sci USA. 2008;105(51):20380-5. [DOI] [PMC free article] [PubMed]
- 16.Guan, B.; Rahmanto, Y.S.; Wu, R.C.; Wang, Y.; Wang, Z.; Wang, T.L.; Shih Ie, M. Roles of deletion of arid1a, a tumor suppressor, in mouse ovarian tumorigenesis. J Natl Cancer Inst. 2014;106(7). [DOI] [PMC free article] [PubMed]
- 17.Wiegand KC, Shah SP, Al-Agha OM, Zhao Y, Tse K, Zeng T, Senz J, McConechy MK, Anglesio MS, Kalloger SE, et al. Arid1a mutations in endometriosis-associated ovarian carcinomas. N Engl J Med. 2010;363:1532–1543. doi: 10.1056/NEJMoa1008433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jones S, Wang TL, Shih Ie M, Mao TL, Nakayama K, Roden R, Glas R, Slamon D, Diaz LA, Jr, Vogelstein B, et al. Frequent mutations of chromatin remodeling gene arid1a in ovarian clear cell carcinoma. Science. 2010;330:228–231. doi: 10.1126/science.1196333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pearce CL, Templeman C, Rossing MA, Lee A, Near AM, Webb PM, Nagle CM, Doherty JA, Cushing-Haugen KL, Wicklund KG, et al. Association between endometriosis and risk of histological subtypes of ovarian cancer: a pooled analysis of case–control studies. Lancet Oncol. 2012;13:385–394. doi: 10.1016/S1470-2045(11)70404-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chene G, Ouellet V, Rahimi K, Barres V, Provencher D, Mes-Masson AM. The arid1a pathway in ovarian clear cell and endometrioid carcinoma, contiguous endometriosis, and benign endometriosis. Int J Gynaecol Obstet. 2015;130:27–30. doi: 10.1016/j.ijgo.2015.02.021. [DOI] [PubMed] [Google Scholar]
- 21.Kobayrashi H. Molecular pathogenesis of endometriosis-associated clear cell carcinoma of the ovary (review). Oncol Rep. 2009;22(2):233-40. [PubMed]
- 22.Yamamoto S, Tsuda H, Takano M, Tamai S, Matsubara O. Loss of arid1a protein expression occurs as an early event in ovarian clear-cell carcinoma development and frequently coexists with pik3ca mutations. Mod Pathol. 2012;25:615–624. doi: 10.1038/modpathol.2011.189. [DOI] [PubMed] [Google Scholar]
- 23.Yamamoto S, Tsuda H, Takano M, Tamai S, Matsubara O. Pik3ca mutations and loss of arid1a protein expression are early events in the development of cystic ovarian clear cell adenocarcinoma. Virchows Arch. 2012;460:77–87. doi: 10.1007/s00428-011-1169-8. [DOI] [PubMed] [Google Scholar]
- 24.Guan B, Mao TL, Panuganti PK, Kuhn E, Kurman RJ, Maeda D, Chen E, Jeng YM, Wang TL, Shih Ie M. Mutation and loss of expression of arid1a in uterine low-grade endometrioid carcinoma. Am J Surg Pathol. 2011;35:625–632. doi: 10.1097/PAS.0b013e318212782a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wiegand KC, Lee AF, Al-Agha OM, Chow C, Kalloger SE, Scott DW, Steidl C, Wiseman SM, Gascoyne RD, Gilks B, et al. Loss of baf250a (arid1a) is frequent in high-grade endometrial carcinomas. J Pathol. 2011;224:328–333. doi: 10.1002/path.2911. [DOI] [PubMed] [Google Scholar]
- 26.Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H, Robertson AG, Pashtan I, Shen R, Benz CC, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497:67–73. doi: 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Luchini C, Veronese N, Solmi M, Cho H, Kim JH, Chou A, Gill AJ, Faraj SF, Chaux A, Netto GJ, et al. Prognostic role and implications of mutation status of tumor suppressor gene arid1a in cancer: a systematic review and meta-analysis. Oncotarget. 2015;6:39088–39097. doi: 10.18632/oncotarget.5142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang ZM, Xiao S, Sun GY, Liu YP, Zhang FH, Yang HF, Li J, Qiu HB, Liu Y, Zhang C, et al. The clinicopathologic significance of the loss of baf250a (arid1a) expression in endometrial carcinoma. Int J Gynecol Cancer. 2014;24:534–540. doi: 10.1097/IGC.0000000000000092. [DOI] [PubMed] [Google Scholar]
- 29.Fadare O, Gwin K, Desouki MM, Crispens MA, Jones HW, 3rd, Khabele D, Liang SX, Zheng W, Mohammed K, Hecht JL, et al. The clinicopathologic significance of p53 and baf-250a (arid1a) expression in clear cell carcinoma of the endometrium. Mod Pathol. 2013;26:1101–1110. doi: 10.1038/modpathol.2013.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fadare O, Renshaw IL, Liang SX. Does the loss of arid1a (baf-250a) expression in endometrial clear cell carcinomas have any clinicopathologic significance? A pilot assessment. J Cancer. 2012;3:129–136. doi: 10.7150/jca.4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yokoyama Y, Matsushita Y, Shigeto T, Futagami M, Mizunuma H. Decreased arid1a expression is correlated with chemoresistance in epithelial ovarian cancer. J Gynecol Oncol. 2014;25:58–63. doi: 10.3802/jgo.2014.25.1.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Itamochi H, Oumi N, Oishi T, Shoji T, Fujiwara H, Sugiyama T, Suzuki M, Kigawa J, Harada T. Loss of arid1a expression is associated with poor prognosis in patients with stage i/ii clear cell carcinoma of the ovary. Int J Clin Oncol. 2015;20:967–973. doi: 10.1007/s10147-015-0811-x. [DOI] [PubMed] [Google Scholar]
- 33.Nagl NG, Jr, Patsialou A, Haines DS, Dallas PB, Beck GR, Jr, Moran E. The p270 (arid1a/smarcf1) subunit of mammalian swi/snf-related complexes is essential for normal cell cycle arrest. Cancer Res. 2005;65:9236–9244. doi: 10.1158/0008-5472.CAN-05-1225. [DOI] [PubMed] [Google Scholar]
- 34.Nagl NG, Jr, Zweitzig DR, Thimmapaya B, Beck GR, Jr, Moran E. The c-myc gene is a direct target of mammalian swi/snf-related complexes during differentiation-associated cell cycle arrest. Cancer Res. 2006;66:1289–1293. doi: 10.1158/0008-5472.CAN-05-3427. [DOI] [PubMed] [Google Scholar]
- 35.Yoo KH, Hennighausen L. Ezh2 methyltransferase and h3k27 methylation in breast cancer. Int J Biol Sci. 2012;8:59–65. doi: 10.7150/ijbs.8.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, Zhang Y. Role of histone h3 lysine 27 methylation in polycomb-group silencing. Science. 2002;298:1039–1043. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- 37.Choi JH, Song YS, Yoon JS, Song KW, Lee YY. Enhancer of zeste homolog 2 expression is associated with tumor cell proliferation and metastasis in gastric cancer. APMIS. 2010;118:196–202. doi: 10.1111/j.1600-0463.2009.02579.x. [DOI] [PubMed] [Google Scholar]
- 38.Fan T, Jiang S, Chung N, Alikhan A, Ni C, Lee CC, Hornyak TJ. Ezh2-dependent suppression of a cellular senescence phenotype in melanoma cells by inhibition of p21/cdkn1a expression. Mol Cancer Res. 2011;9:418–429. doi: 10.1158/1541-7786.MCR-10-0511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu C, Han HD, Mangala LS, Ali-Fehmi R, Newton CS, Ozbun L, Armaiz-Pena GN, Hu W, Stone RL, Munkarah A, et al. Regulation of tumor angiogenesis by ezh2. Cancer Cell. 2010;18:185–197. doi: 10.1016/j.ccr.2010.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rao ZY, Cai MY, Yang GF, He LR, Mai SJ, Hua WF, Liao YJ, Deng HX, Chen YC, Guan XY, et al. Ezh2 supports ovarian carcinoma cell invasion and/or metastasis via regulation of tgf-beta1 and is a predictor of outcome in ovarian carcinoma patients. Carcinogenesis. 2010;31:1576–1583. doi: 10.1093/carcin/bgq150. [DOI] [PubMed] [Google Scholar]
- 41.Guo J, Cai J, Yu L, Tang H, Chen C, Wang Z. Ezh2 regulates expression of p57 and contributes to progression of ovarian cancer in vitro and in vivo. Cancer Sci. 2011;102:530–539. doi: 10.1111/j.1349-7006.2010.01836.x. [DOI] [PubMed] [Google Scholar]
- 42.Li H, Cai Q, Godwin AK, Zhang R. Enhancer of zeste homolog 2 promotes the proliferation and invasion of epithelial ovarian cancer cells. Mol Cancer Res. 2010;8:1610–1618. doi: 10.1158/1541-7786.MCR-10-0398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bachmann IM, Halvorsen OJ, Collett K, Stefansson IM, Straume O, Haukaas SA, Salvesen HB, Otte AP, Akslen LA. Ezh2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J Clin Oncol. 2006;24:268–273. doi: 10.1200/JCO.2005.01.5180. [DOI] [PubMed] [Google Scholar]
- 44.Eskander RN, Ji T, Huynh B, Wardeh R, Randall LM, Hoang B. Inhibition of enhancer of zeste homolog 2 (ezh2) expression is associated with decreased tumor cell proliferation, migration, and invasion in endometrial cancer cell lines. Int J Gynecol Cancer. 2013;23:997–1005. doi: 10.1097/IGC.0b013e318296a265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jia N, Li Q, Tao X, Wang J, Hua K, Feng W. Enhancer of zeste homolog 2 is involved in the proliferation of endometrial carcinoma. Oncol Lett. 2014;8:2049–2054. doi: 10.3892/ol.2014.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou J, Roh JW, Bandyopadhyay S, Chen Z, Munkarah AR, Hussein Y, Alosh B, Jazaerly T, Hayek K, Semaan A, et al. Overexpression of enhancer of zeste homolog 2 (ezh2) and focal adhesion kinase (fak) in high grade endometrial carcinoma. Gynecol Oncol. 2013;128:344–348. doi: 10.1016/j.ygyno.2012.07.128. [DOI] [PubMed] [Google Scholar]
- 47.McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, Liu Y, Graves AP, Della Pietra A, 3rd, Diaz E, et al. Ezh2 inhibition as a therapeutic strategy for lymphoma with ezh2-activating mutations. Nature. 2012;492:108–112. doi: 10.1038/nature11606. [DOI] [PubMed] [Google Scholar]
- 48.Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, Sacks JD, Raimondi A, Majer CR, Song J, et al. A selective inhibitor of ezh2 blocks h3k27 methylation and kills mutant lymphoma cells. Nat Chem Biol. 2012;8:890–896. doi: 10.1038/nchembio.1084. [DOI] [PubMed] [Google Scholar]
- 49.Qi W, Chan H, Teng L, Li L, Chuai S, Zhang R, Zeng J, Li M, Fan H, Lin Y, et al. Selective inhibition of ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc Natl Acad Sci U S A. 2012;109:21360–21365. doi: 10.1073/pnas.1210371110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bitler BG, Aird KM, Garipov A, Li H, Amatangelo M, Kossenkov AV, Schultz DC, Liu Q, Shih Ie M, Conejo-Garcia JR, et al. Synthetic lethality by targeting ezh2 methyltransferase activity in arid1a-mutated cancers. Nat Med. 2015;21:231–238. doi: 10.1038/nm.3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goff BA, Sainz de la Cuesta R, Muntz HG, Fleischhacker D, Ek M, Rice LW, Nikrui N, Tamimi HK, Cain JM, Greer BE, et al. Clear cell carcinoma of the ovary: a distinct histologic type with poor prognosis and resistance to platinum-based chemotherapy in stage iii disease. Gynecol Oncol. 1996;60:412–417. doi: 10.1006/gyno.1996.0065. [DOI] [PubMed] [Google Scholar]
- 52.Hu S, Yu L, Li Z, Shen Y, Wang J, Cai J, Xiao L, Wang Z. Overexpression of ezh2 contributes to acquired cisplatin resistance in ovarian cancer cells in vitro and in vivo. Cancer Biol Ther. 2010;10:788–795. doi: 10.4161/cbt.10.8.12913. [DOI] [PubMed] [Google Scholar]
- 53.Li T, Cai J, Ding H, Xu L, Yang Q, Wang Z. Ezh2 participates in malignant biological behavior of epithelial ovarian cancer through regulating the expression of brca1. Cancer Biol Ther. 2014;15:271–278. doi: 10.4161/cbt.27306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim KH, Roberts CW. Targeting ezh2 in cancer. Nat Med. 2016;22:128–134. doi: 10.1038/nm.4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Knutson SK, Warholic NM, Johnston LD, Klaus CR, Wigle TJ, Iwanowicz D, Littlefield BA, Porter-Scott M, Smith JJ, Moyer MP, et al. Synergistic anti-tumor activity of ezh2 inhibitors and glucocorticoid receptor agonists in models of germinal center non-hodgkin lymphomas. PLoS One. 2014;9:e111840. doi: 10.1371/journal.pone.0111840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kirk JS, Schaarschuch K, Dalimov Z, Lasorsa E, Ku S, Ramakrishnan S, Hu Q, Azabdaftari G, Wang J, Pili R, et al. Top2a identifies and provides epigenetic rationale for novel combination therapeutic strategies for aggressive prostate cancer. Oncotarget. 2015;6:3136–3146. doi: 10.18632/oncotarget.3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fillmore CM, Xu C, Desai PT, Berry JM, Rowbotham SP, Lin YJ, Zhang H, Marquez VE, Hammerman PS, Wong KK, et al. Ezh2 inhibition sensitizes brg1 and egfr mutant lung tumours to topoii inhibitors. Nature. 2015;520:239–242. doi: 10.1038/nature14122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vincent Ribrag, J.-C.S, Jean-Marie Michot, et al. Phase 1 study of tazemetostat (epz-6438), an inhibitor of enhancer of zeste-homolog 2 (ezh2): Preliminary safety and activity in relapsed or refractory nonhodgkin lymphoma (nhl) patients. Blood. 2015;126:473.
- 59.Timothy A Yap, J.N.W, John P. Leonard, et al. A phase i study of gsk2816126, an enhancer of zeste homolog 2(ezh2) inhibitor, in patients (pts) with relapsed/refractory diffuse large b-cell lymphoma (dlbcl), other non-hodgkin lymphomas (nhl), transformed follicular lymphoma (tfl), solid tumors and multiple myeloma (mm). Blood. 2016;128:4203.
- 60.Eytan M Stein, G.G.-M, David A Rizzieri et al. A phase 1 study of the dot1l inhibitor, pinometostat (epz-5676), in adults with relapsed or refractory leukemia: Safety, clinical activity, exposure and target inhibition. Blood.2015:126:2547.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as not datasets were generated or analysed during the current study.