

Abstract
Objective
To describe the various mechanisms of liver disease in patients with HIV infection, and to link these mechanisms to disease states which may utilise them.
Background
Non-AIDS causes of morbidity and mortality are becoming increasingly common in patients chronically infected with HIV. In particular, liver-related diseases have risen to become one of the leading causes of non-AIDS-related death. A thorough understanding of the mechanisms driving the development of liver disease in these patients is essential when evaluating and caring for these patients.
Methods
The literature regarding mechanisms of liver disease by which different disease entities may cause hepatic injury and fibrosis was reviewed and synthesised.
Results
A number of discrete mechanisms of injury were identified, to include: oxidative stress, mitochondrial injury, lipotoxicity, immune-mediated injury, cytotoxicity, toxic metabolite accumulation, gut microbial translocation, systemic inflammation, senescence and nodular regenerative hyperplasia. Disease states may use any number of these mechanisms to exert their effect on the liver.
Conclusions
The mechanisms by which liver injury may occur in patients with HIV infection are numerous. Most disease states use multiple mechanisms to cause hepatic injury and fibrosis.
Keywords: HIV, hepatitis, liver fibrosis, cirrhosis
Introduction
Since the introduction of highly active antiretroviral therapy (HAART) in the mid-1990s, HIV has become a manageable (though incurable) chronic disease.1 As HIV-infected patients live longer, non-AIDS illnesses are becoming increasingly important sources of morbidity and mortality in the HIV-infected population.1 In particular, liver-related diseases are becoming increasingly prominent in HIV-infected patients. For example, 5%–25% of patients may be coinfected with hepatitis B virus (HBV),2 30% with hepatitis C virus (HCV)3 and 30%–40% of patients may exhibit signs of non-alcoholic fatty liver disease (NAFLD).4 Liver-related disease has been estimated to account for 13%–18% of all-cause mortality in HIV-infected patients and is one of the leading causes of non-AIDS-related death.5–7 Therefore, early and prompt recognition and diagnosis of liver disease in its early stages is an essential component of ensuring continued improvements in morbidity and mortality in this population.
The mechanisms of liver diseases in those living with HIV somewhat depend on the function of the immune system. Data from Center for AIDS Research (CFAR) network of Integrated Clinical Systems study cohort demonstrated that poorly controlled HIV monoinfection is an independent risk factor for liver fibrosis.8 The development of liver fibrosis represents the most clinically relevant final common pathway of hepatic injury. A detailed description of the mechanisms of hepatic fibrogenesis is beyond the scope of this review. However, numerous publications that provide a detailed synthesis of this final pathway are available.9 HIV-infected patients with controlled disease (suppressed HIV RNA and restored CD4 counts) are prone to developing liver diseases from simple and common causes such as alcoholic and NAFLD, viral hepatitis, and ageing in addition to more HIV-specific processes such as HAART-related toxicity and direct injury to the liver by the HIV virus itself. While the mechanisms that cause liver injury and fibrosis in the HIV-infected patient are distinct, the patterns with which they are used by various disease states is not, with a single disease state often using a number different mechanisms in its pathogenesis. With this in mind, a broad understanding of these mechanisms of liver injury, followed by a linking of these mechanisms to the diseases that use them, may be of utility to both clinicians and investigators caring for the HIV-infected patient with liver disease. Typical mechanisms of liver disease in these patients include oxidative stress, mitochondrial injury, lipotoxicity, immune-mediated injury, cytotoxicity, toxic metabolite accumulation, gut microbial translocation, systemic inflammation, senescence and nodular regenerative hyperplasia (figure 1).
Figure 1.
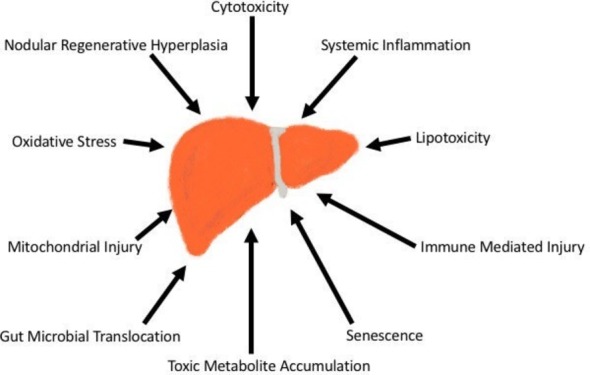
Mechanisms of liver injury in HIV-infected patients.
Oxidative stress
Oxidative stress is a process by which free reactive oxygen species (ROS) cause increased activation of Kupffer cells in the liver. These activated immune cells promote stellate cell activation via nuclear factor kappa-beta (NF-kB) and activator protein 1, leading to increased production of proinflammatory and profibrotic cytokines. Left unchecked, liver damage, fibrosis and cirrhosis may result.10 Alcohol, viral hepatitis, NAFLD, HIV and medications have all been shown to cause liver damage via this mechanism. Alcohol increases concentrations of ROS throughout the body by inducing a systemic hyperhomocysteinaemia.11 In the case of HBV and HCV, the immune response associated with active infection leads to increased concentrations of ROS, with the effect of HCV appearing to be potentiated by comorbid HIV infection.12 In HBV, accumulation of HBV proteins in the endoplasmic reticulum (ER) can lead to DNA damage, and the Negative Regulatory Factor (NEF) protein can affect metabolism of lipid droplets causing an increase in ROS.13 A similar mechanism is seen in NAFLD as a result of increased hepatic free fatty acid (FFA) oxidation.14 The HIV directly activates hepatic stellate cells (HSCs) via the gp120 receptor, activating metabolic pathways resulting in ROS.12 Finally, HIV medications, most notably nucleoside reverse transcriptase inhibitors (NRTIs) such as didanosine can cause mitochondrial toxicity and oxidative stress.15
Mitochondrial injury
Mitochondrial products serve as the primary source of useful energy to the hepatocyte. As such, any process that impairs mitochondrial function can lead to hepatic injury. The primary mechanism by which mitochondrial injury occurs in patients with HIV is increased stress on the ER. This increased ER stress may occur due to a variety of mechanisms, with a final common pathway of triggering increased production of inflammatory cytokines and a mitochondrial ‘alarm response’ that culminates in increased macrophage activation and beta-oxidation of accumulated fatty acids within the liver. In the case of NAFLD, viral hepatitis, HIV and alcohol, this ER stress is initiated by activation of the IRE1/TRAF 2 (Inositol REquiring 1 / TNF receptor-associated factor 2) pathway.11 14
In addition, antiretroviral drugs, in particular older NRTIs and protease inhibitors (PIs), can directly cause mitochondrial toxicity. In the case of the older NRTIs, this is primarily via a mechanism of increased lipid content of cell membranes leading to ER stress and subsequent mitochondrial dysfunction.16 In the case of the PIs (primarily indinavir and ritonavir), a PI-mediated decrease in sacroplasmic/ER calcium ATPase can lead to ER stress by decreasing levels of Ca2+ within the ER. This PI-mediated effect may be potentiated by concomitant alcohol use.11
Lipotoxicity
The primary mechanism of lipotoxicity on the liver is the result of increased FFAs in the liver. Peroxidation of these FFAs leads to increased ROS and ER stress, with resultant fibrosis occurring via mechanisms discussed in corresponding sections above.14 In the HIV-infected patient, a number of mechanisms can be responsible for this increased hepatic FFA accumulation. The prevalence of NAFLD may be up to 30%–40% in patients with HIV.4 The pathophysiology of NAFLD is complex, with accumulation of FFAs in the liver leading to metabolic dysregulation resulting in (and being reinforced by) the development of insulin resistance, dyslipidaemia and obesity, features of the metabolic syndrome that are commonly, but not always, associated with NAFLD.14 While genetic variants such as PNPLA3 polymorphisms17 and endocrinopathies that may predispose a patient to NAFLD or Nonalcoholic steatohepatitis (NASH) may be presumed to be evenly distributed in HIV and non-HIV patients, there are circumstances unique to the HIV-infected patient that may contribute to the development of NAFLD and NASH. For example, medications specific to treatment of HIV may lead to fatty acid accumulation via a variety of pathways. PIs have been noted to alter hepatic FFA composition by a variety of mechanisms such as inducing insulin resistance and dyslipidaemia. PI-mediated alteration of adiponectin and resistin levels leads to increased body fat composition and decreased insulin sensitivity, resulting in increased interleukin (IL)-6 and HSC activation, as well as increased leptin, which in turn upregulates transforming growth factor-beta (TGF-B) production and creation of ROS.12 Additionally, indinavir may increase activity of hydroxymethylglutaryl coenzyme A (HMG CoA) synthase, leading to increased cholesterol in cell membranes, as well as increasing activity of fatty acid synthase, which leads to increasing levels of monounsaturated fatty acids in hepatocytes.16
Immune-mediated injury
The two primary immune cells of the liver are the HSC and the Kupffer cell. Kupffer cells activate local inflammatory responses and hepatocellular repair,18 while HSCs act as the primary drivers of hepatic fibrogenesis and deposition of extracellular matrix proteins.19 20 An upset of balance between the activities of these two cell lines can lead to increased hepatic cell death and fibrosis. Viral agents, primarily HBV, HCV and HIV are the primary diseases that affect the liver via immune-mediated mechanisms. In the case of HBV, the mechanism is primarily via interference of the dendritic cells, via alteration of toll-like receptor signalling leading to inefficient activation of dendritic cells and, by extension, hepatic immune function.13 HCV can cause natural killer cell dysregulation via its E2 protein, leading to overexpression of profibrotic cytokines.21 HIV can interact with HSCs via gp120 producing inappropriate activation22 and increased HSC production of collagen and monocyte chemoattractant protein (MCP-1) (a macrophage chemoattractant).23 HIV also functions to decrease the number of Kupffer cells in the liver, and in doing so significantly impairs the ability of the liver to clear products of microbial translocation from the portal blood.24–26 The imbalance between CD4 and CD8 cells seen in HIV infection can lead to alteration in the cytokine profiles, with reduction in antifibrotic cytokines mediated by a decrease in interferon (IFN)-gamma from Th1 cells and an increase in profibrotic cytokines (IL-4, IL-5, IL-10 and IL-13) due to a relative increase in TH2 signal.27 Finally, autoimmune hepatitis, while rare in the HIV population, exerts its effect by local immune-mediated injury. Literature is limited primarily to case reports, with one reported case of autoimmune hepatitis presenting as a component of immune reconstitution inflammatory syndrome (IRIS).28
IRIS is a unique entity to HIV-infected patients and as such offers a novel avenue for liver injury. IRIS is characterised by paradoxical worsening of a pre-existing infection or the emergence of a new infection after initiation of treatment of HAART, typically within 4–8 weeks after initiation.29 While a number of infectious agents may be implicated, those of most interest to hepatologists are HBV and HCV. In the case of chronic HBV, reconstitution of a previously suppressed immune system may cause a patient to transition from an immune tolerant status characterised by normal alanine aminotransferase (ALT) and minimal liver injury/fibrosis to active hepatitis (chronic or episodic) with resulting liver injury and fibrosis.29 In HIV/HCV coinfection, treatment with HAART may precipitate flares of hepatitis in up to 18% of patients29 and appears to lead to transient increases in liver enzymes, HCV replication rates and viral loads on initiation of HAART.30 Though often self-limited, these flares may lead to hepatic decompensation in patients with pre-existing cirrhosis.29
Cytotoxicity
Direct cytotoxicity as a cause of liver disease in the HIV-infected patient is primarily seen as a result of infection with HCV or HIV. HCV may trigger hepatocyte apoptosis via its E2 protein.31 HIV also has a directly cytopathic effect on hepatocytes, primarily triggering apoptosis via the HIV gp120 protein-receptor signalling pathway.32 These directly cytotoxic effects appear to be enhanced in patients coinfected with HIV and HCV, with each virus having significant effects on the other’s replication, immune dysregulation and cytotoxicity, even with complete control of HIV replication by HAART.12 33 An additional cytotoxic mechanism of injury unique to the HIV/HCV coinfected patient is fibrosing cholestatic hepatitis. First reported in 2002, fibrosing cholestatic HCV is characterised by extensive, dense portal fibrosis and cholestasis with rapid deterioration of liver function and eventual graft failure in patients with HIV/HCV undergoing immunosuppressive therapy after solid organ transplant.34 Incidence and severity are variable. In one relatively large (n=59) cohort, incidence of FCH was 19%, with 9 of the 11 patients affected progressing to graft failure and death within 26.3 months of liver transplant (LT) despite treatment with pegylated IFN and ribavirin.35 Of note, this cohort was collected prior to the widespread use of Direct Acting Antivirals (DAAs) to treat HCV (cohort 1999–2008), and more recent cohorts of patients with post-transplant FCH have shown significantly better outcomes with more modern HCV treatments.36
Accumulation of toxic metabolites
Hepatotoxic metabolites are myriad and responsible for a significant portion of liver disease in non-HIV patients. While acetaminophen is the prototypical agent for toxin-induced liver injury, there are specific mechanisms more unique to the HIV-infected population. Medications used with greater frequency in HIV-infected persons, including both HAART components and medications used to treat opportunistic infections, may lead to accumulation of toxic metabolites. For example, agents such as ketoconazole and erythromycin inhibit cytochrome P450 function in addition to having direct effects on liver chemistries, while drugs such as isoniazid are well known as a direct cause of hepatotoxicity and idiosyncratic liver injury.15 In addition, patients with HIV appear to have increased risk of hypersensitivity reactions when compared with the general population,15 possibly predisposing them to higher likelihood of Drug induced liver injury (DILI) from medications such as trimethoprim-sulfamethoxazole and acyclovir.15 Additionally, drug–drug interactions seen in these patients who are typically burdened by polypharmacy may potentiate hepatotoxic effects of other agents. For example, ribavirin (typically used as a component of treatment in HCV/HIV coinfected patients), if coadministered with didanosine, can decrease didanosine phosphorylation and increase cellular concentrations of didanosine, leading to mitochondrial toxicity.15 In the case of alcohol, PIs appear to cause downregulation of the P450 enzyme CYP2E1 (typically upregulated in response to alcohol consumption to aid in clearance of the toxic alcohol metabolite acetaldehyde), leading to increased susceptibility to alcohol-related hepatotoxicity.37
Gut microbial translocation
Gut microbial translocation leads to hepatic injury primarily via increased hepatic levels of bacterial lipopolysaccharides (LPS) causing hepatic inflammation by one of three mechanisms: (1) recruitment and activation of inflammatory cells (Kupffer cells and HSCs), (2) indirectly inducing systemic immune responses and promoting hepatocyte cell death38 39 and (3) inducing production of proinflammatory cytokines and acute phase reactants such as transforming growth factor beta 1 (TGFB1), IL-6 and IL-10.40 41 As our understanding of gut–liver axis improves, this pathological translocation of products of bacterial degradation is becoming recognised as an increasingly significant mechanism of liver dysfunction in a number of disease states. In the case of HIV, acute infection directly targets gut lymphocyte tissue and preferentially depletes CD22, CD4 and TH17.42–45 In addition, HIV viral proteins increase production of inflammatory cytokines by gut epithelium, leading to increased apoptosis of epithelial cells and breakdown of tight junctions.42 46–48 Of note, this gut barrier dysfunction appears to persist even after successful treatment with HAART, with sCD14 (a surrogate marker for the presence of bacterial LPS) remaining elevated even after systemic markers of infection such as viral load and IL-6 have normalised.48 49
In both cases, the end result is increased intestinal permeability and hepatic exposure to LPS. LPS exerts its effect in the liver primarily by modulating the activity and phenotypes of HSCs via the toll-Like-receptor 4 (TLR4)-mediated signalling pathway.50 Once activated, this HSC TLR4 pathway results in activation of three major transcriptional complexes including nuclear factor kappa beta (NF-KB), activator protein 1 (AP-1) and IFN regulatory factors. The specific actions of these transcriptional complexes are poorly understood, but the net effect of their activation is upregulation of the inflammatory/fibrotic HSC phenotype and potentiation/increased longevity of HSC cell lines. Profibrotic and proinflammatory mediators upregulated by the TLR4 pathway include tumour necrosis factor-alpha (TNF-α), IL-1, IL-6, chemotactic cytokines and macrophage chemoattractants, increased activity of inducible nitric oxygen synthase leading to increased ROS, cyclooxygenase 2 (COX-2) and IFN-gamma.50 HSC cell lines appear to be preserved primarily by alterations in proteins involved in regulating cell-cycle progression and apoptotic thresholds.50
Gut microbial translocation has also been recognised as a possible cause of both alcoholic and non-alcoholic liver disease. In the case of alcohol, both chronic and binge consumption have been shown to increase gut permeability. While the precise mechanism is unclear, it is theorised that alterations in the gut microbiome or bile acid composition may be to blame.51 In the case of NAFLD, there appears to be impairment of gut barrier dysfunction, possibly due to gut dysbiosis (lower concentration of bacteriodetes relative to prevotella) and increased expression of TLR4, TLR9 and TNF receptors.51 52
Systemic inflammation
Systemic inflammation may cause fibrosis via a number of mechanisms including oxidative stress, mitochondrial dysfunction as a result of ER stress or accelerated senescence. States causing systemic inflammation in HIV-infected patients are comprised primarily of chronic infectious processes (HBV, HCV and HIV) and NAFLD, with the specifics of these mechanisms varying with disease state. In NAFLD, decreased insulin sensitivity and increasing levels of FFAs in the liver leads to increased lipid peroxidation, ROS, ER stress and systemic inflammation as well as upregulation of IL-6 and TNF-α, all of which contribute to creating a profibrotic state in the liver.14
Chronic viral infection (HBV, HCV and HIV) generates a systemic inflammatory response via similar mechanisms. NF-kB activity, decreased in hepatitis B and C infections, leads to decreased expression of hepatoprotective genes leading to increased cell death and hepatic inflammation. TGF-B, upregulated in chronic HBV and HCV infection, leads to activation of HSC. Overexpression of TNF-α, IL-1, and IL-6 increase expression of fibrotic phenotypes and impairs fibrosis of HSCs. Additionally, the CD4/CD8 imbalances seen in viral hepatitis and HIV can lead to underexpression of the antifibrotic cytokine IFN-gamma. This decreases IFN-gamma-mediated induction of apoptosis of activated HSCs and serves to potentiate a profibrotic state in the liver.12 13 53 54
Senescence
Senescence is a progressive process by which shortening of telomeres during DNA transcription leads to expression of a senescent cellular phenotype. These senescent cells display numerous derangements, most notably disproportionate secretion of proinflammatory cytokines such as IL-6 and IL-8.55 While this process of cellular ageing is present in patients without HIV, it has been observed that a number of diseases typically associated with ageing including liver disease, renal disease, bone loss, diabetes and non-AIDS cancers occur with greater frequency and at a younger age in HIV-infected patients.56–60 In the HIV-infected patient, normal ageing, possibly accelerated by chronic inflammation related to ongoing infection, is the primary disease state that acts by this mechanism.
Nodular regenerative hyperplasia
Nodular regenerative hyperplasia is a rare condition in which diffuse transformation of liver parenchyma into micronodules without intervening fibrosis leads to non-cirrhotic portal hypertension in patients with HIV.61 62 Liver synthetic function is typically preserved,63 with diagnosis made by biopsy showing histological presence of micronodules not greater than 3 mm without intervening fibrosis.62 Pathophysiology is thought to be due to a ‘two hit’ model in which gut bacterial translocation leads to vascular endothelial damage, stenosis and portal hypertension. The endothelial damage is thought to be either immune mediated or possibly related to direct viral damage by HIV.63 Potentiation by HAART components such as didanosine has also been suggested.63
Summary
Table 1 illustrates the diversity of the mechanisms by which various disease states cause hepatic injury and fibrosis in the HIV-infected patient. While all of the above-mentioned mechanisms are important, some, such as oxidative stress, mitochondrial injury, immune-mediated injury and systemic inflammation, manifest with greater frequency and across a greater spectrum of disease states than others. Finally, as shown in table 1, a single disease state may use a number of mechanisms, with each disease effecting pathogenesis via a complex system of complimentary mechanisms of injury.
Table 1.
Mechanisms of hepatic injury/fibrosis and corresponding disease states
| Alcoholic liver disease | NAFLD | HBV | HCV | Drug effects | HIV-specific effects | Autoimmune disease | Senescence | |
| Oxidative stress | + | ++ | +++ | +++ | + | ++ | ||
| Mitochondrial injury | ++ | + | ++ | ++ | ||||
| Lipotoxicity | +++ | + | ||||||
| Immune mediated | + | + | ++ | +++ | ||||
| Cytotoxic | + | |||||||
| Accumulation of toxic metabolite | +++ | +++ | ||||||
| Gut microbial translocation | + | + | +++ | |||||
| Systemic inflammation | ++ | ++ | ++ | +++ | ||||
| Senescence | +++ | |||||||
| Nodular regenerative hyperplasia | + | +++ |
+, mild contribution; ++, moderate contribution; +++, significant contribution.
HBV, hepatitis B virus; HCV, hepatitis C virus; NAFLD, non-alcoholic fatty liver disease.
While our understanding of the pathophysiology of hepatic fibrosis has advanced significantly, a number of questions regarding the interaction between these various mechanisms, as well as possible regulatory mechanisms and possible therapeutic targets remain lacking. While there have been significant advances in treating HIV, HBV and HCV, there has also been a dramatic increase in the metabolic syndrome in those living with HIV,64 which suggests that NAFLD will become an increasing problem in this patient population.65 Therefore, future studies need to focus on ways to modulate gut microbial translocation, oxidative stress and systemic inflammation. Until then, providers of those with HIV need to be mindful of the various mechanisms of liver injury.
Footnotes
Contributors: MBK drafted manuscript. RKS reviewed and edited manuscript for final content and is primarily responsible for overall content as guarantor.
Competing interests: None declared.
Provenance and peer review: Commissioned; externally peer reviewed.
References
- 1. Palella FJ, Delaney KM, Moorman AC, et al. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV outpatient study investigators. N Engl J Med 1998;338:853–60. doi:10.1056/NEJM199803263381301 [DOI] [PubMed] [Google Scholar]
- 2. Konopnicki D, Mocroft A, de Wit S, et al. Hepatitis B and HIV: prevalence, AIDS progression, response to highly active antiretroviral therapy and increased mortality in the EuroSIDA cohort. AIDS 2005;19:593–601. doi:10.1097/01.aids.0000163936.99401.fe [DOI] [PubMed] [Google Scholar]
- 3. Shepard CW, Finelli L, Alter MJ. Global epidemiology of hepatitis C virus infection. Lancet Infect Dis 2005;5:558–67. doi:10.1016/S1473-3099(05)70216-4 [DOI] [PubMed] [Google Scholar]
- 4. Lemoine M, Serfaty L, Capeau J. From nonalcoholic fatty liver to nonalcoholic steatohepatitis and cirrhosis in HIV-infected patients. Curr Opin Infect Dis 2012;25:10–16. doi:10.1097/QCO.0b013e32834ef599 [DOI] [PubMed] [Google Scholar]
- 5. Palella FJ, Baker RK, Moorman AC, et al. Mortality in the highly active antiretroviral therapy era: changing causes of death and disease in the HIV outpatient study. J Acquir Immune Defic Syndr 2006;43:27–34. doi:10.1097/01.qai.0000233310.90484.16 [DOI] [PubMed] [Google Scholar]
- 6. Smith CJ, Ryom L, Weber R, et al. Trends in underlying causes of death in people with HIV from 1999 to 2011 (D:A:D): a multicohort collaboration. Lancet 2014;384:241–8. doi:10.1016/S0140-6736(14)60604-8 [DOI] [PubMed] [Google Scholar]
- 7. Smith C, Sabin CA, Lundgren JD, et al. Factors associated with specific causes of death amongst HIV-positive individuals in the D:A:D Study. AIDS 2010;24:1537–48. doi:10.1097/QAD.0b013e32833a0918 [DOI] [PubMed] [Google Scholar]
- 8. Kim HN, Nance R, Van Rompaey S, et al. Poorly controlled HIV infection: An independent risk factor for liver fibrosis. J Acquir Immune Defic Syndr 2016;72:437–43. doi:10.1097/QAI.0000000000000992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol 2017;14:397–411. doi:10.1038/nrgastro.2017.38 [DOI] [PubMed] [Google Scholar]
- 10. Fattovich G, Stroffolini T, Zagni I, et al. Hepatocellular carcinoma in cirrhosis: incidence and risk factors. Gastroenterology 2004;127:S35–S50. doi:10.1053/j.gastro.2004.09.014 [DOI] [PubMed] [Google Scholar]
- 11. Kao E, Shinohara M, Feng M, et al. Human immunodeficiency virus protease inhibitors modulate Ca2+ homeostasis and potentiate alcoholic stress and injury in mice and primary mouse and human hepatocytes. Hepatology 2012;56:594–604. doi:10.1002/hep.25702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mastroianni CM, Lichtner M, Mascia C, et al. Molecular mechanisms of liver fibrosis in HIV/HCV coinfection. Int J Mol Sci 2014;15:9184–208. doi:10.3390/ijms15069184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Suhail M, Abdel-Hafiz H, Ali A, et al. Potential mechanisms of hepatitis B virus induced liver injury. World J Gastroenterol 2014;20:12462–72. doi:10.3748/wjg.v20.i35.12462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Machado MV, Diehl AM. Pathogenesis of nonalcoholic fatty liver disease : Zakim and boyer’s hepatology: Elsevier Health Sciences, 2016:369–90. [Google Scholar]
- 15. Sherman KE, Sterling RK. HIV and the liver : Zakim and boyer’s hepatology: Elsevier Health Sciences, 2016:553. [Google Scholar]
- 16. Williams K, Rao YP, Natarajan R, et al. Indinavir alters sterol and fatty acid homeostatic mechanisms in primary rat hepatocytes by increasing levels of activated sterol regulatory element-binding proteins and decreasing cholesterol 7alpha-hydroxylase mRNA levels. Biochem Pharmacol 2004;67:255–67. doi:10.1016/j.bcp.2003.08.044 [DOI] [PubMed] [Google Scholar]
- 17. Valenti L, Bugianesi E, Pajvani U, et al. Nonalcoholic fatty liver disease: cause or consequence of type 2 diabetes? Liver Int 2016;36:1563–79. doi:10.1111/liv.13185 [DOI] [PubMed] [Google Scholar]
- 18. Dixon LJ, Barnes M, Tang H, et al. Kupffer cells in the liver. Compr Physiol 2013;3:785–97. doi:10.1002/cphy.c120026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev 2008;88:125–72. doi:10.1152/physrev.00013.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hui AY, Friedman SL. Molecular basis of hepatic fibrosis. Expert Rev Mol Med 2003;5:1–23. doi:10.1017/S1462399403005684 [DOI] [PubMed] [Google Scholar]
- 21. Glässner A, Eisenhardt M, Kokordelis P, et al. Impaired CD4⁺ T cell stimulation of NK cell anti-fibrotic activity may contribute to accelerated liver fibrosis progression in HIV/HCV patients. J Hepatol 2013;59:427–33. doi:10.1016/j.jhep.2013.04.029 [DOI] [PubMed] [Google Scholar]
- 22. Del Cornò M, Cappon A, Donninelli G, et al. HIV-1 gp120 signaling through TLR4 modulates innate immune activation in human macrophages and the biology of hepatic stellate cells. J Leukoc Biol 2016;100:599–606. doi:10.1189/jlb.4A1215-534R [DOI] [PubMed] [Google Scholar]
- 23. Tuyama AC, Hong F, Saiman Y, et al. Human immunodeficiency virus (HIV)-1 infects human hepatic stellate cells and promotes collagen I and monocyte chemoattractant protein-1 expression: implications for the pathogenesis of HIV/hepatitis C virus-induced liver fibrosis. Hepatology 2010;52:612–22. doi:10.1002/hep.23679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Housset C, Boucher O, Girard PM, et al. Immunohistochemical evidence for human immunodeficiency virus-1 infection of liver Kupffer cells. Hum Pathol 1990;21:404–8. doi:10.1016/0046-8177(90)90202-G [DOI] [PubMed] [Google Scholar]
- 25. Hufert FT, Schmitz J, Schreiber M, et al. Human kupffer cells infected with HIV-1 in vivo. J Acquir Immune Defic Syndr 1993;6:772–7. [PubMed] [Google Scholar]
- 26. Schmitt MP, Steffan AM, Gendrault JL, et al. Multiplication of human immunodeficiency virus in primary cultures of human Kupffer cells-possible role of liver macrophage infection in the physiopathology of AIDS. Res Virol 1990;141:143–52. doi:10.1016/0923-2516(90)90016-C [DOI] [PubMed] [Google Scholar]
- 27. Mehal WZ, Friedman SL. The Role of Inflammation and Immunity in the Pathogenesis of Liver Fibrosis Liver Immunology. Clifton, NJ, USA: Humana Press, 2007:111–21. [Google Scholar]
- 28. Roszkiewicz J, Smolewska E. Kaleidoscope of autoimmune diseases in HIV infection. Rheumatol Int 2016;36:1481–91. doi:10.1007/s00296-016-3555-7 [DOI] [PubMed] [Google Scholar]
- 29. Novak RM, Richardson JT, Buchacz K, et al. Immune reconstitution inflammatory syndrome: incidence and implications for mortality. AIDS 2012;26:721–30. doi:10.1097/QAD.0b013e3283511e91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sherman KE, Guedj J, Shata MT, et al. Modulation of HCV replication after combination antiretroviral therapy in HCV/HIV co-infected patients. Sci Transl Med 2014;6:246ra98 doi:10.1126/scitranslmed.3008195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liberto MC, Zicca E, Pavia G, et al. Virological mechanisms in the coinfection between HIV and HCV. Mediators Inflamm 2015;2015:1–7. doi:10.1155/2015/320532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Blackard JT, Sherman KE. HCV/ HIV co-infection: time to re-evaluate the role of HIV in the liver? J Viral Hepat 2008;15:323–30. doi:10.1111/j.1365-2893.2008.00970.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hernandez MD, Sherman KE. HIV/hepatitis C coinfection natural history and disease progression. Curr Opin HIV AIDS 2011;6:478–82. doi:10.1097/COH.0b013e32834bd365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rosenberg PM, Farrell JJ, Abraczinskas DR, et al. Rapidly progressive fibrosing cholestatic hepatitis-hepatitis C virus in HIV coinfection. Am J Gastroenterol 2002;97:478–83. doi:10.1111/j.1572-0241.2002.05459.x [DOI] [PubMed] [Google Scholar]
- 35. Antonini TM, Sebagh M, Roque-Afonso AM, et al. Fibrosing cholestatic hepatitis in HIV/HCV co-infected transplant patients-usefulness of early markers after liver transplantation. Am J Transplant 2011;11:1686–95. doi:10.1111/j.1600-6143.2011.03608.x [DOI] [PubMed] [Google Scholar]
- 36. Giard JM, Terrault NA. Severe Cholestatic Hepatitis C in Transplant Recipients: No Longer a Threat to Graft Survival. Clin Gastroenterol Hepatol 2015;13:2002–4. doi:10.1016/j.cgh.2015.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Manzo-Avalos S, Saavedra-Molina A. Cellular and mitochondrial effects of alcohol consumption. Int J Environ Res Public Health 2010;7:4281–304. doi:10.3390/ijerph7124281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sacchi P, Cima S, Corbella M, et al. Liver fibrosis, microbial translocation and immune activation markers in HIV and HCV infections and in HIV/HCV co-infection. Dig Liver Dis 2015;47:218–25. doi:10.1016/j.dld.2014.11.012 [DOI] [PubMed] [Google Scholar]
- 39. Page EE, Nelson M, Kelleher P. HIV and hepatitis C coinfection: pathogenesis and microbial translocation. Curr Opin HIV AIDS 2011;6:472–7. doi:10.1097/COH.0b013e32834bbc71 [DOI] [PubMed] [Google Scholar]
- 40. Szabo G. Gut-liver axis in alcoholic liver disease. Gastroenterology 2015;148:30–6. doi:10.1053/j.gastro.2014.10.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wright SD, Ramos RA, Tobias PS, et al. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science 1990;249:1431–3. doi:10.1126/science.1698311 [DOI] [PubMed] [Google Scholar]
- 42. Dandekar S, George MD, Bäumler AJ. Th17 cells, HIV and the gut mucosal barrier. Curr Opin HIV AIDS 2010;5:173–8. doi:10.1097/COH.0b013e328335eda3 [DOI] [PubMed] [Google Scholar]
- 43. Mehandru S, Poles MA, Tenner-Racz K, et al. Primary HIV-1 infection is associated with preferential depletion of CD4+ T lymphocytes from effector sites in the gastrointestinal tract. J Exp Med 2004;200:761–70. doi:10.1084/jem.20041196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kim CJ, McKinnon LR, Kovacs C, et al. Mucosal Th17 cell function is altered during HIV infection and is an independent predictor of systemic immune activation. J Immunol 2013;191:2164–73. doi:10.4049/jimmunol.1300829 [DOI] [PubMed] [Google Scholar]
- 45. Kim CJ, Nazli A, Rojas OL, et al. A role for mucosal IL-22 production and Th22 cells in HIV-associated mucosal immunopathogenesis. Mucosal Immunol 2012;5:670–80. doi:10.1038/mi.2012.72 [DOI] [PubMed] [Google Scholar]
- 46. Canani RB, Cirillo P, Mallardo G, et al. Effects of HIV-1 Tat protein on ion secretion and on cell proliferation in human intestinal epithelial cells. Gastroenterology 2003;124:368–76. doi:10.1053/gast.2003.50056 [DOI] [PubMed] [Google Scholar]
- 47. Nazli A, Chan O, Dobson-Belaire WN, et al. Exposure to HIV-1 directly impairs mucosal epithelial barrier integrity allowing microbial translocation. PLoS Pathog 2010;6:e1000852 doi:10.1371/journal.ppat.1000852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hattab S, Guihot A, Guiguet M, et al. Comparative impact of antiretroviral drugs on markers of inflammation and immune activation during the first two years of effective therapy for HIV-1 infection: an observational study. BMC Infect Dis 2014;14:122 doi:10.1186/1471-2334-14-122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wada NI, Jacobson LP, Margolick JB, et al. The effect of HAART-induced HIV suppression on circulating markers of inflammation and immune activation. AIDS 2015;29:463–71. doi:10.1097/QAD.0000000000000545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Guo J, Friedman SL. Toll-like receptor 4 signaling in liver injury and hepatic fibrogenesis. Fibrogenesis Tissue Repair 2010;3:21 doi:10.1186/1755-1536-3-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Betrapally NS, Gillevet PM, Bajaj JS. Changes in the Intestinal Microbiome and Alcoholic and Nonalcoholic Liver Diseases: Causes or Effects? Gastroenterology 2016;150:1745–55. doi:10.1053/j.gastro.2016.02.073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Farhadi A, Gundlapalli S, Shaikh M, et al. Susceptibility to gut leakiness: a possible mechanism for endotoxaemia in non-alcoholic steatohepatitis. Liver Int 2008;28:1026–33. doi:10.1111/j.1478-3231.2008.01723.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bertoletti A, Lucifora J, Zoulim F. Virology and pathogenesis of hepatitis B : Zakim and boyer’s hepatology : Elsevier health sciences, 2016:369–90. [Google Scholar]
- 54. Tang L, Meissner EG, Kottilil S. Virology and pathogenesis of hepatitis B : Zakim and boyer’s hepatology : Elsevier health sciences, 2016:369–90. [Google Scholar]
- 55. Dubrow R, Silverberg MJ, Park LS, et al. HIV infection, aging, and immune function: implications for cancer risk and prevention. Curr Opin Oncol 2012;24:506–16. doi:10.1097/CCO.0b013e328355e131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Aberg JA. Aging, inflammation, and HIV infection. Top Antivir Med 2012;20:101–5. [PMC free article] [PubMed] [Google Scholar]
- 57. Abraham AG, Althoff KN, Jing Y, et al. End-stage renal disease among HIV-infected adults in North America. Clin Infect Dis 2015;60:941–9. doi:10.1093/cid/ciu919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Althoff KN, McGinnis KA, Wyatt CM, et al. Comparison of risk and age at diagnosis of myocardial infarction, end-stage renal disease, and non-AIDS-defining cancer in HIV-infected versus uninfected adults. Clin Infect Dis 2015;60:627–38. doi:10.1093/cid/ciu869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ipp H, Zemlin AE, Erasmus RT, et al. Role of inflammation in HIV-1 disease progression and prognosis. Crit Rev Clin Lab Sci 2014;51:98–111. doi:10.3109/10408363.2013.865702 [DOI] [PubMed] [Google Scholar]
- 60. Vance DE, Mugavero M, Willig J, et al. Aging with HIV: a cross-sectional study of comorbidity prevalence and clinical characteristics across decades of life. J Assoc Nurses AIDS Care 2011;22:17–25. doi:10.1016/j.jana.2010.04.002 [DOI] [PubMed] [Google Scholar]
- 61. Chang PE, Miquel R, Blanco JL, et al. Idiopathic portal hypertension in patients with HIV infection treated with highly active antiretroviral therapy. Am J Gastroenterol 2009;104:1707–14. doi:10.1038/ajg.2009.165 [DOI] [PubMed] [Google Scholar]
- 62. Wanless IR. Micronodular transformation (nodular regenerative hyperplasia) of the liver: a report of 64 cases among 2,500 autopsies and a new classification of benign hepatocellular nodules. Hepatology 1990;11:787–97. doi:10.1002/hep.1840110512 [DOI] [PubMed] [Google Scholar]
- 63. Sood A, Castrejón M, Saab S. Human immunodeficiency virus and nodular regenerative hyperplasia of liver: A systematic review. World J Hepatol 2014;6:55–63. doi:10.4254/wjh.v6.i1.55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Worm SW, Friis-Møller N, Bruyand M, et al. High prevalence of the metabolic syndrome in HIV-infected patients: impact of different definitions of the metabolic syndrome. AIDS 2010;24:427–35. doi:10.1097/QAD.0b013e328334344e [DOI] [PubMed] [Google Scholar]
- 65. Lemoine M, Serfaty L, Capeau J. From nonalcoholic fatty liver to nonalcoholic steatohepatitis and cirrhosis in HIV-infected patients: diagnosis and management. Curr Opin Infect Dis 2012;25:10–16. doi:10.1097/QCO.0b013e32834ef599 [DOI] [PubMed] [Google Scholar]