

Abstract
Dinitrosyl iron complexes (DNIC) spontaneously form in aqueous solutions of Fe(II), nitric oxide (NO), and various anions. They exist as an equilibrium between diamagnetic, dimeric (bi-DNIC) and paramagnetic, monomeric (mono-DNIC) forms. Thiolate groups (e.g., on glutathione or protein cysteine residues) are the most biologically relevant anions to coordinate to Fe(II). Low molecular weight DNIC have previously been suggested to be important mediators of NO biology in cells, and emerging literature supports their role in the control of iron-dependent cellular processes. Recently, it was shown that DNIC may be one of the most abundant NO-derived products in cells and may serve as intermediates in the cellular formation of S-nitrosothiols. In this work, we examined the stability of low molecular weight DNIC and investigated issues with their detection in the presence of other NO-dependent metabolites such as S-nitrosothiols. By using spectrophotometric, Electron Paramagnetic Resonance, ozone-based chemiluminesence, and HPLC techniques we established that at neutral pH, bi-DNIC remain stable for hours, whereas excess thiol results in decomposition to form nitrite. NO was also detected during the decomposition, but no S-nitrosothiol formation was observed. Importantly, mercury chloride accelerated the degradation of DNIC; thus, the implications of this finding for the diagnostic use of mercury chloride in the detection of S-nitrosothiols were determined in simple and complex biological systems. We conclude S-nitrosothiol levels may have been substantially overestimated in all methods where mercury chloride has been used.
Keywords: DNIC, nitric oxide, S-nitrosothiols, NO-dependent chemiluminescence
Graphical Abstract
INTRODUCTION
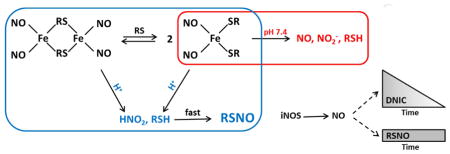
Dinitrosyl iron complexes (DNIC) are biologically relevant bioinorganic complexes of nitric oxide (NO). Such complexes spontaneously form in solutions of NO, ferrous iron and many anionic species. In biological systems, thiolate is thought to be the major anion involved in DNIC formation. DNIC can be generated from both low molecular weight (lmw) thiolates and from protein thiolates and have been shown to be present in inflammatory cells [1, 2], tissues expressing inducible nitric oxide synthase (iNOS) [3] and upon exposure to NO-releasing compounds [4, 5]. In chemical systems, they have been shown to exist as an equilibrium between diamagnetic, dimeric (bi-DNIC) and paramagnetic, monomeric (mono-DNIC) forms (Scheme 1 based on [6]), the position of which depends on the concentration of thiolate and pH. Both forms possess potential biological importance [7].
Scheme 1.

Structure of mono- and bi DNIC.
Biological DNIC have been a challenge to study and have been almost exclusively characterized by their electron paramagnetic resonance (EPR) spectra which has a diagnostic maximum at g = 2.04. However, only mono-DNIC are paramagnetic, while bi-DNIC are EPR silent. Recently, Hickock et al [2] carefully quantified the EPR signals in cells exposed to NO donor molecules and in cells activated to express iNOS, and reached the surprising conclusion that DNIC were present in much greater abundance than the more commonly-appreciated NO metabolite, S-nitrosothiol. Interestingly, others have suggested that DNIC may be precursors of S-nitrosothiol formation [8, 9].
In this study, we attempted to provide alternative methods than EPR for the study of DNIC and also to examine the possible interference of DNIC with S-nitrosothiol detection methods. We have developed an HPLC method to examine low molecular weight DNIC and to study their decomposition mechanism. The major conclusion of these studies is that DNIC decay at pH 7.4 does not result in the formation of S-nitrosothiols.
In addition, we have examined the possible interference of DNIC with chemiluminescence methods for S-nitrosothiol detection and show that DNIC give a mercury(II)-inhibitable signal when using triiodide as a reducing agent, a condition thought to be diagnostic for the presence of S-nitrosothiols. This interference can be partially overcome by allowing the more transient DNIC to decay before S-nitrosothiol analysis. As an alternative, copper (II) and ascorbate can be used in place of triiodide as the reducing mixture, in which case only S-nitrosothiols are observed (albeit with a much poorer detection limit). The clear distinction between these different NO adducts is essential to fully appreciate their unique contributions to biological processes.
EXPERIMENTAL SECTION
Materials
ProliNONOate (Proli/NO) and spermineNONOate (Sper/NO) were purchased from Cayman Chemical. All other chemicals were purchased from Sigma-Aldrich and used without further purification.
Synthesis of DNIC
DNIC were synthesized using a Coy anaerobic chamber from deoxygenated solutions of FeSO4 and cysteine or glutathione (GSH). Typically, 2 ml solution of FeSO4 (100 μM) and thiol (200 μM or 2 mM for ratios of 1:2 and 1:20) were mixed with 2 μl of Proli/NO (100 mM dissolved in 0.1 N NaOH). The light yellowish-green color of DNIC appeared immediately. While the 1:2 Fe:thiol ratio largely forms bi-DNIC, the higher ratio gives a mixture of mono and bi forms with up to 50% of the latter [10]. We achieved 14 % and 50 % mono-DNIC at Fe:thiol ratios of 1:20, and 1:1000, respectively. We carried out all the experiments in 15 mM Hepes pH 7.4, since we found that unlike phosphate, the sulfonium anion of this buffer cannot compete with the thiolate anion for binding sites of the iron (See Supplementary Figure 1).
Synthesis of S-nitrosocysteine
S-Nitrosocysteine (CysNO) was freshly synthesized [11] from L-cysteine (200 mM) and NaNO2 (210 mM) in HCl. After 10 min incubation at room temperature, protected from light, the CysNO solution was neutralized with NaOH and diluted with buffer to the proper concentration (extinction coefficient ε334nm=900 M−1cm−1).
Cell culture
MCF7 human mammary adenocarcinoma cells were a generous gift from Dr. B. Kalyanaraman and were maintained in MEMα media (Invitrogen; Carlsbad, CA) supplemented with 10% fetal bovine serum, 200 U/mL penicillin, 200 μg/mL streptomycin, and non-essential amino acids (10 μM). Cultures were incubated at 37°C in a humidified atmosphere of 5% carbon dioxide and 95% air. For treatments, cells were exposed to Sper/NO (100 μM) or L-CysNO (100 μM) for 2 h in Hank’s Balanced Salt Solution (HBSS), and then cells were harvested by scraping into 15 mM Hepes or into 50 mM phosphate/1 mM diethylene triamine pentaacetic acid (DTPA)/1 mM N-ethylmaleimide (NEM) buffer as noted in figure legends (pH 7.4 for both), sonicated and centrifuged at 10,000 x g for 10 min. The supernatant was used for chemiluminsecence analysis, and values were normalized to protein concentration. The protein concentration was determined by using the BCA assay (Pierce; Rockford, IL).
Murine RAW 264.7 macrophage-like cells were routinely cultured as previously described [12]. Cells were seeded at density of 1 × 106/ml in 100 mm dishes and grown overnight to reach 70–80% confluence. Lipopolysaccharide (LPS, 0.5 μg/ml) was added to medium and incubated for 13 h, and then harvested as described above, using 50 mM phosphate/1 mM DTPA/1 mM NEM buffer, pH 7.4. In additional experiments, L-NG-Nitroarginine methyl ester (L-NAME, 5 mM) was added to inhibit NO production during iNOS induction.
For EPR measurements, cells were scraped into 15 mM Hepes buffer, pH 7.4, and then put into quartz EPR tube and immediately snap-froze in liquid nitrogen.
EPR spectroscopy
A Bruker EMX spectrometer at 100K was used with the following parameters: 1 mW microwave power, 9.3 GHz microwave frequency, 10 G modulation amplitude, 20.94 ms time constant, and 41.94 s conversion time. DNIC were quantified by comparing the double integral of spectra with those of known concentrations of nitrosyl hemoglobin.
Griess and Saville assay
These assays were used to test the mercury(II)-stability of DNIC. Assays were performed according to Griess [13] and Saville [14]. Briefly, for the Saville assay, 200 μl of sample was mixed with 10 μl of mercury chloride (HgCl2) (10 mM in double distilled water), and then 10 μl of sulfanilamide (30 mM in 2 N HCl) and naphthyl ethylene diamine (10 μl of 30 mM in 0.1 N HCl) were added consecutively. The absorbance was read at 540 nm. Griess assay was performed the same way, but without HgCl2. Results were quantified based on standard curves generated from S-nitrosoglutathione (GSNO) for Saville assay and from sodium nitrite for Griess assay. The difference of values obtained with both assays refers for the mercury(II)-depletable content.
Ozone-based chemiluminescence
NO-derivatives were analyzed with triiodide-dependent, ozone-based chemiluminescence [15] using a Sievers Model 280 NO analyzer. Briefly, the reaction solution was made daily from KI and I2 in glacial acetic acid and double distilled water and placed in the purge vessel of the analyzer. Sulfanilamide (100 mM in 2 N HCl, added in 10% vol/vol) was added to samples to remove nitrite, while HgCl2 (5 mM, added in 10% vol/vol) was added to nominally verify the presence of S-nitrosothiols. Quantification was performed based on the detector response for known amounts of GSNO.
S-nitrosothiols were also analyzed with Cu2+/ascorbate-dependent, ozone-based chemiluminescence [16, 17]. After adding a solution of ascorbate (10 mM or 100 mM in 15 mM Hepes, pH 7.4) to the purge vessel of the analyzer and purging with argon, Cu(II)SO4 (final concentration 10 μM or 100 μM) was injected to establish the reducing system.
HPLC analysis
The analysis was performed using an Agilent 1100 chromatograph. Samples (50 μl) were injected onto a Kromasil C-18 column (250 × 4.6 mm, 0.5 μM particle size) and eluted with mobile phases of methanol and 0.05% tetraflouroaceticacid (TFA) using a gradient method [18]. The effluent was monitored with a diode array spectrophotometer detector at 310 nm (DNIC) and at 336 nm (GSNO and nitrite). Integration of peaks was done by Agilent ChemStation software. Known amounts of standards were used for quantification. Samples were not acidified unless indicated.
Absorption spectroscopy
Spectra were taken on an Agilent 8452 diode array UV-Visible spectrophotometer in a 1 cm quartz cuvette.
Statistics
A Student’s two-tailed t-test was used for statistics, and p≤0.05 was considered statistically significant. All data are reported as mean ± standard error unless otherwise indicated.
RESULTS
Analysis of DNIC with HPLC
In order to expand the methodology available to study DNIC and also to examine their decomposition, we developed an HPLC method to detect these species and their degradation products. This method was essentially the same as the reverse phase HPLC method used routinely for analysis of GSNO and nitrite [19]. The separated compound had a spectrum (Figure 1) identical to that reported [10] for bi-DNIC and the published extinction coefficient [10, 20] of bi-GSH-DNIC (ε310nm=4600 M−1cm−1 based on iron) was used for quantification. We determined the detector response (slope of calibration curve = 336 ± 27 peak area/μmol), and estimated a detection limit of ~0.5 nmol. Interestingly, we only observed bi-DNIC by HPLC regardless of the initial thiol:iron ratio, strongly suggesting that as the compound is separated from excess thiol on the HPLC column, any mono-DNIC converts to bi-DNIC (Scheme 1). Figure 1 shows that GSNO, nitrite, and GSH-DNIC can be resolved using these chromatographic conditions. We were not successful in detecting the DNIC formed from cysteine by this method, so all experiments were performed with GSH as the thiolic ligand.
Figure 1. Detection of DNIC and its degradation products with HPLC.

A representative chromatogram of DNIC (Fe:GSH=1:20) after 15 h incubation in closed vials at room temperature. Spectra of GSNO (tret=7.4 min), nitrite (tret=9.4 min), and DNIC (tret=11.7 min) are marked with arrows. Table shows the detector response to monitored species.
To examine stability, GSH-DNIC was synthesized at pH 7.4 in Hepes buffer, immediately transferred into an HPLC vial, and repeat-injected every 80 min. When a 1:2 Fe/GSH ratio was used, the observed bi-DNIC remained stable for 17 hours (Figure 2A); however, at the higher ratio (1:20) both bi and mono-DNIC decomposed completely within 12 hours. As bi-DNIC is EPR silent, the decay of mono-DNIC trace of the mixture was followed with EPR, see Supplementary Figure 2. The DNIC degradation was accompanied by extensive nitrite formation (Figure 2B). The yield of nitrite from DNIC decay (based on Fe content) was approximately 1:1, half of the expected 2:1 if nitrite formation was the only fate of the nitrosyl groups. In contrast to Borudulin et al [20] we did not observe substantial GSNO formation at neutral pH. The GSNO level remained under 2% of the initial DNIC concentration.
Figure 2. Decomposition of DNIC at neutral and at acidic pH.

DNIC was synthesized from 100 μM of FeSO4, and 100 μm of Proli/NO with GSH in 15 mM Hepes pH 7.4, and analyzed with reverse phase HPLC/UV-Vis detector. Fe:thiol=1:2 (A, C, E), and 1:20 (B, F). (A, B): DNIC decay, and accumulation of nitrite at pH 7.4. (C): Nitrite accumulation in the presence of HgCl2. (D): HPLC traces of the first sample of (C). Spectrum of intermediate species is indicated with arrow at tret=7.7 min. (E, F): DNIC decay, and accumulation of GSNO in the presence of 10% 2N HCl. Diamonds: DNIC, triangles: nitrite, circles: GSNO. Closed triangles: DNIC was supplemented with HgCl2. Data are reported as mean±standard error, N=3.
Mercury(II) chloride is used as a diagnostic for the presence of S-nitrosothiols in chemiluminescence methods, as it will rapidly destroy S-nitrosothiols liberating nitrite, which can then be removed with sulfanilamide under acidic conditions. We were therefore interested to see if mercury(II) could enhance the decay of DNIC. The addition of HgCl2 (5 mM) to DNIC (1:2 Fe/GSH) immediately (by the time of first injection at ~10 minutes after mixing) resulted in destruction of DNIC with the concomitant formation of nitrite (Figure 2C). An identical profile was also observed with DNIC (1:20 Fe/GSH) (Supplementary Figure 3). Nitrite yields compared to the iron concentration were again approximately 1:1. At the initial time point, the nitrite yield was much lower, and we detected an HPLC peak (Figure 2D), suggestive of an intermediate in the decay. We also monitored the mercury(II)-dependent decomposition of mono-GSH-DNIC with EPR, and the DNIC spectrum was quickly replaced by a fast decaying symmetrical EPR signal of an intermediate species at g=2.01, line width 190 G. (Supplementary Figure 4). This suggests the presence of a transient NO-containing intermediate in the mercury(II)-dependent decomposition of DNIC to nitrite. A possible identity of this species could be a one-electron reduced dimeric DNIC that has a similar EPR spectrum [21].
The decomposition of DNIC was also monitored under acidic conditions (2 N HCl) to approximate conditions used in the triiodide based chemiluminescence assays (see below). In this case, both DNIC (1:2) and DNIC (1:20) decayed, with DNIC (1:20) decaying more slowly than DNIC (1:2). In both cases GSNO was formed with a yield of approximately 1:1 with respect to iron (Figure 2 E, F).
In total, these data suggest that at physiological pH, bi-DNIC are stable for many hours but the presence of excess thiol results in slow generation of nitrite as the major NO-derived product from one of the bound NO molecules. Under acidic conditions, DNIC decay yields S-nitrosothiols. It is possible that GSNO is formed directly and/or from the reaction between nitrite/nitrous acid and glutathione at acid pH. Importantly, HgCl2 rapidly decomposes DNIC generating nitrite.
We next sought to examine if this observation had implications for the detection of S-nitrosothiols by chemiluminesence.
Detection of DNIC in the Saville Assay
The Saville assay for S-nitrosothiols involves the mercury(II)-dependent formation of nitrite, followed by acidic diazotization with sulfanilamide and naphthyl ethylene diamine to form an intensely-colored purple product. To examine if DNIC report as positive in this assay, we tested DNIC at both Fe/GSH ratios in the Griess/Saville assay, and observed the mercury(II)-sensitive formation of purple azo-compound (30.1±2.5 μM for DNIC (1:2) and 39.8±2.4 μM for DNIC (1:20), n=3 from solutions 100 μM in Fe). This finding suggested that accurate detection and quantification of S-nitrosothiols might be compromised if DNIC are also present.
Detection of DNIC with ozone-based chemiluminescence
Chemiluminescence methods for the detection of NO and its metabolites work by selectively converting metabolites back to NO and detecting the nascent NO by its reaction with ozone, which generates the chemiluminescence. The method is both sensitive and specific for NO; however, metabolite identification depends on the selectivity of the conversion process. Triiodide/acetic acid can convert S-nitrosothiols, nitrite, and iron nitrosyl species, as well as C-, O- and N- nitroso species, to NO with varying efficiency [22]. Nitrite is removed with sulfanilamide at low pH, and the heme nitrosyls and C, O, and N- nitroso species are mercury(II)-resistant. Thus, in the presence of sulfanilamide, the mercury-sensitive signals are attributed to the presence of S-nitrosothiols. DNIC have been largely ignored as a potential interference in this method; however, the HPLC data above suggest that DNIC could also contribute to the mercury(II)-sensitive signal.
Using triiodide/acetic acid-based chemiluminescence, we observed that GSH-DNIC are readily detected, and the signal intensity, either at pH 7.4 or acidified, was stable during 2 hours of repeat injection. The presence of acid/sulfanilamide resulted in a loss of signal, that was enhanced by the presence of mercury(II) (Figure 3A). The results are consistent with the instability of DNIC in acid and in the presence of mercury(II). Similar decay kinetics were observed with Cysteine-DNIC (Supplementary Figure 5), although the chemiluminescence peaks of Cysteine-DNIC were narrower than those of GSH-DNIC, demonstrating that the former is more facile in triiodide (Supplementary Figure 5, lower panel). The raw data from chemiluminescence analysis consists of a photomultiplier voltage trace, and quantification is achieved by obtaining the area under the peaks, by integration. The width of the peak is an indication of how easily the specific metabolite is converted to NO by the reducing media. When mercury(II) was added to DNIC at pH 7.4, the chemiluminescence peak sharpened (Figure 3B), suggestive of conversion of the DNIC to nitrite. This was confirmed by the addition of acid/sulfanilamide which largely removed this signal. The peak for GSH-DNIC was much broader than that observed for nitrite or GSNO, indicating slower NO formation in the triiodide medium (Figure 3B). These results are in good agreement with our HPLC data (Figure 2A, C, E) showing the mercury(II)-dependent liberation of nitrite from DNIC.
Figure 3. Detection of DNIC with ozone-based chemiluminescence.

DNIC was synthesized from 100 μM of FeSO4, and 100 μM of Proli/NO with GSH (Fe:thiol=1:2 or 1:20) in 15 mM Hepes pH 7.4, and analyzed with ozone-based chemiluminescence. (A): Time dependence of detected NO as GSNO equivalent, Fe:thiol=1:2. The experiment was completed with triiodide reducing medium. Diamonds: no additive, neutral pH, squares: addition of 10 % 2 N HCl, triangles: addition of 10 % sulfanilamide, circles: addition of 10 % HgCl2 solution and 10 % sulfanilamide
(B): Chemiluminescence traces detected in triiodide reducing medium, HgCl2 was added under neutral pH, SA (dissolved in 2 N HCl) was added to the samples 20 min after HgCl2 addition.
(C, D): Chemiluminescence traces of GSNO (0.05, 0.1, and 0.15, 0.2 nmol, respectively) and DNIC (Fe:GSH=1:2, and 1:20, respectively, 0.20 nmol NO in both), sodium nitrite (0.1 nmol). Reducing medium: (C) 10 mM ascorbate/10 μM in CuSO4 15mM Hepes pH 7.4 (D): 100 mM ascorbate/100 μM CuSO4 in 15 mM Hepes pH 7.4.
Inset: Peak area as a function of injected GSNO amount.
Data are reported as mean±standard error, N=3.
We next compared our results using triiodide/acetic acid chemiluminescence to a similar method utilizing copper and ascorbate [23] at pH 7.4 as the reducing medium. Ascorbate reduces Cu(II) to Cu(I) which subsequently reduces S-nitrosothiol to thiol and NO. Unlike triiodide, this mixture does not detect nitrite, so nitrite removal by acid/sulfanilamide is unnecessary. We were able to detect GSNO when CuSO4 (10 μM)/ascorbate (10 mM) was used as the reductant (Figure 3C). The chemiluminescence signal peaks were broader than observed with triiodide/acetic acid, but the detector response (area under the curve) was similar with either reducing solution. Also, as expected, HgCl2 abolished the chemiluminescence signal from GSNO. In contrast, DNIC gave no signal in Cu2+/ascorbate unless the samples were treated with HgCl2. Similar signals were observed when mercury(II)-treated DNIC samples were injected into Hepes buffer (15 mM, pH 7.4) in the purge vessel (i.e. in the absence of any reducing agent, Supplementary Figure 6), indicating direct NO formation from the mercury(II)-dependent DNIC degradation at neutral pH. If the copper and ascorbate concentrations were increased to 100 μM and 100 mM, respectively, to accelerate the GSNO reduction in the purge vessel, the GSNO peaks sharpened, but a slight signal from DNIC was now observed (Figure 3D).
Together, these data indicate that DNIC could give a mercury(II)-inhibitable signal in triiodide-based chemiluminescence methods that is indiscriminative from the S-nitrosothiol signal. The Cu2+/ascorbate method is unambiguous for S-nitrosothiol detection, but due to peak broadness, has a much higher limit of detection than triiodide. This is particularly problematic with biological samples as discussed below.
Analysis of S-nitrosothiols and DNIC in cells with ozone-based chemiluminescence
In order to test whether the two chemiluminescence methods are able to discriminate between S-nitrosothiols and DNIC in a cellular system, we treated MCF7 human breast adenocarcinoma cells with Sper/NO (100 μM for 2 h). The cells were then washed and lysed in Hepes buffer (15 mM, pH 7.4). Lysate was analyzed by both triiodide- and copper/ascorbate-dependent chemiluminescence simultaneously. Sulfanilamide-supplemented samples gave a robust signal using the triiodide method, which was abolished after addition of HgCl2 (Figure 4A). In contrast, much smaller signals were observed with the copper/ascorbate system (Figure 4B). The quantification of the traces (Figure 4C) revealed the formation of 60 ± 2.4 pmol/mg protein S-nitrosothiol equivalents (with triiodide method), and 6.8 ± 2.2 pmol/mg protein of S-nitrosothiol equivalents (with copper/ascorbate). The presence of DNIC in these samples was confirmed with EPR (Figure 4D). As mentioned above, Cu2+/ascorbate-dependent signals are broader and more challenging to integrate. In addition, injection of protein-containing samples to Cu2+/ascorbate results in more extensive bubbling than with triiodide/acetic acid. This makes the Cu2+/ascorbate method far from ideal for the detection of S-nitrosothiols. Despite these limitations, the magnitude of the difference between the two methods strongly suggests that DNIC are being detected by the triiodide method.
Figure 4. Detection of S-nitrosothiols and DNIC with triiodide-based and copper/ascorbate-based chemiluminescence methods.

MCF7 cells were treated with 100 μM Sper/NO (A–D, G) or 100 μM CysNO (E, F, H) for 2 h. Lysates were simultaneously analyzed in both reducing media. (A, B): Chemiluminescence traces of control and Sper/NO-treated cells after lysed in 15 mM Hepes. (C): Quantification of (A) and (B). (D): EPR signal detected in cells after Sper/NO treatment. (E): Chemiluminescence traces of control and CysNO-treated cells detected in copper/ascorbate system. Cells were lysed in 15 mM Hepes. (F): Quantification of (E) and the chemiluminescence signal of the same samples detected with tri-iodide method (raw data in Supplementary Figure 4). (G) Quantification of chemiluminescence signal of control and Sper/NO-treated cells after lysed in 50 mM phosphate/1 mM DTPA/1 mM NEM. (H): Quantification of chemiluminescence signal of control and CysNO-treated cells after lysed in 50 mM phosphate/1 mM DTPA/1 mM NEM.
Quantified data are reported as mean±standard error, N=3. *p≤0.01, **p≤0.005.
ND means not detectable.
As a positive control, we treated the cells with CysNO (100 μM for 2 h), a condition in which a fairly high level of S-nitrosothiol was anticipated, due to CysNO transport into the cells and NO-independent formation of protein S-nitrosothiols [24], with a much smaller DNIC component [5, 25]. Indeed, 470 ± 58 pmol/mg protein S-nitrosothiol was detected using the copper/ascorbate method (Figure 4E, F). The integration of triiodide-dependent signal revealed liberation of 790 ± 68 pmol/mg protein NO, a major part of which was mercury sensitive (Figure 4F and supplementary Figure 7). We hesitate to suggest the difference between the two methods corresponds to DNIC as this would be much higher than previously published EPR quantification [5, 25] of DNIC and accurate quantification of Cu/Asc is problematic. However, EPR may underestimate DNIC levels as it only reports on paramagnetic complexes.
Although these result indicate that Cu2+/ascorbate is able to more specifically detect cellular S-nitrosothiols, the broad peaks and sample bubbling give less confidence in the use of this method for absolute quantification of low endogenous levels. It is of course possible that part of the difference in Figure 4F is due to CysNO-dependent formation of DNIC (detectable with triiodide) in the cells as has been previously demonstrated [26].
Additional experiments were performed in the lysis buffer that we conventionally use when measuring cellular S-nitrosothiols. This consists of 50 mM phosphate/1 mM DTPA/50 mM NEM. In this study, samples were analyzed immediately and also after 20 hours incubation at 4 °C, a time we estimated to be long enough to allow the substantial decay of any DNIC present. In the case of Sper/NO treatment, the results did not show meaningful differences between cells lysed in Hepes or in phosphate/DTPA/NEM (Figure 4C vs. 4G), and 88% of the chemiluminescence signal decayed after 20 h (Figure 4G). On the other hand, when the cells were treated with CysNO, about 7 times more product was detected when phosphate/DTPA/NEM lysis buffer was used (Figure 4F vs. 4H), a condition expected to protect S-nitrosothiols from degradation. Importantly, there was no significant decay of signal after 20 hours (Figure 4H). These results support previous findings that CysNO-treatment results in mostly S-nitrosothiol formation. In contrast, Sper/NO-treatment appeared to almost exclusively form DNIC in cells, with little S-nitrosothiol formed [2].
Effect of endogenous NO production on DNIC and S-nitrosothiol formation
To extend these findings to a system in which NO is produced endogenously, we exposed RAW 264.7 macrophages to LPS for 13 hours to induce iNOS-derived NO production. Figure 5 shows that after treatment, the cells were harvested in phosphate/DTPA/NEM, and the lysate was analyzed with chemiluminescence using both triiodide and copper/ascorbate reducing solutions. Very small signals were found with copper/ascorbate, while a sizable triiodide-based signal was detected. The latter were completely eliminated after addition of HgCl2 and decayed over time. The lack of copper/ascorbate-based detection and the time-dependent decay of the triiodide-sensitive product suggest that LPS treatment led mainly to DNIC formation in agreement with EPR data [2]. We confirmed the formation of DNIC by EPR in LPS-treated cells (Supplementary Figure 8). These data demonstrate that rapid processing of samples from LPS-treated RAW cells reveals a signal using triiodide reduction that decays to a level of about 20 pmol/mg protein after 20 h, which is similar to the level we previously attributed to S-nitrosothiols under these conditions [12, 27]. This implies that DNIC are present at > 4 times higher levels than S-nitrosothiols at the time of cell lysate preparation, in qualitative but not quantitative agreement with Hickok et al [2] who reported much greater DNIC levels, and the DNIC signal is lost over time to reveal only the more stable S-nitrosothiol signal. It should be noted that Hickok et all observed a much faster decay of intracellular RSNO compared to DNIC after the exposure of RAW cells to exogenous NO [2], however once lysed and ‘stabilized’ with DTPA and NEM, the RSNO are stable and the DNIC decay.
Figure 5. DNIC formation in LPS-treated RAW 264.7 macrophages.

Cells were treated with LPS (final concentration 0.5 μg/ml) and incubated for 13 h, then washes and lysed 50 mM phosphate/1 mM DTPA/1 mM NEM pH 7.4. (A): Lysates were simultaneously analyzed with triiodide-based, and copper/ascorbate-based NO-dependent chemiluminescence methods. (B): The nitrite concentration in the medium was determined with Griess assay.
Quantified data are reported as mean±standard error, N=3. *p≤0.005.
DISCUSSION
Thiolate-based dinitrosyl iron complexes, both low molecular weight and protein-based, have been reported to be important in diverse biological functions such as vasorelaxation, platelet aggregation, and wound healing [1]. Their formation may also play role in NO-dependent regulation of DNA methylation [28]. They have also been proposed to be efficient mediators of S-nitrosothiol synthesis [8, 26]. Detection of DNIC is mainly based on their characteristic EPR spectrum at g=2.04, although this technique is limited to determination of only paramagnetic species (e. g. mono-DNIC), and is insensitive to non-paramagnetic DNIC (including bi-DNIC) may exist. DNIC can be identified by optical spectra [10], but prior separation is required for proper assignment of these species.
In this study, we investigated the detectability and stability of cysteine- and GSH-based DNIC with spectrophotometric, EPR, chemiluminescence, and HPLC methods. Based on our HPLC measurements under neutral conditions, bi-GSH-DNIC is fairly stable, while the presence of excess thiol induces a decay resulting in nitrite as a major product. We did not detect quantitative conversion of DNIC to nitrite which implies that additional nitrogen species are also produced. Indeed, extensive nitric oxide formation was observed when partially decomposed DNIC was injected into the purge vessel of the NO analyzer containing only buffer. GSNO was not found among the degradation products by either HPLC or chemiluminescence methods. These results are inconsistent with the mechanism suggested by Vanin [1] in which hydrolysis of DNIC leads to nitrite and NO, but excess thiol prevents hydrolysis by coordinating to the NO moiety. As an alternative, it has also been proposed that DNIC are in equilibrium with Fe(II), NO, thiolate, and S-nitrosothiol [1, 10]. Our observations do not support either hypothesis, but at least partially support the work of Tsou and Liaw [29] who showed that one, but not two, thiol-containing DNIC can produce S-nitrosothiol, but only under acidic conditions. These authors did not consider that this could occur through the intermediacy of nitrite/nitrous acid. On the other hand, we observed that under strongly acidic conditions, extensive GSNO was produced which was accompanied the GSH-DNIC deterioration. This is consistent with reaction between nascent thiol and nitrous acid [30] during DNIC decay. We can conclude that although S-nitrosothiols may play a role in the formation of DNIC [31] they are not primary decomposition products.
A major method for quantification of S-nitrosothiols is based on the sulfanilamide/mercury-depletable signal of triiodide-dependent chemiluminescence, and DNIC being a nitrosyl compound potentially might interfere with this analysis. We previously found that cytochrome c heme nitrosyl gives a decaying mercury-resistant chemiluminescence signal making it imperative that the mercury-treated and non-treated samples are analyzed in close temporal proximity to avoid artifacts caused by instability of iron-nitrosyl compounds [32]. In the current study we show that unlike heme nitrosyls, DNIC are mercury sensitive and so cannot be distinguished from S-nitrosothiols in assays that rely on mercury-dependence of the observable for specificity. As DNIC will decay over time, while S-nitrosothiols can be stabilized by careful buffer selection (e. g. the presence of NEM and DTPA), it suggests a potential way to distinguish the two species to avoid DNIC contamination in S-nitrosothiol measurements. Unfortunately, care taken to inject samples rapidly after preparation, is more likely to lead to a greater degree of contamination with DNIC, whereas samples stored overnight likely gives more time for the DNIC to decay. What is abundantly clear is that, if anything, the triiodide method will overestimate, but not underestimate, the level of S-nitrosothiol in the sample. As the levels of DNIC may be much higher than S-nitrosothiols, detection of a small percentage of DNIC could have large effects on the putative S-nitrosothiol signal. Simply utilizing this difference in decomposition rate can provide a reasonable way to distinguish between these two species, although this is somewhat unsatisfactory as it assumes that all S-nitrosothiols are stabilized by NEM/DTPA containing buffer. Although we have no evidence to suggest that they are not, this is nevertheless an assumption. Copper/ascorbate-dependent chemiluminescence could be a promising alternative to discriminating between S-nitrosothiol and DNIC, however, the broad peaks and extensive bubbling make this unworkable for the low levels of S-nitrosothiols found in biological systems.
From a biological perspective the observations made here have some profound consequences that are generally in agreement with a recent study by Hickock et al [2]. On a quantitative basis, DNIC appear to be a more robust NO-dependent thiol modification than S-nitrosothiols with in some cases a greater than 4:1 ratio of DNIC to S-nitrosothiol in the systems studied here. This is likely an underestimate because of the instability of DNIC and significant decay will have occurred before analysis. In an LPS-activated macrophage generating high levels of NO from iNOS, the S-nitrosothiol content is about 20 pmol/mg protein in agreement with our previous studies. However, the DNIC content is about 80 pmol/mg protein. Of course, quantity does not imply importance, and whereas a wealth of pathways has been implicated to be modulated by S-nitrosation there is little direct evidence for the involvement of DNIC in cellular signaling processes. This may be, however, because this NO-dependent protein adduct has been largely overlooked. In addition to analytical methods, S-nitrosothiols have also been detected through switch methods which involve the selective reduction of S-nitrosothiols followed by labeling of the nascent thiol. How or if the presence of DNIC affects these methods is currently unknown.
In conclusion, DNIC are abundant cellular products of NO that can be misidentified as S-nitrosothiols using some current methodology. Consequently, S-nitrosothiol content may have been overestimated in the past. The difference in stability of DNIC and S-nitrosothiols can be used to separate these two metabolites although this procedure has limitations and is based on some assumptions. Better methods are needed to clearly distinguish these two important NO-dependent protein modifications.
Supplementary Material
Supplementary Figure 1. Influence of anion ligands and pH on EPR spectra of DNIC. (A): Spectra of DNIC from FeSO4, Proli/NO in 10 mM PBS (green), 15 mM Hepes (blue), and with cysteine in 15 mM Hepes (Fe:Cys=1:20) (red); pH 7.4 in each case. (B): Same as in (A) with 10 % of 2 N HCl addition to the DNIC samples. (C): Intensity of DNIC (Fe:Cys=1:20, in 15 mM Hepes) spectra as a function of pH. (D): Spectra of DNIC from FeSO4, Proli/NO and cysteine (Fe:Cys=1:20) in 10 mM PBS at pH 7.4 (black), and with 10 % of 2 N HCl (grey).
The DNIC formed from Hepes or PBS in the absence of cysteine exhibited an altered shape of the EPR spectrum, but did not lose paramagnetic intensity. When Cys-DNIC was prepared in PBS, acidification resulted in change of shape, but not intensity (D). The shape change was consistent with a transformation from axial to rhombic symmetry [8] indicative of thiolate ligand displacement by phosphate at low pH. The results suggest that the binding of thiolate is much stronger than that of the sulfonate ligand in Hepes, while phosphate is competitive with thiolate at low pH. The practical conclusion from these observations is that the effect of acidification on DNIC is buffer–dependent, and the paramagnetic nature of these compounds is preserved in phosphate, but decreased in Hepes. For these reasons, all further experiments were performed in Hepes buffer.
Supplementary Figure 2. Decay of mono-DNIC followed by EPR. DNIC was synthesized from 100 μM of FeSO4, and 100 μm of Proli/NO with GSH (Fe:GSH=1:20) in 15 mM Hepes pH=7.4, and the kinetics of DNIC was followed with EPR.
Supplementary Figure 3. Decomposition of DNIC made with excess thiol in the presence of HgCl2 at neutral pH. DNIC was synthesized from 100μM of FeSO4, and 100 μm of Proli/NO with GSH in 15 mM Hepes pH 7.4, Fe:thiol=1:20. Nitrite was detected with reverse phase HPLC/UV-Vis detector.
Supplementary Figure 4. Effect of mercury chloride on mono-DNIC. DNIC (Fe:GSH=1:20) in 15 mM Hepes pH 7.4 was monitored with EPR before and after addition of HgCl2 solution. Spectra were recorded from freshly synthesized DNIC (black), immediately after HgCl2 (red) addition, and 20 min later (blue). Time points are indicated with arrows.
Supplementary Figure 5. Detection of DNIC with triiodide-dependent ozone-based chemiluminescence. DNIC was synthesized from 100 μM of FeSO4, and 100 μm of Proli/NO with cysteine (Fe:Cys=1:2) in 15 mM Hepes pH=7.4. Upper panel: Time dependence of detected NO as GSNO equivalent, Diamonds: no additive, squares: addition of 10% 2N HCl, triangles: addition of 10% sulfanilamide, circles: addition of 10% HgCl2 solution and 10% sulfanilamide. Lower panel: Chemiluminescence traces. Data are reported as mean±standard error, N=3.
Supplementary Figure 6: Nitric oxide formation during mercury-assisted decay of DNIC. Chemiluminescence traces after injection of (1) DNIC (1:2) and (2) DNIC (1:2) after addition of HgCl2, and (3) DNIC (1:20) and (4) DNIC (1:20) after addition of HgCl2 The purge vessel contains only 15 mM Hepes pH 7.4.
Supplementary Figure 7: Chemiluminescence traces of MCF7 cells (control and treated with 100 μm CysNO for 2 h). Quantification of peaks is shown in Figure 4F.
Supplementary Figure 8: EPR spectrum detected in RAW 264.7 macrophages. Cells were treated with LPS for 13 h, and then after scraping into 15 mM Hepes were put into EPR tube and snap-froze in liquid nitrogen.
Low molecular weight dinitrosyl iron complexes decay to nitric oxide and nitrite and not S-nitrosothiol at neutral pH
Detection of dinitrosyl iron complexes by tri-iodide-depndent ozone-based chemiluminescence gives a mercury-inhibitable signal, suggesting possible mis-identification as S-nitrosothiols.
Differentiation between S-nitrosothiols and dinitrosyl iron complexes can be made on the basis of differential stability once cells are lyzed.
Acknowledgments
Financial support from NIH GM55792
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Vanin AF. Dinitrosyl iron complexes with thiolate ligands: physico-chemistry, biochemistry and physiology. Nitric Oxide. 2009;21(1):1–13. doi: 10.1016/j.niox.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 2.Hickok JR, Sahni S, Shen H, Arvind A, Antoniou C, Fung LW, Thomas DD. Dinitrosyliron complexes are the most abundant nitric oxide-derived cellular adduct: biological parameters of assembly and disappearance. Free Radic Biol Med. 2011;51(8):1558–1566. doi: 10.1016/j.freeradbiomed.2011.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Muller B, Kleschyov AL, Stoclet JC. Evidence for N-acetylcysteine-sensitive nitric oxide storage as dinitrosyl-iron complexes in lipopolysaccharide-treated rat aorta. Br J Pharmacol. 1996;119(6):1281–5. doi: 10.1111/j.1476-5381.1996.tb16034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Watts RN, Hawkins C, Ponka P, Richardson DR. Nitrogen monoxide (NO)-mediated iron release from cells is linked to NO-induced glutathione efflux via multidrug resistance-associated protein 1. Proc Natl Acad Sci U S A. 2006;103(20):7670–5. doi: 10.1073/pnas.0602515103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Toledo J, Bosworth CA, Hennon SW, Mahtani HA, Bergonia HA, Lancaster JR. Nitric Oxide-induced Conversion of Cellular Chelatable Iron into Macromolecule-bound Paramagnetic Dinitrosyliron Complexes. Journal of Biological Chemistry. 2008;283(43):28926–28933. doi: 10.1074/jbc.M707862200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McDonald CC, Phillips WD, Mower HF. An Electron Spin Resonance Study of Some Complexes of Iron, Nitric Oxide, and Anionic Ligands. J Am Chem Soc. 1965;87:3319–3326. [Google Scholar]
- 7.Strube K, de Vries S, Cramm R. Formation of a dinitrosyl iron complex by NorA, a nitric oxide-binding di-iron protein from Ralstonia eutropha H16. J Biol Chem. 2007;282(28):20292–300. doi: 10.1074/jbc.M702003200. [DOI] [PubMed] [Google Scholar]
- 8.Boese M, Mordvintcev P, Vanin AF, Busse R, Mülsch A. S-Nitrosation of serum albumin by dinitrosyl-iron complex. Journal of Biological Chemistry. 1995;270(49):29244–29249. doi: 10.1074/jbc.270.49.29244. [DOI] [PubMed] [Google Scholar]
- 9.Bosworth CA, Toledo JC, Zmijewski JW, Li Q, Lancaster JR. Dinitrosyliron complexes and the mechanism(s) of cellular protein nitrosothiol formation from nitric oxide. Proceedings of the National Academy of Sciences. 2009;106(12):4671–4676. doi: 10.1073/pnas.0710416106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vanin AF, Poltorakov AP, Mikoyan VD, Kubrina LN, Burbaev DS. Polynuclear water-soluble dinitrosyl iron complexes with cysteine or glutathione ligands: electron paramagnetic resonance and optical studies. Nitric Oxide. 2010;23(2):136–49. doi: 10.1016/j.niox.2010.05.285. [DOI] [PubMed] [Google Scholar]
- 11.Mathews WR, Kerr SW. Biological activity of S-nitrosothiols: the role of nitric oxide. Journal of Pharmacology and Experimental Therapeutics. 1993;267(3):1529–1537. [PubMed] [Google Scholar]
- 12.Zhang Y, Hogg N. The Formation and Stability of S-Nitrosothiols in RAW 264.7 Cells. AJP - Lung Cellular and Molecular Physiology. 2004;287:L467–L474. doi: 10.1152/ajplung.00350.2003. [DOI] [PubMed] [Google Scholar]
- 13.Griess P. Bemerkungen zu der Abhandlung der HH. Weselky und Benedikt Ueber einige Azoverbindungen. Chemische Berichte. 1879;12:426–428. [Google Scholar]
- 14.Saville B. A scheme for the colorimetric determination of microgram amounts of thiols. Analyst. 1958;83:670–672. [Google Scholar]
- 15.Samouilov A, Zweier JL. Development of chemiluminescence-based methods for specific quantitation of nitrosylated thiols. Analytical Biochemistry. 1998;258(2):322–330. doi: 10.1006/abio.1998.2609. [DOI] [PubMed] [Google Scholar]
- 16.Nagababu E, Ramasamy S, Abernethy DR, Rifkind JM. Active Nitric Oxide Produced in the Red Cell under Hypoxic Conditions by Deoxyhemoglobin-mediated Nitrite Reduction. Journal of Biological Chemistry. 2003;278(47):46349–46356. doi: 10.1074/jbc.M307572200. [DOI] [PubMed] [Google Scholar]
- 17.Wang X, Kettenhofen NJ, Hogg N, Gladwin MT. Copper dependence of the biotin switch assay: modified assay for measuring cellular and blood nitrosated proteins. Free Radical Biology and Medicine. 2008;44(7):1362–1372. doi: 10.1016/j.freeradbiomed.2007.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singh RJ, Hogg N, Joseph J, Kalyanaraman B. Mechanism of nitric oxide release from S-nitrosothiols. Journal of Biological Chemistry. 1996;271(31):18596–18603. doi: 10.1074/jbc.271.31.18596. [DOI] [PubMed] [Google Scholar]
- 19.Keszler A, Zhang Y, Hogg N. The Reaction between Nitric Oxide, Glutathione and Oxygen in the Presence and Absence of Protein: How are S-Nitrosothiols Formed? Free Radical Biology and Medicine. 2009;48:55–64. doi: 10.1016/j.freeradbiomed.2009.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Borodulin RR, Kubrina LN, Mikoyan VD, Poltorakov AP, Shvydkiy Vcapital C, Burbaev OD, Serezhenkov VA, Yakhontova ER, Vanin AF. Dinitrosyl iron complexes with glutathione as NO and NO(+) donors. Nitric Oxide. 2013;29:4–16. doi: 10.1016/j.niox.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 21.Lu TT, Tsou CC, Huang HW, Hsu IJ, Chen JM, Kuo TS, Wang Y, Liaw WF. Anionic Roussin’s red esters (RREs) syn-/anti-[Fe(mu-SEt)(NO)2]2(−): the critical role of thiolate ligands in regulating the transformation of RREs into dinitrosyl iron complexes and the anionic RREs. Inorg Chem. 2008;47(13):6040–50. doi: 10.1021/ic800360m. [DOI] [PubMed] [Google Scholar]
- 22.Basu S, Wang X, Gladwin MT, Kim-Shapiro DB. Chemiluminescent detection of S-nitrosated proteins: comparison of tri-iodide, copper/CO/cysteine, and modified copper/cysteine methods. Methods Enzymol. 2008;440:137–156. doi: 10.1016/S0076-6879(07)00808-7. [DOI] [PubMed] [Google Scholar]
- 23.Nagababu E, Ramasamy S, Rifkind JM. S-Nitrosohemoglobin: A mechanism for its formation in conjunction with nitrite reduction by deoxyhemoglobin. Nitric Oxide. 2006;15:20–9. doi: 10.1016/j.niox.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Y, Hogg N. The mechanism of transmembrane S-nitrosothiol transport. Proceedings of the National Academy of Sciences. 2004;101(21):7891–7896. doi: 10.1073/pnas.0401167101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hickok JR, Vasudevan D, Thatcher GR, Thomas DD. Is S-nitrosocysteine a true surrogate for nitric oxide? Antioxid Redox Signal. 2012;17(7):962–8. doi: 10.1089/ars.2012.4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bosworth CA, Toledo J, Zmijewski JW, Li Q, Lancaster JR., Jr The mechanism of cellular nitrosothiol formation from nitric oxide. Free Radical Biology and Medicine. 2008;45(S1):S110. [Google Scholar]
- 27.Broniowska KA, Keszler A, Basu S, Kim-Shapiro DB, Hogg N. Cytochrome c-mediated formation of S-nitrosothiol in cells. Biochem J. 2012;442(1):191–197. doi: 10.1042/BJ20111294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hickok JR, Vasudevan D, Antholine WE, Thomas DD. Nitric oxide modifies global histone methylation by inhibiting Jumonji C domain-containing demethylases. J Biol Chem. 2013;288(22):16004–16015. doi: 10.1074/jbc.M112.432294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsou CC, Liaw WF. Transformation of the {Fe(NO)2}9 dinitrosyl iron complexes (DNICs) into S-nitrosothiols (RSNOs) triggered by acid-base pairs. Chemistry. 2011;17(47):13358–66. doi: 10.1002/chem.201100253. [DOI] [PubMed] [Google Scholar]
- 30.Hogg N. The biological chemistry and clinical potential of S-nitrosothiols. Free Radical Biology and Medicine. 2000;28(10):1478–1486. doi: 10.1016/s0891-5849(00)00248-3. [DOI] [PubMed] [Google Scholar]
- 31.Costanzo S, Menage S, Purrello R, Bonomo RP, Fontecave M. Re-examination of the formation of dinitrosyl-iron complexes during reaction of S-nitrosothiols with Fe(II) Inorg Chim Acta. 2001;318(1–2):1–7. [Google Scholar]
- 32.Diers AR, Keszler A, Hogg N. Detection of S-nitrosothiols. Biochim Biophys Acta. 2014;1840(2):892–900. doi: 10.1016/j.bbagen.2013.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Influence of anion ligands and pH on EPR spectra of DNIC. (A): Spectra of DNIC from FeSO4, Proli/NO in 10 mM PBS (green), 15 mM Hepes (blue), and with cysteine in 15 mM Hepes (Fe:Cys=1:20) (red); pH 7.4 in each case. (B): Same as in (A) with 10 % of 2 N HCl addition to the DNIC samples. (C): Intensity of DNIC (Fe:Cys=1:20, in 15 mM Hepes) spectra as a function of pH. (D): Spectra of DNIC from FeSO4, Proli/NO and cysteine (Fe:Cys=1:20) in 10 mM PBS at pH 7.4 (black), and with 10 % of 2 N HCl (grey).
The DNIC formed from Hepes or PBS in the absence of cysteine exhibited an altered shape of the EPR spectrum, but did not lose paramagnetic intensity. When Cys-DNIC was prepared in PBS, acidification resulted in change of shape, but not intensity (D). The shape change was consistent with a transformation from axial to rhombic symmetry [8] indicative of thiolate ligand displacement by phosphate at low pH. The results suggest that the binding of thiolate is much stronger than that of the sulfonate ligand in Hepes, while phosphate is competitive with thiolate at low pH. The practical conclusion from these observations is that the effect of acidification on DNIC is buffer–dependent, and the paramagnetic nature of these compounds is preserved in phosphate, but decreased in Hepes. For these reasons, all further experiments were performed in Hepes buffer.
Supplementary Figure 2. Decay of mono-DNIC followed by EPR. DNIC was synthesized from 100 μM of FeSO4, and 100 μm of Proli/NO with GSH (Fe:GSH=1:20) in 15 mM Hepes pH=7.4, and the kinetics of DNIC was followed with EPR.
Supplementary Figure 3. Decomposition of DNIC made with excess thiol in the presence of HgCl2 at neutral pH. DNIC was synthesized from 100μM of FeSO4, and 100 μm of Proli/NO with GSH in 15 mM Hepes pH 7.4, Fe:thiol=1:20. Nitrite was detected with reverse phase HPLC/UV-Vis detector.
Supplementary Figure 4. Effect of mercury chloride on mono-DNIC. DNIC (Fe:GSH=1:20) in 15 mM Hepes pH 7.4 was monitored with EPR before and after addition of HgCl2 solution. Spectra were recorded from freshly synthesized DNIC (black), immediately after HgCl2 (red) addition, and 20 min later (blue). Time points are indicated with arrows.
Supplementary Figure 5. Detection of DNIC with triiodide-dependent ozone-based chemiluminescence. DNIC was synthesized from 100 μM of FeSO4, and 100 μm of Proli/NO with cysteine (Fe:Cys=1:2) in 15 mM Hepes pH=7.4. Upper panel: Time dependence of detected NO as GSNO equivalent, Diamonds: no additive, squares: addition of 10% 2N HCl, triangles: addition of 10% sulfanilamide, circles: addition of 10% HgCl2 solution and 10% sulfanilamide. Lower panel: Chemiluminescence traces. Data are reported as mean±standard error, N=3.
Supplementary Figure 6: Nitric oxide formation during mercury-assisted decay of DNIC. Chemiluminescence traces after injection of (1) DNIC (1:2) and (2) DNIC (1:2) after addition of HgCl2, and (3) DNIC (1:20) and (4) DNIC (1:20) after addition of HgCl2 The purge vessel contains only 15 mM Hepes pH 7.4.
Supplementary Figure 7: Chemiluminescence traces of MCF7 cells (control and treated with 100 μm CysNO for 2 h). Quantification of peaks is shown in Figure 4F.
Supplementary Figure 8: EPR spectrum detected in RAW 264.7 macrophages. Cells were treated with LPS for 13 h, and then after scraping into 15 mM Hepes were put into EPR tube and snap-froze in liquid nitrogen.