

Abstract
Background & objectives:
Hearing impairment is a common and heterogeneous sensory disorder in humans. Among about 90 genes, which are known to be associated with hearing impairment, mutations in the GJB2 (gap junction protein beta 2) gene are the most prevalent in individuals with hereditary hearing loss. Contribution of the other deafness-causing genes is relatively poorly understood. Here, we present our findings on two families with transmembrane channel like 1 (TMC1) gene variants of the 47 families with nonsyndromic hearing loss (NSHL) studied.
Methods:
Forty seven families including 26 consanguineous families with at least two hearing impaired children and one normal hearing child and 21 non-consanguineous families having at least three hearing impaired children and one normal hearing child were enrolled for this study. Genetic linkage studies were carried out in 41 families that were GJB2 (Connexin 26) negative. Seven polymorphic short tandem repeat markers at the DFNB7/11 locus were studied employing fluorescently labelled markers.
Results:
A novel homozygous missense mutation c.1283C>A (p.Ala428Asp) was identified co-segregating with hearing loss. This change results in substitution of a highly conserved polar alanine to a charged aspartic acid and is predicted to be deleterious. In addition, a previously reported nonsense mutation, p.R34X in TMC1, was found.
Interpretation & conclusions:
While mutations in TMC1 are not as common a cause of NSHL as those in GJB2, TMC1 should be considered for diagnostic investigations in cases of NSHL in GJB2-negative families.
Keywords: DFNB7/11 locus, DNA sequencing, hearing impairment, India, linkage study, short tandem repeat markers, transmembrane channel like 1 gene
Hearing impairment is a common and genetically heterogeneous sensory disorder in humans. The frequency of congenital hearing loss is estimated to be 155 per 100,000 live births or around 1 in 650 newborns1. In 15-30 per cent of cases, hearing impairment is syndromic in its manifestation, and among the remaining, non-syndromic. It is estimated that more than 200 genes may be associated with hearing impairment in humans. To date, more than 90 genes have been identified2. The gap junction protein beta 2 (GJB2) gene is the most common gene responsible for congenital nonsyndromic hearing impairment across different ethnic populations of the world including India3,4,5,6,7,8,9,10. Other frequently involved genes include SLC26A4, MYO15A, TMC1, CDH23 and TRIOBP. Mutations in the MT-RNR1 gene increase the risk of developing nonsyndromic hearing impairment due to antibiotic exposure11,12,13. Transmembrane channel like 1 or transmembrane cochlear-expressed gene 1 (TMC1) is one of the hearing loss genes which is implicated in both autosomal recessive (DFNB7/11, MIM: 606705) and dominant (DFNA36, MIM: 600974) hearing impairment. TMC1-linked dominant hearing loss is not very common, and only three different missense mutations, p.D572N14, p.D572H15 from North American families and p.M418K16 in a Chinese family have been reported. However, recessive mutations in TMC1 have been identified in many ethnic populations of the world including Sudan17, Iran18, Pakistan15,19, Turkey20,21,22 and Tunisia23. Two previous reports from the Indian subcontinent have also described mutations in the TMC1 gene14,24. The objective of the study was to uncover the contribution of mutations in genes other than GJB2 for nonsyndromic hearing loss (NSHL) in India.
Material & Methods
Family enrolment and clinical evaluation: This study was conducted between February 2008 and February 2014 at the department of Pediatrics, All India Institute of Medical Sciences, New Delhi, and Molecular Biology and Genetics Unit, Jawaharlal Nehru Centre for Advanced Scientific Research, Bengaluru, India. The study was approved by the Institute's Ethics Committee. Written informed consent was obtained from all families.
Families with consanguineous parents, two hearing impaired children and at least one normal hearing child, and non-consanguineous parents, three hearing impaired children and at least one normal hearing child, were enrolled for the study by home visit. Before enrolling the families, detailed medical history of all the affected individuals was obtained to exclude syndromic and environmental causes of hearing loss. Families were examined for abnormalities of skin and hair pigmentation and problems relating to balance, vision, night blindness, heart, kidney and thyroid. In addition, clinical histories of infectious diseases such as rubella, mumps, meningitis, typhoid, trauma and antibiotic/ototoxic drugs usage were also considered. Families were asked about the onset of hearing loss in each affected individual to confirm the hearing impairment to be congenital. Clinical information was collected in a proforma, and the hearing loss was evaluated by an audiologist. All affected individuals had severe-to-profound hearing impairment. Twenty six families were consanguineous and had two or more hearing impaired children. Twenty one families were non-consanguineous and had three or more hearing impaired children. History of hearing loss in the extended family was present in some families. A total of 47 families were enrolled for the study through deaf schools in Delhi, Uttar Pradesh, Madhya Pradesh, Gujarat and 50 healthy individuals having normal hearing with no family history of hearing problems were enrolled as controls. Peripheral blood sample (5 ml) was collected from all the affected children, normal children and parents by making home visits. Genomic DNA was extracted by the salting out method25.
Genotyping: Two hearing impaired children from each family were screened for mutations in exon 2 of GJB2 (Transcript: ENST00000382848), exon 3 and 309 kb deletion of GJB6 gene (Transcript: ENST00000241124) by polymerase chain reaction (PCR) and sequencing. Six families harbouring mutations (p.W24X, p.W77X, p.Q124X, c.303-316del14, IVS1+1G>A) in GJB2 were excluded from linkage study for TMC1. In the remaining 41 families negative for GJB2 and GJB6 mutations, linkage analysis was performed using seven fluorescently labelled polymorphic short tandem repeat markers for the DFNB7/11 locus. The seven microsatellite markers used were D9S301, D9S1822, D9S1124, D9S1837, D9S1876, D9S237 and D9S175 between 66.32 cM (D9S301) and 70.33 cM (D9S175) within the DFNB7/11 locus. PCR (ABI 2720 Thermal Cycler, Applied Biosystems, USA) amplification was carried out in a total 10 μl of reaction volume, containing 50 ng of genomic DNA, 1 μl 10X PCR buffer, 1 μl 2 mM dNTPs each (G Biosciences, Geno Technology, Inc., USA), 0.5U Taq polymerase (Merck Bioscience, India) and 3 pmol primers. Forward primer was labelled with 6-FAM (6-carboxyfluorescein), HEX (hexachlorofluorescein) and NED. PCR amplification included 5 min denaturing step at 95°C followed by 10 cycles of 95°C for 30 sec, 55°C for 30 sec and 72°C for 30 sec followed by the second step of 25 cycles of 89°C for 30 sec, 56°C for 30 sec and 72°C for 30 sec and a final extension at 72°C for 30 min. Post-PCR products were pooled and diluted. One microlitre of pooled PCR product was mixed with 8.7 μl of 100 per cent Hi Di formamide (Applied Biosystems) and 0.3 μl of the internal size standard (GeneScan™ 500 ROX™ Size Standard, Applied Biosystems), denatured at 95°C for 5 min and separated by capillary electrophoresis on an ABI 3130 Genetic Analyzer (Applied Biosystems). The allele sizes were analyzed using Gene Mapper v.4.0 (Applied Biosystems, USA).
Sequence analysis: PCR primers for TMC1 were designed using Primer3 software (http://bioinfo.ut.ee/primer3-0.4.0/). Direct sequencing of TMC1 was performed by amplifying its coding regions including the exon-intron boundaries. PCR-amplified products were incubated with Exonuclease I and shrimp alkaline phosphatase (Fermentas, Thermo Fisher Scientific, USA) to remove unincorporated primers and nucleotides. Cycle sequencing was done using ABI prism BigDye Terminator cycle sequencing Ready Reaction Kit v3.1 (Applied Biosystems) and was followed by ethanol/EDTA/sodium acetate precipitation and resuspension in 10 μl Hi Di formamide, denaturation and capillary electrophoresis using an ABI 3130 Genetic Analyzer. Sequence analysis was performed using Chromas Pro software (http://technelysium.com.au) and compared with reference sequence of TMC1 in the NCBI database (http://www.ncbi.nlm.nih.gov/). The 1000 genomes database (http://www.1000genomes.org/), dbSNP database (http://www.ncbi.nlm.nih.gov/snp/) and Human gene mutation database (HGMD, http://www.hgmd.cf.ac.uk/ac/index.php) were used to check the pathogenicity and novelty of the variations identified in TMC1.
In silico analysis: PolyPhen2 (http://genetics.bwh.harvard.edu/pph2) and Sorting Intolerant from Tolerant (SIFT) (http://sift.jcvi.org) programs were used to predict the qualitative effect of missense change on the protein function. Multiple sequence alignments were performed to check the conservation of amino acid across species.
Results
As sequencing of the entire TMC1 gene was expensive and time taking, linkage studies were done to screen the families. Seven microsatellite markers were analysed encompassing the DFNB7/11 locus in 41 families. Families were selected for TMC1 gene sequencing on the basis of concordance of markers. In two unrelated families DDL4 and LKO5, concordance with all seven markers in all the affected children was observed. In the non-consanguineous family DDL4 with three affected siblings, all the seven markers co-segregated with hearing loss exhibiting homozygosity between D9S1124 and D9S237 markers which were at an interval of 0.54 cM (Fig. 1). In the consanguineous family LKO5 with two affected siblings, a small homozygous region was found between D9S1822 and D9S1837 which were 0.54 cM apart (Fig. 2). DNA sequencing of TMC1 revealed mutations in both the families. In DDL4 (Fig. 1), a homozygous mutation, c.100C>T (p.R34X), segregated with hearing loss. Both parents and normal hearing child were heterozygous for c.100C>T. In LKO5 (Fig. 2), a novel homozygous change c.1283C>A (p. Ala428Asp) was found in the affected children. Parents and normal hearing children were heterozygous for this change. In silico analysis of p. Ala428Asp showed that the change was damaging and probably damaging by SIFT and PolyPhen2, respectively.
Fig. 1.
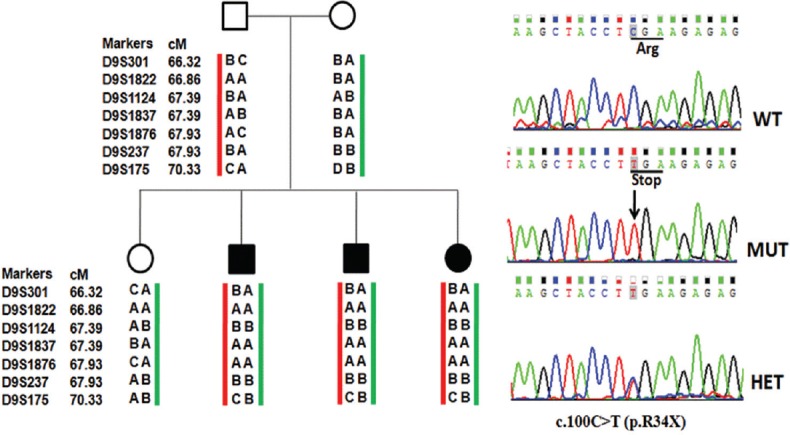
Pedigree and haplotypes (family DDL4), co-segregating nonsyndromic autosomal recessive hearing loss (filled symbols) with short tandem repeat markers at the DFNB7/11 locus. Sequence chromatogram of transmembrane channel like 1 gene showing homozygous change c.100C>T (p.R34X) in hearing impaired children, parents and normal hearing child are heterozygous for c.100C>T.
Fig. 2.
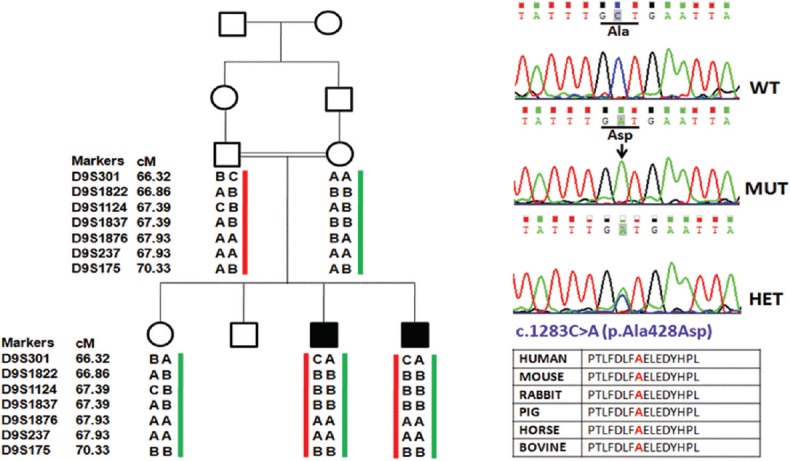
Pedigree and haplotypes (family LKO5), co-segregating nonsyndromic autosomal recessive hearing loss (filled symbols) with short tandem repeat markers at the DFNB7/11 locus. Sequence chromatogram of TMC1 gene show a novel homozygous change c.1283C>A (p.Ala428Asp) in hearing impaired children, parents and normal hearing child are heterozygous for c.1283C>A.
Discussion
Homozygosity mapping is a useful method of linkage analysis for autosomal recessive inherited disorders leading to the identification of hundreds of genes responsible for autosomal recessive genetic diseases. The TMC1 gene is one of the hearing loss genes which is implicated in both autosomal recessive (DFNB7/11) and dominant (DFNA36) hearing impairment. The chromosomal position of DFNB7/11 locus (OMIM 606706) is 9q13–q21. TMC1 contains 24 exons (NCBI Reference Sequence: NM_138691.2, Transcript: ENST00000297784), encodes 760 amino acids, 87.8 kDa multipass transmembrane protein, with six transmembrane domains. However, the exact function of TMC1 protein in the inner ear is unknown but is predicted to have a role in maintaining the electrochemical homeostasis, structure and function of neurosensory hair cells of the inner ear.
More than 53 mutations in TMC1 (missense/nonsense, splicing, small and large deletions) have been reported in HGMD (http://www.hgmd.cf.ac.uk/) in different ethnic populations of the world, in which c.100C>T (p.R34X) is the most common recessive mutation in the gene and is most frequently reported in Pakistani and Tunisian populations and is attributed to founder effect15,23. Kurima et al14 reported an incidence of TMC1 mutations in India and Pakistan to be 5.4 per cent in the 230 families that were screened. c.1960A>G and c.295delA mutations were identified in the Indian families studied. In another study of 374 Indian families negative for the GJB2 gene mutations, c.100C>T, c.237-6T>G, c.453+2T>C, c.628_630del, c.800G>A, c.1114G>A, c.1333C>T and c.1566+1G>A mutations were reported and the prevalence of TMC1 gene mutations was 1.6 per cent16.
We identified a novel homozygous mutation c.1283C>A (p. Ala428Asp) in a consanguineous family, LKO5, having change involving a transversion from cytosine to adenine at nucleotide position of 1283 (NCBI Reference Sequence: NM_138691.2, Transcript: ENST00000297784) which causes substitution of a highly conserved polar amino acid alanine, at codon 428, to a charged aspartic acid (p. Ala428Asp). This change was predicted as damaging and probably damaging by SIFT and PolyPhen2, respectively. This novel mutation was absent in 50 healthy normal controls. c.1283C>A (p. Ala428Asp) mutation is located in the extracellular topological domain and amino acid sequence alignment using MEGA software 9 (http://www.megasoftware.net/previousVersions.php) of different vertebrates (mouse, rabbit, pig, horse and bovine) shows that the Ala428 residue is conserved across species.
In the DDL4 family, with three hearing impaired children with recessive, prelingual, severe-to-profound hearing loss and one normal hearing child, a nonsense mutation c.100C>T (p.R34X), previously reported in a Pakistani family14, was identified. The parents and the normal hearing child were carriers for this mutation. Numerous studies indicate that p.R34X is a common mutation of TMC1 reported in several hearing impaired individuals across many ethnic populations. It has been suggested that p.R34X arose from a common founder and the estimated age of mutation was calculated to be between 1075 and 1900 yr by setting the marker mutation26 rate at 10−3 to 10−6.
The genetic aetiology of NSHL is heterogeneous in India as in the rest of the world. Except GJB2, only a few genes have been reported from India; these are TMIE24,27, TMC114,24, USH1C24,28,29, TMPRSS324, CDH2324, MYO15A30 and SLC26A431 and can be considered to be second-tier genes. None of these have the frequency similar to GJB2 mutations, hence it is difficult to specify the second most common causative gene.
In conclusion, the identification of a previously identified c.100C>T mutation, and a novel homozygous mutation, c.1283C>A in TMC1, in this study supports TMC1 gene as one of the second-tier hearing loss genes, after GJB2 in India. Testing for TMC1 may be considered in all GJB2-negative NSHL cases.
Acknowledgment
Authors thank the families for their cooperation and participation and the Department of Biotechnology, Government of India, for financial support.
References
- 1.Mehl AL, Thomson V. The Colorado newborn hearing screening project, 1992-1999: On the threshold of effective population-based universal newborn hearing screening. Pediatrics. 2002;109:E7. doi: 10.1542/peds.109.1.e7. [DOI] [PubMed] [Google Scholar]
- 2.Hereditary Hearing Loss Homepage. [accessed on March 30, 2017]. Available from: http://hereditaryhearingloss.org/main.aspx .
- 3.Maheshwari M, Vijaya R, Ghosh M, Shastri S, Kabra M, Menon PS. Screening of families with autosomal recessive non-syndromic hearing impairment (ARNSHI) for mutations in GJB2 gene: Indian scenario. Am J Med Genet A. 2003;120A:180–4. doi: 10.1002/ajmg.a.20014. [DOI] [PubMed] [Google Scholar]
- 4.RamShankar M, Girirajan S, Dagan O, Ravi Shankar HM, Jalvi R, Rangasayee R, et al. Contribution of connexin26 (GJB2) mutations and founder effect to non-syndromic hearing loss in India. J Med Genet. 2003;40:e68. doi: 10.1136/jmg.40.5.e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ghosh M, Vijaya R, Kabra M. Genetics of deafness in India. Indian J Pediatr. 2004;71:531–3. doi: 10.1007/BF02724296. [DOI] [PubMed] [Google Scholar]
- 6.Ramchander PV, Nandur VU, Dwarakanath K, Vishnupriya S, Padma T. Study of families of nonsyndromic hearing impairment segregating with mutations in Cx26 gene. Indian J Hum Genet. 2004;10:58–64. [Google Scholar]
- 7.Ramchander PV, Nandur VU, Dwarakanath K, Vishnupriya S, Padma T. Prevalence of Cx26 (GJB2) gene mutations causing recessive nonsyndromic hearing impairment in India. Int J Hum Genet. 2005;5:241–6. [Google Scholar]
- 8.Padma G, Ramchander PV, Nandur VU, Padma T. A rare event of maternal UPD in a proband with congenital non-syndromic hearing impairment with homozygosity for GJB2 p.W24X mutation. Int J Genet Mol Biol. 2009;2:125–9. [Google Scholar]
- 9.Padma G, Ramchander PV, Nandur UV, Padma T. GJB2 and GJB6 gene mutations found in Indian probands with congenital hearing impairment. J Genet. 2009;88:267–72. doi: 10.1007/s12041-009-0039-5. [DOI] [PubMed] [Google Scholar]
- 10.Pavithra A, Jeffrey JM, Chandru J, Ramesh A, Srisailapathy CR. High incidence of GJB2 gene mutations among assortatively mating hearing impaired families in Kerala: Future implications. J Genet. 2014;93:207–13. doi: 10.1007/s12041-014-0338-3. [DOI] [PubMed] [Google Scholar]
- 11.Padma G, Ramchander PV, Nandur VU, Kumar KR, Padma T. Novel mutations affecting the secondary structure of MT-RNR1 gene: A causal relationship with profound nonsyndromic hearing impairment. Genet Test Mol Biomarkers. 2012;16:1092–7. doi: 10.1089/gtmb.2012.0036. [DOI] [PubMed] [Google Scholar]
- 12.Subathra M, Selvakumari M, Ramesh A, Ramakrishnan R, Karan KR, Kaur M, et al. Complete mitochondrial genome analysis and clinical documentation of a five-generational Indian family with mitochondrial 1555A>G mutation and postlingual hearing loss. Ann Hum Genet. 2014;78:217–34. doi: 10.1111/ahg.12061. [DOI] [PubMed] [Google Scholar]
- 13.Pavithra A, Harini PT, Chandru J, Sarvani C, Rastogi A, Selvakumari M, et al. Is screening for mitochondrial A1555G mutation among assortative mating hearing impaired families important. A prefatory quest? Res Otolaryngol. 2014;3:1–7. [Google Scholar]
- 14.Kurima K, Peters LM, Yang Y, Riazuddin S, Ahmed ZM, Naz S, et al. Dominant and recessive deafness caused by mutations of a novel gene, TMC1, required for cochlear hair-cell function. Nat Genet. 2002;30:277–84. doi: 10.1038/ng842. [DOI] [PubMed] [Google Scholar]
- 15.Kitajiri S, Makishima T, Friedman TB, Griffith AJ. A novel mutation at the DFNA36 hearing loss locus reveals a critical function and potential genotype-phenotype correlation for amino acid-572 of TMC1. Clin Genet. 2007;71:148–52. doi: 10.1111/j.1399-0004.2007.00739.x. [DOI] [PubMed] [Google Scholar]
- 16.Zhao Y, Wang D, Zong L, Zhao F, Guan L, Zhang P, et al. A novel DFNA36 mutation in TMC1 orthologous to the Beethoven (Bth) mouse associated with autosomal dominant hearing loss in a Chinese family. PLoS One. 2014;9:e97064. doi: 10.1371/journal.pone.0097064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meyer CG, Gasmelseed NM, Mergani A, Magzoub MM, Muntau B, Thye T, et al. Novel TMC1 structural and splice variants associated with congenital nonsyndromic deafness in a Sudanese pedigree. Hum Mutat. 2005;25:100. doi: 10.1002/humu.9302. [DOI] [PubMed] [Google Scholar]
- 18.Hildebrand MS, Kahrizi K, Bromhead CJ, Shearer AE, Webster JA, Khodaei H, et al. Mutations in TMC1 are a common cause of DFNB7/11 hearing loss in the Iranian population. Ann Otol Rhinol Laryngol. 2010;119:830–5. doi: 10.1177/000348941011901207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Santos RL, Wajid M, Khan MN, McArthur N, Pham TL, Bhatti A, et al. Novel sequence variants in the TMC1 gene in Pakistani families with autosomal recessive hearing impairment. Hum Mutat. 2005;26:396. doi: 10.1002/humu.9374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kalay E, Karaguzel A, Caylan R, Heister A, Cremers FP, Cremers CW, et al. Four novel TMC1 (DFNB7/DFNB11) mutations in Turkish patients with congenital autosomal recessive nonsyndromic hearing loss. Hum Mutat. 2005;26:591. doi: 10.1002/humu.9384. [DOI] [PubMed] [Google Scholar]
- 21.Hilgert N, Alasti F, Dieltjens N, Pawlik B, Wollnik B, Uyguner O, et al. Mutation analysis of TMC1 identifies four new mutations and suggests an additional deafness gene at loci DFNA36 and DFNB7/11. Clin Genet. 2008;74:223–32. doi: 10.1111/j.1399-0004.2008.01053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sirmaci A, Duman D, Oztürkmen-Akay H, Erbek S, Incesulu A, Oztürk-Hismi B, et al. Mutations in TMC1 contribute significantly to nonsyndromic autosomal recessive sensorineural hearing loss: A report of five novel mutations. Int J Pediatr Otorhinolaryngol. 2009;73:699–705. doi: 10.1016/j.ijporl.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 23.Tlili A, Rebeh IB, Aifa-Hmani M, Dhouib H, Moalla J, Tlili-Chouchène J, et al. TMC1 but not TMC2 is responsible for autosomal recessive nonsyndromic hearing impairment in Tunisian families. Audiol Neurootol. 2008;13:213–8. doi: 10.1159/000115430. [DOI] [PubMed] [Google Scholar]
- 24.Ganapathy A, Pandey N, Srisailapathy CR, Jalvi R, Malhotra V, Venkatappa M, et al. Non-syndromic hearing impairment in India: High allelic heterogeneity among mutations in TMPRSS3, TMC1, USHIC, CDH23 and TMIE. PLoS One. 2014;9:e84773. doi: 10.1371/journal.pone.0084773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ben Saïd M, Hmani-Aifa M, Amar I, Baig SM, Mustapha M, Delmaghani S, et al. High frequency of the p.R34X mutation in the TMC1 gene associated with nonsyndromic hearing loss is due to founder effects. Genet Test Mol Biomarkers. 2010;14:307–11. doi: 10.1089/gtmb.2009.0174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Naz S, Giguere CM, Kohrman DC, Mitchem KL, Riazuddin S, Morell RJ, et al. Mutations in a novel gene, TMIE, are associated with hearing loss linked to the DFNB6 locus. Am J Hum Genet. 2002;71:632–6. doi: 10.1086/342193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jain PK, Lalwani AK, Li XC, Singleton TL, Smith TN, Chen A, et al. A gene for recessive nonsyndromic sensorineural deafness (DFNB18) maps to the chromosomal region 11p14-p15.1 containing the Usher syndrome type 1C gene. Genomics. 1998;50:290–2. doi: 10.1006/geno.1998.5320. [DOI] [PubMed] [Google Scholar]
- 29.Ahmed ZM, Smith TN, Riazuddin S, Makishima T, Ghosh M, Bokhari S, et al. Nonsyndromic recessive deafness DFNB18and Usher syndrome type IC are allelic mutations of USHIC. Hum Genet. 2002;110:527–31. doi: 10.1007/s00439-002-0732-4. [DOI] [PubMed] [Google Scholar]
- 30.Liburd N, Ghosh M, Riazuddin S, Naz S, Khan S, Ahmed Z, et al. Novel mutations of MYO15A associated with profound deafness in consanguineous families and moderately severe hearing loss in a patient with Smith-Magenis syndrome. Hum Genet. 2001;109:535–41. doi: 10.1007/s004390100604. [DOI] [PubMed] [Google Scholar]
- 31.Park HJ, Shaukat S, Liu XZ, Hahn SH, Naz S, Ghosh M, et al. Origins and frequencies of SLC26A4 (PDS) mutations in East and South Asians: Global implications for the epidemiology of deafness. J Med Genet. 2003;40:242–8. doi: 10.1136/jmg.40.4.242. [DOI] [PMC free article] [PubMed] [Google Scholar]