

Abstract
Tumor suppressor p53 plays a central role in tumor suppression. p53 is the most frequently mutated gene in human cancer, and over half of human cancers contain p53 mutations. Majority of p53 mutations in cancer are missense mutations, leading to the expression of full-length mutant p53 protein. While the critical role of wild type p53 in tumor suppression has been firmly established, mounting evidence has demonstrated that many tumor-associated mutant p53 proteins not only lose tumor suppressive function of wild type p53, but also gain new activities to promote tumorigenesis independently of wild type p53, termed gain-of-function. Mutant p53 protein often accumulates to very high levels in tumors, contributing to malignant progression. Recently, mutant p53 has become an attractive target for cancer therapy. Further understanding of the mechanisms underlying mutant p53 protein accumulation and gain-of-function will accelerate the development of targeted therapies for human cancer harboring mutant p53. In this review, we summarize the recent advances in the studies on mutant p53 protein accumulation and gain-of-function as well as targeted therapies for mutant p53 in human cancer.
Keywords: p53, mutant p53, protein stabilization, gain-of-function, targeted therapy
Graphical abstract
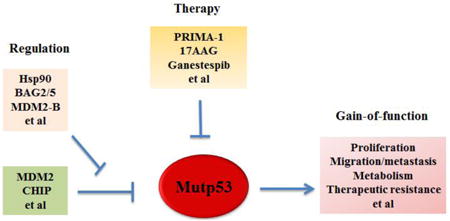
Introduction of mutant p53
Tumor suppressor p53 plays a central role in human cancer [1-4]. p53 is a transcription factor, and mainly exerts its role in tumor suppression through its transcriptional regulation of its downstream target genes [1-4]. p53 has a very short protein half-life and its protein levels are low in normal cells and tissues under non-stressed conditions. p53 can sense and be activated by a wide variety of extra- and intra-cellular stress signals, including DNA damage, oncogene activation, hypoxia, ROS level change, etc [1-4]. These stress signals activate p53 protein through post-translational modifications, including phosphorylation, acetylation, methylation, etc., to stabilize p53 protein and rapidly increase p53 protein levels in cells [1-5]. Upon activation, p53 binds to the p53 consensus DNA-binding elements usually located in the promoter and/or introns of its target genes to selectively regulate the transcriptional expression of its target genes in a cell/tissue type-specific and stress signal type-specific manner. These products of p53 target genes are involved in a variety of important cellular processes, including apoptosis, cell cycle arrest, senescence, DNA repair, anti-oxidant function and metabolic regulation, contributing to the tumor suppressive function of p53 [1-4, 6]. As the “guardian of the genome”, p53 ensures the replication fidelity and genomic stability to prevent tumor initiation and progression. Loss of p53 function is often a prerequisite for tumor initiation and progression, which has been most clearly demonstrated by the increased cancer risk in Li-Fraumeni syndrome patients with germline p53 mutations and p53 knockout mouse models [7-10]. Approximately 50% of Li-Fraumeni syndrome patients develop different types of cancers, including sarcoma, breast cancer, and brain cancer, by the age of 30, and almost 100% of p53 knockout mice develop tumors, primarily lymphomas and sarcomas, by the age of 10 months [7-10]. In human cancer, p53 is the most frequently mutated gene. Around 50% of human cancers harbor p53 mutations [11-14]. In addition to p53 mutations, p53 function is frequently attenuated and the p53 signaling is dysfunctional in human cancers through multiple mechanisms, including overexpression and/or amplification of different p53 negative regulators, such as MDM2, MDM4, Cop1, Pirh2, Trim32 and LIF [2, 15-21].
While the critical role of wild type p53 in tumor suppression has been firmly established, mounting evidence has demonstrated that many tumor-associated mutant p53 proteins not only lose tumor suppressive function of wild type p53, but also gain tumor-promoting function through dominant-negative regulation of remaining wild type p53 or independently of wild type p53 [11, 22, 23]. While many other tumor suppressor genes that are predominantly inactivated through deletion or truncating mutations in cancer, majority of p53 mutations in human cancer are missense mutations, which leads to the expression of full-length mutant p53 proteins with the substitution of a single amino acid [11, 22, 23]. Further, majority of p53 mutations cluster in the DNA binding domain (DBD), a region which is required for wild type p53 to bind to its target genes and function as a transcription factor. Interestingly, more than 25% of p53 mutations in human cancers fall within 6 “hotspots”, including amino acid residues R175, G245, R248, R249, R273 and R282, although p53 mutations have been found in almost every codon within the p53 DBD in cancer. p53 mutations have two major categories: DNA-contact mutations (e.g. R248 and R273) and conformational mutations (e.g. R175, G245, R282 and R249) [11-13, 24]. DNA contact mutations occur in the amino acid residues that make direct contact with p53 target DNA sequences and are critical for DNA binding, which impair the transcriptional activity of wild type p53 without dramatically affecting the conformation of the p53 protein. Conformational mutations usually result in a more dramatic alteration of p53 protein structure compared with DNA contact mutants. These p53 mutations lead to the loss of affinity to majority of p53 consensus DNA-binding elements in wild type p53 target genes. In addition, many mutant p53 proteins exhibit dominant-negative (DN) effects on the remaining wild type p53 allele. Mutant p53 proteins can form a tetramer with wild type p53, which can block the function of the remaining wild type p53 in tumor suppression [25-28]. In addition to its DN effect on wild type p53, mutant p53 proteins often exhibit oncogenic activity in cells lacking wild type p53. p53 missense mutations in human cancer are usually followed by loss of heterozygosity (LOH) at the corresponding locus, suggesting that there is a selective advantage conferred by losing the remaining wtp53, even after one allele has been mutated [11, 23, 29]. The discovery of p53 as a “proto-oncogene” shortly after p53 discovery was in part due to the cloning of mutant p53 cDNA from cancer cells; ectopic expression of mutant p53 promoted cell transformation and increased tumorigenicity in p53-null cells [4, 30-32]. While these early studies masked the true function of wild type p53, they hinted the gain-of-function (GOF) of mutant p53 in tumorigenesis.
Mutant p53 gain-of-function
Mutant p53 GOF has been demonstrated by numerous cell-based experiments. This set of evidence has been mainly obtained through ectopically expressing mutant p53 in p53-null human tumor cells or knockdown of endogenous mutant p53 in cells containing only one allele of mutant p53 [11, 22, 23, 33]. So far, many mutant p53 GOFs have been identified, including promoting tumor cell proliferation, survival, migration and invasion, enhancing chemoresistance, disrupting proper tissue architecture, and promoting cancer metabolism (both Warburg effect and lipid metabolism) [11, 22, 23, 33-37] (Figure 1). Mutant p53 GOF has also been clearly demonstrated in mutant p53 knock-in mouse models, engineered by introducing tumor-associated hotspot p53 mutations into the endogenous Trp53 locus in mice using homologous recombination [38-40]. These mice develop a different tumor spectrum, more aggressive tumors or earlier tumors compared with p53 null mice [38, 39, 41]. In human Li-Fraumeni syndrome patients, those with germline missense p53 mutations have an early age onset of cancer development compared with those with germline p53 deletion mutations [8]. In many different types of human cancer, p53 mutations are frequently associated with more aggressive cancer, poorer response towards therapy and prognosis [12, 42-44]. It needs to point out that recent studies also showed that different p53 mutants can exhibit different GOF phenotypes or have same GOF phenotypes but to different extents [11, 12, 40, 45]. Collectively, these results from cell-based experiments, mouse models and human studies support the concept of mutant p53 GOF.
Figure 1. Mutant p53 GOF in tumorigenesis.
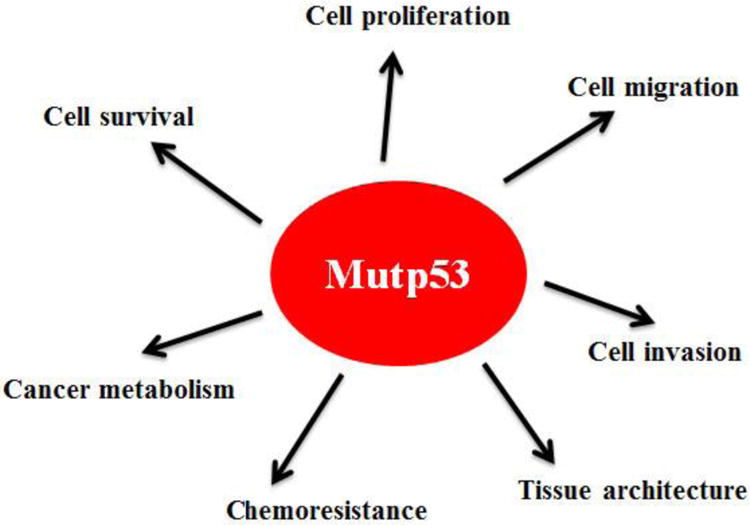
Mutant p53 (mutp53) regulates many cellular processes, including cell proliferation, cell migration, cell invasion, cell survival, cell metabolism, chemoresistance and tissue architecture, to promote tumor progression independently of wild type p53.
Mutant p53 in general can no longer recognize and bind to p53 DNA binding elements and lose the transcriptional activity towards wild type p53-regulated target genes [11, 22, 23]. However, many tumor-associated mutant p53 proteins are mainly localized in the nucleus and can regulate the transcription of some genes through mechanisms different from wild type p53. For example, mutant p53 can bind to TAp63 and TAp73 and inhibit their transcriptional activities [46-49]. TAp63 and TAp73 are two important p53 family members which can regulate some of wild type p53 target genes and compensate for some of p53 tumor suppressive functions [50, 51]. Mutant p53 can also interact with other transcriptional factors and cofactors, including NF-Y, SREBP, VDR, Sp1, ETS2 and NRF2, and enhance or decrease their transcription activities to promote tumor progression [11, 52-56]. Mutant p53 can also participate in the regulation of chromatin structure to regulate the expression of some genes. For instance, mutant p53 was reported to interact with chromatin remodeling complex SWI/SNF to cooperate their chromatin remodeling function [57]. Mutant p53 can also recognize and bind with high affinity to DNA regions of matrix attachment region DNA elements (MARs), which have high potential for DNA base-unpairing [58]. In addition to its nuclear function, mutant p53 has cytoplasmic function that contributes to its GOF. For example, mutant p53 promotes translocation of glucose transporter 1 (GLUT1) to plasma membrane to stimulate the Warburg effect, a characterized metabolic change in tumor cells and a key contributor to tumor development [37].
Mutant p53 protein accumulation in tumors
One unique feature of mutant p53 is that mutant p53 proteins often become stable and accumulate to very high levels in tumors [11, 22, 25, 59]. Based on this characteristic of mutant p53 protein, positive immunohistochemical (IHC) staining of p53 in tumor tissues has been widely used as a surrogate for p53 mutation detection [11, 60, 61]. Importantly, mutant p53 accumulation in tumors is critical for mutant p53 to exert its GOF in tumorigenesis and contributes to more advanced tumors [11, 22, 25, 62]. Destabilizing mutant p53 can greatly reduce mutant p53 GOF in tumorigenesis, which is a promising strategy for cancer therapy that is currently under active investigation [11, 22, 25, 63, 64].
The mechanism for mutant p53 protein accumulation in tumors is not well-understood. Under non-stressed conditions, wild type p53 protein levels are kept low in cells through efficient proteasomal degradation mainly mediated by E3 ubiquitin ligase MDM2 [5, 15, 20, 65]. MDM2 is a key negative regulator for wild type p53; MDM2 binds to and degrades wild type p53 through ubiquitination. Meanwhile, MDM2 itself is a p53-regulated gene. Thus, MDM2-forms a negative feedback loop with wild type p53 to tightly regulate wild type p53 protein levels and functions [5, 15, 20]. Mutant p53 can no longer induce MDM2. MDM2 interacts with multiple regions of wild type p53. The conformation change of p53 protein due to mutations may decrease the interaction efficiency of MDM2 towards these regions. The overall efficiency of mutant p53 ubiquitination is reduced compared with that of wild type p53 [66]. The inability of mutant p53 to induce MDM2 and reduced ability of MDM2 to degrade mutant p53 can disrupt the MDM2-p53 loop, and had long been thought to be the main underlying mechanism for the accumulation of mutant p53 protein in tumors [11, 22, 25]. However, this notion has been challenged by recent studies showing that mutant p53 proteins are accumulated exclusively in tumor tissues but not normal tissues in mutant p53 knock-in mice [38, 39]. Furthermore, MDM2 loss in mutant p53 knock-in mice leads to mutant p53 accumulation in normal tissues, which in turn promotes mutant p53 GOF in tumor development and reduces survival [62]. These observations provide in vivo evidence that: 1) MDM2 can negatively regulate mutant p53 protein levels; and 2) mutant p53 protein accumulation promotes mutant p53 GOF. Consistently, recent studies showed that MDM2 can ubiquitinate and degrade mutant p53 in vitro [66, 67]. These results suggest that MDM2 maintains mutant p53 protein levels low in normal tissues, whereas some changes in tumors disrupt MDM2-mediated mutant p53 degradation, thereby leading to mutant p53 protein accumulation in tumors.
Inhibition of MDM2-mediated mutant p53 degradation by MDM2 short isoforms
MDM2 has multiple spliced short isoforms in addition to the full-length MDM2 [68-70]. MDM2 isoforms are frequently overexpressed in different types of human tumors [67-74]. Overexpression of MDM2 short isoforms often correlated with tumor malignancy, metastasis, and poor prognosis in human cancer, including colorectal, breast, lung and brain cancers [68-74]. A common feature of majority of MDM2 isoforms is that they lack the central region which contains several critical function domains for p53 regulation, including the p53 binding domain and nuclear localization signal (NLS) [68, 69]. Unlike full-length MDM2, MDM2 isoforms cannot bind to p53 (due to the lack of p53 binding domain), and is mainly localized in the cytoplasm (due to the lack of NLS), and cannot degrade p53 [68, 69]. A recent study from our group revealed that MDM2 isoform B (MDM2-B), the most frequently overexpressed MDM2 isoform in human tumors, can promote mutant p53 protein accumulation in cancer [67]. MDM2-B interacts with full-length MDM2 and sequesters full-length MDM2 in the cytoplasm, which in turn reduces the interaction of full-length MDM2 with mutant p53 in the nucleus. The interaction of MDM2 isoform with full-length MDM2 also disrupts the dimerization and oligomerization of MDM2, which is critical for its activity to degrade mutant p53 [67]. Therefore, MDM2 isoforms inhibit MDM2-mediated mutant p53 protein degradation, which in turn promotes mutant p53 accumulation and GOF in tumorigenesis [67]. In a cohort of human colorectal tumor samples, we observed a significant correlation of MDM2-B overexpression with mutant p53 accumulation [67]. Interestingly, in mutant p53 knock-in mice, the overexpression of a MDM2 short isoform similar to human MDM2 short isoforms in structure was observed in the majority of tumors that display mutant p53 protein accumulation [67]. This mouse MDM2 short isoform also promotes mutant p53 accumulation and the growth of xenograft tumors in a largely mutant p53-dependent manner [67]. Thus, overexpression of MDM2 isoforms in tumors is an important mechanism to compromise the function of MDM2 in mutant p53 degradation, promoting mutant p53 accumulation and GOF in tumorigenesis (Figure 2).
Figure 2. Regulation of mutant p53 accumulation and GOF.
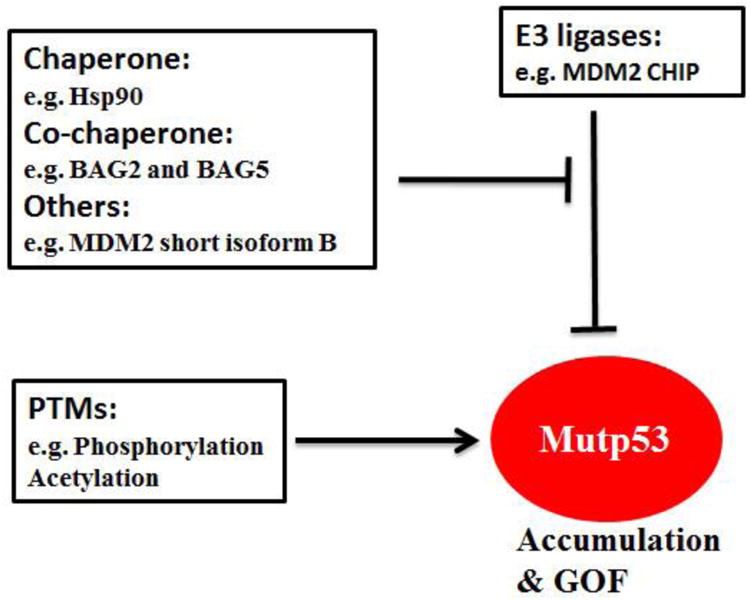
Hsp90, BAG2, BAG5 and MDM2 short isoform B inhibit ubiquitination and degradation of mutant p53 (mutp53) by E3 ligases MDM2 and CHIP. In addition, mutant p53 can also be stabilized by posttranslational modifications (PTMs), including phosphorylation and acetylation. However, specific phosphorylations can also down-regulate mutant p53 protein levels and activity.
Regulation of mutant p53 stabilization by chaperon proteins
Heat shock chaperone proteins stabilize newly synthesized proteins to ensure correct folding and help to refold damaged proteins [75, 76]. Hsp90, a family member of heat shock chaperone proteins, is a core protein of chaperone machinery and plays a critical role in the conformational stabilization of many mutant onco-proteins [77, 78]. Hsp90 is frequently overexpressed in many human tumors [77, 78]. Hsp90 can interact with mutant p53 to inhibit the ubiquitination and degradation of mutant p53 [79-82] (Figure 2). Disruption of the Hsp90-mutant p53 interaction or employing SAHA to inhibit HDAC6, a positive regulator of Hsp90, destabilizes mutant p53 and promotes mutant p53 degradation [63, 79]. In addition to MDM2, a chaperone-dependent E3 ubiquitin ligase, CHIP (C-terminus of Hsp70-interacting protein) is another major E3 ubiquitin ligase for mutant p53 ubiquitination and degradation [66, 79, 83, 84]. Hsp90 inhibits the ubiquitination and degradation of mutant p53 mediated by both MDM2 and CHIP [84, 85]. Unlike Hsp90, Hsp70 partially inhibits MDM2-mediated mutant p53 degradation and promotes CHIP-mediated mutant p53 degradation [84, 85].
Regulation of mutant p53 stabilization by BAG family proteins
BAG family proteins are a group of multifunctional proteins containing six family members (BAG1-BAG6) [86, 87]. They all contain at least one BAG (Bcl-2-associated athanogene) domain. BAG family proteins function as adapter or co-chaperone proteins through the BAG domain which mediates direct interaction with the ATPase domain of Hsp70/Hsc70 molecular chaperones. BAG proteins interact with a variety of proteins and take part in diverse cellular processes, including cell division, cell death and differentiation [86, 87]. Recent studies from our group revealed that BAG2 and BAG5, two BAG family proteins, preferentially interact with mutant p53 proteins through the BAG domain [83, 88]. This interaction inhibits mutant p53 degradation to increase mutant p53 protein stabilization and accumulation, which in turn promotes mutant p53 GOF in tumorigenesis [83, 88] (Figure 2). Furthermore, BAG2 and BAG5 proteins exhibit a cooperative effect on promoting mutant p53 protein accumulation and GOF in cancer cells [83]. Both BAG2 and BAG5 proteins are overexpressed in many types of human cancers, including breast cancer, lung cancer, skin cancer and colorectal cancer, suggesting that their overexpression contributes to mutant p53 accumulation in cancer [83, 88].
Regulation of mutant p53 levels and activity by post-translational modifications
Post-translational modifications are critical for proteins to exert their biological functions. The regulation of the stability and activity of wild type p53 in response to many stress signals is mainly achieved by post-translational modifications, including phosphorylation, acetylation, methylation, sumoylation, etc., which have been extensively summarized by many excellent reviews [2, 3, 5, 89]. For example, genotoxic stress such as UV and ionizing radiation (IR) leads to p53 phosphorylation on Serine 15 and Serine 20, which contributes to p53 stabilization and activation through inhibition of MDM2-mediated ubiquitination and degradation [90, 91]. It has been reported that there are ∼60 residues of wild type 53 that can be post-translationally modified [92]. Majority of these post-translational modification sites are not located in the p53 DBD and the mutation of these post-translational modification sites is relatively infrequent. Therefore, majority of post-translational modification sites remain wild type in mutant p53 and the post-translational modifications of p53 are non-discriminatory between wild type and mutant p53. A number of reports showed that many stress conditions that cause wild type p53 stabilization by post-translational modifications, such as UV, IR and oxidative stress, can also lead to mutant p53 stabilization and accumulation [62, 93, 94] (Figure 2). DNA damage was also reported to lead to the phosphorylation of mutant p53 on Serine 15 and Serine 20 [79, 94, 95]. In some cancer cells, there is constitutive phosphorylation of mutant p53 on Serine 15, which may protect mutant p53 from MDM2-mediated degradation and contribute to mutant p53 stabilization and accumulation in these cells [96].
Although the post-translational modification sites did not change too much between wild type and mutant p53, some post-translational modifications of mutant p53 may regulate mutant p53 levels and functions through mechanisms depending on specific cellular oncogenic context. For example, oncogenic Ras was reported to phosphorylate mutant p53 at serine 6 and serine 9. These phosphorylation modifications promote the formation of mutant p53/Smads complex. In turn, Smads serve as essential platforms to induce the assembly of mutant p53/p63 complex and inhibit the function of p63 in preventing metastasis [53]. Plk2 (polo-like kinase 2) is a member of polo-like kinase family that is frequently overexpressed in human cancer [97, 98]. Plk2 can phosphorylate mutant p53 at Threonine 377 to promote the mutant p53-p300 interaction and enhance the transcriptional activity of mutant p53, which in turn enhances mutant p53 GOF in tumorigenesis [99]. The promyelocytic leukemia (PML) can interact with mutant p53 and facilitate the phosphorylation of mutant p53 at Serine 20 and Threonine 18 to enhance the transcriptional activities of mutant p53 and promote its GOF [100]. Phosphorylation of p53 at Serine 392 can stabilize p53 tetramer formation. It was reported that Serine 392 phosphorylation frequently occurs in mutant p53 proteins in urothelial transitional cell carcinomas (TCCs), which may promote the dominant-negative effects of mutant p53 by forming hetero-oligomerization with wild type p53 [101]. Interestingly, mutant p53 Serine 392 phosphorylation levels are associated with advanced tumor stage, high tumor grade and poor prognosis in ovarian cancer patients [102].
Besides phosphorylation, acetylation on lysine residues is another important post-translational modification for p53 to regulate its levels and function [103, 104]. Acetylation of Lys320, Lys373 and Lys382 have been observed in mutant p53 [105]. Deacetylation of mutant p53 by glucose restriction or activation of SIRT1 can reduce mutant p53 protein levels [106-108]. These observations suggest a role of acetylation in regulating stability of mutant p53 protein.
It is worth noting that there are several post-translational modification sites located in the p53 DBD, including Lys132, Thr155, Lys 164, Ser215, Glu258, Asp259 and Cys277. Although they do not belong to tumor-associated p53 mutation hotspots, they have relative higher mutation rates compared with other non-mutation hotspot residues in p53 DBD, suggesting that these post-translational modification sites may have physiological roles in regulation of p53 activity and tumorigenesis [92]. While many post-translational modifications of mutant p53 positively regulate mutant p53 protein levels and functions, some post-translational modifications negatively regulate mutant p53 protein levels and functions. For instance, phosphorylation of Thr155 and Ser215, two post-translational modification sites in DBD with higher mutation rates, were reported to negatively regulate mutant p53 protein levels and functions; phosphorylation of Thr155 promotes degradation of mutant p53, and phosphorylation of Ser215 inhibits mutant p53 activity in DNA binding and transactivation of its target genes [109, 110].
Regulation of mutant p53 transcriptional activity through protein interactions
The activity of mutant p53, especially its activity in gene transcriptional regulation can be regulated by additional proteins/complexes in addition to above-mentioned mechanisms. A recent study reported by Carol Prives and her colleagues showed that mutant p53 protein interacts and cooperates with the SWI/SNF chromatin remodeling complex to enhance SWI/SNF-dependent chromatin remodeling in the promoter region of its regulated gene [57]. The promoter regions with the binding of mutant p53 and SWI/SNF complex can be restructured into an “open” conformation, which leads to increased expression of its target genes, such as VEGFR2. Mutant p53 transcriptional profiles have ∼40% overlap with SWI/SNF-regulated genes, suggesting that many mutant p53-regulated genes are co-regulated by SWI/SNF complex [57]. Recently, we identified Pontin, an AAA+ ATPase and potent helicase, as a novel mutant p53 binding protein [111]. The interaction of Pontin and mutant p53 is critical for mutant p53 to transcriptionally regulate a subgroup of its regulated genes, which in turn promotes mutant p53 GOF. The interaction of Pontin and mutant p53 promotes the binding of mutant p53 in the promoter region of its regulated genes. Knockdown of Pontin in cancer cells containing R175H mutant p53 greatly reduced transcriptional regulation of mutant p53 towards a group of genes involved in oncogenic functions [111]. We further found that the ATPase activity of Pontin is crucial for Pontin to regulate the transcriptional activity of mutant p53. Blocking the ATPase activity of Pontin greatly diminishes mutant p53 GOF [111]. Interestingly, Pontin interacts with SWI/SNF complex [112]. It remains to be elucidated whether Pontin plays a cooperative role with SWI/SNF complex in regulating mutant p53's transcriptional activity.
Therapeutic strategies to target mutant p53
Given that mutant p53 proteins often accumulate to high levels and exert GOF to promote malignant progression in human cancer, targeting mutant p53 has become an attractive therapeutic strategy for cancer containing mutant p53 [11, 22, 23, 113]. The main strategies to target mutant p53 are restoring the wild type p53 activity and depleting mutant p53 in cancer (Figure 3).
Figure 3. Strategies to target mutant p53 in cancer.
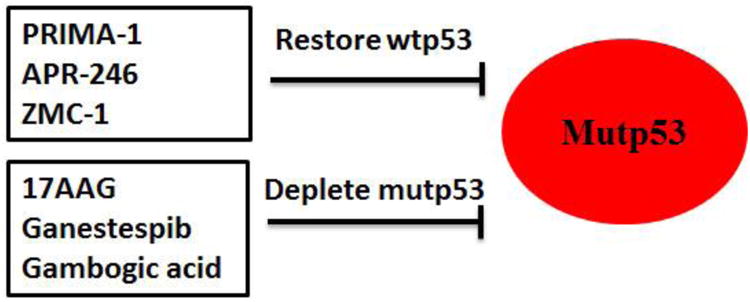
The strategies to target mutant p53 in cancer include restoring wild type p53 (wtp53) by PRIMA-1, APR-246, ZMC-1, etc, and depleting mutant p53 (mutp53) by 17AAG, Ganestespib, Gambogic acid, etc, in cancer containing mutant p53.
Restoring wild type p53 activity
Mutant p53 proteins stabilize and accumulate only in tumors but not in normal tissues as shown by recent mutant p53 knock-in mouse models [38, 39, 41]. Therefore, restoring wild type p53 activity can have the targeted effect on tumor cells through inducing cell apoptosis and inhibiting cell proliferation and migration in tumor cells containing mutant p53 with limited side effects to normal tissues containing wild type p53. This strategy is especially useful for the treatment of the late stage tumors, which often have one allele of mutant p53 and lose the remaining wild type p53 allele [114, 115]. These tumors usually are more aggressive and resistant to treatment. The compounds that can restore wild type p53 activity have been summarized in several recent reviews [12, 116, 117]. Among all the compounds, PRIMA-1 [2,2-bis (hydroxymethyl)-3-quinuclidinone] is the most well-advanced small molecule [117, 118]. PRIMA-1 was identified from a screening showing preferential inhibitory effect on proliferation of p53-null Saos2 cells with ectopic expression of mutant p53 R175H compared with control Saos2 cells [118]. PRIMA-1 and its methyl analog APR-246 can restore wild type p53 activity by inducing conformation change of mutant p53 proteins and refolding accumulated unfolded mutant p53 proteins [117, 118]. In addition, PRIMA-1 also restores unfolded wild type p53, which can promote tumor invasion just like mutant p53 proteins [118-120]. It is therefore possible that PRIMA-1 will be beneficial to cancer patients with either mutant p53 or wild type p53 in tumors. Currently, PRIMA-1 has shown a favorable safety profile in phase I clinical trials, and APR-246 has entered a phase II clinical trial [117, 121, 122].
ZMC-1 (zinc metallochaperone-1) is another recently identified small molecule that restores the proper protein folding and transcriptional activity of mutant p53 R175H [123, 124]. Zinc is required for the proper folding of wild type p53 protein to ensure its stabilization and correct DNA binding domain structure. Some mutant p53 proteins have the impaired zinc binding ability that prevents their proper protein folding and function. ZMC-1 buffers the intracellular free zinc levels to promote the binding of mutant p53 to zinc, and therefore facilitates the proper folding of mutant p53 [123, 124]. In addition, ZMC1 treatment generates ROS and induces DNA oxidation which can activate newly rescued mutant p53 to induce expression of wild type p53 target genes [123, 124]. ZMC-1 exhibited strong toxicity to cells containing mutant p53 R175H and a potent antitumor activity in p53 R175H tumors, but showed limited effects on cells and tumors containing wild type p53 or other mutant p53 (R248Q and R273H) [124]. In addition to p53 R175H mutant, there are some other zinc-binding p53 mutants, including C176, C238, C242, H179 and M237. It is possible that ZMC-1 can reactivate these zinc-binding mutant p53 like R175H, which should be elucidated by future studies.
Depleting mutant p53
Growing evidence further suggest that many tumor cells became addicted to mutant p53 protein and mutant p53 GOF. Knockdown of mutant p53 in cancer cells greatly reduce cell proliferation, tumor growth, chemo-response and metastasis. For example, ablation of mutant p53 in a conditional mouse model expressing R248Q mutant p53 induces the regression of advanced tumors and greatly extends animal survival [41]. These observations on mutant p53 addiction indicate that tumors containing mutant p53 depend on the sustained expression of mutant p53 for continued growth, and strongly support depleting mutant p53 proteins as a promising therapeutic strategy for tumors carrying mutant p53.
Hsp90 can promote the stabilization and accumulation of mutant p53 by inhibiting the degradation of mutant p53 mediated by MDM2 and CHIP [79-81, 84]. Inhibitors for Hsp90, including geldanamycin, 17AAG and Ganetespib, have been tested as therapeutic agents for cancer containing mutant p53. Treatment of cancer cells with 17AAG leads to the degradation of different mutant p53, including R175H, L194F, R273H and R280K, and the reduced viability of cancer cells containing these mutant p53 [79]. Ganestespib, the highly potent synthetic HSP90 inhibitor, has a much higher efficacy (more than 50-fold) than 17AAG in degrading mutant p53 (but not wild type p53), and killing cancer cells containing mutant p53 [41]. Results from in vivo animal studies employing mutant p53 knock-in mice and p53 null mice showed that ganestespib treatment suppressed the tumor growth and increased the survival in a mutant p53-dependent manner [41]. Ganestespib is currently being evaluated in the clinical trial, including a phase III lung cancer trial, and has demonstrated a favorable safety profile in cancer patients [125-127]. In addition to Hsp90, inhibiting the activity of HDAC6, an essential positive regulator of Hsp90 has also been shown to degrade mutant p53 [63]. Histone deacetylase inhibitors (HDACi), specifically, suberoylanilide hydroxamic acid (SAHA), can degrade mutant p53 proteins, exhibit higher cytotoxicity to cells carrying mutant p53 compared with cells containing wild type p53 or cells deficient for p53 [63]. Gambogic Acid (GA), a natural product from Garcinia hanburyi tree, has been shown to promote mutant p53 degradation [128]. It has been suggested that GA promotes mutant p53 degradation through preventing mutant p53-Hsp90 interaction, enhancing mutant p53-Hsp70 interaction and promoting mutant p53 nuclear exportation for degradation [129].
Summary and remaining questions
p53 is the most frequently-mutated gene in human cancer, and is one of the most extensively studied proteins. In this review, we focused on the functions and regulation of mutant p53 in cancer. Many tumor-associated missense mutant p53 proteins not only abrogate tumor suppressor functions, but also gain new oncogenic functions to promote tumor progression. While the concept of GOF has been well-established, it is still unclear whether p53 mutations at different residues have same GOFs. p53 mutations are scattered throughout the DNA binding domain. So far, majority of studies on mutant p53 GOF have focused on several mutational hotspots in human cancer. Further studies on mutant p53 at different residues will help address this question. It is still not well-understood how mutant p53 proteins exert GOFs. While mutant p53 proteins lose transcriptional regulation functions of wild type p53, they interact with several transcriptional factors (NF-Y, Sp-1, SREBP, Ets-1, VDR and NRF2) to regulate a different set of genes to exert their GOFs [11, 52-56]. It is possible that mutant p53 proteins exhibit different GOFs in different tissue/cell types due to different levels and activity of these transcriptional factors in different tissue/cell types.
Mutant p53 proteins are often stabilized and accumulated to high levels in tumors, which is crucial for their GOFs [11, 22, 25, 59]. However, mutant p53 proteins do not possess inherent stability in normal cells/tissues [38, 39, 41]. It is cancer cells that provide an environment that promotes the stability and accumulation of mutant p53 protein. In this review, we discussed many factors that promoted mutant p53 stabilization and accumulation in tumors, including chaperone proteins (Hsp90), co-chaperone BAG family proteins, MDM2 short isoforms, etc. Many of these factors have oncogenic activity and are overexpressed in human cancer. Further, their overexpression is often associated with poor prognosis of cancer patients. It is possible that promoting mutant p53 protein accumulation is an important mechanism for these proteins' oncogenic activities. Targeting these proteins could have great potential to improve the prognosis and extend the survival of patients with tumors containing mutant p53. Hsp90 inhibitors have been tested in clinical trial [125-127]. It is unclear whether different tumor types have different factors that promote mutant p53 stabilization and accumulation. Further understanding of the mechanism of mutant p53 accumulation in tumors will help develop effective therapeutic strategies targeting tumors containing mutant p53.
Highlights.
Following please find highlights of the review article:
p53 is the most commonly mutated gene in human cancer.
Mutant p53 is frequently accumulated to high levels in cancer.
Mutant p53 often displays gain-of-function oncogenic activities.
Targeting mutant p53 is a promising therapeutic strategy for cancer.
Acknowledgments
W.H. is supported by National Institutes of Health (NIH) Grants 1R01CA160558-01, 1R01CA203965 and DOD W81XWH-16-1-0358. Z. F. is supported by NIH grant R01CA143204 and Bush Medical Research Award. X.Y. is supported by NJCCR Postdoctoral Fellowship Award.
Abbreviations used
- wtp53
wild type p53
- mutp53
mutant p53
- GOF
gain-of-function
- DBD
DNA binding domain
- MDM2-B
MDM2 isoform B
- NLS
nuclear localization signal
- BAG
Bcl-2 –associated athanogene
- IR
ionizing radiation
- CHIP
C-terminus of Hsp70-interacting protein
- HDACi
Histone deacetylase inhibitors
- SAHA
suberoylanilide hydroxamic acid
- PRIMA-1
2,2-bis(hydroxymethyl)-3-quinuclidinone
- ZMC-1
zinc metallochaperone-1
Footnotes
Authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–10. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 2.Levine AJ, Hu W, Feng Z. The P53 pathway: what questions remain to be explored? Cell Death Differ. 2006;13:1027–36. doi: 10.1038/sj.cdd.4401910. [DOI] [PubMed] [Google Scholar]
- 3.Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009;137:413–31. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 4.Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. 2009;9:749–58. doi: 10.1038/nrc2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brooks CL, Gu W. p53 ubiquitination: Mdm2 and beyond. Molecular cell. 2006;21:307–15. doi: 10.1016/j.molcel.2006.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feng Z, Levine AJ. The regulation of energy metabolism and the IGF-1/mTOR pathways by the p53 protein. Trends in cell biology. 2010;20:427–34. doi: 10.1016/j.tcb.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Srivastava S, Zou ZQ, Pirollo K, Blattner W, Chang EH. Germ-line transmission of a mutated p53 gene in a cancer-prone family with Li-Fraumeni syndrome. Nature. 1990;348:747–9. doi: 10.1038/348747a0. [DOI] [PubMed] [Google Scholar]
- 8.Bougeard G, Sesboue R, Baert-Desurmont S, Vasseur S, Martin C, Tinat J, et al. Molecular basis of the Li-Fraumeni syndrome: an update from the French LFS families. J Med Genet. 2008;45:535–8. doi: 10.1136/jmg.2008.057570. [DOI] [PubMed] [Google Scholar]
- 9.Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, et al. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- 10.Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA, Jr, Butel JS, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–21. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 11.Freed-Pastor WA, Prives C. Mutant p53: one name, many proteins. Genes Dev. 2012;26:1268–86. doi: 10.1101/gad.190678.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muller PA, Vousden KH. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell. 2014;25:304–17. doi: 10.1016/j.ccr.2014.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harris CC, Hollstein M. Clinical implications of the p53 tumor-suppressor gene. The New England journal of medicine. 1993;329:1318–27. doi: 10.1056/NEJM199310283291807. [DOI] [PubMed] [Google Scholar]
- 14.Olivier M, Hussain SP, Caron de Fromentel C, Hainaut P, Harris CC. TP53 mutation spectra and load: a tool for generating hypotheses on the etiology of cancer. IARC Sci Publ. 2004:247–70. [PubMed] [Google Scholar]
- 15.Harris SL, Levine AJ. The p53 pathway: positive and negative feedback loops. Oncogene. 2005;24:2899–908. doi: 10.1038/sj.onc.1208615. [DOI] [PubMed] [Google Scholar]
- 16.Wade M, Wahl GM. Targeting Mdm2 and Mdmx in cancer therapy: better living through medicinal chemistry? Molecular cancer research : MCR. 2009;7:1–11. doi: 10.1158/1541-7786.MCR-08-0423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dornan D, Wertz I, Shimizu H, Arnott D, Frantz GD, Dowd P, et al. The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature. 2004;429:86–92. doi: 10.1038/nature02514. [DOI] [PubMed] [Google Scholar]
- 18.Leng RP, Lin Y, Ma W, Wu H, Lemmers B, Chung S, et al. Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell. 2003;112:779–91. doi: 10.1016/s0092-8674(03)00193-4. [DOI] [PubMed] [Google Scholar]
- 19.Liu J, Zhang C, Wang XL, Ly P, Belyi V, Xu-Monette ZY, et al. E3 ubiquitin ligase TRIM32 negatively regulates tumor suppressor p53 to promote tumorigenesis. Cell Death Differ. 2014;21:1792–804. doi: 10.1038/cdd.2014.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wade M, Li YC, Wahl GM. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer. 2013;13:83–96. doi: 10.1038/nrc3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu H, Yue X, Zhao Y, Li X, Wu L, Zhang C, et al. LIF negatively regulates tumour-suppressor p53 through Stat3/ID1/MDM2 in colorectal cancers. Nature communications. 2014;5:5218. doi: 10.1038/ncomms6218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muller PA, Vousden KH. p53 mutations in cancer. Nature cell biology. 2013;15:2–8. doi: 10.1038/ncb2641. [DOI] [PubMed] [Google Scholar]
- 23.Brosh R, Rotter V. When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer. 2009;9:701–13. doi: 10.1038/nrc2693. [DOI] [PubMed] [Google Scholar]
- 24.Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science. 1994;265:346–55. doi: 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- 25.Oren M, Rotter V. Mutant p53 gain-of-function in cancer. Cold Spring Harbor perspectives in biology. 2010;2:a001107. doi: 10.1101/cshperspect.a001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Milner J, Medcalf EA, Cook AC. Tumor suppressor p53: analysis of wild-type and mutant p53 complexes. Mol Cell Biol. 1991;11:12–9. doi: 10.1128/mcb.11.1.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Milner J, Medcalf EA. Cotranslation of activated mutant p53 with wild type drives the wild-type p53 protein into the mutant conformation. Cell. 1991;65:765–74. doi: 10.1016/0092-8674(91)90384-b. [DOI] [PubMed] [Google Scholar]
- 28.Sigal A, Rotter V. Oncogenic mutations of the p53 tumor suppressor: the demons of the guardian of the genome. Cancer Res. 2000;60:6788–93. [PubMed] [Google Scholar]
- 29.Baker SJ, Preisinger AC, Jessup JM, Paraskeva C, Markowitz S, Willson JK, et al. p53 gene mutations occur in combination with 17p allelic deletions as late events in colorectal tumorigenesis. Cancer research. 1990;50:7717–22. [PubMed] [Google Scholar]
- 30.Eliyahu D, Raz A, Gruss P, Givol D, Oren M. Participation of p53 cellular tumour antigen in transformation of normal embryonic cells. Nature. 1984;312:646–9. doi: 10.1038/312646a0. [DOI] [PubMed] [Google Scholar]
- 31.Jenkins JR, Rudge K, Currie GA. Cellular immortalization by a cDNA clone encoding the transformation-associated phosphoprotein p53. Nature. 1984;312:651–4. doi: 10.1038/312651a0. [DOI] [PubMed] [Google Scholar]
- 32.Parada LF, Land H, Weinberg RA, Wolf D, Rotter V. Cooperation between gene encoding p53 tumour antigen and ras in cellular transformation. Nature. 1984;312:649–51. doi: 10.1038/312649a0. [DOI] [PubMed] [Google Scholar]
- 33.Dittmer D, Pati S, Zambetti G, Chu S, Teresky AK, Moore M, et al. Gain of function mutations in p53. Nat Genet. 1993;4:42–6. doi: 10.1038/ng0593-42. [DOI] [PubMed] [Google Scholar]
- 34.Muller PA, Caswell PT, Doyle B, Iwanicki MP, Tan EH, Karim S, et al. Mutant p53 drives invasion by promoting integrin recycling. Cell. 2009;139:1327–41. doi: 10.1016/j.cell.2009.11.026. [DOI] [PubMed] [Google Scholar]
- 35.Blandino G, Deppert W, Hainaut P, Levine A, Lozano G, Olivier M, et al. Mutant p53 protein, master regulator of human malignancies: a report on the Fifth Mutant p53 Workshop. Cell Death Differ. 2012;19:180–3. doi: 10.1038/cdd.2011.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Freed-Pastor WA, Mizuno H, Zhao X, Langerod A, Moon SH, Rodriguez-Barrueco R, et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell. 2012;148:244–58. doi: 10.1016/j.cell.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang C, Liu J, Liang Y, Wu R, Zhao Y, Hong X, et al. Tumour-associated mutant p53 drives the Warburg effect. Nature communications. 2013;4:2935. doi: 10.1038/ncomms3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell. 2004;119:861–72. doi: 10.1016/j.cell.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 39.Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 2004;119:847–60. doi: 10.1016/j.cell.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 40.Hanel W, Marchenko N, Xu S, Yu SX, Weng W, Moll U. Two hot spot mutant p53 mouse models display differential gain of function in tumorigenesis. Cell death and differentiation. 2013;20:898–909. doi: 10.1038/cdd.2013.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alexandrova EM, Yallowitz AR, Li D, Xu S, Schulz R, Proia DA, et al. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature. 2015;523:352–6. doi: 10.1038/nature14430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Elledge RM, Fuqua SA, Clark GM, Pujol P, Allred DC, McGuire WL. Prognostic significance of p53 gene alterations in node-negative breast cancer. Breast Cancer Res Treat. 1993;26:225–35. doi: 10.1007/BF00665800. [DOI] [PubMed] [Google Scholar]
- 43.Olivier M, Langerod A, Carrieri P, Bergh J, Klaar S, Eyfjord J, et al. The clinical value of somatic TP53 gene mutations in 1,794 patients with breast cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2006;12:1157–67. doi: 10.1158/1078-0432.CCR-05-1029. [DOI] [PubMed] [Google Scholar]
- 44.Petitjean A, Achatz MI, Borresen-Dale AL, Hainaut P, Olivier M. TP53 mutations in human cancers: functional selection and impact on cancer prognosis and outcomes. Oncogene. 2007;26:2157–65. doi: 10.1038/sj.onc.1210302. [DOI] [PubMed] [Google Scholar]
- 45.Mello SS, Attardi LD. Not all p53 gain-of-function mutants are created equal. Cell Death Differ. 2013;20:855–7. doi: 10.1038/cdd.2013.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Muller PA, Trinidad AG, Caswell PT, Norman JC, Vousden KH. Mutant p53 regulates Dicer through p63-dependent and -independent mechanisms to promote an invasive phenotype. J Biol Chem. 2014;289:122–32. doi: 10.1074/jbc.M113.502138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stindt MH, Muller PA, Ludwig RL, Kehrloesser S, Dotsch V, Vousden KH. Functional interplay between MDM2, p63/p73 and mutant p53. Oncogene. 2015;34:4300–10. doi: 10.1038/onc.2014.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gaiddon C, Lokshin M, Ahn J, Zhang T, Prives C. A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol Cell Biol. 2001;21:1874–87. doi: 10.1128/MCB.21.5.1874-1887.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Y, Prives C. Are interactions with p63 and p73 involved in mutant p53 gain of oncogenic function? Oncogene. 2007;26:2220–5. doi: 10.1038/sj.onc.1210311. [DOI] [PubMed] [Google Scholar]
- 50.Belyi VA, Ak P, Markert E, Wang H, Hu W, Puzio-Kuter A, et al. The origins and evolution of the p53 family of genes. Cold Spring Harbor perspectives in biology. 2010;2:a001198. doi: 10.1101/cshperspect.a001198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hu W. The role of p53 gene family in reproduction. Cold Spring Harbor perspectives in biology. 2009;1:a001073. doi: 10.1101/cshperspect.a001073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Di Agostino S, Strano S, Emiliozzi V, Zerbini V, Mottolese M, Sacchi A, et al. Gain of function of mutant p53: the mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell. 2006;10:191–202. doi: 10.1016/j.ccr.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 53.Dupont S, Mamidi A, Cordenonsi M, Montagner M, Zacchigna L, Adorno M, et al. FAM/USP9x, a deubiquitinating enzyme essential for TGFbeta signaling, controls Smad4 monoubiquitination. Cell. 2009;136:123–35. doi: 10.1016/j.cell.2008.10.051. [DOI] [PubMed] [Google Scholar]
- 54.Sampath J, Sun D, Kidd VJ, Grenet J, Gandhi A, Shapiro LH, et al. Mutant p53 cooperates with ETS and selectively up-regulates human MDR1 not MRP1. J Biol Chem. 2001;276:39359–67. doi: 10.1074/jbc.M103429200. [DOI] [PubMed] [Google Scholar]
- 55.Walerych D, Lisek K, Sommaggio R, Piazza S, Ciani Y, Dalla E, et al. Proteasome machinery is instrumental in a common gain-of-function program of the p53 missense mutants in cancer. Nature cell biology. 2016;18:897–909. doi: 10.1038/ncb3380. [DOI] [PubMed] [Google Scholar]
- 56.Stambolsky P, Tabach Y, Fontemaggi G, Weisz L, Maor-Aloni R, Siegfried Z, et al. Modulation of the vitamin D3 response by cancer-associated mutant p53. Cancer Cell. 2010;17:273–85. doi: 10.1016/j.ccr.2009.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pfister NT, Fomin V, Regunath K, Zhou JY, Zhou W, Silwal-Pandit L, et al. Mutant p53 cooperates with the SWI/SNF chromatin remodeling complex to regulate VEGFR2 in breast cancer cells. Genes & development. 2015;29:1298–315. doi: 10.1101/gad.263202.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Will K, Warnecke G, Wiesmuller L, Deppert W. Specific interaction of mutant p53 with regions of matrix attachment region DNA elements (MARs) with a high potential for base-unpairing. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:13681–6. doi: 10.1073/pnas.95.23.13681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu J, Zhang C, Hu W, Feng Z. Tumor suppressor p53 and its mutants in cancer metabolism. Cancer letters. 2013 doi: 10.1016/j.canlet.2013.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bartek J, Iggo R, Gannon J, Lane DP. Genetic and immunochemical analysis of mutant p53 in human breast cancer cell lines. Oncogene. 1990;5:893–9. [PubMed] [Google Scholar]
- 61.Alsner J, Jensen V, Kyndi M, Offersen BV, Vu P, Borresen-Dale AL, et al. A comparison between p53 accumulation determined by immunohistochemistry and TP53 mutations as prognostic variables in tumours from breast cancer patients. Acta Oncol. 2008;47:600–7. doi: 10.1080/02841860802047411. [DOI] [PubMed] [Google Scholar]
- 62.Terzian T, Suh YA, Iwakuma T, Post SM, Neumann M, Lang GA, et al. The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev. 2008;22:1337–44. doi: 10.1101/gad.1662908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li D, Marchenko ND, Moll UM. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. 2011;18:1904–13. doi: 10.1038/cdd.2011.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Alexandrova EM, Yallowitz AR, Li D, Xu S, Schulz R, Proia DA, et al. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature. 2015 doi: 10.1038/nature14430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hu W, Feng Z, Levine AJ. The Regulation of Multiple p53 Stress Responses is Mediated through MDM2. Genes Cancer. 2012;3:199–208. doi: 10.1177/1947601912454734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lukashchuk N, Vousden KH. Ubiquitination and degradation of mutant p53. Mol Cell Biol. 2007;27:8284–95. doi: 10.1128/MCB.00050-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zheng T, Wang J, Zhao Y, Zhang C, Lin M, Wang X, et al. Spliced MDM2 isoforms promote mutant p53 accumulation and gain-of-function in tumorigenesis. Nature communications. 2013;4:2996. doi: 10.1038/ncomms3996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bartel F, Taubert H, Harris LC. Alternative and aberrant splicing of MDM2 mRNA in human cancer. Cancer Cell. 2002;2:9–15. doi: 10.1016/s1535-6108(02)00091-0. [DOI] [PubMed] [Google Scholar]
- 69.Harris LC. MDM2 splice variants and their therapeutic implications. Current cancer drug targets. 2005;5:21–6. doi: 10.2174/1568009053332654. [DOI] [PubMed] [Google Scholar]
- 70.Sigalas I, Calvert AH, Anderson JJ, Neal DE, Lunec J. Alternatively spliced mdm2 transcripts with loss of p53 binding domain sequences: transforming ability and frequent detection in human cancer. Nat Med. 1996;2:912–7. doi: 10.1038/nm0896-912. [DOI] [PubMed] [Google Scholar]
- 71.Okoro DR, Arva N, Gao C, Polotskaia A, Puente C, Rosso M, et al. Endogenous human MDM2-C is highly expressed in human cancers and functions as a p53-independent growth activator. PloS one. 2013;8:e77643. doi: 10.1371/journal.pone.0077643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lukas J, Gao DQ, Keshmeshian M, Wen WH, Tsao-Wei D, Rosenberg S, et al. Alternative and aberrant messenger RNA splicing of the mdm2 oncogene in invasive breast cancer. Cancer Res. 2001;61:3212–9. [PubMed] [Google Scholar]
- 73.Matsumoto R, Tada M, Nozaki M, Zhang CL, Sawamura Y, Abe H. Short alternative splice transcripts of the mdm2 oncogene correlate to malignancy in human astrocytic neoplasms. Cancer Res. 1998;58:609–13. [PubMed] [Google Scholar]
- 74.Jacob AG, O'Brien D, Singh RK, Comiskey DF, Jr, Littleton RM, Mohammad F, et al. Stress-induced isoforms of MDM2 and MDM4 correlate with high-grade disease and an altered splicing network in pediatric rhabdomyosarcoma. Neoplasia. 2013;15:1049–63. doi: 10.1593/neo.13286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Doyle SM, Genest O, Wickner S. Protein rescue from aggregates by powerful molecular chaperone machines. Nature reviews Molecular cell biology. 2013;14:617–29. doi: 10.1038/nrm3660. [DOI] [PubMed] [Google Scholar]
- 76.Liberek K, Lewandowska A, Zietkiewicz S. Chaperones in control of protein disaggregation. EMBO J. 2008;27:328–35. doi: 10.1038/sj.emboj.7601970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Proia DA, Bates RC. Ganetespib and HSP90: translating preclinical hypotheses into clinical promise. Cancer research. 2014;74:1294–300. doi: 10.1158/0008-5472.CAN-13-3263. [DOI] [PubMed] [Google Scholar]
- 78.Shrestha L, Bolaender A, Patel HJ, Taldone T. Heat Shock Protein (HSP) Drug Discovery and Development: Targeting Heat Shock Proteins in Disease. Current topics in medicinal chemistry. 2016;16:2753–64. doi: 10.2174/1568026616666160413141911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li D, Marchenko ND, Schulz R, Fischer V, Velasco-Hernandez T, Talos F, et al. Functional inactivation of endogenous MDM2 and CHIP by HSP90 causes aberrant stabilization of mutant p53 in human cancer cells. Mol Cancer Res. 2011;9:577–88. doi: 10.1158/1541-7786.MCR-10-0534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Blagosklonny MV, Toretsky J, Bohen S, Neckers L. Mutant conformation of p53 translated in vitro or in vivo requires functional HSP90. Proc Natl Acad Sci U S A. 1996;93:8379–83. doi: 10.1073/pnas.93.16.8379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Whitesell L, Sutphin PD, Pulcini EJ, Martinez JD, Cook PH. The physical association of multiple molecular chaperone proteins with mutant p53 is altered by geldanamycin, an hsp90-binding agent. Mol Cell Biol. 1998;18:1517–24. doi: 10.1128/mcb.18.3.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Peng Y, Chen L, Li C, Lu W, Chen J. Inhibition of MDM2 by hsp90 contributes to mutant p53 stabilization. The Journal of biological chemistry. 2001;276:40583–90. doi: 10.1074/jbc.M102817200. [DOI] [PubMed] [Google Scholar]
- 83.Yue X, Zhao Y, Huang G, Li J, Zhu J, Feng Z, et al. A novel mutant p53 binding partner BAG5 stabilizes mutant p53 and promotes mutant p53 GOFs in tumorigenesis. Cell Discov. 2016;2:16039. doi: 10.1038/celldisc.2016.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Muller P, Hrstka R, Coomber D, Lane DP, Vojtesek B. Chaperone-dependent stabilization and degradation of p53 mutants. Oncogene. 2008;27:3371–83. doi: 10.1038/sj.onc.1211010. [DOI] [PubMed] [Google Scholar]
- 85.Pratt WB, Morishima Y, Peng HM, Osawa Y. Proposal for a role of the Hsp90/Hsp70-based chaperone machinery in making triage decisions when proteins undergo oxidative and toxic damage. Exp Biol Med (Maywood) 2010;235:278–89. doi: 10.1258/ebm.2009.009250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kabbage M, Dickman MB. The BAG proteins: a ubiquitous family of chaperone regulators. Cell Mol Life Sci. 2008;65:1390–402. doi: 10.1007/s00018-008-7535-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Takayama S, Reed JC. Molecular chaperone targeting and regulation by BAG family proteins. Nat Cell Biol. 2001;3:E237–41. doi: 10.1038/ncb1001-e237. [DOI] [PubMed] [Google Scholar]
- 88.Yue X, Zhao Y, Liu J, Zhang C, Yu H, Wang J, et al. BAG2 promotes tumorigenesis through enhancing mutant p53 protein levels and function. Elife. 2015;4 doi: 10.7554/eLife.08401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Meek DW, Anderson CW. Posttranslational modification of p53: cooperative integrators of function. Cold Spring Harb Perspect Biol. 2009;1:a000950. doi: 10.1101/cshperspect.a000950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shieh SY, Ikeda M, Taya Y, Prives C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell. 1997;91:325–34. doi: 10.1016/s0092-8674(00)80416-x. [DOI] [PubMed] [Google Scholar]
- 91.Chehab NH, Malikzay A, Stavridi ES, Halazonetis TD. Phosphorylation of Ser-20 mediates stabilization of human p53 in response to DNA damage. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:13777–82. doi: 10.1073/pnas.96.24.13777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nguyen TA, Menendez D, Resnick MA, Anderson CW. Mutant TP53 posttranslational modifications: challenges and opportunities. Hum Mutat. 2014;35:738–55. doi: 10.1002/humu.22506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Midgley CA, Lane DP. p53 protein stability in tumour cells is not determined by mutation but is dependent on Mdm2 binding. Oncogene. 1997;15:1179–89. doi: 10.1038/sj.onc.1201459. [DOI] [PubMed] [Google Scholar]
- 94.Suh YA, Post SM, Elizondo-Fraire AC, Maccio DR, Jackson JG, El-Naggar AK, et al. Multiple stress signals activate mutant p53 in vivo. Cancer Res. 2011;71:7168–75. doi: 10.1158/0008-5472.CAN-11-0459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Alsheich-Bartok O, Haupt S, Alkalay-Snir I, Saito S, Appella E, Haupt Y. PML enhances the regulation of p53 by CK1 in response to DNA damage. Oncogene. 2008;27:3653–61. doi: 10.1038/sj.onc.1211036. [DOI] [PubMed] [Google Scholar]
- 96.Melnikova VO, Santamaria AB, Bolshakov SV, Ananthaswamy HN. Mutant p53 is constitutively phosphorylated at Serine 15 in UV-induced mouse skin tumors: involvement of ERK1/2 MAP kinase. Oncogene. 2003;22:5958–66. doi: 10.1038/sj.onc.1206595. [DOI] [PubMed] [Google Scholar]
- 97.Holtrich U, Wolf G, Brauninger A, Karn T, Bohme B, Rubsamen-Waigmann H, et al. Induction and down-regulation of PLK, a human serine/threonine kinase expressed in proliferating cells and tumors. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:1736–40. doi: 10.1073/pnas.91.5.1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ando K, Ozaki T, Yamamoto H, Furuya K, Hosoda M, Hayashi S, et al. Polo-like kinase 1 (Plk1) inhibits p53 function by physical interaction and phosphorylation. J Biol Chem. 2004;279:25549–61. doi: 10.1074/jbc.M314182200. [DOI] [PubMed] [Google Scholar]
- 99.Valenti F, Fausti F, Biagioni F, Shay T, Fontemaggi G, Domany E, et al. Mutant p53 oncogenic functions are sustained by Plk2 kinase through an autoregulatory feedback loop. Cell Cycle. 2011;10:4330–40. doi: 10.4161/cc.10.24.18682. [DOI] [PubMed] [Google Scholar]
- 100.Haupt S, di Agostino S, Mizrahi I, Alsheich-Bartok O, Voorhoeve M, Damalas A, et al. Promyelocytic leukemia protein is required for gain of function by mutant p53. Cancer Res. 2009;69:4818–26. doi: 10.1158/0008-5472.CAN-08-4010. [DOI] [PubMed] [Google Scholar]
- 101.Furihata M, Kurabayashl A, Matsumoto M, Sonobe H, Ohtsuki Y, Terao N, et al. Frequent phosphorylation at serine 392 in overexpressed p53 protein due to missense mutation in carcinoma of the urinary tract. J Pathol. 2002;197:82–8. doi: 10.1002/path.1082. [DOI] [PubMed] [Google Scholar]
- 102.Bar JK, Slomska I, Rabczynki J, Noga L, Grybos M. Expression of p53 protein phosphorylated at serine 20 and serine 392 in malignant and benign ovarian neoplasms: correlation with clinicopathological parameters of tumors. Int J Gynecol Cancer. 2009;19:1322–8. doi: 10.1111/IGC.0b013e3181b70465. [DOI] [PubMed] [Google Scholar]
- 103.Brooks CL, Gu W. Ubiquitination, phosphorylation and acetylation: the molecular basis for p53 regulation. Curr Opin Cell Biol. 2003;15:164–71. doi: 10.1016/s0955-0674(03)00003-6. [DOI] [PubMed] [Google Scholar]
- 104.Dai C, Gu W. p53 post-translational modification: deregulated in tumorigenesis. Trends Mol Med. 2010;16:528–36. doi: 10.1016/j.molmed.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Minamoto T, Buschmann T, Habelhah H, Matusevich E, Tahara H, Boerresen-Dale AL, et al. Distinct pattern of p53 phosphorylation in human tumors. Oncogene. 2001;20:3341–7. doi: 10.1038/sj.onc.1204458. [DOI] [PubMed] [Google Scholar]
- 106.Rodriguez OC, Choudhury S, Kolukula V, Vietsch EE, Catania J, Preet A, et al. Dietary downregulation of mutant p53 levels via glucose restriction: mechanisms and implications for tumor therapy. Cell Cycle. 2012;11:4436–46. doi: 10.4161/cc.22778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yi YW, Kang HJ, Kim HJ, Kong Y, Brown ML, Bae I. Targeting mutant p53 by a SIRT1 activator YK-3-237 inhibits the proliferation of triple-negative breast cancer cells. Oncotarget. 2013;4:984–94. doi: 10.18632/oncotarget.1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhang ZY, Hong D, Nam SH, Kim JM, Paik YH, Joh JW, et al. SIRT1 regulates oncogenesis via a mutant p53-dependent pathway in hepatocellular carcinoma. J Hepatol. 2015;62:121–30. doi: 10.1016/j.jhep.2014.08.007. [DOI] [PubMed] [Google Scholar]
- 109.Bech-Otschir D, Kraft R, Huang X, Henklein P, Kapelari B, Pollmann C, et al. COP9 signalosome-specific phosphorylation targets p53 to degradation by the ubiquitin system. EMBO J. 2001;20:1630–9. doi: 10.1093/emboj/20.7.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Liu Q, Kaneko S, Yang L, Feldman RI, Nicosia SV, Chen J, et al. Aurora-A abrogation of p53 DNA binding and transactivation activity by phosphorylation of serine 215. J Biol Chem. 2004;279:52175–82. doi: 10.1074/jbc.M406802200. [DOI] [PubMed] [Google Scholar]
- 111.Zhao Y, Zhang C, Yue X, Li X, Liu J, Yu H, et al. Pontin, a new mutant p53-binding protein, promotes gain-of-function of mutant p53. Cell Death Differ. 2015;22:1824–36. doi: 10.1038/cdd.2015.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Shen X, Mizuguchi G, Hamiche A, Wu C. A chromatin remodelling complex involved in transcription and DNA processing. Nature. 2000;406:541–4. doi: 10.1038/35020123. [DOI] [PubMed] [Google Scholar]
- 113.Gurpinar E, Vousden KH. Hitting cancers' weak spots: vulnerabilities imposed by p53 mutation. Trends in cell biology. 2015;25:486–95. doi: 10.1016/j.tcb.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 114.Cavenee WK, Scrable HJ, James CD. Molecular genetics of human cancer predisposition and progression. Mutat Res. 1991;247:199–202. doi: 10.1016/0027-5107(91)90015-g. [DOI] [PubMed] [Google Scholar]
- 115.Gonzalez MV, Pello MF, Lopez-Larrea C, Suarez C, Menendez MJ, Coto E. Loss of heterozygosity and mutation analysis of the p16 (9p21) and p53 (17p13) genes in squamous cell carcinoma of the head and neck. Clin Cancer Res. 1995;1:1043–9. [PubMed] [Google Scholar]
- 116.Oren M, Tal P, Rotter V. Targeting mutant p53 for cancer therapy. Aging (Albany NY) 2016;8:1159–60. doi: 10.18632/aging.100992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bykov VJ, Zhang Q, Zhang M, Ceder S, Abrahmsen L, Wiman KG. Targeting of Mutant p53 and the Cellular Redox Balance by APR-246 as a Strategy for Efficient Cancer Therapy. Front Oncol. 2016;6:21. doi: 10.3389/fonc.2016.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bykov VJ, Issaeva N, Shilov A, Hultcrantz M, Pugacheva E, Chumakov P, et al. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nature medicine. 2002;8:282–8. doi: 10.1038/nm0302-282. [DOI] [PubMed] [Google Scholar]
- 119.Rieber M, Strasberg-Rieber M. Hypoxia, Mn-SOD and H(2)O(2) regulate p53 reactivation and PRIMA-1 toxicity irrespective of p53 status in human breast cancer cells. Biochem Pharmacol. 2012;84:1563–70. doi: 10.1016/j.bcp.2012.09.003. [DOI] [PubMed] [Google Scholar]
- 120.Trinidad AG, Muller PA, Cuellar J, Klejnot M, Nobis M, Valpuesta JM, et al. Interaction of p53 with the CCT complex promotes protein folding and wild-type p53 activity. Mol Cell. 2013;50:805–17. doi: 10.1016/j.molcel.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Lehmann BD, Pietenpol JA. Targeting mutant p53 in human tumors. J Clin Oncol. 2012;30:3648–50. doi: 10.1200/JCO.2012.44.0412. [DOI] [PubMed] [Google Scholar]
- 122.Lehmann S, Bykov VJ, Ali D, Andren O, Cherif H, Tidefelt U, et al. Targeting p53 in vivo: a first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. J Clin Oncol. 2012;30:3633–9. doi: 10.1200/JCO.2011.40.7783. [DOI] [PubMed] [Google Scholar]
- 123.Blanden AR, Yu X, Loh SN, Levine AJ, Carpizo DR. Reactivating mutant p53 using small molecules as zinc metallochaperones: awakening a sleeping giant in cancer. Drug Discov Today. 2015;20:1391–7. doi: 10.1016/j.drudis.2015.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yu X, Vazquez A, Levine AJ, Carpizo DR. Allele-specific p53 mutant reactivation. Cancer Cell. 2012;21:614–25. doi: 10.1016/j.ccr.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ramalingam S, Goss G, Rosell R, Schmid-Bindert G, Zaric B, Andric Z, et al. A randomized phase II study of ganetespib, a heat shock protein 90 inhibitor, in combination with docetaxel in second-line therapy of advanced non-small cell lung cancer (GALAXY-1) Ann Oncol. 2015;26:1741–8. doi: 10.1093/annonc/mdv220. [DOI] [PubMed] [Google Scholar]
- 126.Goyal L, Wadlow RC, Blaszkowsky LS, Wolpin BM, Abrams TA, McCleary NJ, et al. A phase I and pharmacokinetic study of ganetespib (STA-9090) in advanced hepatocellular carcinoma. Invest New Drugs. 2015;33:128–37. doi: 10.1007/s10637-014-0164-8. [DOI] [PubMed] [Google Scholar]
- 127.Jhaveri K, Chandarlapaty S, Lake D, Gilewski T, Robson M, Goldfarb S, et al. A phase II open-label study of ganetespib, a novel heat shock protein 90 inhibitor for patients with metastatic breast cancer. Clin Breast Cancer. 2014;14:154–60. doi: 10.1016/j.clbc.2013.12.012. [DOI] [PubMed] [Google Scholar]
- 128.Gu H, Wang X, Rao S, Wang J, Zhao J, Ren FL, et al. Gambogic acid mediates apoptosis as a p53 inducer through down-regulation of mdm2 in wild-type p53-expressing cancer cells. Mol Cancer Ther. 2008;7:3298–305. doi: 10.1158/1535-7163.MCT-08-0212. [DOI] [PubMed] [Google Scholar]
- 129.Wang J, Zhao Q, Qi Q, Gu HY, Rong JJ, Mu R, et al. Gambogic acid-induced degradation of mutant p53 is mediated by proteasome and related to CHIP. J Cell Biochem. 2011;112:509–19. doi: 10.1002/jcb.22941. [DOI] [PubMed] [Google Scholar]