

Abstract
Surfactant protein A (SP-A) is a collagenous C-type lectin (collectin) that is critical for pulmonary defense against inhaled microorganisms. Bifunctional avidity of SP-A for pathogen associated molecular patterns (PAMPs) such as lipid A and for dipalmitoylphosphatidylcholine (DPPC), the major component of surfactant membranes lining the air liquid interface of the lung, ensures that the protein is poised for first line interactions with inhaled pathogens. To better understand the motifs that are required for interactions with microbes and surfactant structures, we explored the role of the tyrosine-rich binding surface on the carbohydrate recognition domain of SP-A in the interaction with DPPC and lipid A using crystallography, site-directed mutagenesis, and molecular dynamics simulations. Critical binding features for DPPC binding include a three-walled tyrosine cage that binds the choline head group through cation-π interactions and a positively charged cluster that binds the phosphoryl group. This basic cluster is also critical for binding of lipid A, a bacterial PAMP and target for SP-A. Molecular dynamics simulations further predict that SP-A binds lipid A more tightly than DPPC. These results suggest that the differential binding properties of SP-A favor transfer of the protein from surfactant DPPC to pathogen membranes containing appropriate lipid PAMPs to effect key host defense functions.
Graphical Abstract
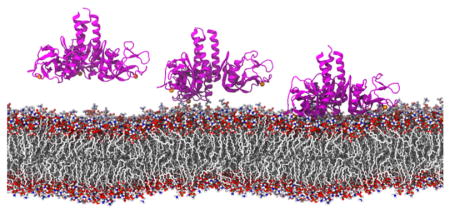
Pulmonary surfactant performs dual physiological roles, reducing surface pressure in the lung to permit effortless respiration and effecting host defense. By weight, surfactant is 90% phospholipids and 10% protein. Surfactant protein A (SP-A), a collagenous C-type lectin, is the most abundant of the four surfactant-associated proteins, which also includes surfactant proteins B (SP-B), C (SP-C) and D (SP-D). SP-A is known to rapidly associate with secreted lamellar bodies and to occupy the corners of the unfolding phospholipid membranes that ultimately constitute tubular myelin,1,2 the lattice-like structure that serves as the reservoir for replenishment of the surfactant monolayer. The critical role of SP-A in the structure of surfactant aggregates became apparent when it was found that Sftpa−/− mice, which are deficient in SP-A, lacked tubular myelin.3 Surprisingly, these animals did not have obvious alterations in surfactant function, content or secretion that had been predicted by more than a decade of in vitro experiments, but were found to have defects in host defense when challenged with infectious microorganisms.4,5 Data collected from studying these animals and other experiments suggests that the association of SP-A with surfactant phospholipids primarily functions to position the protein at the air-liquid interface as the first line of defense against inhaled pathogens.1,6–11 It is therefore important to understand the molecular interactions that determine differential binding of SP-A to phospholipid and microbial surfaces.
Studies of SP-A binding to phospholipids12,13 have shown that SP-A binds to phosphatidylcholine (PC), but not to phosphatidylglycerol (PG), phosphatidylinositol (PI), phosphatidylethanolamine (PE) or phosphatidylserine (PS), demonstrating that the polar head group influences binding. The protein binds most avidly to dipalmitoylphosphatidylcholine (DPPC), the major lipid component of surfactant, but not to other dipalmitoylglycerophospholipids such as dipalmitoylphosphatidylglycerol (DPPG) or dipalmitoylphosphatidylethanolamine (DPPE). The length and saturation state of the fatty acid groups in the sn-1 and sn-2 positions also influences binding; SP-A binds avidly to distearoylphosphatidylcholine but not dimyristoylphosphatidylcholine, and fails to bind to l-palmitoyl-2-linoleoyl-dilinoleoylphosphatidylcholine or lysoPC. The binding of SP-A to phospholipids typically requires calcium. In addition to its key role in the structure of tubular myelin, binding of SP-A to surfactant membranes has been reported to enhance the resistance of surfactant to foreign protein-mediated inhibition of its surface tension lowering properties, and to the recycling of surfactant into type II cells.14
SP-A also recognizes pathogen-associated molecular patterns (PAMPs) such as the lipid A portion of lipopolysaccharide of gram-negative bacteria.15 This interaction is required for the key roles of the SP-A-mediated immune response against these microorganisms, which includes direct antimicrobial activity, agglutination, and opsonization. SP-A-sensitive PAMPs typically contain polar domains and hydrophobic microbial components, such as are found in lipid A.16 The binding sites on SP-A for DPPC, lipid A or other lipids are unknown, but the structural features of lipid ligands suggest that lectin-type activity is not involved. In the alveoli, SP-A must alternately interact with surfactant membrane interfaces and with hydrophobic PAMPs. The extent of physical overlap between these ligand binding sites is a major knowledge gap. Multiple lines of evidence have shown that the carbohydrate recognition domain (CRD) of SP-A is capable of binding to lipid.17–21 Earlier mapping studies using chimeric SP-A proteins that contained homologous regions from surfactant protein D implicated broad domains in PC binding.22 The objective of the current work is to refine our molecular understanding of SP-A/lipid interactions and how these features influence SP-A function.
In previous crystallographic studies of SP-A,23,24 we studied a construct containing the entire CRD and neck domain (NCRD), which recapitulates many of the binding properties of the intact protein. In the initial report,23 we observed that the tyrosine-rich CRD surface included three surface-exposed tyrosine residues (Y164, Y208, and Y192) arranged in part as a box-like structure, and a proximal basic cluster containing two arginines (R216 and R222). Our proposal that these two features together form part of a PC binding site was supported by the nearby presence of a bound MES (2-(N morpholino)ethanesulfonic acid) buffer molecule, a close structural mimetic of phosphocholine, the hydrophilic component of PC. In the crystal structure, the MES binding site was not contiguous with the lectin calcium-binding site, and therefore was inconsistent with a lectin-mediated binding mechanism. The MES site was notable for binding of the positively charged morpholino group to Y164, one of the aforementioned tyrosines, via an apparent π-cation interaction. We postulated that for PC, the choline group would associate primarily with the tyrosine box and that the phosphoryl group would bind to the basic cluster. Unfortunately, analysis of binding of other ligand molecules to this region was precluded by the presence of crystal contacts and the use of sulfate salts in the crystallization medium, as the sulfate bound strongly to the basic cluster. We therefore sought to crystallize an SP-A NCRD/phosphocholine complex in the absence of sulfate, and these results are reported herein. We further tested the validity of the phosphocholine site using mutagenesis and binding assays. Finally, we extended these studies to investigate lipid A/SP-A complexes, for which there are no crystallographic data, utilizing large-scale molecular dynamics simulations to investigate DPPC and lipid A binding in a membrane milieu. These combined studies present a cohesive view of the molecular interactions, on an atomic level, of SP-A and two of its major lipid ligands as occur in the presence of membranes.
MATERIALS AND METHODS
Expression and purification of mutant recombinant proteins
A baculovirus expression system was used to generate WT and mutant, trimeric peptides comprised of the neck and carbohydrate recognition domain (NCRD) regions of rat SP-A. The isolation and sequencing of the 1.6kb cDNA for rat SP-A was previously reported.21 Site-directed mutagenesis of the cDNA was performed using a QuikChange Lightning site-directed mutagenesis kit (ThermoFisher). Inserts were ligated into the unique Eco R1 site of the PVL 1392 vector (Invitrogen),25 and the proper orientation was confirmed by restriction mapping with Kpn I. Sf-9 cells used for recombinant virus production, plaque assays and viral amplifications were maintained in 150 mL spinner cultures at 25–28 °C in an air atmosphere in media composed of IPL-41 insect culture media, tryptose phosphate broth, 0.1% pluronic (shear-reducing surfactant compound) (Sigma), antibiotics, and 10% fetal calf serum.18,26 Recombinant baculoviruses containing mutant cDNAs for SP-A were produced by homologous recombination in Sf-9 cells following cotransfection with linear viral DNA and the recombinant PVL 1392- SP-A constructs (Baculogold, Pharmingen). Trichoplusia ni (T. ni) cells passaged in adherent cultures were used for the production of recombinant proteins. Fresh monolayers of 107 T. ni cells were infected with plaque purified recombinant viruses and incubated with serum-free media (EX-CELL 405) supplemented antibiotics for 72 h. Recombinant SP-A was purified from the culture media by adsorption to mannose-Sepharose 6B columns in the presence of 1 mM calcium and elution with 2 mM EDTA.27 The purified recombinant SP-A was dialyzed against 5 mM Tris (pH 7.4) and stored at − 20 ° C. Bovine serum albumin (BSA) and disodium EDTA were from Sigma-Aldrich (St. Louis, MO, USA).
Preparation of multilamellar liposomes
DPPC/PG (w/w 85:15) and PI/lipid A (w/w 85:15) lipid mixtures were dissolved at a concentration of 1 mg/ml in chloroform, dried to a film under nitrogen in a depyrogenated glass tube, and rehydrated for 0.5 h at 37 °C in TBS (140 mM NaCl and 20 mM Tris-HCl (pH 7.5)) with intermittent, vigorous vortexing. Centrifugation at 12K × g room temperature for 10 min was used to pellet the multilamellar liposomes. 1,2-Dipalmitoyl-sn-glycero-3-phosphatidylcholine (DPPC), L-α-phosphatidylglycerol (egg, chicken(PG), and L-α-phosphatidylinositol (Soy PI) were from Avanti Polar Lipids, Inc. (Alabaster, AL). Diphosphoryl Lipid A from Escherichia coli F583 (Rd mutant) was from Sigma-Aldrich. All other reagents used were analytical grade.
SP-A/liposome binding assay
Mutant SP-A NCRD proteins were preincubated for 15 min at room temperature in the presence of 5mM CaCl2 or 5mM EDTA. A suspension of DPPC/PG or lipid A multilamellar liposomes was added, and the mixture was incubated for 60 min at room temperature with shaking. The liposomes and bound proteins were collected by centrifugation. The supernatant and pellet were coated onto 96 well plates overnight, blocked, and washed. Bound proteins were detected by sandwich ELISA using an anti-rat SP-A. Data are mean ± S.E., n=3.
Crystallography
SP-A NCRD wild-type and mutants were expressed and purified as described previously.23,24 Crystals were grown in hanging drops over reservoirs containing 0.5 ml of reservoir solution (10 mM sodium cacodylate (pH 6.0), 10 mM calcium acetate, and 10% (w/v) polyethylene glycol, MW 20,000). Phosphocholine (POC) was purchased from Sigma-Aldrich. For the phosphocholine complex, the crystals were soaked prior to X-ray data collection in reservoir solution with 50 mM POC and 5%, 10%, and 20% v/v 2-methyl-2,5-pentanediol for approximately 5 minutes each to provide cryoprotection. Data were collected on a RAXIS-IV image plate using a Rigaku RU-300 rotating anode X-ray source. Indexing and processing of X-ray data were performed using DENZO and Scalepack.28 Structures were solved by difference Fourier using the published wild type/mannose complex coordinates (PDB code 3PAK)24 less waters and ligands as a starting model. Initial models were rebuilt using the AutoBuild function in Phenix29 with the input model excluded to reduce phase bias. Afterward, structures were subjected to iterative cycles of manual rebuilding in Coot30 and refinement in Phenix. TLS refinement of the B factors was used with 2 zones defined – residues 87-109 (neck) and residues 100-228 (CRD).
System preparation for MD simulations
The SP-A trimer was obtained by using the three-fold symmetry properties of the monomer. Two Cysteine residues C135-C226 and C204-C218 were cross-linked using psfgen in VMD31. An equilibrated structure of DPPC membrane was obtained from a previous study.32 To accommodate a sufficiently large binding area for the SP-A, four identical membrane bilayers were merged into a DPPC membrane bilayer of area 142 Å × 142 Å. The resulting membrane, with 648 DPPC lipid molecules in total, was further equilibrated for 20 ns under NPT-γ ensemble. The structure of the lipid A monomer was obtained from Wu et al33; 288 lipid A monomers were arranged into a bilayer configuration of an area of 142 Å × 156 Å and the membrane was equilibrated for a total of 30 ns under NPT-γ ensemble. The phosphate group of the lipid A was protonated such that each lipid A had a -2 charge to mimic the physiological condition of the bacterial membrane. The shape of the carbohydrates of lipid A were maintained in the chair conformation by imposing additional restraints on the glucosamine rings. The modified topology and parameter files can be downloaded in the supplementary materials.
MD simulations
All coordinated ligands of the calcium ion were restrained to maintain the expected lectin site structure. The 6 coordinated atoms of the calcium were constrained with a harmonic potential of the form U(x) = k(x− xref)2, where k was 1000 kcal mol−1 Å−2 and xref was set to 2.41 Å34 throughout minimization, heating, equilibration, and production simulations.35 SP-A was placed ~15 Å above the membrane bilayer. The whole system was minimized, heated up and equilibrated for 5 ns with the backbone of SP-A constrained. Simulations were performed under NPT-γ ensemble using the program NAMD 2.936 assuming the CHARMM36 force field for the protein and lipid37 and assuming the TIP3P model for water molecules.38 Periodic boundary conditions were assumed, and the particle-mesh-Ewald (PME) summation method was employed for the evaluation of Coulomb forces. The van der Waals energy was calculated using a cutoff of 12 Å. Temperature and pressure were maintained at 323 K and 1 atm using a Langevin thermostat with a damping constant of 1 ps−1 and Nosé-Hoover Langevin piston methods.39 The integration time step was 2 fs with all bonds involving hydrogen atoms constrained using SHAKE algorithm.40
Steered Molecular Dynamics (SMD) simulations
With SP-A bound to membranes after 200 ns of simulation, SMD was used to apply a force perpendicular to the membrane to pull the SP-A off the membranes. The phosphate atoms of the bottom layer of the membrane bilayer were restrained such that the vertical motion is subjected to a harmonic potential with a force constant of 1 kcal mol−1 Å−2. Note that the lipids can diffuse freely in the lateral direction. The last 10 Cα atoms of the N-terminus of SP-A trimer were pulled with a constant velocity of 1.25 Å/ns and a spring constant of 0.1 Nm−1 for 41 – 68 ns.
Analysis of MD trajectory
PME electrostatic potential maps and the binding footprint of SP-A were calculated using the PME electrostatics and Volmap plugins in VMD.31 The number of hydrogen bonds formed between SP-A and lipid molecules and their occupancies were calculated using the Hydrogen-Bonds plugin in VMD. Residue-based interaction energies between SP-A and the lipid molecules were calculated using a modified generalized-Born (GB) model.35,41 This model considers van der Waals interaction, electrostatics, and a solvation energy component that takes solvent-polarization into account. All the graphs were plotted using Matlab versions R2015a.
Statistics
Differences between groups were analyzed by the Student’s t-test, p values of <0.05 were considered significant.
RESULTS
Crystal structure of SP-A complex with phosphocholine (POC)
To date there have been no crystal structures of complexes of pulmonary collectins bound to lipid. In order to determine whether SP-A specifically recognizes the polar head group of PC, we soaked SP-A crystals in mother liquor supplemented with phosphocholine (POC); cocrystallization was unsuccessful presumably due to neighboring crystal contacts. The complex of SP-A with POC was solved at 2.2 Å resolution. The final model shows good geometry and agreement with the observed X-ray diffraction data (Table S1). The structure is very similar to the SP-A/mannose complex;24 the superimposed structures have a root-mean-square deviation of 0.25 Å calculated using all α-carbons. There is one molecule of POC found in the structure (Figure 1). The phosphate group of the ligand is making salt bridging interactions with R216 and R222, as well as a hydrogen bond to the side chain of Q220. The choline group is not making any hydrogen bonds, however the positively charged group is well positioned to make cation-π interactions with the aromatic ring of Y164.
Figure 1.

Crystal structure of SP-A in complex with POC. The phosphate group of POC forms hydrogen bonds with Q220, R216 and R222. The choline group of POC interacts with Y164 via cation-π interaction.
Mutational Analysis
In order to test the importance of the contact area features observed in the SPA/POC crystallographic complex, we produced alanine mutants of N162, Y164, R216, L219, Q220, Y221, and R222. Y221was not part of the POC complex but is in the same region and highly conserved in SP-A sequences, so it was included in the analysis. The binding of these mutants to phospholipids is shown in Figure 2A and 2B. The data show that both alanine substitutions for tyrosine at Y164A and Y221A effectively eliminated SP-A binding to DPPC liposomes. In contrast, hydrophobic or neutral mutants of neighboring residues showed little or no effect on DPPC binding compared with wild-type. In particular, L219A and Q220A showed no significant loss of binding, while N162A exhibited an intermediate range of binding. R216 and R222 also were tested for DPPC and lipid A binding. Alanine mutation of both of these arginines similarly reduced both DPPC and lipid A binding. For both mutants, alanine substitution had a greater effect in R222A than in R216A.
Figure 2.

Effect of mutations on binding of SP-A to DPPC and lipid A. Wild type and N162A, Y164A, L219A, Q220A, Y221A mutant NCRD forms of SP-A were incubated with DPPC/PG (panel A) liposomes for 60 min at room temperature. Wild type and R216A, R222A mutant NCRDs were incubated with DPPC/PG (panel B) and lipid A (panel C) liposomes, respectively, for 60 minutes at room temperature. The liposomes and bound proteins were sedimented by centrifugation. Supernatant and pellet fractions were coated onto plates, blocked, and washed. Bound fusion proteins were detected by ELISA using an anti-rat SP-A antibody, and expressed as a fraction of total SP-A (bound + unbound) in the reaction. Data are mean ± S.E. n=3.
Crystal structures of SP-A mutants
In order to determine whether any loss of lipid binding could be due to structural destabilization of the tyrosine mutants, we crystallized Y164A and Y221A. The structures were very similar to wild-type (root mean square deviation over all Cα atoms was 0.21Å and 0.33Å for Y164A and Y221A, respectively), indicating that the observed effects were due solely to loss of side chain functionality rather than structural destabilization.
Molecular Dynamics (MD) simulation results
To obtain both atomic-level description and the dynamical properties of the membrane binding events, a series of MD simulations were performed on two systems, namely, SP-A on DPPC membrane and SP-A on lipid A membrane (Table 1). SP-A, initially placed ~15 Å above the membrane, binds firmly to both membranes after 200 ns of MD simulations, respectively (Figure 3).
Table 1.
MD simulations performed in this study
| Simulation | Description | Time (ns) | No. of runs |
|---|---|---|---|
| EPC | Equilibrium simulation of SP-A on DPPC membrane | 200 | 2 |
| ELA | Equilibrium simulation of SP-A on lipid A membrane | 200 | 2 |
| SPC | SMD simulation of SP-A on DPPC membrane | 41 | 2 |
| SLA | SMD simulation of SP-A on lipid A membrane | 53–68 | 2 |
Figure 3.

Lipid membrane binding by SP-A simulated using MD simulations. SP-A (ribbon representation in magenta) diffuses and binds firmly to the membrane bilayer within 100ns and stayed bound for another 100ns.
The tyrosine residues bind to choline of DPPC lipid via cation-π interaction
Cation-π interaction, although not being calculated explicitly in a classical force field, is present. Despite classical force field underestimation of the cation-π interaction,42 we consistently observed binding events of choline by several tyrosine residues. Therefore, we employed a geometry criterion to count the interaction in a more qualitative approach.43
Four tyrosine residues (Y164, Y192, Y208, and Y221) bind frequently to the choline of DPPC (Figure 4B), especially after 100 ns of simulation where all three chains of the SP-A trimer are bound to the membrane. To estimate the relative strengths of the tyrosine-choline interactions between different tyrosine residues, durations of the binding events were calculated and only the events that lasted for more than 5 ns were presented (Figure 4C). In simulations EPC, the tyrosine residues (Y164, Y192, Y208) that form the tyrosine box bind to choline substantially longer than does an isolated tyrosine residue (Y221). The isolated Y221 never binds to a choline for more than 10 ns while Y208, the center of the tyrosine box, showed a binding duration of 30 ns, two times longer than the reported cation-π duration of arginine and tryptophan.44 Since the only way Y208 can bind to choline is in the tyrosine box (Figure 4A), we suspect the box-like arrangement of tyrosine molecules amplifies the cation-π interaction.
Figure 4.

Tyrosine box amplifies the cation-π interaction. (A, B) Three tyrosine residues form a box-like conformation to bind with choline, part of the DPPC lipid head group. Occupancy plot (C) and histogram (D) show the frequency and duration of a choline bound to the key tyrosine residues of SP-A in simulation ELPC, respectively. SP-A is represented in transparent magenta ribbon, calcium ion is shown as orange sphere, and DPPC lipid, which comprises nitrogen (blue), oxygen (red), carbon (cyan), and phosphorus (tan) atoms, is shown in ball-and-stick representation.
Comparison of lipid A and DPPC interfacial binding surfaces in SP-A as visualized through molecular dynamics simulations of explicit membranes
MD simulations also estimate the effect of SP-A binding to multiple lipid molecules. Trimeric SP-A interacts with more lipid A molecules (13.9 ± 2.0) than with DPPC molecules (9.8 ± 4.4), as measured from the number of lipid molecules forming hydrogen bonds with SP-A in the last 10 ns of the MD simulations. SP-A also forms more hydrogen bonds with lipid A (Figure 5) than with DPPC because SP-A can bind not only to phosphate groups of lipid A, but also to the hydroxyl groups of its carbohydrate rings. Additionally, hydrogen bonds that involve the basic residues (particularly R216 and R222) and Y221 are the most consistently formed. The occupancy values for most of the residues listed in Table 2 are larger in simulations ELA (Table 1), indicating that SP-A forms more consistent hydrogen bonds on lipid A. Some basic residues at certain chains have unusually low hydrogen bond occupancy (<10%) (for full table refer Table S2) due to lack of access to the hydroxyl or the phosphate groups of the lipids.
Figure 5.

Hydrogen bonding plays a more important role for SP-A binding to lipid A than to DPPC. The hydrogen bonds curves plotted are the average hydrogen bond numbers of the two independent simulations ELA (black curve) and simulations EPC (gray curve), respectively. See Table 1.
Table 2.
Evaluation of the strength of interaction between SP-A and lipid membrane. Important SP-A residues were identified by calculating their average interaction energies with DPPC or lipid A membranes for the last 50 ns of the simulations. R216, Y221 and R222 form the most consistent hydrogen bonds with the lipid molecules. Only residues that have the highest hydrogen bond occupancy among the three SP-A monomers are listed. The occupancy values (in percentage) were calculated for the last 100 ns of the simulations. For some residues, occupancy of >100% was observed because two residues can form more than one bond simultaneously. For a full table of hydrogen bond occupancy and interaction energies, please refer to Tables S2 and S3.
| Residue of SP-A | Average interaction energies (kcal/mol) | Hydrogen bond occupancy (%) | ||
|---|---|---|---|---|
|
| ||||
| SP-A/DPPC | SP-A/lipid A | SP-A/DPPC | SP-A/lipid A | |
|
| ||||
| N162 | −6.4 ± 4.8 | −6.6 ± 3.7 | 20 | 41 |
| N163 | −3.3 ± 1.6 | −2.8 ± 1.6 | 3 | 9 |
| Y164 | −5.3 ± 5.9 | −7.5 ± 4.2 | 43 | 78 |
| Y192 | −7.8 ± 6.9 | −4.2 ± 2.9 | 47 | 65 |
| R197 | −21.4 ± 16.1 | −21.4 ± 12.5 | 84 | 112 |
| K201 | −6.1 ± 10.9 | −30.3 ± 12.4 | 40 | 148 |
| Y208 | −2.9 ± 3.0 | −2.8 ± 2.4 | 19 | 63 |
| R216 | −30.0 ± 16.9 | −27.1 ± 7.7 | 82 | 172 |
| Q220 | −14.0 ± 7.6 | −10.4 ± 2.9 | 57 | 93 |
| Y221 | −9.4 ± 7.0 | −10.0 ± 2.9 | 67 | 96 |
| R222 | −22.6 ± 10.6 | −22.3 ± 6.2 | 81 | 177 |
| Calcium | −11.7 ± 7.9 | −8.9 ± 2.2 | - | - |
The basic residues contribute the majority of the interaction energies for the protein-lipid interfaces of both DPPC and lipid A membranes (Table 2). K201 plays an important role in forming strong hydrogen bonds with lipid A, although less so with DPPC. Q220 and Y221 also form hydrogen bonds with the lipid A phosphate group. Not as prominent as the other basic residues, the hydrogen bonds formed by these two polar residues are not negligible. Based on the hydrogen bond occupancy, trajectory plots and interaction energy analysis, SP-A forms stronger interactions, including more hydrogen bonds, more consistently with lipid A than with DPPC, suggesting that SP-A binds stronger to lipid A than to DPPC.
Differences in the properties of lipid A and DPPC
Figure 6 shows that the vertical (perpendicular to the membrane surface) fluctuation amplitude of DPPC is almost twice as large as that of lipid A. When SP-A binds to the DPPC membrane, the higher vertical fluctuation of DPPC increases the frequency of bond breaking and may effectively weaken the binding of SP-A and DPPC. Evidently, the SP-A DPPC interaction is more intermittent than that with lipid A. SP-A moves from one DPPC lipid to another throughout the 200 ns simulation (Supplemental movie), whereas such movement was rarely observed in SP-A/lipid A binding.
Figure 6.

The lipid head groups of DPPC have larger vertical fluctuations than lipid A. The normalized histograms and their corresponding fitted Gaussian curves show that the fluctuation amplitude of DPPC lipid (gray) is almost twice of that of lipid A (black).
Although lipid A is negatively charged and DPPC is neutral, the electrostatic potentials of both membrane surfaces are similar because the sodium ions from bulk water screens the negative charges of the phosphate group of lipid A. Figure S1 shows that the electrostatic potentials of both lipid membrane surfaces are nearly indistinguishable.
Estimation of relative lipid binding strength of SP-A via Steered Molecular Dynamics simulations
To compare the binding strengths of SP-A on DPPC and lipid A membranes, steered molecular dynamics (SMD) simulations were performed to pull SP-A away from the membrane with constant velocity. The speed of pulling was chosen to be identical for both systems. The force-extension curves are plotted in Figure 7. The pulling forces of SP-A are consistently larger for lipid A than for DPPC.
Figure 7.
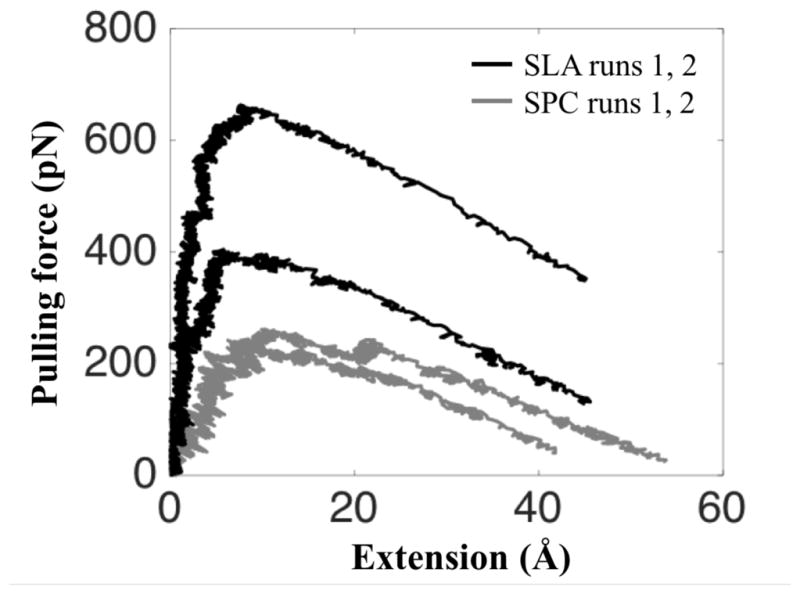
A larger force is required to pull SP-A off a lipid A than off a DPPC membrane. The force-extension curves for simulations SLA (black) show that the applied pulling forces are at least 60% larger than that of simulations SPC (gray). See Table 1.
Despite the long durations of our SMD simulations (41–68 ns), relatively long for system of this size,45 the time lengths of the simulations, t0 ~ 40 ns, and the typical membrane binding distances (here referred to as extensions), d0 ~ 50 A, permitted only pulling velocities of 1.25 Å/ns, which are a few orders of magnitude higher than the velocities in a typical atomic force microscopy (AFM) experiment; as a result, the pulling forces arising in our simulations are larger than experimentally measured ones.46 Nevertheless, as is typical in such cases, the qualitative force-extension profile gleaned from SMD simulations is consistent with in AFM and optical tweezers pulling experiments.47,48 Given the consistent difference of the force extension curves shown in Figure 7, namely forces in SLA runs being higher than forces in SPC runs, our simulations suggest that SP-A binds to lipid A more tightly than to DPPC as tighter binding implies larger pulling forces.
DISCUSSION
The soluble surfactant protein SP-A undergoes binding to various membrane lipids, including phosphatidylcholine from host membranes and bacterial lipids such as lipid A. The binding of peripheral proteins to such membrane lipids is as yet poorly described on a molecular level. While there are many examples of peripheral membrane proteins binding to anionic phospholipids, little is known about those binding neutral phospholipids such as phosphatidylcholine. Still less is known about peripheral binding to microbial lipids such as lipid A. The techniques used in this present study, i.e. x-ray crystallography, mutagenesis, and large-scale molecular dynamics, offer an atomic resolution glimpse into the less known interactions and allow one to construct a detailed model of SP-A interactions for the cases of two key ligands, namely DPPC and lipid A.
DPPC Binding by SP-A
The present studies reveal that peripheral membrane interactions between SP-A and DPPC rely significantly upon two structural features of the protein surface: (1) surface tyrosine residues that facilitate cation-π interactions between protein and phospholipid and (2) basic residues, primarily arginines, which stabilize binding to lipid phosphoryl groups. Cation-π interactions occur frequently in proteins, where they offer structural stability and often participate in ligand binding.49 Opportunities for cation-π interactions are numerous at the membrane interface, an environment often rich in aromatic residues that can serve as π electron donors.44 The crystal structure of a phosphatidylcholine transfer protein complex described an unusual structural motif in which the tetramethylammonium (choline) group of the PC lipid ligand is held within a three-tyrosine cage via cation-π interactions with the tyrosines.50 More recently has such a tyrosine box motif been found in B. thuringiensis phosphatidylinositol-specific phospholipase C (PI-PLC) and has been identified as a phosphatidylcholine specific membrane targeting motif.43
We propose that SP-A also contains a cation-π box motif that promotes selective binding of phosphatidylcholine. In initial crystallographic studies of a construct based on the SP-A NCRD,23 a belt of surface-exposed tyrosine residues had been identified, three of which are arranged as an open box. In SP-A sequences, the three box tyrosines (Y164, Y192, and Y208) are conserved whereas a fourth belt tyrosine is conserved at position 220 or 221. The invariance of these residues suggest that they play key functional roles in SP-A, and indeed it has been previously proposed that the site is important for binding to PC.23 Unfortunately, the earlier studies could not confirm this proposal crystallographically using soaked crystals since critical crystal contacts occluded the entrance of lipid to the box. However, the initial crystal structure suggested that there exists another PC binding site adjacent to the box and involving one of the three box tyrosines, Y164.23 In this structure, which contained a MES molecule, the positively charged morpholino group, analogous to a PC choline, bound to Y164 via an apparent cation-π interaction. To follow up on this observation, a crystal structure of a complex with POC was obtained, as presented above (see Figure 1). The choline group also bound similarly to Y164 while the phosphoryl group was bound at a highly conserved basic cluster consisting of R216, Q220, and R222.
In order to support and extend the crystallographic data, SP-A was subjected to alanine mutational analysis. The mutagenesis results confirmed the critical importance of the tyrosines and the basic cluster in binding to DPPC. While Y192 or Y208, two box residues, were not tested due to the presence of critical interactions stabilizing the tertiary structure of the protein, the more solvent exposed Y164A and Y221A mutants abrogated DPPC binding. The two arginines in the basic cluster also lost significant DPPC binding activity when mutated to alanine.
To gain further structural and mechanistic information, large–scale molecular dynamics simulations using explicit DPPC membrane bilayer models were undertaken. The advantage of using MD simulations to study SP-A binding is that multiple binding sites can be identified in a single simulation trajectory. Figure 8 demonstrates the accuracy of MD simulations in reproducing the PC binding site observed in the SP-A/POC crystal structure. More importantly, the MD simulations of SP-A binding to the DPPC membrane revealed that all four tyrosines participate in PC binding. The binding site that has been suggested by the POC complex crystal structure (Figure 1) and supported by the mutational analysis (Figure 2A) was confirmed by MD simulations. However, the tyrosine box itself also contained a choline group from DPPC, stabilized by cation-π interactions. Y221, while not part of the tyrosine box, also exhibits cation-π interactions with DPPC choline. Amplification of the cation-π interaction by the box motif was clearly observed as the tyrosine residues that form the tyrosine box were observed to interact with choline for a much longer duration than Y221, the latter side group not participating in any such structural motif. Such amplification of cation-π interactions due to geometrical arrangement of aromatic residues was also observed in molecular dynamics studies of PI-PLC.43 In addition to the cation-π interactions, tyrosines 164, 192 and 221 were also seen to form hydrogen bonds with the phosphate groups of the lipid molecules.
Figure 8.

Binding site of POC in SP-A (purple ribbon). The lectin site calcium (orange sphere), key side chains (licorice), and POC and DPPC (ball and stick) are shown. (A) The final, refined model of the complex between SP-A and POC obtained from X-ray crystallography. (B) The same binding site as observed in the MD simulations.
Lipid A binding by SP-A
Lipopolysaccharide (LPS), the major component of the outer leaflet of the outer membrane of gram-negative bacteria, is a major microbial lipid target for SP-A. Full-length LPS consists of three regions: lipid A, which is the membrane component, a conserved core oligosaccharide region, and a highly variable O-antigen region. Studies of LPS of various lengths indicate that SP-A binds specifically to the lipid A component,15 and that the high saccharide content of the O-antigen region actually inhibits SP-A binding. Therefore, studies of lipid A binding to SP-A15,51 additionally have utilized Re-LPS, a naturally occurring mutant form of LPS containing only lipid A and two Kdo (3-Deoxy-D-manno-oct-2-ulosonic acid) saccharides from the conserved core region. For the MD simulations, the pure lipid A bilayer was used because it is a simple and well-established model,33 and because SP-A specifically targets lipid A.
The greater number of hydrogen bonds between SP-A and lipid A suggests that lipid A binds more tightly to SP-A than does DPPC. Supporting this idea, experimental measurements have yielded Kd values of 5μM52 and 28nM53 for DPPC and Re-LPS, respectively. Although lipid A does not bind in the tyrosine box, strong hydrogen bonds are made with the same tyrosine side chains (Table 2). Additionally, the basic cluster which binds DPPC phosphoryl groups also form key electrostatic interactions with the phosphoryl groups of lipid A. Accordingly, the R216A and R222A mutants show significant reduction in lipid A binding (Figure 2C).
Despite the observation from MD simulations that SP-A utilizes a similar group of residues for DPPC and lipid A binding, SP-A was estimated to bind 60% more tightly to lipid A than to DPPC according to the peak pulling forces obtained from SMD simulations. Such large discrepancy in pulling forces could largely be attributed to the dynamic properties of the lipid membranes and the number of lipid molecules bound to SP-A. Lipid A, approximately twice the size of DPPC, is therefore less mobile than DPPC as clearly shown by the slower lateral diffusion (Figure S2) and the lower vertical fluctuations (Figure 6) of the lipid A membrane bilayer. Therefore, the thermal perturbation on the SP-A/lipid A interactions is effectively damped. Additionally, the fact that SP-A binds to more lipid A molecules results in further damping of the dynamics of SP-A when bound to lipid A membrane. As a result, once bound, SP-A does not ‘scoot’ on the membrane surface of lipid A, and SP-A forms more consistent hydrogen bonds with lipid A than with DPPC.
In their studies of mixed monolayers of DPPC and lipid A or LPS of varying lengths, including Re-LPS, Casals and coworkers found that the highly miscible mixed monolayers interact strongly with SP-A.51 Such monolayers could appear in the lung as LPS from inhaled bacteria becomes incorporated into DPPC-rich monolayers. Studies from the McCormack lab suggest that binding of SP-A to LPS initiates membrane permeabilization and direct killing of the bacteria by SP-A.54,55 Other studies show that SP-A destabilizes LPS membrane structure.56 It is tempting to speculate on the basis of the present MD simulations that SP-A remains bound to surfactant DPPC until close proximity of the bacterial cell and bacterial lipids, at which point SP-A transfers to LPS due to its higher affinity binding and initiates bactericidal activity. However, additional studies are needed to investigate this scenario.
In summary, the present studies utilized three complementary approaches to probe, at atomic resolution, the interactions between SP-A and the lipid surfaces of DPPC or lipid A. The crystal structure of a complex of SP-A with POC mimicked specific interactions with the PC polar head, thus identifying two key components to the SP-A binding site, the choline box and the basic cluster. We further tested hypotheses regarding the phosphocholine site using alanine mutagenesis and binding assays. We also extended the present studies to investigate lipid A/SP-A complexes, for which there are no crystallographic data. For this purpose we utilized large-scale molecular dynamics approaches to investigate DPPC and lipid A binding in an explicit membrane milieu. The combined studies present a collective view of the molecular interactions, on an atomic level, of SP-A binding to two of its major lipid ligands, suggesting furthermore a molecular mechanism underlying the bactericidal activity of SPA.
Supplementary Material
Acknowledgments
This work was supported by NIH 9P41GM104601 (K.S., B.C.G.), VA Merit Review Grant (F.X.M), and NIH PO1AI0833222 (B.A.S. and F.X.M.). This work used computer time on Stampede at the Texas Advanced Computing Center (TACC), provided by grant MCA93S028 from the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by National Science Foundation grant number OCI-1053575. We thank Dr. Wei Han and Dr. Zhe Wu for helpful discussions. We also thank Prof. Wonpil Im for sharing the topology and coordinate files of lipid A.
Abbreviations
- BSA
bovine serum albumin
- CHARMM
Chemistry at HARvard Macromolecular Mechanics
- CRD
carbohydrate recognition domain
- Cα
alpha carbon
- DPPC
dipalmitoylphosphatidylcholine
- DPPE
dipalmitoylphosphatidylethanolamine
- DPPG
dipalmitoylphosphatidylglycerol
- EDTA
ethylenediaminetetraacetic acid
- ELISA
enzyme-linked immunosorbent assay
- GB
generalized-Born
- Kd
dissociation constant
- Kdo
3-deoxy-D-manno-oct-2-ulosonic acid
- LPS
lipopolysaccharide
- MD
molecular dynamics
- MES
2-(N morpholino)ethanesulfonic acid
- NAMD
NAnoscale Molecular Dynamics
- NCRD
neck and carbohydrate recognition domain
- NPT-γ
constant number of particle, pressure, and temperature while keeping the ratio of the unit cell in the x-y plane constant
- PAMP
pathogen associated molecular pattern
- PE
phosphatidylethanolamine
- PG
phosphatidylglycerol
- PI-PLC
phosphatidylinositol-specific phospholipase C
- PI
phosphatidylinositol
- PME
particle-mesh-Ewald
- POC
phosphocholine
- PS
phosphatidylserine
- SMD
steered molecular dynamics
- SP-A
surfactant protein A
- SP-B
surfactant protein B
- SP-C
surfactant protein C
- SP-D
surfactant protein D
- Tris
tris(hydroxymethyl)aminomethane
- VMD
Visual Molecular Dynamics
- WT
wild type
Footnotes
Financial support: NIH PO1AI0833222 (B.A.S. and F.X.M.), VA Merit Review Grant (F.X.M), NIH 9P41GM104601 (K.S.), and XSEDE MCA93S028 (K.S.).
Accession Codes
Atomic coordinates have been deposited as Protein Data Bank entries 5FFR (SP-A/POC complex), 5FFS (SP-A Y164A mutant) and 5FFT (SP-A Y221A mutant).
Supporting Information Available:
The Supporting Information is available free of charge via the ACS Publication Website at http://pubs.acs.org.
Electrostatic potential maps of the membrane surfaces (Figure S1), the plots of the lateral diffusion of DPPC and lipid A (Figure S2). The table containing crystallographic data collection and refinement statistics (Table S1), the full lists of hydrogen bond occupancies (Table S2) and interaction energies of SP-A binding to membranes (Table S3). Method of the lipid lateral diffusion (Supplemental Method). Videos of the binding and unbinding events of SP-A on DPPC and lipid A membranes (Supplemental Movies). Topology and parameter files for protonated lipid A (Supplemental files).
References
- 1.Kingma PS, Whitsett JA. In defense of the lung: surfactant protein A and surfactant protein D. Curr Opin Pharmacol. 2006;6:277–283. doi: 10.1016/j.coph.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 2.Voorhout WF, Veenendaal T, Haagsman HP, Verkleij AJ, Vangolde LMG, Geuze HJ. Surfactant Protein A Is Localized At The Corners Of The Pulmonary Tubular Myelin Lattice. J Histochem Cytochem. 1991;39:1331–1336. doi: 10.1177/39.10.1940306. [DOI] [PubMed] [Google Scholar]
- 3.Korfhagen TR, Bruno MD, Ross GF, Huelsman KM, Ikegami M, Jobe AH, Wert SE, Stripp BR, Morris RE, Glasser SW, Bachurski CJ, Iwamoto HS, Whitsett JA. Altered surfactant function and structure in SP-A gene targeted mice. Proc Natl Acad Sci. 1996;93:9594–9599. doi: 10.1073/pnas.93.18.9594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.LeVine AM, Kurak KE, Wright JR, Watford WT, Bruno MD, Ross GF, Whitsett JA, Korfhagen TR. Surfactant Protein-A Binds Group B Streptococcus Enhancing Phagocytosis and Clearance from Lungs of Surfactant Protein-A–Deficient Mice. Am J Respir Cell Mol Biol. 1999;20:279–286. doi: 10.1165/ajrcmb.20.2.3303. [DOI] [PubMed] [Google Scholar]
- 5.LeVine AM, Whitsett JA, Gwozdz JA, Richardson TR, Fisher JH, Burhans MS, Korfhagen TR. Distinct effects of surfactant protein A or D deficiency during bacterial infection on the lung. J Immunol. 2000;165:3934–3940. doi: 10.4049/jimmunol.165.7.3934. [DOI] [PubMed] [Google Scholar]
- 6.Waters P, Vaid M, Kishore U, Madan T. Lung surfactant proteins A and D as pattern recognition proteins. Adv Exp Med Biol. 2009;653:74–97. doi: 10.1007/978-1-4419-0901-5_6. [DOI] [PubMed] [Google Scholar]
- 7.McCormack FX, Whitsett JA. The pulmonary collectins, SP-A and SP-D, orchestrate innate immunity in the lung. J Clin Invest. 2002;109:707–712. doi: 10.1172/JCI15293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crouch EC, Wright JR. Surfactant proteins A and D and pulmonary host defense. Annu Rev Physiol. 2001;63:521–554. doi: 10.1146/annurev.physiol.63.1.521. [DOI] [PubMed] [Google Scholar]
- 9.Kuroki Y, Takahashi M, Nishitani C. Pulmonary collectins in innate immunity of the lung. Cell Microbiol. 2007;9:1871–1879. doi: 10.1111/j.1462-5822.2007.00953.x. [DOI] [PubMed] [Google Scholar]
- 10.Haagsman HP, Hogenkamp A, Van Eijk M, Veldhuizen EJA. Surfactant collectins and innate immunity. Neonatology. 2008;93:288–294. doi: 10.1159/000121454. [DOI] [PubMed] [Google Scholar]
- 11.McCormack FX. New concepts in collectin-mediated host defense at the air-liquid interface of the lung. Respirology. 2006;11:S7–S10. doi: 10.1111/j.1440-1843.2006.00798.x. [DOI] [PubMed] [Google Scholar]
- 12.Kuroki Y, Akino T. Pulmonary surfactant protein A (SP-A) specifically binds dipalmitoylphosphatidylcholine. J Biol Chem. 1991;266:3068–3073. [PubMed] [Google Scholar]
- 13.Casals C. Role of surfactant protein A (SP-A)/lipid interactions for SP-A functions in the lung. Pediatr Pathol Mol Med. 2001;20:249–268. [PubMed] [Google Scholar]
- 14.Cockshutt AM, Weitz J, Possmayer F. Pulmonary surfactant-associated protein A enhances the surface activity of lipid extract surfactant and reverses inhibition by blood proteins in vitro. Biochemistry. 1990;29:8424–8429. doi: 10.1021/bi00488a032. [DOI] [PubMed] [Google Scholar]
- 15.Van Iwaarden JF, Pikaar JC, Storm J, Brouwer E, Verhoef J, Oosting RS, van Golde LMG, van Strijp JAG. Binding of surfactant protein A to the lipid A moiety of bacterial lipopolysaccharides. Biochem J. 1994;303:407–411. doi: 10.1042/bj3030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seaton BA, Crouch EC, McCormack FX, Head JF, Hartshorn KL, Mendelsohn R. Review: Structural determinants of pattern recognition by lung collectins. Innate Immun. 2010;16:143–150. doi: 10.1177/1753425910368716. [DOI] [PubMed] [Google Scholar]
- 17.Kuroki Y, McCormack FX, Ogasawara Y, Mason RJ, Voelker DR. Epitope mapping for monoclonal antibodies identifies functional domains of pulmonary surfactant protein A that interact with lipids. J Biol Chem. 1994;269:29793–29800. [PubMed] [Google Scholar]
- 18.McCormack FX, Kuroki Y, Stewart JJ, Mason RJ, Voelker DR. Surfactant protein A amino acids Glu195 and Arg197 are essential for receptor binding, phospholipid aggregation, regulation of secretion, and the facilitated uptake of phospholipid by type II cells. J Biol Chem. 1994;269:29801–29807. [PubMed] [Google Scholar]
- 19.Honma T, Kuroki Y, Tsunezawa W, Ogasawara Y, Sohma H, Voelker DR, Toyoaki Akino. The Mannose-Binding Protein A Region of Glutamic Acid185–Alanine221 Can Functionally Replace the Surfactant Protein A Region of Glutamic Acid195–Phenylalanine228 without Loss of Interaction with Lipids and Alveolar Type II Cells. Biochemistry. 1997;36:7176–7184. doi: 10.1021/bi962967e. [DOI] [PubMed] [Google Scholar]
- 20.McCormack FX, Stewart J, Voelker DR, Damodarasamy M. Alanine Mutagenesis of Surfactant Protein A Reveals That Lipid Binding and pH-Dependent Liposome Aggregation Are Mediated by the Carbohydrate Recognition Domain. Biochemistry. 1997;36:13963–13971. doi: 10.1021/bi970745q. [DOI] [PubMed] [Google Scholar]
- 21.Sano H, Kuroki Y, Honma T, Ogasawara Y, Sohma H, Voelker DR, Akino T. Analysis of Chimeric Proteins Identifies the Regions in the Carbohydrate Recognition Domains of Rat Lung Collectins That Are Essential for Interactions with Phospholipids, Glycolipids, and Alveolar Type II Cells. J Biol Chem. 1998;273:4783–4789. doi: 10.1074/jbc.273.8.4783. [DOI] [PubMed] [Google Scholar]
- 22.Ogasawara Y, McCormack FX, Mason RJ, Voelker DR. Chimeras of surfactant proteins A and D identify the carbohydrate recognition domains as essential for phospholipid interaction. J Biol Chem. 1994;269:29785–29792. [PubMed] [Google Scholar]
- 23.Head JF, Mealy TR, McCormack FX, Seaton BA. Crystal Structure of Trimeric Carbohydrate Recognition and Neck Domains of Surfactant Protein A. J Biol Chem. 2003;278:43254–43260. doi: 10.1074/jbc.M305628200. [DOI] [PubMed] [Google Scholar]
- 24.Shang F, Rynkiewicz MJ, McCormack FX, Wu H, Cafarella TM, Head JF, Seaton BA. Crystallographic Complexes of Surfactant Protein A and Carbohydrates Reveal Ligand-induced Conformational Change. J Biol Chem. 2011;286:757–765. doi: 10.1074/jbc.M110.175265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luckow VA, Summers MD. Signals important for high-level expression of foreign genes in Autographa californica nuclear polyhedrosis virus expression vectors. Virology. 1988;167:56–71. doi: 10.1016/0042-6822(88)90054-2. [DOI] [PubMed] [Google Scholar]
- 26.McCormack FX, Calvert HM, Watson PA, Smith DL, Mason RJ, Voelker DR. The structure and function of surfactant protein A. Hydroxyproline- and carbohydrate-deficient mutant proteins. J Biol Chem. 1994;269:5833–5841. [PubMed] [Google Scholar]
- 27.Fornstedt N, Porath J. Characterization studies on a new lectin found in seeds of Vicia ervilia. FEBS Lett. 1975;57:187–191. doi: 10.1016/0014-5793(75)80713-7. [DOI] [PubMed] [Google Scholar]
- 28.Otwinowski Z, Minor W. Macromolecular Crystallography Part A. Academic Press; 1997. [20] Processing of X-ray diffraction data collected in oscillation mode; pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 29.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr Sect D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr Sect D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 31.Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 32.Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD, Pastor RW. Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J Phys Chem B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu EL, Engström O, Jo S, Stuhlsatz D, Yeom MS, Klauda JB, Widmalm G, Im W. Molecular Dynamics and NMR Spectroscopy Studies of E. coli Lipopolysaccharide Structure and Dynamics. Biophys J. 2013;105:1444–1455. doi: 10.1016/j.bpj.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zheng H, Chruszcz M, Lasota P, Lebioda L, Minor W. Data mining of metal ion environments present in protein structures. J Inorg Biochem. 2008;102:1765–1776. doi: 10.1016/j.jinorgbio.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goh BC, Rynkiewicz MJ, Cafarella TR, White MR, Hartshorn KL, Allen K, Crouch EC, Calin O, Seeberger PH, Schulten K, Seaton BA. Molecular Mechanisms of Inhibition of Influenza by Surfactant Protein D Revealed by Large-Scale Molecular Dynamics Simulation. Biochemistry. 2013;52:8527–8538. doi: 10.1021/bi4010683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Best RB, Zhu X, Shim J, Lopes PEM, Mittal J, Feig M, MacKerell AD. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ,ψ and Side-Chain χ1 and χ2 Dihedral Angles. J Chem Theory Comput. 2012;8:3257–3273. doi: 10.1021/ct300400x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926. [Google Scholar]
- 39.Martyna GJ, Tobias DJ, Klein ML. Constant pressure molecular dynamics algorithms. J Chem Phys. 1994;101:4177. [Google Scholar]
- 40.Ryckaert JP, Ciccotti G, Berendsen HJ. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 41.Onufriev A, Bashford D, Case DA. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins Struct Funct Bioinforma. 2004;55:383–394. doi: 10.1002/prot.20033. [DOI] [PubMed] [Google Scholar]
- 42.Minoux H, Chipot C. Cation-π interactions in proteins: can simple models provide an accurate description? J Am Chem Soc. 1999;277:10366–10372. [Google Scholar]
- 43.Grauffel C, Yang B, He T, Roberts MF, Gershenson A, Reuter N. Cation-π Interactions As Lipid-Specific Anchors for Phosphatidylinositol-Specific Phospholipase C. J Am Chem Soc. 2013;135:5740–5750. doi: 10.1021/ja312656v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aliste MP, MacCallum JL, Tieleman DP. Molecular Dynamics Simulations of Pentapeptides at Interfaces: Salt Bridge and Cation-π Interactions. Biochemistry. 2003;42:8976–8987. doi: 10.1021/bi027001j. [DOI] [PubMed] [Google Scholar]
- 45.Lumb CN, He J, Xue Y, Stansfeld PJ, Stahelin RV, Kutateladze TG, Sansom MSP. Biophysical and Computational Studies of Membrane Penetration by the GRP1 Pleckstrin Homology Domain. Structure. 2011;19:1338–1346. doi: 10.1016/j.str.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Izrailev S, Stepaniants S, Balsera M, Oono Y, Schulten K. Molecular Dynamics Study of Unbinding of the Avidin-Biotin Complex. 1997;72:1568–1581. doi: 10.1016/S0006-3495(97)78804-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sotomayor M, Schulten K. Single-Molecule Experiments in Vitro and in Silico. Science. 2007;316:1144–1148. doi: 10.1126/science.1137591. [DOI] [PubMed] [Google Scholar]
- 48.Lee EH, Hsin J, Sotomayor M, Comellas G, Schulten K. Discovery Through the Computational Microscope. Structure. 2009;17:1295–1306. doi: 10.1016/j.str.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mahadevi AS, Sastry GN. Cation-π interaction: Its role and relevance in chemistry, biology, and material science. Chem Rev. 2013;113:2100–2138. doi: 10.1021/cr300222d. [DOI] [PubMed] [Google Scholar]
- 50.Roderick SL, Chan WW, Agate DS, Olsen LR, Vetting MW, Rajashankar KR, Cohen DE. Structure of human phosphatidylcholine transfer protein in complex with its ligand. Nat Struct Biol. 2002;9:507–511. doi: 10.1038/nsb812. [DOI] [PubMed] [Google Scholar]
- 51.García-Verdugo I, Cañadas O, Taneva SG, Keough KMW, Casals C. Surfactant Protein A Forms Extensive Lattice-Like Structures on 1,2-Dipalmitoylphosphatidylcholine/Rough-Lipopolysaccharide- Mixed Monolayers. Biophys J. 2007;93:3529–3540. doi: 10.1529/biophysj.107.109793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Meyboom A, Maretzki D, Stevens PA, Hofmann KP. Interaction of pulmonary surfactant protein A with phospholipid liposomes: a kinetic study on head group and fatty acid specificity. Biochim Biophys Acta. 1999;1441:23–35. doi: 10.1016/s1388-1981(99)00142-0. [DOI] [PubMed] [Google Scholar]
- 53.García-Verdugo I, Sánchez-Barbero F, Soldau K, Tobias PS, Casals C. Interaction of SP-A (surfactant protein A) with bacterial rough lipopolysaccharide (Re-LPS), and effects of SP-A on the binding of Re-LPS to CD14 and LPS-binding protein. Biochem J. 2005;391:115–124. doi: 10.1042/BJ20050529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu H, Kuzmenko A, Wan S, Schaffer L, Weiss A, Fisher JH, Kim KS, McCormack FX. Surfactant proteins A and D inhibit the growth of Gram-negative bacteria by increasing membrane permeability. J Clin Invest. 2003;111:1589–1602. doi: 10.1172/JCI16889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kuzmenko AI, Wu H, McCormack FX. Pulmonary collectins selectively permeabilize model bacterial membranes containing rough lipopolysaccharide. Biochemistry. 2006;45:2679–2685. doi: 10.1021/bi0522652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cañadas O, García-Verdugo I, Keough KMW, Casals C. SP-A permeabilizes lipopolysaccharide membranes by forming protein aggregates that extract lipids from the membrane. Biophys J. 2008;95:3287–3294. doi: 10.1529/biophysj.108.137323. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.