

SUMMARY
Little is known about the cellular events promoting metastasis. We show that knockout of phospholipase D2 (PLD2), which generates the signaling lipid phosphatidic acid (PA), inhibits lung metastases in the mammary tumor virus (MMTV)-Neu transgenic mouse breast cancer model. PLD2 promotes local invasion through the regulation of the plasma membrane targeting of MT1-MMP and its associated invadopodia. A liposome pulldown screen identifies KIF5B, the heavy chain of the motor protein kinesin-1, as a new PA-binding protein. In vitro assays reveal that PA specifically and directly binds to the C-terminus of KIF5B. The binding between PLD2-generated PA and KIF5B is required for the vesicular association of KIF5B, surface localization of MT1-MMP, invadopodia, and invasion, in cancer cells. Taken together, these results identify a role of PLD2-generated PA in the regulation of kinesin-1 motor functions and breast cancer metastasis, and suggest PLD2 as a potential therapeutic target for metastatic breast cancer.
Keywords: PLD2, phosphatidic acid, breast cancer, metastasis, MT1-MMP, invadopodia, KIF5B
eTOC BLURB
The cellular events that regulate cancer metastasis are poorly understood. Wang et al. demonstrate that PLD2, through generating the signaling lipid phosphatidic acid (PA), promotes breast cancer lung metastasis. Direct PA binding to the motor protein KIF5B controls MT1-MMP plasma membrane trafficking, invadopodia formation, and local invasion.
INTRODUCTION
Metastasis is the major cause of mortality in cancer patients (Chaffer and Weinberg, 2011). One intrinsic property of metastatic tumor cells that allows them to break tissue barriers is their ability to degrade the proteins of the extracellular matrix (ECM) by matrix-degrading proteases, including matrix metalloproteinases (MMPs) (Chaffer and Weinberg, 2011). In particular, a subgroup of membrane-anchored MMPs, including membrane type 1-matrix metalloproteinase (MT1)-MMP (also known as MMP14), have been recognized as important proteases involved in local invasion and metastasis (Castro-Castro et al., 2016). During cancer cell invasion, MT1-MMP accumulates in specialized matrix-degrading plasma-membrane domains called invadopodia (Castro-Castro et al., 2016; Murphy and Courtneidge, 2011), which have been shown to be critical for the invasion of many types of cancer cells, such as melanoma, breast cancer, colon cancer, lung cancer and prostate cancer (Murphy and Courtneidge, 2011; Paz et al., 2014). It has been reported that the delivery of MT1-MMP to invadopodia is critical to cancer cell invasion (Castro-Castro et al., 2016; Monteiro et al., 2013). However, it remains unclear how MT1-MMP-containing vesicles are delivered to invadopodia.
It has been reported that a lipid messenger, phosphatidic acid (PA), modulates several cellular processes, such as membrane trafficking, actin cytoskeletal reorganization, cell motility, and cell proliferation (Bruntz et al., 2014; Zhang and Du, 2009). Signaling PA is primarily generated by two families of enzymes, including two members of the phospholipase D (PLD) family and ten members of the diacylglycerol kinase (DGK) family (Bruntz et al., 2014; Frohman, 2015). Two PLD family members, PLD1 and PLD2, perform distinct functions due to different subcellular localization and regulation. While PLD1 has been recently reported to promote cancer progression using genetically engineered mouse models (Chen et al., 2012; Kang et al., 2015), the role of PLD2 in cancer remains to be defined. PLD2 is mainly localized to the plasma membrane and on the endosomes, and has been reported to regulate both cell motility and membrane trafficking in a variety of cell types (Du et al., 2004; Frohman, 2015). The expression and activity of PLD2 are upregulated in some types of cancer including breast cancer (Bruntz et al., 2014; Gomez-Cambronero, 2014). In addition, increasing PLD2 expression enhanced, whereas inhibition of PLD2 blocked, cancer cell proliferation, invasion and metastasis, in cell lines and a mouse xenograft model (Henkels et al., 2013; Zhang et al., 2014).
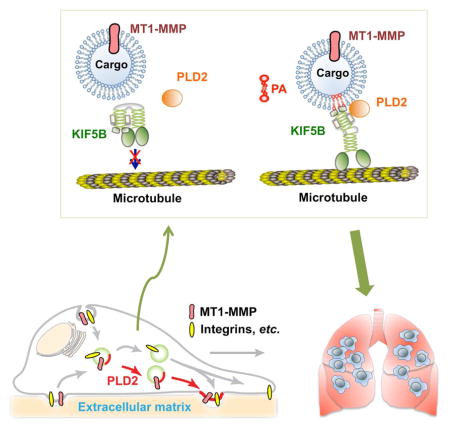
We herein report that breast cancer cell invasion and metastasis are driven by a lipid signaling regulated invadopodia formation. We show that PLD2 deficiency blocks the lung metastasis of breast tumors in MMTV-Neu transgenic mice. Mechanistically, the direct interaction of PLD2-generated PA with KIF5B is required for the plasma membrane localization of MT1-MMP, invadopodia formation, and invasion, both in vitro and in vivo. Our data demonstrate an exocytic pathway by which PLD2-generated PA selectively couples vesicles carrying MT1-MMP to KIF5B for their anterograde plasma membrane transportation, and suggest a critical role of PLD2 in breast cancer metastasis.
RESULTS
PLD2 is required for the lung metastasis of mammary tumors in the MMTV-Neu breast cancer mouse model
To evaluate the function of PLD2 in mammary tumor progression, we employed the transgenic MMTV-Neu mouse model, which overexpresses the rat NEU (human ERBB2 homologue) in mammary glands (Guy et al., 1992). We bred the Pld2+/− mice with MMTV-Neu mice after 10 generations of backcrossing the Pld2+/− mice from the mixed C57BL/6J-129/SvJ background to the FVB/N background. Similar to those on C57BL/6 background, PLD2-deficient mice on the FVB/N background show no obvious defects (Oliveira et al., 2010). PLD2 deficiency had no significant effect on the time of the appearance of the palpable tumors, the growth rate or the size of the primary tumors (Figures 1A–C). Examination of the histology of the primary tumors by Ki67, activated caspase-3, CD68 and CD31 staining, did not reveal any effect of Pld2 ablation on cell proliferation, apoptosis, macrophage infiltration and angiogenesis (Figures S1A–S1H). Similarly, there is also no difference in Ki67 staining in PLD2 inhibitor-treated highly metastatic MDA-MB-231 breast cancer cells (Figures S1I & S1J). These results are consistent with our recent finding that PLD2 knockdown or inhibitor treatment did not affect the proliferation of the same cells in the normal culture condition (Cai et al., 2016).
Figure 1.

PLD2 promotes lung metastasis in the MMTV-Neu breast cancer mouse model. (A) Tumorigenesis is not affected by PLD2 deficiency. The appearance of mammary tumors was examined weekly in MMTV-Neu;Pld2+/+ (n=22) and MMTV-Neu;Pld2−/− mice (n=25). (B) PLD2 deficiency does not affect tumor size. Tumor size was measured weekly after the first appearance of a palpable tumor in MMTV-Neu;Pld2+/+ (n=27) and MMTV-Neu;Pld2−/− (n=24) mice. (C) Weight of mammary tumors in MMTV-Neu;Pld2+/+ (n=24) and MMTV-Neu;Pld2−/− (n=21) mice collected at 9 weeks after the first appearance of a palpable tumor. (D) Macroscopic images of the lungs of tumor-bearing in MMTV-Neu;Pld2+/+ mice and MMTV-Neu;Pld2−/− mice. Metastases are indicated by arrows. Scale bar = 1.5 mm. (E) Quantification of macroscopic lung metastasis in D. MMTV-Neu;Pld2+/+ (n=26), MMTV-Neu;Pld2−/− (n=22). (F) Representative H&E-stained lung histological sections. Metastases are indicated by arrows. Scale bar = 100 μm. (G) Quantification of tumor foci in the lung of tumor-bearing mice. n=12 per group. Quantifications are presented as mean ± SD; t-test, **p < 0.01, NS (not significant, p> 0.05). See also Figure S1.
At later stages of tumor progression, MMTV-Neu mammary tumors progress from hyperplasia to metastatic carcinoma (Guy et al., 1992). Examination of the lungs revealed that 54% of wild-type mice exhibited macroscopically visible lung metastases, whereas only 27% of Pld2−/− mice showed visible lung metastases (Figures 1D and 1E). Histological analysis confirmed a striking decrease in the number of metastases in the lung of Pld2−/− mice with a 77% decrease in metastatic foci (Figures 1F and 1G). These results indicate that PLD2 is not involved in tumorigenesis or tumor growth, but is required for the lung metastasis of breast tumor.
PLD2 deficiency in mice reduced the number of circulating tumor cells and the invasion of cancer cells
To dissect the steps of the metastatic cascade affected by PLD2 deficiency, we assessed the ability of the cells to enter the circulation. We found that the number of circulating tumor cells (CTCs) was significantly decreased in PLD2-deficient mice (Figure 2A). The decreased CTCs in Pld2−/− mice might be caused by anoikis, a form of cell death induced by detachment from normal ECM (Cristofanilli et al., 2004). We measured the number of viable primary mouse tumor cells grown in suspension, and found no difference in cell viability between primary tumor cells from wild-type and Pld2−/− mice (Figure 2B). Similarly, PLD2 inhibitor did not alter anoikis in MDA-MB-231 breast cancer cells (Figure 2C). Alternatively, the reduction in CTCs could also be caused by the reduction of cancer cell local invasion (Chaffer and Weinberg, 2011). One the other hand, primary Pld2−/− tumor cells, and primary wild-type tumor cells or MDA-MB-231 cells treated with PLD2 inhibitor showed a significant decrease in invasion ability as compared to control cells (Figures 2D–F), suggesting that the decrease in CTCs in PLD2-deficient mice is caused by a defect of local invasion.
Figure 2.

PLD2 deficiency reduces the number of circulating tumor cells and invasion of cultured cancer cells. (A) Decreased circulating tumor cells in PLD2-deficient mice. n=19 per group. (B) The numbers of viable primary mammary tumor cells cultured in suspension. n=3. (C) The numbers of viable MDA-MB-231 cells cultured in suspension in the presence of DMSO or PLD2 inhibitor (5μM). n=3. (D) Invasion of primary mammary tumor cells from MMTV-Neu;Pld2+/+ and MMTV-Neu;Pld2−/− mice. n=3. (E) Invasion of primary mammary tumor cells from MMTV-Neu mice in the presence of DMSO or PLD2 inhibitor (5μM). n=3. (F) Invasion of MDA-MB-231 cells in the presence of DMSO or PLD2 inhibitor (5μM). n=3. Quantifications are presented as mean ± SD; t-test, ***p < 0.001.
PLD2 deficiency inhibited invadopodia formation in breast cancer cells
Since cancer cells use invadopodia to invade into ECM (Eckert et al., 2011; Murphy and Courtneidge, 2011; Paz et al., 2014), we examined invadopodia in primary tumors by measuring the co-localization of two essential invadopodia proteins, TKS5 and cortactin (Blouw et al., 2015; Eckert et al., 2011). PLD2 deficiency greatly reduced the colocalization of TKS5 and cortactin, indicating the reduction of invadopodia formation (Figures 3A and 3B), but did not affect their expression (Figure 3C). To confirm that the impairment of invadopodia in PLD2-deficient mice is intrinsic to tumor cells, we performed gelatin degradation assays in cultured cells (Artym et al., 2006; Paz et al., 2014; Wang et al., 2016). We observed that invadopodia formation was significantly decreased in both PLD2-deficient primary mouse cancer cells (Figures 3D and 3E) and PLD2 inhibitor-treated MDA-MB-231 cells (Figures 3F and 3G).
Figure 3.

PLD2 deficiency blocks invadopodia formation in cancer cells. (A) PLD2 deficiency reduces the invadopodia formation in vivo. Invadopodia were identified by the colocalization of cortactin and Tks5 in the cryosections of mouse mammary tumors (indicated by arrows). n=4 for each genotype. At least 200 cells from six different fields were analyzed. (B) Quantification of invadopodia cell in A. (C) PLD2 does not regulate the expression of TKS5 and cortactin. (D) Pld2 knockout blocks invadopodia formation in primary mammary tumor cells in vitro. Invadopodia were identified by removal of the fluorescently labeled gelatin (black spots) as marked by arrow heads. (E) Quantification of invadopodia in D. n=3. (F) PLD2 inhibitor (5μM) blocks invadopodia formation in MDA-MB-231 cells. (G) Quantification of invadopodia in F. n=3. Scale bars = 10 μm. Quantifications are presented as mean ± SD; t-test, **p < 0.01, ***p < 0.001.
The plasma membrane localization of MT1-MMP is disrupted in PLD2-deficient cells
To investigate the mechanisms of PLD2 action, we examined the activity of several commonly altered oncogenic signaling pathways in primary tumors, including EGFR, ERBB2, ERK, and AKT, by Western blotting (Figure S2). Consistent with the lack of effect on tumor size, PLD2 deficiency also did not change the activity of these pathways, suggesting that PLD2 regulates the invasion and metastasis of breast cancer cells through a different mechanism.
Functional invadopodia require ECM degradation by MT1-MMP (Castro-Castro et al., 2016; Monteiro et al., 2013; Murphy and Courtneidge, 2011; Paz et al., 2014). MT1-MMP undergoes endo-exocytic cycling, and the plasma membrane localization of MT1-MMP is critical for the formation of invadopodia (Castro-Castro et al., 2016; Monteiro et al., 2013; Yu et al., 2012). In mouse tumors and primary cells, MT1-MMP was predominantly localized to the plasma membrane that was reduced after Pld2 deletion, as shown by either confocal microscopy or flow cytometry (Figures 4A–C). In MDA-MB-231 cells, MT1-MMP was localized to both the plasma membrane and intracellular vesicles (Figure 4D), most of which represent late endosomes and lysosomes (Monteiro et al., 2013; Yu et al., 2012). PLD2 inhibitor treatment inhibited the plasma membrane localization of MT1-MMP while increased its vesicle localization (Figure 4D). Similar to the endogenous protein, the plasma membrane localization of MT1-MMP-GFP was also disrupted by PLD2 inhibitor treatment in an MDA-MB-231 cell line stably expressing a low level of MT1-MMP-GFP (Figure 4E). To get better understanding of PLD2 regulation of MT1-MMP localization, we plated MDA-MB-231 cells expressing MT1-MMP-GFP on gelatin-coated coverslips and performed X-Z axis re-construction of confocal images. MT1-MMP-GFP was mainly localized to invading invadopodia; but this localization was disrupted by PLD2 inhibitor treatment (Figure 4F). In contrast to localization, PLD2 knockout or inhibition did not change the protein levels of MT1-MMP in both tumors and MDA-MB-231 cells (Figures 4G and 4H). It is worthy to note that PLD2 inhibitor or knockout only impaired the plasma membrane localization of MT1-MMP but not other transmembrane proteins we have examined, including EGFR, integrin β1 and ERBB2/HER2 in primary tumors or MDA-MD-231 cells (Figure S3). The regulation of MT1-MMP localization is specific to PLD2, because PLD1 knockdown or inhibitor treatment did not change the plasma membrane localization of MT1-MMP (Figure S4).
Figure 4.

Inhibition of PLD2 leads to the loss of MT1-MMP plasma membrane localization and accumulation of intracellular vesicles. (A) Immunofluorescent staining of MT1-MMP in cancer cells using cryosections of primary tumors. Pictures are representatives of tumors from 4 mice for each genotype. (B) Histograms of fluorescent intensity analyzed by flow cytometry showing MT1-MMP cell surface expression in primary tumor cells. (C) Statistic results of the median MT1-MMP fluorescence intensity. Background IgG fluorescence was subtracted. n=3. (D and E) Localization of endogenous MT1-MMP (D) or MT1-MMP-GFP (E) in MDA-MB-231 cells treated with DMSO or PLD2 inhibitor (5μM). (F) Localization of MT1-MMP-GFP in MDA-MB-231 cells plated on Alexa 594-labeled gelatin in the presence of DMSO or PLD2 inhibitor (5μM). X-Z images were reconstructed from a series of X-Y confocal images. The dash lines outline cell board in X-Z reconstruction. (G) PLD2 deficiency did not affect MT1-MMP expression in tumors. Protein samples were collected from tumors from 4 individual mice for each genotype. (H) PLD2 inhibitor had no effect on MT1-MMP expression in MDA-MB-231 cells. The control and PLD2 inhibitor treatment were performed in duplicate. (I) PLD2 regulates the recycling of MT1-MMP to the cell surface. Cell surface proteins were labeled with (lane 2–7) or without biotin (lane 1) on ice for 15 min. The biotinylated cells were incubated at 37°C for 30 min to allow endocytosis, then treated with or without MESNA (1st MESNA) on ice to measure intracellular (lane 3) and total (lane 2) labeled proteins, respectively. After internalization, the biotinylated cells were incubated at 37°C for 15 (lanes 4 and 5) or 30 min (lanes 6 and 7) to allow recycling, and then treated with or without MESNA (second) to measure intracellular (lanes 5 and 7) and total (lanes 4 and 6) labeled proteins, respectively. Biotinylated proteins were recovered by streptavidin beads and analyzed by Western blotting using an MT1-MMP antibody. (J) Quantification of results in I. The intracellular labeled MT1-MMP was normalized to total labeled MT1-MMP. n=3. Scale bars = 10 μm. Quantifications are presented as mean ± SD; t-test, *p < 0.05, ***p < 0.001, NS (p> 0.05). See also Figures S2–S4.
To determine how PLD2 inhibition reduces MT1-MMP levels on the plasma membrane, we examined the trafficking of MT1-MMP by surface biotin labeling. Short-term treatment of PLD2 inhibitor (30 min) did not lead to a detectable change in either total or intracellular levels of biotin-labeled MT1-MMP (Figures 4I and 4J), suggesting that PLD2 is not involved in the internalization of MT1-MMP. In contrast, PLD2 inhibitor led to significant accumulation of intracellular biotinylated MT1-MMP after its internalization (Figures 4I and 4J), suggesting a defect in recycling back to the plasma membrane.
PA specifically and directly binds to the C-terminal of KIF5B
We hypothesized that PLD2 functions through its catalytic product, PA, because invadopodia formation and the plasma membrane localization of MT1-MMP were inhibited by the PLD2 enzymatic activity inhibitor (Scott et al., 2009) (Figures 3 and 4B–D). PA is known to activate type I phosphatidylinositol 4-phosphate 5-kinases (PIPKIs) that generate PI4,5P2 (Ishihara et al., 1998; Tay et al., 2017). We examined the levels and distribution of phosphatidylinositol-4,5-bisphosphate (PI4,5P2) using high-performance liquid chromatography (HPLC) and subcellular localization using GFP-PLCδ-PH, a PI4,5P2 biosensor (Varnai and Balla, 1998). We did not observe any difference in the abundance of PI4,5P2 or the membrane association and subcellular distribution of GFP-PLCδ-PH between control and PLD2-inhibited cells (Figure S5).
To discover the mechanism, we performed a screen for PA-binding proteins using a liposome-pulldown assay using two types of liposomes: control phosphatidylserine (PS) liposomes [containing phosphatidylcholine (PC), phosphatidylethanolamine (PE), and PS] and PA-liposomes (containing PC, PE and PA), and followed by reverse-phase liquid chromatography-tandem mass spectrometry (LC-MS/MS) (Figure 5A). Among 1,183 proteins pulled down by liposomes (Table S1), label-free quantification (Bai et al., 2013) revealed that 63 proteins displayed selective or preferential binding to PA-liposomes (Figure 5B, Tables S2 and S3, < 5% false discovery rate). Recovery of known PA-binding proteins such as RAC1, PLCD3 and Lipin3 (see reviews in (Raghu et al., 2009; Stace and Ktistakis, 2006; Zhang and Du, 2009)) validated our method. Many of these proteins are involved in the regulation of membrane trafficking and actin reorganization, or both (Figure 5B), supporting the previously known functions of PA.
Figure 5.

KIF5B binds to PA directly and specifically. (A) The screening strategy for PA-binding proteins. (B) PA-biding proteins identified by liposome pulldown followed by mass spectrometry. The number in the brackets next to protein names indicates the spectral counts. (C). Domain structure of a kinesin-1 motor protein that composed of two KIF5B and two kinesin light chain (KLC). The C-terminus of KIF5B (KIF5B-C) is the cargo-binding domain. (D). The purified C-terminus of KIF5B (KIF5B-C) specifically bound to PA, and to a less extent to other phospholipids in a lipid strip binding assay. (E) The purified KIF5B-C specifically bound to PA in a liposome pulldown assay. KIF5B-C bound to liposomes containing indicated phospholipids was detected by Western blot using a GST antibody. (F) Identification of the PA-binding sites on KIF5B using saturated liposomes (1 mM). Up, candidate PA-binding sites on KIF5B-C and corresponding alanine mutants. Low, Western blot analysis of PA binding ability of KIF5B-C wild-type and mutants. (G) Identification of A5 as the key PA-binding site on KIF5B using serially diluted liposomes. The mutant A4/A5 did not further reduce the PA-binding of KIF5B. (H) Quantitation of PA binding results in G. n=3. Quantifications are presented as mean ± SD; t-test, **p < 0.01, ***p < 0.001. See also Figure S5 and Tables S1–S3.
Among these candidates, we focused on KIF5B, the heavy chain of kinesin-1, for further characterization since it is known to regulate anterograde trafficking of many proteins including MT1-MMP (Hirokawa et al., 2009; Verhey and Hammond, 2009; Wiesner et al., 2010). Since kinesin-1 interacts with cargo through its carboxyl terminus (KIF5B-C) (Figure 5C)(Hirokawa et al., 2009; Skoufias et al., 1994; Verhey and Hammond, 2009), we purified KIF5B-C from E. coli and tested its binding with a variety of lipids spotted on a nitrocellulose membrane. KIF5B-C showed differential binding to PA, phosphatidylinositol (PI), PI4,5P2 and phosphatidylinositol-3,4,5-trisphosphate PI3,4,5P3, with the strongest binding to PA (Figure 5D). In the liposome pulldown assay, KIF5B-C also bound strongly to liposomes containing PA, but much weaker or not to other phospholipids including PC, PS, PI4,5P2 and PI3,4,5P3 (Figure 5E). Since proteins often bind to PA through a stretch of positively charged amino acids (Raghu et al., 2009; Stace and Ktistakis, 2006), five regions rich in lysine and arginine in KIF5B-C are likely candidate sites of PA interaction (Figure 5F). We mutated all lysine and arginine residues to alanine in these five sites, expressed and purified the resulting mutants from E. coli, and tested their PA-binding ability using liposome pulldown assay. Two of the mutants, A4 and A5, significantly lost their binding to PA liposomes in the primary screening using a high concentration of liposomes (1 mM) (Figure 5F). To further characterize these two mutants, we tested their binding to serially diluted liposomes. While wild-type KIF5B-C bond to PA liposomes in a concentration dependent manner, both A4 and A5 mutants significantly lost the PA binding; among them, A5 showed the greater loss of PA binding (Figures 5G and 5H). The combination of mutant A4 and A5 did not lead to a greater loss in PA binding (Figures 5G and 5H). Together, these data show that KIF5B directly and selectively binds to PA through both A4 and A5 sites, and that the A5 site mutation suffices to disrupt binding of KIF5B to PA.
PLD2 and PA are required for the association of KIF5B with MT1-MMP vesicles and invasion
The specificity of PA regulation of KIF5B is further supported by localization studies in cells. KIF5B is evenly distributed on intracellular vesicles in both tumor sections (Figure 6A) and MDA-MB-231 cells (Figures 6B and S6A), consistent with its involvement of many types of intracellular organelles (Hirokawa et al., 2009; Verhey and Hammond, 2009; Wiesner et al., 2010). PLD2 knockout and inhibitor treatment led to a coalescing of KIF5B-associated vesicles (Figures 6A and 6B), presumably due to the loss of KIF5B association with selected vesicles or inhibition of KIF5B-mediated trafficking processes. Super-resolution microscopy revealed that KIF5B was localized on MT1-MMP-positive vesicles; and this localization was inhibited by the PLD2 inhibitor (Figures 6C and 6D). In contrast to localization, PLD2 knockout or inhibitor had no effect on the expression of KIF5B (Figure S6B). The requirement of PLD2-generated PA for recruitment of KIF5B to MT1-MMP vesicles is different from ARF6, a known PLD2 activator (Ghossoub et al., 2014; Honda et al., 1999). ARF6 promotes the fusion of MT1-MMP vesicles with the plasma membrane, and is not involved in the recruitment of KIF5B to MT1-MMP vesicles (Castro-Castro et al., 2016; Marchesin et al., 2015). We also found that knockdown of ARF6 had no effect on the localization of KIF5B to MT1-MMP vesicles (Figures 6E–G), supporting the previous finding (Marchesin et al., 2015). Furthermore, expression of either the exogenous ARF6 wild-type or a PLD-deficient ARF6 mutant, N48R (Jovanovic et al., 2006; Moreau et al., 2012), had no effect on the recruitment of KIF5B to MT1-MMP vesicles (Figures 6E–G). Taken together, KIF5B recruitment to MT1-MMP vesicles is a PLD2-dependent but ARF6-independent process.
Figure 6.

PLD2-generated PA couples KIF5B to selected vesicles and is critical for MT1-MMP plasma membrane localization, invadopodia formation, and invasion. (A) Coalescing of KIF5B-associated vesicles in PLD2-deficient cancer cells in cryosections of mammary tumors. (B) PLD2 inhibitor treatment reduced KIF5B membrane association in MDA-MB-231 cells. (C) PLD2 inhibitor disrupted the association of KIF5B with MT1-MMP vesicles. (D) Quantification of the co-localization of KIF5B and MT1-MMP in C. (E) Western blotting showing knocking down of ARF6 and re-expression of exogenous ARF6 wild-type and N48R. (F) ARF6 is not required for the association of KIF5B with MT1-MMP vesicles. (G) Quantification of the co-localization of KIF5B and MT1-MMP in F. (H) PLD2 interacts with KIF5B. PLD2 was IPed in MDA-MB-231 cells with a PLD2 antibody (#226) and association of KIF5B was examined by immunoblotting. WCL, whole cell lysates. (I) GFP-KIF5B was IPed in MDA-MB-231 cells stably expressing GFP-KIF5B with a GFP antibody and association of endogenous PLD2 was examined by immunoblotting. (J) Co-localization of PLD2 and KIF5B on MT1-MMP vesicles. MDA-MB-231 cells expressing Flag-PLD2 and MT1-MMP-GFP were stained with mouse KIF5B (SUK 4-c)/Alexa 568 goat anti-mouse secondary antibodies and rabbit Flag/Alexa 647 goat anti-rabbit secondary antibodies. (K) Disruption of PA binding blocked the membrane association of KIF5B in MDA-MB-231 cells. (L) Western blot showed the knocking down of endogenous KIF5B and re-expression of the exogenous KIF5B and KIF5B-PA− mutant in MDA-MB-231 cells. (M-O) Wild-type KIF5B but not the KIF5B-PA− mutant rescued the cellular defects in MT1-MMP plasma membrane localization (M), invadopodia formation (N), and invasion (O), in MDA-MB-231 cells expressing a KIF5B shRNA. n=3. Pictures in A, B, K and M, were taken by confocal microscopy (scale bars = 10 μm). Pictures in C, F and J, were taken by N-SIM super-resolution microscopy (scale bars = 1 μm). Quantifications are presented as mean ± SD; t-test, **p < 0.01, ***p < 0.001, NS (p> 0.05). See also Figure S6.
PIPKIs bind to the effectors of PI4,5P2 to assure localized and highly specific PI4,5P2 functions (Choi et al., 2015). To determine if PLD2 also binds to KIF5B like PIPKIs, we performed a co-immunoprecipitation (co-IP) experiment. Endogenous KIF5B co-IPed with endogenous PLD2 (Figure 6H). Vice versa, PLD2 co-IPed with GFP-KIF5B (Figure 6I). Furthermore, co-localization study using super-resolution microscopy revealed that KIF5B and PLD2 are co-localized on MT1-MMP-positive vesicles (Figure 6J).
To directly evaluate PA regulation of KIF5B, we generated a KIF5B PA-binding deficient mutant (KIF5B-PA−) by mutating all of the lysine and arginine residues to alanine on the A5 site identified above (Figures 5F–H). Like the endogenous KIF5B, GFP-KIF5B was localized to vesicles; however, GFP-KIF5B-PA− completely lost vesicle association (Figure 6K). The discrepancy between the localization of wild-type KIF5B in PLD2 deficient cells and KIF5B-PA− is likely because PLD2 inhibitor or knockout only inhibits PA production from PLD2 but not the other PA-generating enzymes whereas KIF5B-PA− completely loses PA binding.
We next examined the functional importance of the PA-KIF5B interaction. Knockdown of KIF5B in MDA-MB-231 cells by an shRNA phenocopied the cellular defects related to cell invasion observed in PLD2-deficient cells, i.e., loss of MT1-MMP plasma membrane localization, reduced invadopodia formation and invasion (Figures 6M–O). Importantly, all of these defects were restored by the expression of an RNAi-resistant wild-type KIF5B, but not by the KIF5B-PA− (Figures 6M–O). Together, these data support a model that binding to PLD2-generated PA couples KIF5B to vesicles carrying MT1-MMP for its anterograde trafficking to the plasma membrane to promote breast cancer cell invasion.
DISCUSSION
PLD2 regulates breast tumor lung metastasis in the MMTV-Neu transgenic mouse model
The current understanding of PLD in cancer is mainly based on studies in cancer cell lines (Bruntz et al., 2014; Frohman, 2015). The two PLD family members, PLD1 and PLD2, mostly function in distinct manners. Studies in Pld1 knockout mice suggest that PLD1 activity is required for both tumor growth and metastasis. Xenografts generated by implantation of wild-type cancer cells into Pld1−/− mice grew slowly and exhibited reduced angiogenesis in a hypoxic tumor environment (Chen et al., 2012). In addition, aberrant interaction of the tumor cells with the Pld1−/− platelets also reduced lung metastasis of intravenously injected melanoma cells (Chen et al., 2012). In ApcMin/+ and azoxymethane/dextran sodium sulfate mice models, genetic and pharmacological targeting of PLD1 disrupted spontaneous and colitis-associated intestinal tumorigenesis by reducing self-renewal capacity of colon cancer–initiating cells and intestinal tumorigenesis (Kang et al., 2015). The role of PLD2 in cancer has not been reported in genetically modified mouse models. Using the genetically modified MMTV-Neu breast cancer model, we demonstrated that PLD2 is dispensable for tumorigenesis and growth of breast tumors, but is critical for lung metastasis (Figure 1). The impairment of metastasis is correlated with reduced invadopodia formation and invasion in PLD2-deficient cancer cells (Figures 2 and 3). Taken together, these results demonstrate that PLD1 and PLD2 promote tumor progression through distinct mechanisms. PLD1 is critical for both cancer and stromal cells (Chen et al., 2012; Kang et al., 2015), whereas PLD2-dependent cancer metastasis is intrinsic to cancer cells.
It remains unknown whether the expression level of PLD2 protein is directly associated with the metastasis of breast cancer. Unfortunately, there is no reliable PLD2 antibody for immunohistochemical staining. It is possible that the catalytic activity of PLD2 can be activated by a range of mechanisms, some of which do not required increased expression levels of mRNA and protein. Development of assays that can measure the activated status of PLD2, e.g., an antibody that can measure the posttranslational modification associated with PLD2 activation, would be of great help to access the correlation of the activity of PLD2 and metastasis in human breast cancer patients.
PLD2-regulated plasma membrane localization of MT1-MMP is critical for invadopodia formation and invasion
The precisely controlled trafficking of surface molecules to and from the plasma membrane is a fundamental process of migrating and invading cells (Caswell and Norman, 2008). Many studies have demonstrated the important role of invadopodia and MT1-MMP in local matrix degradation during cell invasion. Our data show that both PLD2 inhibitor and genetic knockout block the plasma membrane targeting of MT1-MMP (Figure 4), and PLD2-controled MT1-MMP plasma membrane localization is critical for invadopodia formation, matrix degradation, tumor cell invasion, and metastasis (Figures 1–4). The PLD2 deficiency phenotype in our study is very similar to that in the xenograft mouse model implanted with MDA-MB-231 cells expressing a MT1-MMP shRNA, e.g., reduced lung metastasis but having minimal effect on tumor growth (Perentes et al., 2011). Interestingly, inhibition of PLD2 only alters the plasma membrane localization of MT1-MMP, and has no apparent effect on the localization of EGFR, NEU/ERBB2, and integrin β1 (Figures and S3), suggesting that the molecular machinery governing the membrane trafficking of MT1-MMP vesicles is different from other surface proteins. Elucidating the underlying mechanisms through which different membrane trafficking regulators selectively couple to certain cargoes may provide insights into selectively targeting a particular route of membrane trafficking in cancer cells, therefore reduce the toxicity caused by inhibition of the general membrane trafficking routes. Given the importance of MT1-MMP in tumor cell invasion, there has been a great interest in targeting this enzyme with inhibitors. However, no MMP inhibitor has been successfully used in clinic yet (Vandenbroucke and Libert, 2014). Our results suggest an alternative strategy of preventing invasion and metastasis by blocking the delivery of MT1-MMP to invasive structures.
PA binding couples KIF5B to its cargoes
It has been generally believed that the kinesin-1 family is committed to multiple cargoes indirectly by recognizing scaffold or adaptor proteins on the cargo surface, such as fasciculation and elongation protein-ζ1 (FEZ1) and JNK-interacting protein 1 (JIP1), as part of a protein complex (Hirokawa et al., 2009; Verhey and Hammond, 2009). However, an early study suggested that the C-terminal domain of kinesin-1 binds directly to membranes (Skoufias et al., 1994). We find that PLD2-generated PA directly binds to KIF5B and recruits it to MT1-MMP vesicles (Figures 5 and 6). It is therefore plausible to hypothesize that different cargoes are selectively linked to kinesin-1 through different mechanisms, e.g., indirect binding of adaptor protein, direct binding of PA and/or other phospholipids, or both. PA can be generated at various subcellular compartments by different PLD and DGK family members (Bruntz et al., 2014; Frohman, 2015; Shulga et al., 2011); and these spatially segregated PA may selectively couple KIF5B to different cargoes for its diverse functions (Hirokawa et al., 2009; Verhey and Hammond, 2009). This concept is supported by the fact that PLD2 deficiency only disrupted the association of KIF5B to certain vesicles, whereas the PA-binding deficient mutant completely lost the vesicle association (Figure 6). Other motor proteins, such as myosin-1b and myosin-VI, were also found to bind PA in our liposome pulldown screen (Figure 5B), suggesting that PA binding may function as a general mechanism for the attachment of membranes or vesicles to motor proteins. It is worth noting that certain phosphoinositide species, such as PI4,5P2 and PI3,4,5P3, have been reported to couple vesicles to motor proteins (McConnell and Tyska, 2010; Sweeney and Houdusse, 2010). However, PA has not been shown to perform such a role before. The requirement of different phospholipids for different motor proteins might be a way to assure the specific recruitment of selected cargoes to certain motor proteins. Further investigation of PA binding of other motors identified in our screen (Figure 5B) will strengthen our concept that PA regulation of motor protein is a general mechanism of regulating membrane trafficking, cell motility, and invasion.
PLD2 functions downstream of ARF6 to regulate actin cytoskeleton, endocytic recycling and exosome biogenesis (Ghossoub et al., 2014; Honda et al., 1999). Our current study (Figures 6E–G) supports the previous finding that ARF6 is not required for the recruitment of KIF5B to MT1-MMP vesicles (Castro-Castro et al., 2016; Marchesin et al., 2015). Therefore, the recruitment of KIF5B to MT1-MMP on late endosomes/lysosomes for its recycling is a PLD2-dependent but ARF6-independent process, which is different from the biogenesis of syntenin exosomes and HIV-1 budding. The ARF6-PLD2 pathway regulates the biogenesis of syntenin exosomes by promoting the budding of intraluminal vesicles (ILVs) into multivesicular bodies (MVBs), whereas it has no effect on the budding of HIV-1 (Ghossoub et al., 2014). These results suggest that the dependence of ARF6 and PLD2 on either sorting into ILVs for secretion or remaining on endosomes or lysosomes for recycling may depend on individual cargoes or cellular contexts. One mechanism may be through PA binding. It has been shown that direct interaction between syntenin and PA supports the sorting of syntenin to PA-enriched lipid microdomains that is critical to the budding of ILVs into MVBs (Ghossoub et al., 2014). In contrast, the recycling of MT1-MMP depends on PLD2 and PA binding with the motor protein KIF5B, rather than MT1-MMP itself. The specific roles of ARF6 and PLD2-PA in the determination of trafficking routes for different cargoes remain further clarification.
In conclusion, we demonstrate a critical role of PLD2-regulated membrane trafficking process in the invasion and metastasis of breast cancer cells. The direct bindings between KIF5B and PLD2-generated PA couple the cargo carrying MT1-MMP to the microtubule for its plasma membrane transportation. KIF5B also binds to PLD2, which might increase efficiency and specificity of the recruitment of KIF5B to MT1-MMP vesicles. Proper plasma membrane localization of MT1-MMP regulated by the PA-KIF5B interaction is required for the invadopodia formation, invasion, and the lung metastasis of breast cancer cells. PLD2 knockout mice are developmentally normal (Oliveira et al., 2010), and PLD2-isoform specific inhibitors have not been associated with off-target or toxic effect in mice (Frohman, 2015), strongly supporting that PLD2 may be a target for treating metastatic breast cancer patients.
STAR METHODS
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-CD31 (1:200 for IF) | Abcam | Cat# AB28364; RRID:AB_726362 |
| Rabbit anti-CD68 (1:200 for IHC) | Abcam | Cat# AB125212; RRID:AB_10975465 |
| Rabbit anti-GST (1:500 for WB) | Abcam | Cat# AB111947; RRID:AB_10861266 |
| Mouse anti-Kinesin Heavy Chain, KIF5B (IgM) (1:500 for IF) | Abcam | Cat# AB9097; RRID:AB_2132377 |
| Rabbit anti-KIF5B (1:1000 for WB) | Abcam | Cat# AB167429 |
| Mouse anti-KIF5B (1:200 for IF in N-SIM super resolution microscopy) | DSHB | Cat# SUK 4-c |
| Rabbit anti-MMP14 (MT1-MMP) (1:200 for IF, 1:100 for FACS) | Abcam | Cat# AB38971; RRID:AB_776422 |
| Rabbit anti-ARF6 (1:500 for WB) | EMD Millipore | Cat# MABN1170 |
| Mouse anti-AKT (1:1000 for WB) | Cell Signaling | Cat# 2920; RRID:AB_1147620 |
| Rabbit anti-pAKT (Thr308) (1:1000 for WB) | Cell Signaling | Cat# 2965; RRID:AB_2255933 |
| Rabbit anti-pAKT (ser473) (1:1000 for WB) | Cell Signaling | Cat# 4060; RRID:AB_2315049 |
| Rabbit anti-cleaved caspase-3 (1:100 for IHC) | Cell Signaling | Cat# 9661; RRID:AB_2341188 |
| Rabbit anti-EGFR (1:500 for IF, 1:1000 for WB) | Cell Signaling | Cat# 4267; RRID:AB_1554875 |
| Rabbit anti-pEGFR (Tyr1068) (1:1000 for WB) | Cell Signaling | Cat# 3777; RRID:AB_2277657 |
| Rabbit anti-p-ERBB2 (Tyr1248) (1:1000 for WB) | Cell Signaling | Cat# 2247; RRID:AB_331725 |
| Rabbit anti-Ki67 (1:400 for IF or IHC) | Cell Signaling | Cat# 12202; RRID:AB_2620142 |
| Rabbit anti-pSRC (Tyr416) (1:500 for WB) | Cell Signaling | Cat# 6943; RRID:AB_10860257 |
| Rabbit anti-Flag (1:1000 for WB, 1:300 for IF) | Cell Signaling | Cat# 14793; RRID:AB_2572291 |
| Rabbit anti-ERBB2 (1:1000 for IF, 1:2000 for WB) | Epitomics | Cat# 2064-1; RRID:AB_991707 |
| Mouse anti-GFP (1:200 for IP, for WB, 1:1000 for WB) | Thermo Fisher | Cat# MA5-15256; RRID:AB_10979281 |
| Rabbit anti-PLD1 (1:500 for WB) | Abcam | Cat# AB68150; RRID:AB_10621407 |
| Rabbit Anti-PLD2 (#226) (1:200 for IP) | Dr. Yoshiko Banno | N/A |
| Rabbit Anti-PLD2 (1:500 for WB) | Cell Signaling | Cat# 13904 |
| Rabbit Anti-TKS5 (1:100 for IF, 1:500 for WB) | EMD Millipore | Cat# 09-403; RRID:AB_1587226 |
| Mouse anti-Cortactin (1:200 for IF) | Santa Cruz | Cat# SC-55579; RRID:AB_831187 |
| Rabbit anti-Cortactin (1:500 for WB) | Cell Signaling | Cat#3502S; RRID:AB_10693785 |
| Mouse anti-Integrin β1 (1:200 for IF) | Santa Cruz | Cat# SC-18887; RRID:AB_627006 |
| Mouse anti-alpha tubulin (1:5000 for WB) | Sigma-Aldrich | Cat# T5168; RRID:AB_477579 |
| Mouse anti-MAPK-YT (activated ERK, 1:40000 for WB) | Sigma-Aldrich | Cat# M9692; RRID:AB_260729 |
| Rabbit anti-MAPK (ERK, 1:40000 for WB) | Sigma-Aldrich | Cat# M5670; RRID:AB_477216 |
| Mouse anti-SRC (1:500 for WB) | Upstate | Cat# 05-184; RRID:AB_2302631 |
| Alexa Fluor 488, goat anti-rabbit IgG (H+L) (1:300 for IF) | Thermo Fisher | Cat# A-11034; RRID:AB_2576217 |
| FITC, goat anti-rabbit IgG for FACS (1:400 for FACS) | Thermo Fisher | Cat# F-2765; RRID:AB_2536525 |
| Alexa Fluor 594, goat anti-mouse IgM (1:500 for IF) | Thermo Fisher | Cat# A-21044; RRID:AB_2535713 |
| Alexa Fluor 488, goat anti-mouse IgM (1:500 for IF) | Thermo Fisher | Cat# A-21042; RRID:AB_2535711 |
| Alexa Fluor 568, goat anti-mouse IgG (H+L) (1:300 for IF) | Thermo Fisher | Cat# A-11004; RRID:AB_2534072 |
| Alexa Fluor 647, goat anti-rabbit IgG (H+L) (1:300 for IF) | Thermo Fisher | Cat# A-21244; RRID:AB_2535812 |
| Alexa Fluor 680, goat anti-rabbit IgG (H+L) (1:5000 for WB) | Thermo Fisher | Cat# A-21076; RRID:AB_2535736 |
| IRDye 800CW, goat anti-mouse IgG (H+L) (1:5000 for WB) | LI-COR | Cat# 92632210; RRID:AB_621842 |
| Bacterial and Virus Strains | ||
| Rosetta™ 2 E. coli strain | EMD Millipore | Cat# 71400 |
| NEB Stable | New England BIolabs | Cat# C3040H |
| Chemicals, Peptides, and Recombinant Proteins | ||
| PLD1 inhibitor (VU0359595) | Avanti Polar Lipids | Cat# 857371P |
| PLD2 inhibitor (VU0285655-1) | Avanti Polar Lipids | Cat# 857372P |
| Protease inhibitor cocktail | Roche | Cat# 05892970001 |
| MESNA (2-Mercaptoethane Sulfonic Acid Sodium Salt) | MP Biomedicals, Inc | Cat# 0215160205 |
| Gelatin | Sigma-Aldrich | Cat# G2500 |
| IPTG | Sigma-Aldrich | Cat# I6758-10G |
| Poly-HEMA | Sigma-Aldrich | Cat# 192066-10G |
| Collagenase A | Sigma-Aldrich | Cat# C1639-50MG |
| Streptavidin Agarose | Sigma-Aldrich | Cat# S1638 |
| Fetal bovine serum (FBS) | Thermo Fisher | Cat# 10437-036 |
| Alexa Fluor 488, phalloidin (1:300) | Thermo Fisher | Cat# A-12379 |
| Dylight 594, phalloidin (1:300) | Cell Signaling | Cat# 12877 |
| Alexa Fluor 647, phalloidin (1:300) | Thermo Fisher | Cat# A-22287 |
| DAPI (4′,6-diamidino-2-phenylindole) (50 ng/ml) | Thermo Fisher | Cat# D-1306 |
| PLUS Reagent | Thermo Fisher | Cat# 10964-021 |
| Lipofectamine Reagent | Thermo Fisher | Cat# 50470 |
| Pierce ECL Western Blotting Substrate | Thermo Fisher | Cat# 32106 |
| Odyssey blocking buffer | LI-COR | Cat# 927-40000 |
| Harris Hematoxylin With Glacial Acetic Acid | Poly Scientific R&D Corp | Cat# S212A-1GL |
| Eosin Phloxine Alcoholic Working Solution | Poly Scientific R&D Corp | Cat# S176-1GL |
| EZ-Link™ Sulfo-NHS-SS-Biotin | Thermo Scientific | Cat# PI21328 |
| Glutathione Agarose | Thermo Scientific | Cat# 16100 |
| Protein A/G PLUS-Agarose | Santa Cruz | Cat# SC-2003 |
| Nuclepore Track-Etch Membrane (pore-size: 100nm) | Whatman | Cat# 110605 |
| Nitrocellulose Membrane, 0.2 μm | Bio-Rad | Cat# 1620112 |
| Critical Commercial Assays | ||
| NEBuilder HiFi DNA Assembly Cloning Kit | NEB | Cat# E5520 |
| Matrigel Invasion Chambers | Corning | Cat# 354480 |
| DyLight 488 Labeling Kit | Thermo Scientific | Cat# 53024 |
| DyLight 594 Labeling Kit | Thermo Scientific | Cat# 53044 |
| Deposited Data | ||
| N/A | ||
| Experimental Models: Cell Lines | ||
| MDA-MB-231 | ATCC | Cat# HTB-26 |
| MCF-7 | ATCC | Cat# HTB-22 |
| TLA-HEK293T | GE Healthcare BioSciences | Cat# HCL4517 |
| Experimental Models: Organisms/Strains | ||
| MMTV-Neu transgenic mice (FVB/N-Tg(MMTVneu)202Mul/J) | the Jackson Laboratory | Cat# 002376 |
| Pld2+/− in the C57BL/6J-129/SvJ background | (Oliveira et al., 2010) | N/A |
| Oligonucleotides | ||
| See Table S4 | ||
| Recombinant DNA | ||
| pMDLg/pRRE | Addgene | Cat# 12251 |
| pRSV-Rev | Addgene | Cat# 12253 |
| pMD2.G | Addgene | Cat# 12259 |
| LentiCas9-Blast | Addgene | Cat# 52962 |
| LentiGuide-Puro | Addgene | Cat# 52963 |
| KIF5B-shA in pLKO vector | Sigma-Aldrich | Cat# TRCN0000113874 |
| KIF5B-shE in pLKO vector | Sigma-Aldrich | Cat# TRCN0000338652 |
| ARF6-sh1301 in pLKO vector | Sigma-Aldrich | Cat# TRCN0000048005 |
| ARF6-sh1302 in pLKO vector | Sigma-Aldrich | Cat# TRCN0000048003 |
| pQCXIP-MT1-MMP-GFP | Dr. Jian Cao | N/A |
| human KIF5B cDNA | Dr. Paulo A Ferreira | N/A |
| pGEX-4T-1-KIF5B-C wt, A1, A2, A3, A4, A5, and A4/A5 mutants | This paper | N/A |
| pCDH-KIF5B-GFP | This paper | N/A |
| pCDH-KIF5B-PA−-GFP | This paper | N/A |
| pCDH-KIF5B wobble mutant (no tag) | This paper | N/A |
| pCDH-KIF5B-PA− wobble mutant (no tag) | This paper | N/A |
| pHAGE-ARF6 | This paper | N/A |
| pHAGE-ARF6-N48R | This paper | N/A |
| LentiGuide-PLD2-puro | This paper | N/A |
| pLKO.1-Luc shRNA | (Cai et al., 2016) | N/A |
| pLKO.1-PLD1-599 shRNA | (Cai et al., 2016) | N/A |
| pLKO.1-PLD1-2478 shRNA | (Cai et al., 2016) | N/A |
| Software and Algorithms | ||
| Other | ||
| 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) | Avanti Polar Lipids | Cat# 850375C |
| 1,2-didecanoyl-sn-glycero-3-phospho-L-serine (DOPS) | Avanti Polar Lipids | Cat# 840036C |
| 1,2-dioleoyl-sn-glycero-3-phosphate (DOPA) | Avanti Polar Lipids | Cat# 840875C |
| 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) | Avanti Polar Lipids | Cat# 850725C |
| Phosphatidylinositol-4,5-bisphosphate (PI(4,5)P2) | Avanti Polar Lipids | Cat# 850155P |
| D-(+)-sn-1-O-oleoyl-glyceryl-3-phosphate (Oleoyl LPA) | Echelon | Cat# L0181 |
| Sphingosine 1-Phosphate (S1P) | Echelon | Cat# S2000 |
| Phosphatidylinositol diC16 (PI) | Echelon | Cat# P0016 |
| Phosphatidylinositol 3-phosphate diC16 [PI(3)P] | Echelon | Cat# P3016 |
| Phosphatidylinositol 3,4-bisphosphate diC16 [PI(3,4)P2] | Echelon | Cat# P3416 |
| Phosphatidylinositol 3,5-bisphosphate diC16 [PI(3,5)P2] | Echelon | Cat# P3516 |
| Phosphatidylinositol 4-phosphate diC16 [PI(4)P] | Echelon | Cat# P4016 |
| Phosphatidylinositol 5-phosphate diC16 [PI(5)P] | Echelon | Cat# P5016 |
| 1-Palmitoyl-2-hydroxy-sn-glycero-3-phosphocholine (LPC) | Echelon | Cat# L1516 |
| Phosphatidylinositol-tris-3,4,5-phosphate [PI(3,4,5)P3] | Matreya LLC | Cat# 1775-1 |
| 5 μm magic C18AQ; pore size, 200 Å | Michrom Bioresources | Cat# c18aq-5μm-200A |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Guangwei Du (guangwei.du@uth.tmc.edu).
Experimental Model and Subject Details
Mouse Strains and Genotyping
All mouse experiments were performed in accordance with AAALAC guidelines and with University of Texas Health Science Center Institutional Animal Care and Use Committee approval. Pld2+/− mice generated on a mixed C57BL/6J-129/SvJ background were backcrossed 10 generations to the FVB/N background prior to crossing to the MMTV-Neu transgenic mice on an inbred FVB/N background (the Jackson Laboratory, Bar Harbor, ME). MMTV-Neu;Pld2+/− mice were crossed with Pld2+/− mice to generate the females with the desired genotypes: MMTV-Neu;Pld2+/+, and MMTV-Neu;Pld2−/−, for phenotyping analyses. Genotypes of Pld2 alleles were determined by PCR analysis of genomic DNA from tail biopsy using the primers in Table S4: Pld2-F, Pld2-R, Neomycin-R. The Neu transgenes were determined by PCR using primers and conditions recommended by Jackson Laboratories.
Cell Lines and Primary Cells
Human MDA-MB-231 and MCF-7 breast cancer cells were obtained from American Type Culture Collection (ATCC; Gaithersburg, MD). TLA-HEK293T cells were obtained from GE Healthcare Bio-Sciences (Pittsburgh, PA). Primary mouse tumor cells were isolated from mouse mammary gland tumors as described before (Qin et al., 2008). In brief, individual mammary tumors were isolated and minced in Dulbecco’s modified Eagle’s medium (DMEM) with 1 mg/ml collagenase A. After overnight digestion at 37°C and resuspension, the cells were cultured in DMEM with 10% FBS. To count circulating tumor cells, cells isolated from 200 μl blood were grown on regular tissue culture dishes until individual colonies formed. Circulating tumor cells were measured as described earlier (Wang et al., 2009). In brief, 200 μl blood was extracted from each mouse using heparin-dipped syringe and added to 6–7 ml DMEM medium (containing 10% serum) immediately. The mixture was centrifuged for 3 minutes at 350 × g. The cell pellet was washed with above medium once and placed on 10-cm dish to culture for 2 days. The suspended blood cells were removed by washing with PBS twice, and tumor cells were continuous cultured for 10 days. Tumor cells started to grow out around 3rd ~7th days after seeding on the dish. The colony number was counted at 10th day. Tumor cells derived from mammary glands were verified by the expression of Neu transgene and epithelial marker cytokeratin 8 using immunofluorescent staining. All cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS).
Method Details
Analysis of Breast Tumors
Age-matched littermates were monitored for mammary tumor by weekly palpation. Tumor-free survival was measured as the date that first mammary tumor was palpable. Primary tumor burden was determined by caliper measurements once a week and the total tumor burden per mouse was calculated as the sum of the volume of each individual mammary tumor as calculated by ½ × (Length × Weight × Height). Mice were euthanized 9 weeks after the first palpable tumor, or total tumor burden reached 3 cm in the diameter. Tumors and lungs were collected for Western blot analysis or immunochemical staining after formalin fixed and paraffin embedded. The number of metastatic nodules presented on the surface of lung was counted under a dissecting microscope.
Histological and Immunofluorescent Analyses
Mammary gland tumors and lung were harvested at the indicated time points, and fixed in formalin. Paraffin embedding and sectioning of tissue samples were performed by Breast Care Center at Baylor College of Medicine. Hematoxylin and eosin (H & E) staining were performed using standard protocol. After rehydration, immunohistochemical or immunofluorescent staining of lung sections was performed for proliferation, apoptosis, macrophage infiltration, and angiogenesis using Ki67, capspase-3, CD68, and CD31, respectively. The results were visualized by the incubation of fluorescently labeled secondary antibodies, or appropriate biotinylated secondary antibodies and followed by ABC staining (Vector Lab, CA). For the localization of TKS5, cortactin, MT1-MMP, KIF5B, EGFR, ERBB2, and integrin β1 in tumor sections, tumors were fixed in 4% paraformaldehyde, embedded in optimal cutting temperature compound (OCT), and cryosectioned. The same immunofluorescent staining protocol was used for both tissue sections and cultured cells as described before (Cai et al., 2016; He et al., 2017). Briefly, cells were fixed with 4% paraformaldehyde, and permeabilized with 0.1% Triton X-100 in PBS. After blocking in PBS containing 5% normal goat serum for 1 hr at room temperature, cells were stained with primary antibodies, followed by appropriate fluorescent dye-conjugated secondary antibodies. Coverslips were mounted on to slides with 4% propyl gallate mounting solution and sealed with nail polish along the edge. Unless indicated, the immunofluorescent images were captured by a Nikon A1 Confocal Laser Microscope. The invadopodia in tumor tissues were quantified by the co-localization of TKS5 and cortactin.
For super-resolution microscopy analysis of KIF5B and PLD2 association with MT1-MMP-positive endosomes, MDA-MB-231 cells stably expressing MT1-MMP-GFP and Flag-PLD2 cultured on gelatin-coated coverslips were stained as described above. Images were acquired with a Nikon’s N-SIM microscopy system using a CFI Apo TIRF 100× oil objective and a highly sensitive cooled charge-coupled device (CCD) camera (Andor Technology iXon DU897 EMCCD). To quantify the association of KIF5B to MT1-MMP-positive endosomes, the numbers of positive-stained KIF5B spots associated with MT1-MMP were counted for each cell. The total number of associated KIF5B was divided by the number of MT1-MMP positive endosomes in each cell to make an index of KIF5B and MT1-MMP endosome association. The index was normalized by setting the result from control condition to 100%.
Anoikis Assay
Primary mouse tumor cells (2×105 cells) or MDA-MB-231 cells (1×106 cells) were seeded into each well that was coated by incubating with 1 ml of 6 mg/ml poly (2-hydroxyethyl methacrylate) (pHEMA), and then dried overnight in the hood, as reported early (Brunquell et al., 2012). Cells were cultured in 10% FBS DMEM medium in the presence or absence of DMSO or PLD2 inhibitor (5 μM). The numbers of viable cells were counted by trypan blue staining at different time points.
Flow Cytometry
Mouse primary breast cancer cells were detached by incubation with PBS containing 5 mM EDTA, and then fixed with 4% paraformaldehyde for 15 min at room temperature. After washed with PBS for 3 times, cells were incubated with the anti-MT1-MMP antibody (1:100) at room temperature for 1 h, cells were washed with PBS for three times, followed by incubating with FITC conjugated goat-anti-rabbit secondary antibody (1:400) for 1 h at room temperature. After 3 times wash with PBS, at least 10,000 cells each group were collected by a LSRFortessa Cell Analyzer (BD Biosciences, San Jose, CA, USA), and the fluorescence intensity was analyzed by FlowJo software (Ashland, OR, USA).
Western Blotting
Cultured cells and mechanically disrupted tissues were lysed in cold radioimmunoprecipitation assay (RIPA) buffer containing protease and phosphatase inhibitor cocktails. After brief sonication, samples were load on SDS-polyacrylamide gels and transferred onto nitrocellulose membranes, which was blocked in 1% casein and probed with the indicated primary antibodies and fluorescently labeled secondary antibodies at the concentrations indicated in the Key Resource Table. These primary antibodies include total and/or phospho-specific antibodies for AKT, EGFR, ERBB2, SRC, ERK, Flag tag, GST tag, GFP tag, MT1-MMP, PLD1, PLD2, α-tubulin, TKS5, Cortactin, and ARF6. The blots were detected by the Li-COR Odyssey infrared imaging system from Li-COR Biotechnology (Lincoln, NE).
Invasion Assay and Analysis of Invadopodia Formation in vitro
The invasion ability of MDA-MB-231 and primary tumor cells was measured by transwell assay. 1×105 cells in 0.1% FBS medium were seeded to Matrigel-coated inserts (8 μm pore size) from BD Biosciences (Bedford, MA). Cells were allowed to invade at 37°C for overnight from the upper chamber containing 0.1% FBS to the lower chamber containing 10% FBS, in the presence or absence of the PLD2 inhibitor. The non-migrated cells were removed by cotton swabs. The migrated cells were fixed and permeabilized by cold methanol for 10 minutes, and stained with DAPI solution for 5 minutes. The numbers of migrated cells identified by DAPI-positive staining were presented as the mean cell numbers of eight different fields.
Invadopodia formation was measured using previously described method (Artym et al., 2006; Wang et al., 2016). Gelatin (Sigma, G-2500) was labeled with Dylight 488 or 594 as the previous description. Briefly, the coverslips were treated with 50 μg/mL poly-L-lysine for 20 min, washed with PBS twice, and crosslinked with 0.5% glutaraldehyde for 15 min. After extensive washing, the coverslips were inverted on a 40 μl gelatin mixture on a piece of parafilm film (1 mg/ml of fluorescently labeled gelatin was mixed with unlabeled gelatin at a 1:4 ratio) for 20 min. After washing with PBS, the coverslips were treated with 5 mg/ml sodium borohydride for 3 min to quench the autofluorescence, followed by extensively washing with PBS. Cells were seeded on the gelatin-coated coverslips in growth medium in the presence or absence of the PLD2 inhibitor or DMSO for 4 hr. After incubation, cells were fixed with 4% paraformaldehyde and stained with fluorescently conjugated phalloidin (1:300) at room temperature for 1 h. After washed 3 times with PBS, the slides were mounted on coverslips with 4% n-propyl gallate
Plasmid Construction
The bacterial expression construct carrying the C-terminus of human KIF5B (854-963aa) fused to Glutathione-S-transferase (GST) was cloned into the BamH I and Not I sites of pGEX-4T-1 to generate pGEX-4T-1-KIF5B-C by polymerase chain reaction (PCR) amplification. The pGEX-4T-1-KIF5B-C mutants in which arginine and lysine residues were mutated to alanine, A1, A2, A3, A4, and A5, were generated by PCR site-directed mutagenesis. The expression of KIF5B-EGFP in MDA-MB-231 cells was mediated by lentiviral delivery. The protein coding sequence of KIF5B was amplified from human KIF5B cDNA (Cho et al., 2009), and fused to the N-terminus of EGFP. The resulting KIF5B-EGFP cDNA fragment was then cloned into the Nhe I and Not I sites of pCDH-CMV-MCS-EF1-puro vector. The A5 mutant of KIF5B was introduced by PCR site-directed mutagenesis using the primers listed in Table S4. To prevent the degradation of exogenously expressed KIF5B and PA− (A5) mutant mRNA, wobble mutations that changed the codons but not encoded amino acids at site targeted by KIF5B-shE were introduced by PCR. The ORF of KIF5B and PA− mutant without GFP tag were amplified by PCR and cloned in the Nhe I and Not I sites of pCDH-CMV-MCS-EF1-puro vector using NEBuilder HiFi DNA Assembly Cloning Kit. pcDNA-ARF6-N48R was generated by PCR site-directed mutagenesis using pcDNA-ARF6 (mouse) as a template (Honda et al., 1999). To clone ARF6 and N48R into the lentiviral vector, they are amplified by PCR using primers ARF6-N-F and ARF6-C-R, and the cloned into pHAGE-EF1a-GAW-IRES-Hygro cut with Nhe I and EcoR I sites using NEBuilder HiFi DNA Assembly Cloning Kit. LentiGuide-PLD2-puro was generated by cloning a pair of oligos targeting tattctgtccgcttgactca in the exon 3 of PLD2 inot LentiGuide-Puro cut with BsmB I.
All primers used for molecular cloning and mutagenesis were summarized in Table S4. All plasmid constructs were confirmed by DNA sequencing.
Lentiviral Transduction
shRNA knockdown of gene expression and exogenous cDNA expression of KIF5B, ARF6 or PLD1 in MDA-MB-231 cells were mediated by lentiviral delivery. PLD2 knockout in MDA-MB-231 cells were done by lentiviral delivery of Cas9 and PLD2 small guide RNA (sgRNA) using CRISPR/Cas9 system. Viruses were generated in TLA-293T cells by co-transfection of four plasmids including the lentiviral vector carrying desired shRNAs or cDNAs, pMDLg/pRRE, pRSV-Rev, and pMD2.G, using Lipofectamine and Plus reagent from Thermo Fisher. At 48 hr and 72 hr posttransfection, virus-containing supernatants were collected for infection. The cells were used for experiments 2 to 3 days postinfection.
Surface Protein Biotinylation and Recycling Assay
Surface protein biotinylation and recycling assay were performed as described before (Gabriel et al., 2009; Remacle et al., 2003). MDA-MB-231 cells grown in a six-well plate were washed twice with ice-cold SBS buffer (14.7 mM KH2PO4, 2 mM Na2HPO4, 120 mM Sorbitol, pH 7.8) and incubated for a further 10 min in ice-cold SBS. Surface protein were labeled by incubating with 0.75 ml of freshly prepared ice-cold SBS containing 0.3 mg/ml EZ-Link NHS-SS-biotin (sulfosuccinimidyl-2-(biotinamido) ethyl-1,3-dithiopropionate) per well with shaking in cold room for 15 min. After washing with ice-cold SBS twice, cells were incubated with ice-cold SBS containing 100 mM glycine for 10 min to quench unreacted biotin. Cells were then washed twice with ice-cold SBS and incubated with growth medium at 37 °C to allow the internalization of biotinylated surface proteins. After 30 min, cells were washed with ice-cold SBS and incubated on ice for 10 min. Cells were then incubated with 150 mM of MESNA (2-mercaptoethane sulfonic acid) in ice-cold SBS (pH 8.2) for 25 minutes to remove biotin remained on the plasma membrane. After three washes with ice-cold SBS, cells were lysed in 0.2 ml/well of RIPA buffer (20 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.1% SDS, 1% Triton X-100, 1% deoxycholate) plus proteinase inhibitor cocktail. For recycling assay, MESNA treated cells were washed with ice-cold SBS and incubated in growth medium at 37°C for 15 or 30 min to allow recycling of the internalized proteins. After stopping recycling by washing the cells with ice-cold SBS and placing them on an ice for 10 min, a second MESNA treatment was performed to remove biotin on the proteins recycled back to the cell surface. Finally, the cells were washed three times with ice-cold SBS, and lysed in RIPA buffer as described above. Cell lysates in RIPA were centrifuged at 16,000 g for 10 min at 4 °C. The supernatants were then incubated with 25 μl of streptavidin agarose beads over night at 4 °C, spun down, and washed 5 times with 0.75 ml RIPA buffer. Proteins were solubilized with 30 μl 2xSDS loading buffer and detected with a rabbit MT1-MMP antibody.
Liposome Preparation
Sucrose-loaded liposomes were prepared as described previously (Roach et al., 2012; Tay et al., 2017). In brief, 1,2-dioleoyl-sn-glycero-3-phosphocholine (PC), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (PE), and 1,2-dioleoyl-sn-glycero-3-phosphate (PA), 1,2-didecanoyl-sn-glycero-3-phospho-L-serine (PS), phosphatidylinositol 4,5-bisphosphate [PI(4,5)P2] or Phosphatidylinositol 3, 4, 5-trisphosphate [PI(3,4,5)P3], were mixed at a molar ratio of 6:3:1 for screening for PA-binding proteins, or 6.5:3:0.5 for purified KIF5B-C. After dried by a vacuum, the lipid mixtures were re-suspended with the insider buffer (256 mM sucrose, 20 mM Tris HCl, pH7.4), and subjected to 10 cycles of freezing in liquid nitrogen and thawing in 37°C water bath. The formed multilamellar lipids were extruded through polycarbonate membranes (pore size 100 nm, Whatman) for 10 times to generate small unilamellar liposomes. Liposomes were washed with the binding buffer (150 mM NaCl, 20 mM Tris, pH7.4) and centrifuged at 70,000× g for 1 hr, the pelleted liposomes were re-suspended with the binding buffer and used within 1 week. The buffers used for the purified GST-KIF5B were: inside buffer (471 mM sucrose, 20 mM Tris, pH7.4) and binding buffer (300 mM NaCl, 20 mM Tris, pH7.4).
Identification of PA-binding Proteins by Liposome Pulldown and Mass Spectrometry
The cytosolic and peripheral proteins were prepared from MCF7 breast cancer cells using alkaline carbonate extraction (100 mM Na2CO3, pH11.5) (Fujiki et al., 1982), and were dialyzed with the liposome binding buffer plus 2 mM dithiothreitol (DTT) prior to incubating with liposomes for 1 hr. The bindings to each type of liposomes were performed in duplicate. Proteins bound to liposomes were recovered by centrifugation at 70,000xg for 30 min, and separated by SDS-PAGE. After staining with Coomassie blue, the whole gel lane of each sample was excised to multiple slices. The gel slices were individually digested with trypsin, and the resulting peptides were analyzed by reverse-phase liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) on a LTQ-Orbitrap mass spectrometer (Thermo Finnigan, San Jose, CA) as previously described (Xu et al., 2009). Briefly, the protein samples were run on an SDS gel. The whole gel lane was excised into gel slices, followed by standard in-gel digestion with trypsin. The resulting peptides were extracted, dried and dissolved in sample loading buffer [6% acetic acid, 0.1% trifluoroacetic acid (TFA)]. The peptides were loaded on a 75 μm I.D. × 12 cm fused-silica capillary column (5 μm magic C18AQ; pore size, 200 Å; Michrom Bioresources), and eluted by a gradient [buffer A, 0.4% acetic acid, 0.005% heptafluorobutyric acid (HFBA), and 5% acetonitrile; buffer B, 0.4% acetic acid, 0.005% HFBA, and 95% acetonitrile]. The eluted peptides were analyzed by a hybrid LTQ-Orbitrap MS (Thermo Scientific). The MS settings included MS1 scan (60,000 resolution and 1 ×106 for automatic gain control) and Top10 MS2 scans (isolation width of 2 m/z, 35% normalized collision energy, 5,000 for automatic gain control, 60 sec dynamic exclusion, preview mode enabled with selection of 2+, 3+, and 4+ ions). MS/MS spectra were searched against a concatenated protein target-decoy human reference database for the identification of proteins (Wang et al., 2014). The search results were filtered to reduce protein false discovery rate to less than 0.1%. If matched peptides were shared by a family of proteins, these proteins were clustered into one group. Based on the principle of parsimony, the group was represented by the protein with the highest number of peptides.
Label-free quantification was performed based on spectral counts of identified proteins, and G-test analysis was used to evaluate statistical significance as previously reported (Bai et al., 2013). Proteins were quantified in analyzed samples by detected spectral counts (SC). First, the spectral counts were normalized between samples to allow that the average SC per protein was the same in all datasets. Then G-test was used to judge statistical significance of protein abundance difference. The G-value of each protein was calculated by equation: G = 2 × (S1 × ln[S1 ÷ ((S1+S2)÷2)] + S2 × ln[S2 ÷ ((S1+S2)÷2)]), where S1 and S2 were the spectral counts of a given protein in any of two comparative samples, and “ln” was the natural logarithm. The G values approximately fit to the χ2 distribution (one degree of freedom), enabling the calculation of related p-values. False discovery rate was analyzed by null experiments (e.g. comparison of replicates under the same conditions). Finally, a threshold with balanced sensitivity and specificity was selected to filter data with false discovery rate of ~5%, resulting in the acceptance of 63 PA-binding proteins. The classification of these proteins into different functional groups was done by searching DAVID bioinformatics and PANTHER databases (Huang da et al., 2009; Mi et al., 2010) and manual annotation.
Bacterial Protein Expression and Purification
Rosetta™ host bacteria derived from BL21 carrying pGEX-4T-1-KIF5B-C wild-type or mutants were cultured with 100μg/ml ampicillin at 37°C till the optical density (OD) at wavelength of 600 reached 0.6, and then induced for protein expression with 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) at 18°C overnight. After centrifuged at 5,000 rpm and re-suspended in buffer A (50 mM Tris, 150 mM NaCl, pH8.0, 0.01% Triton X-100, 4 mM DTT, plus protease inhibitor cocktail), the bacteria were lysed by French pressure cell press. The supernatants from centrifugation at 18,000 rpm for 30 minutes were incubated with the glutathione agarose (Thermo Fisher) for 1 hr at 4°C. The protein-agarose mixture was loaded onto a plastic chromatography column and washed with buffer A for 3 times. The protein was eluted by the elution buffer (50 mM Tris, 150 mM NaCl, pH8.0, 10 mM reduced glutathione, 0.01% Triton X-100) and dialyzed witthe binding buffer (300 mM NaCl, 20 mM Tris, pH7.4). The purity of the purified proteins was examined by SDS-PAGE and Coomassie blue staining.
Protein-lipid Overlay Assay
Lipid strip was prepared by spotting 100 pmol of each lipid on a nitrocellulose membrane (0.2 μm, Bio-Rad). The strip was blocked with 5% fatty acid-free BSA in TBST for 2 hrs, followed by incubating with the purified GST-KIF5B-C proteins at the concentration of 1μg/ml in blocking buffer overnight. The lipid strip was then incubated with a rabbit anti-GST antibody for 1 hr and a horseradish peroxidase-conjugated goat anti-rabbit IgG for 1 hr. After, 3 washes with TBS, the lipid-bound protein was visualized with the exposure to X-ray film using enhanced chemiluminescence kit from Thermo Fisher.
Characterization of Phospholipid Binding of the Recombinant KIF5B-C using Liposome Pulldown Assay
0.4 μg purified KIF5B-C proteins were mixed and incubated with the serial diluted liposomes at 4°C for 30 minutes. After centrifuge at 70,000xg for 30 minutes, the supernatants were carefully removed. The pellets were resuspended with 1 × SDS loading buffer and boiled for 5 minutes, and then separated by SDS-PAGE. The binding ability of KIF5B-C wild-type and mutants with liposomes was detected by immunoblotting against an anti-GST antibody.
Co-immunoprecipitation
For immunoprecipitation analysis, GFP-KIF5B was expressed in MDA-MB-231 cells. MDA-MB-231-GFP-KIF5B or parental MDA-MB-231 cells were lysed on ice with lysis buffer (50 mM Tris-HCl, pH 7.6, 150 mM NaCl, 10% glycerol, 1% Triton-X100 and protease inhibitor cocktail) for 30 min and centrifuged at 16,000 g for 15 min at 4°C. The supernatants were incubated with anti-PLD2 antibody (#226) (1:200) or anti-GFP antibody (MA5-15256, Thermo Fisher) (1:200) for 2h at 4°C. Then protein A/G PLUS-agarose was added and rotated for 2h at 4°C. After the mixture was centrifuged at 400 × g for 30 seconds, the supernatant was separated as an input (whole cell lysate) and the beads were washed 4 times with lysis buffer. The immune complexes were then eluted in 2X SDS sample buffer and analyzed by SDS-PAGE and immunoblotting with indicated antibodies.
PI4,5P2 Analysis
The analysis of PI4,5P2 was done as described (Berman et al., 2008; McIntire et al., 2012). Briefly, lipids were extracted with cells grown in 100-mm dishes wer scraped in 1 ml of ice-cold chloroform:methanol:10N HCl (20:40::1) supplemented with 2 mM AlCl3, and transferred to 2-ml polypropylene tubes. Tubes were vortexed for 1 min for 3 times with 5 min interval on ice. After adding 0.3 ml chloroform and 0.5 ml ice-cold water, tubes were vortexed for 10 s twice and spun for 10 min at 4000 rpm at 4°C. The lower solvent phases were collected and transferred to new tubes containing 1 ml ice-cold methanol:2mM oxalic acid in water (1:0.9). After vortexing for 1 min, tubes were spun for 2 min at10000 rpm at 4°C. The lower solvent phases were transferred into new tubes, and dried with a speed vacuum. Lipids were then resuspended and deacylated by incubation with 0.5 ml of 40% methylamine:water:n-butanol:methanol (36:8:9:47) at 50°C for 45 min. The aqueous phase was dried with a speed vacuum, and then resuspended in 0.5 ml of n-butanol:petroleum ether:ethyl formate (20:40:1), and extracted twice with an 0.5 ml of water. Aqueous extracts were dried, resuspended in water, and subjected to anion-exchange HPLC on an Ionpac AS11-HC column (Dionex, Sunnyvale, CA). Negatively charged glycerol head groups were eluted with 1.5–86 mM KOH gradient and detected by suppressed conductivity in a Dionex ion chromatography system equipped with an ASRS-ultra II self-regenerating suppressor. Individual peaks were identified and peak areas calculated using Chromeleon software (Dionex). Lipid masses were calculated and expressed as molar fractions of total anionic phospholipids present in the sample.
Statistics
The statistical differences were evaluated between control and each of the treatments using two-tailed Student’s t test. All data are shown as mean ± standard deviation (SD). *, p < 0.05, **; p < 0.01; and ***, p < 0.001.
Supplementary Material
Table S1, Related to Figures 5. List of PA- and PS-associated proteins identified by label-free proteomics analysis.
Table S2, Related to Figures 5. False Discovery Rate (FDR) analysis based on different p values derived from label-free quantification.
Table S3, Related to Figures 5. Proteins specifically or preferentially bound to PA-liposome revealed by label-free proteomics analysis.
HIGHLIGHTS.
The lung metastasis of breast cancer cells is dependent on PLD2
PLD2 promotes invadopodia formation
PLD2-generated phosphatidic acid binds to and regulates the motor protein KIF5B
The PLD2-PA-KIF5B signaling controls MT1-MMP surface localization and invasion
Acknowledgments
We are grateful to the human KIF5B cDNA provided by Dr. Paulo A Ferreira at Duke University. PLD2 rabbit polyclonal antibody was shared by Dr. Yoshiko Banno at Gifu University Graduate School of Medicine. pQCXIP-MT1-MMP-GFP was from Dr. Jian Cao at Stony Brook University. pLKO.1-ARF6 shRNAs are from Dr. Philippe Chavrier at Institut Curie. W.N. is recipient of a fellowship from the China Scholarship Council. This work was supported by grants from the Cancer Prevention and Research Institute of Texas (CPRIT) (RP130425 and RP160775 to G.D.), National Institutes of Health (R01HL119478 to G.D., R01GM114260 to J.P., and R01CA112403 and R01CA193455 to J.X). The authors claim no conflicts of interest.
Footnotes
AUTHOR CONTRIBUTIONS
Conceptualization, Z.W, F.Z., and G.D.; Methodology, Z.W, F.Z., P.W., and G.D.; Investigation, Z.W, F.Z., P.W., J.H., L.W.R.T., M.C., N.W., Y.W., L.Q., J.T.C, L.B.M, J.P., and G.D; Resources, G.D.P., and J.X.; Formal Analysis, Z.W, F.Z., J.T.C, J.P. and G.D.; Writing, Z.W, F.Z., G.D.P., and G.D.
Supplemental Information includes a PDF file including 6 figures and Table S4 (DNA oligos used for plasmid constructs and genotyping), and 3 excel tables (S1–S3).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Artym VV, Zhang Y, Seillier-Moiseiwitsch F, Yamada KM, Mueller SC. Dynamic interactions of cortactin and membrane type 1 matrix metalloproteinase at invadopodia: defining the stages of invadopodia formation and function. Cancer Res. 2006;66:3034–3043. doi: 10.1158/0008-5472.CAN-05-2177. [DOI] [PubMed] [Google Scholar]
- Bai B, Hales CM, Chen PC, Gozal Y, Dammer EB, Fritz JJ, Wang X, Xia Q, Duong DM, Street C, et al. U1 small nuclear ribonucleoprotein complex and RNA splicing alterations in Alzheimer’s disease. Proc Nat Acad Sci USA. 2013;110:16562–16567. doi: 10.1073/pnas.1310249110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman DE, Dall’Armi C, Voronov SV, McIntire LB, Zhang H, Moore AZ, Staniszewski A, Arancio O, Kim TW, Di Paolo G. Oligomeric amyloid-beta peptide disrupts phosphatidylinositol-4,5-bisphosphate metabolism. Nat Neurosci. 2008;11:547–554. doi: 10.1038/nn.2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blouw B, Patel M, Iizuka S, Abdullah C, You WK, Huang X, Li JL, Diaz B, Stallcup WB, Courtneidge SA. The invadopodia scaffold protein Tks5 is required for the growth of human breast cancer cells in vitro and in vivo. PLoS One. 2015;10:e0121003. doi: 10.1371/journal.pone.0121003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunquell C, Biliran H, Jennings S, Ireland SK, Chen R, Ruoslahti E. TLE1 is an anoikis regulator and is downregulated by Bit1 in breast cancer cells. Mol Cancer Res. 2012;10:1482–1495. doi: 10.1158/1541-7786.MCR-12-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruntz RC, Lindsley CW, Brown HA. Phospholipase D signaling pathways and phosphatidic acid as therapeutic targets in cancer. Pharmacol Rev. 2014;66:1033–1079. doi: 10.1124/pr.114.009217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai M, He J, Xiong J, Tay LW, Wang Z, Rog C, Wang J, Xie Y, Wang G, Banno Y, et al. Phospholipase D1-regulated autophagy supplies free fatty acids to counter nutrient stress in cancer cells. Cell Death Dis. 2016;7:e2448. doi: 10.1038/cddis.2016.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro-Castro A, Marchesin V, Monteiro P, Lodillinsky C, Rosse C, Chavrier P. Cellular and Molecular Mechanisms of MT1-MMP-Dependent Cancer Cell Invasion. Annu Rev Cell Dev Biol. 2016;32:555–576. doi: 10.1146/annurev-cellbio-111315-125227. [DOI] [PubMed] [Google Scholar]
- Caswell P, Norman J. Endocytic transport of integrins during cell migration and invasion. Trends Cell Biol. 2008;18:257–263. doi: 10.1016/j.tcb.2008.03.004. [DOI] [PubMed] [Google Scholar]
- Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331:1559–1564. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- Chen Q, Hongu T, Sato T, Zhang Y, Ali W, Cavallo JA, van der Velden A, Tian H, Di Paolo G, Nieswandt B, et al. Key roles for the lipid signaling enzyme phospholipase d1 in the tumor microenvironment during tumor angiogenesis and metastasis. Sci Signal. 2012;5:ra79. doi: 10.1126/scisignal.2003257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho KI, Yi H, Desai R, Hand AR, Haas AL, Ferreira PA. RANBP2 is an allosteric activator of the conventional kinesin-1 motor protein, KIF5B, in a minimal cell-free system. EMBO Rep. 2009;10:480–486. doi: 10.1038/embor.2009.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S, Thapa N, Tan X, Hedman AC, Anderson RA. PIP kinases define PI4,5P(2)signaling specificity by association with effectors. Biochim Biophys Acta. 2015;1851:711–723. doi: 10.1016/j.bbalip.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Matera J, Miller MC, Reuben JM, Doyle GV, Allard WJ, Terstappen LW, et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N Engl J Med. 2004;351:781–791. doi: 10.1056/NEJMoa040766. [DOI] [PubMed] [Google Scholar]
- Du G, Huang P, Liang BT, Frohman MA. Phospholipase D2 localizes to the plasma membrane and regulates angiotensin II receptor endocytosis. Mol Biol Cell. 2004;15:1024–1030. doi: 10.1091/mbc.E03-09-0673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert MA, Lwin TM, Chang AT, Kim J, Danis E, Ohno-Machado L, Yang J. Twist1-induced invadopodia formation promotes tumor metastasis. Cancer Cell. 2011;19:372–386. doi: 10.1016/j.ccr.2011.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohman MA. The phospholipase D superfamily as therapeutic targets. Trends Pharmacol Sci. 2015;36:137–144. doi: 10.1016/j.tips.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiki Y, Hubbard AL, Fowler S, Lazarow PB. Isolation of intracellular membranes by means of sodium carbonate treatment: application to endoplasmic reticulum. J Cell Biol. 1982;93:97–102. doi: 10.1083/jcb.93.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel L, Stevens Z, Melikian H. Measuring plasma membrane protein endocytic rates by reversible biotinylation. J Vis Exp. 2009 doi: 10.3791/1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghossoub R, Lembo F, Rubio A, Gaillard CB, Bouchet J, Vitale N, Slavik J, Machala M, Zimmermann P. Syntenin-ALIX exosome biogenesis and budding into multivesicular bodies are controlled by ARF6 and PLD2. Nat Commun. 2014;5:3477. doi: 10.1038/ncomms4477. [DOI] [PubMed] [Google Scholar]
- Gomez-Cambronero J. Phospholipase D in cell signaling: from a myriad of cell functions to cancer growth and metastasis. J Biol Chem. 2014;289:22557–22566. doi: 10.1074/jbc.R114.574152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy CT, Webster MA, Schaller M, Parsons TJ, Cardiff RD, Muller WJ. Expression of the neu protooncogene in the mammary epithelium of transgenic mice induces metastatic disease. Proc Nat Acad Sci USA. 1992;89:10578–10582. doi: 10.1073/pnas.89.22.10578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Zhang F, Tay LW, Boroda S, Nian W, Levental KR, Levental I, Harris TE, Chang JT, Du G. Lipin-1 regulation of phospholipid synthesis maintains endoplasmic reticulum homeostasis and is critical for triple-negative breast cancer cell survival. FASEB J. 2017 doi: 10.1096/fj.201601353R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkels KM, Boivin GP, Dudley ES, Berberich SJ, Gomez-Cambronero J. Phospholipase D (PLD) drives cell invasion, tumor growth and metastasis in a human breast cancer xenograph model. Oncogene. 2013;32:5551–5562. doi: 10.1038/onc.2013.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Noda Y, Tanaka Y, Niwa S. Kinesin superfamily motor proteins and intracellular transport. Nat Rev Mol Cell Biol. 2009;10:682–696. doi: 10.1038/nrm2774. [DOI] [PubMed] [Google Scholar]
- Honda A, Nogami M, Yokozeki T, Yamazaki M, Nakamura H, Watanabe H, Kawamoto K, Nakayama K, Morris AJ, Frohman MA, et al. Phosphatidylinositol 4-phosphate 5-kinasea is a Downstream Effector of the Small G protein ARF6 in Membrane Ruffle Formation. Cell. 1999;99:521–532. doi: 10.1016/s0092-8674(00)81540-8. [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Ishihara H, Shibasaki Y, Kizuki N, Wada T, Yazaki Y, Asano T, Oka Y. Type I phosphatidylinositol-4-phosphate 5-kinases. Cloning of the third isoform and deletion/substitution analysis of members of this novel lipid kinase family. J Biol Chem. 1998;273:8741–8748. doi: 10.1074/jbc.273.15.8741. [DOI] [PubMed] [Google Scholar]
- Jovanovic OA, Brown FD, Donaldson JG. An effector domain mutant of Arf6 implicates phospholipase D in endosomal membrane recycling. Mol Biol Cell. 2006;17:327–335. doi: 10.1091/mbc.E05-06-0523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang DW, Choi CY, Cho YH, Tian H, Di Paolo G, Choi KY, Min do S. Targeting phospholipase D1 attenuates intestinal tumorigenesis by controlling beta-catenin signaling in cancer-initiating cells. J Exp Med. 2015;212:1219–1237. doi: 10.1084/jem.20141254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchesin V, Castro-Castro A, Lodillinsky C, Castagnino A, Cyrta J, Bonsang-Kitzis H, Fuhrmann L, Irondelle M, Infante E, Montagnac G, et al. ARF6-JIP3/4 regulate endosomal tubules for MT1-MMP exocytosis in cancer invasion. J Cell Biol. 2015;211:339–358. doi: 10.1083/jcb.201506002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell RE, Tyska MJ. Leveraging the membrane - cytoskeleton interface with myosin-1. Trends Cell Biol. 2010;20:418–426. doi: 10.1016/j.tcb.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntire LB, Berman DE, Myaeng J, Staniszewski A, Arancio O, Di Paolo G, Kim TW. Reduction of synaptojanin 1 ameliorates synaptic and behavioral impairments in a mouse model of Alzheimer’s disease. J Neurosci. 2012;32:15271–15276. doi: 10.1523/JNEUROSCI.2034-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Dong Q, Muruganujan A, Gaudet P, Lewis S, Thomas PD. PANTHER version 7: improved phylogenetic trees, orthologs and collaboration with the Gene Ontology Consortium. Nucleic Acids Res. 2010;38:D204–210. doi: 10.1093/nar/gkp1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro P, Rosse C, Castro-Castro A, Irondelle M, Lagoutte E, Paul-Gilloteaux P, Desnos C, Formstecher E, Darchen F, Perrais D, et al. Endosomal WASH and exocyst complexes control exocytosis of MT1-MMP at invadopodia. J Cell Biol. 2013;203:1063–1079. doi: 10.1083/jcb.201306162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau K, Ravikumar B, Puri C, Rubinsztein DC. Arf6 promotes autophagosome formation via effects on phosphatidylinositol 4,5-bisphosphate and phospholipase D. J Cell Biol. 2012;196:483–496. doi: 10.1083/jcb.201110114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy DA, Courtneidge SA. The ‘ins’ and ‘outs’ of podosomes and invadopodia: characteristics, formation and function. Nat Rev Mol Cell Biol. 2011;12:413–426. doi: 10.1038/nrm3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira TG, Chan RB, Tian H, Laredo M, Shui G, Staniszewski A, Zhang H, Wang L, Kim TW, Duff KE, et al. Phospholipase d2 ablation ameliorates Alzheimer’s disease-linked synaptic dysfunction and cognitive deficits. J Neurosci. 2010;30:16419–16428. doi: 10.1523/JNEUROSCI.3317-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paz H, Pathak N, Yang J. Invading one step at a time: the role of invadopodia in tumor metastasis. Oncogene. 2014;33:4193–4202. doi: 10.1038/onc.2013.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perentes JY, Kirkpatrick ND, Nagano S, Smith EY, Shaver CM, Sgroi D, Garkavtsev I, Munn LL, Jain RK, Boucher Y. Cancer cell-associated MT1-MMP promotes blood vessel invasion and distant metastasis in triple-negative mammary tumors. Cancer Res. 2011;71:4527–4538. doi: 10.1158/0008-5472.CAN-10-4376. [DOI] [PubMed] [Google Scholar]
- Qin L, Liao L, Redmond A, Young L, Yuan Y, Chen H, O’Malley BW, Xu J. The AIB1 oncogene promotes breast cancer metastasis by activation of PEA3-mediated matrix metalloproteinase 2 (MMP2) and MMP9 expression. Mol Cell Biol. 2008;28:5937–5950. doi: 10.1128/MCB.00579-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghu P, Manifava M, Coadwell J, Ktistakis NT. Emerging findings from studies of phospholipase D in model organisms (and a short update on phosphatidic acid effectors) Biochim Biophys Acta. 2009;1791:889–897. doi: 10.1016/j.bbalip.2009.03.013. [DOI] [PubMed] [Google Scholar]
- Remacle A, Murphy G, Roghi C. Membrane type I-matrix metalloproteinase (MT1-MMP) is internalised by two different pathways and is recycled to the cell surface. J Cell Sci. 2003;116:3905–3916. doi: 10.1242/jcs.00710. [DOI] [PubMed] [Google Scholar]
- Roach AN, Wang Z, Wu P, Zhang F, Chan RB, Yonekubo Y, Di Paolo G, Gorfe AA, Du G. Phosphatidic acid regulation of PIPKI is critical for actin cytoskeletal reorganization. J Lipid Res. 2012;53:2598–2609. doi: 10.1194/jlr.M028597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott SA, Selvy PE, Buck JR, Cho HP, Criswell TL, Thomas AL, Armstrong MD, Arteaga CL, Lindsley CW, Brown HA. Design of isoform-selective phospholipase D inhibitors that modulate cancer cell invasiveness. Nat Chem Biol. 2009;5:108–117. doi: 10.1038/nchembio.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulga YV, Topham MK, Epand RM. Regulation and functions of diacylglycerol kinases. Chemical reviews. 2011;111:6186–6208. doi: 10.1021/cr1004106. [DOI] [PubMed] [Google Scholar]
- Skoufias DA, Cole DG, Wedaman KP, Scholey JM. The carboxyl-terminal domain of kinesin heavy chain is important for membrane binding. J Biol Chem. 1994;269:1477–1485. [PubMed] [Google Scholar]
- Stace CL, Ktistakis NT. Phosphatidic acid- and phosphatidylserine-binding proteins. Biochim Biophys Acta. 2006;1761:913–926. doi: 10.1016/j.bbalip.2006.03.006. [DOI] [PubMed] [Google Scholar]
- Sweeney HL, Houdusse A. Myosin VI rewrites the rules for myosin motors. Cell. 2010;141:573–582. doi: 10.1016/j.cell.2010.04.028. [DOI] [PubMed] [Google Scholar]
- Tay LW, Wang Z, Du G. Analysis of Phosphatidic Acid Binding and Regulation of PIPKI In Vitro and in Intact Cells. Methods Enzymol. 2017;583:359–374. doi: 10.1016/bs.mie.2016.09.043. [DOI] [PubMed] [Google Scholar]
- Vandenbroucke RE, Libert C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nature reviews Drug discovery. 2014;13:904–927. doi: 10.1038/nrd4390. [DOI] [PubMed] [Google Scholar]
- Varnai P, Balla T. Visualization of phosphoinositides that bind pleckstrin homology domains: calcium- and agonist-induced dynamic changes and relationship to myo-[3H]inositol-labeled phosphoinositide pools. J Cell Biol. 1998;143:501–510. doi: 10.1083/jcb.143.2.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhey KJ, Hammond JW. Traffic control: regulation of kinesin motors. Nat Rev Mol Cell Biol. 2009;10:765–777. doi: 10.1038/nrm2782. [DOI] [PubMed] [Google Scholar]
- Wang S, Yuan Y, Liao L, Kuang SQ, Tien JC, O’Malley BW, Xu J. Disruption of the SRC-1 gene in mice suppresses breast cancer metastasis without affecting primary tumor formation. Proc Nat Acad Sci USA. 2009;106:151–156. doi: 10.1073/pnas.0808703105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Li Y, Wu Z, Wang H, Tan H, Peng J. JUMP: a tag-based database search tool for peptide identification with high sensitivity and accuracy. Mol Cell Proteomics. 2014;13:3663–3673. doi: 10.1074/mcp.O114.039586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Liang X, Cai M, Du G. Analysis of Invadopodia Formation in Breast Cancer Cells. Methods Mol Biol. 2016;1406:203–210. doi: 10.1007/978-1-4939-3444-7_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesner C, Faix J, Himmel M, Bentzien F, Linder S. KIF5B and KIF3A/KIF3B kinesins drive MT1-MMP surface exposure, CD44 shedding, and extracellular matrix degradation in primary macrophages. Blood. 2010;116:1559–1569. doi: 10.1182/blood-2009-12-257089. [DOI] [PubMed] [Google Scholar]
- Xu P, Duong DM, Peng J. Systematical optimization of reverse-phase chromatography for shotgun proteomics. J Proteome Res. 2009;8:3944–3950. doi: 10.1021/pr900251d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Zech T, McDonald L, Gonzalez EG, Li A, Macpherson I, Schwarz JP, Spence H, Futo K, Timpson P, et al. N-WASP coordinates the delivery and F-actin-mediated capture of MT1-MMP at invasive pseudopods. J Cell Biol. 2012;199:527–544. doi: 10.1083/jcb.201203025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Wang Z, Lu M, Yonekubo Y, Liang X, Zhang Y, Wu P, Zhou Y, Grinstein S, Hancock JF, et al. Temporal production of the signaling lipid phosphatidic acid by phospholipase D2 determines the output of extracellular signal-regulated kinase signaling in cancer cells. Mol Cell Biol. 2014;34:84–95. doi: 10.1128/MCB.00987-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Du G. Phosphatidic acid signaling regulation of Ras superfamily of small guanosine triphosphatases. Biochim Biophys Acta. 2009;1791:850–855. doi: 10.1016/j.bbalip.2009.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1, Related to Figures 5. List of PA- and PS-associated proteins identified by label-free proteomics analysis.
Table S2, Related to Figures 5. False Discovery Rate (FDR) analysis based on different p values derived from label-free quantification.
Table S3, Related to Figures 5. Proteins specifically or preferentially bound to PA-liposome revealed by label-free proteomics analysis.