

Summary
Numerous studies indicate an inflammatory link between obesity and type 2 diabetes. The inflammatory kinases IKKε and TBK1 are elevated in obesity; their inhibition in obese mice reduces weight, insulin resistance, fatty liver and inflammation. Here we studied amlexanox, an inhibitor of IKKε/TBK1, in a proof-of-concept randomized, double blind, placebo-controlled study of 42 obese patients with type 2 diabetes and nonalcoholic fatty liver disease. Treatment of patients with amlexanox produced a statistically significant reduction in Hemoglobin A1c and fructosamine. Interestingly, a subset of drug responders also exhibited improvements in insulin sensitivity and hepatic steatosis. This subgroup was characterized by a distinct inflammatory gene expression signature from biopsied subcutaneous fat at baseline. They also exhibited a unique pattern of gene expression changes in response to amlexanox, consistent with increased energy expenditure. Together, these data suggest that IKKε/TBK1 inhibitors may be effective therapies for metabolic disease in an identifiable subset of patients.
Keywords: Obesity, inflammation, protein kinase
eTOC Blurb
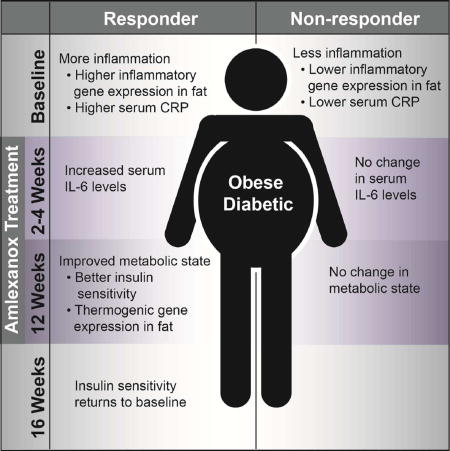
XXX et al investigate the effect of the anti-inflammatory/antiallergic drug amlexanox in a proof-of-concept clinical study of type 2 diabetes and nonalcoholic fatty liver disease. Amlexanox improved blood glucose levels in diabetic patients and a sub-group showed enhanced response, including liver fat loss, associated with increased energy expenditure.
Although the molecular events underlying the relationship between obesity and type 2 diabetes remain uncertain, numerous studies have implicated an inflammatory link (Hotamisligil, 2010; Lackey and Olefsky, 2016; Lumeng and Saltiel, 2011). Obesity produces a state of chronic, low-grade inflammation in liver and fat accompanied by the appearance of M1 polarized proinflammatory macrophages (Lumeng et al., 2007) and local secretion of cytokines and chemokines that attenuate insulin action (Chawla et al., 2011; Glass and Olefsky, 2012; Vandanmagsar et al., 2011; Weisberg et al., 2003). Knockout or pharmacological inhibition of inflammatory pathways in rodents can disrupt the link between genetic- or diet-induced obesity and both insulin and catecholamine resistance, suggesting that local inflammation is a key step in the generation of cellular resistance to important hormones that regulate metabolism. While the role of inflammation in human obesity is less clear, numerous studies have demonstrated a strong association between inflammatory signals and insulin resistance (Lackey and Olefsky, 2016; Lumeng and Saltiel, 2011).
The nuclear factor-κB (NF-κB) transcriptional program is activated in obese fat, and may play an important role in the development of insulin resistance (Arkan et al., 2005; Chiang et al., 2009). We recently reported (Reilly et al., 2013) that the NF-κB sensitive genes, Tbk1 and Ikbke, are increased at both the mRNA and protein level in adipose tissue during high fat diet (HFD) feeding of mice. Moreover, deletion of the Ikbke gene rendered mice partially resistant to the HFD-dependent development of obesity, insulin resistance, hepatic steatosis and inflammation (Chiang et al., 2009). This led us to identify the IKKε/TBK1 inhibitor, amlexanox, which had been previously developed for the treatment of asthma, allergic rhinitis and aphthous ulcers (Makino et al., 1987). Administration of this selective drug to obese mice produced reversible weight loss and improved insulin sensitivity, reduced inflammation and attenuated hepatic steatosis (Reilly et al., 2013). A primary target of amlexanox in rodents is the subcutaneous fat tissue, where it increases energy expenditure partly by enhancing catecholamine sensitivity and inducing the browning of adipocytes, in the process burning energy to restore metabolic flexibility (Mowers et al., 2013; Reilly et al., 2015). The increase in catecholamine sensitivity also resulted in a transient increase in IL-6 synthesis and secretion from adipocytes and preadipocytes, which was responsible for acute lowering of blood sugar via reduced hepatic glucose output (Reilly et al., 2015).
Here, we report a randomized double blind, placebo-controlled clinical study of 42 patients (38 completing the protocol) with type 2 diabetes and nonalcoholic fatty liver disease (NAFLD) treated for 12 weeks with amlexanox or placebo, which reveals a drug-induced statistically significant improvement in Hemoglobin A1c (HbA1c) and fructosamine between placebo and drug-treated patients. In addition, we identify a subset of responders, characterized by a distinct transcriptional profile in subcutaneous fat both at baseline and in response to amlexanox treatment. These responders exhibit improved insulin sensitivity and hepatic steatosis, following a transient increase in serum IL-6 levels at 2 to 4 weeks. These findings, coupled with its long safety record, indicate that amlexanox may be a promising candidate for further investigation in select patients for the treatment of type 2 diabetes with NAFLD and obesity.
Amlexanox improves metabolic parameters in an open label clinical study
An open label trial (NCT 01842282) was first launched to ensure that amlexanox could be safely administered to diabetic patients at doses previously indicated for the treatment of asthma without significant hypoglycemia while also assessing metabolic effects as secondary outcomes. Amlexanox was administered to 6 subjects with type 2 diabetes and NAFLD who were on stable oral therapies at 25 mg three times daily for 2 weeks, and then increased to 50 mg three times daily for an additional 10 weeks (Figure 1a). Subjects were followed for an additional 4 weeks post-treatment to ensure safety. Ages of patients ranged from 24–68 years (Figure 1b). Four of the six subjects were female, and five were Caucasian. The duration of diabetes ranged from 1.5 to 17 years. Owing to inclusion criteria, all subjects were obese, with BMIs ranging from 30.9 to 38.6 kg/m2, and all had hyperlipidemia and a variety of other comorbidities (Figure 1b). None of the patients presented with proteinuria or proliferative retinopathy.
Figure 1. Study design and insulin sensitivity measurements from the open-label study.

a) Summary of the study protocols. † indicates protocol elements that only apply to the open-label study. b) Baseline characteristics of the six open-label study participants. Metabolic data of the six patients included in the study at baseline and 12 weeks are shown: c) Hemoglobin A1c % (HbA1c). d) Fasting glucose. e) Fasting insulin. (f) Insulin resistance calculated by the homeostatic model assessment linear estimation, HOMA-IR. g–i) Hyperinsulinemic euglycemic clamp: g) Rate of glucose disappearance (Glucose Rd). h) body weight-normalized glucose Rd and (i) Hepatic glucose production at fasting state. j) Percent fat in liver, as determined via MRI using Dixon method. k) Glucose (left panel) insulin (middle panel) and FFAs (right panel) during the mixed meal test. Data presented as individual values (c–h) or mean ± s.e.m. (k). n = 6, except in g and h, as the 6th patient did not achieve steady state during the clamp performed at 12 weeks, where n = 5. * indicates p-value ≤ 0.05, # indicates p-value ≤ 0.10 (two-tailed paired t-test). DEXA, Dual Emission X-ray Absorptiometry; t.i.d., ter in die, three times a day. See also Figure S1, and Table S1.
After 12 weeks of treatment, HbA1c was reduced in four of the six patients (Figure 1c). We observed a measurable reduction in fasting blood glucose (Figure 1d) and fasting insulin in some but not all patients (Figure 1e). Insulin resistance was improved as shown by both HOMA-IR (Figure 1f) and rate of glucose disappearance during a hyperinsulinemic euglycemic clamp (Figure 1g and h) after amlexanox treatment. We observed decreased hepatic glucose production during fasting in four of the patients (Figure 1i). One patient (#3) did not exhibit any indication of improved insulin sensitivity, and was also the only patient in whom no reduction in hepatic fat was observed by the MRI-Dixon method (Figure 1j). Patient 5 had the greatest reduction in HbA1c (0.6 %), and showed an improvement in all measures of insulin sensitivity (Figure 1c–h). The mixed meal tolerance test showed an overall improvement in the early hyperglycemic peak after treatment with amlexanox when compared to baseline (Figure 1k). There was a reduction in plasma free fatty acid levels after treatment both at baseline and throughout the mixed meal test in the entire cohort of patients (Figure 1k). No changes were observed in triglycerides, with a slight increase in HDL (not shown). These results pointed towards an improvement of insulin sensitivity in the adipose tissue and liver during this short period, consistent with what was observed in the earlier rodent studies (Reilly et al., 2015; Reilly et al., 2013).
Statistically significant weight loss was observed during the 12-week amlexanox treatment (Figure 2a). Biopsies were obtained from subcutaneous adipose tissue before and after drug treatment, and RNA was prepared for analysis by qPCR. Interestingly, increased expression of several genes associated with increased energy expenditure was observed after the 12-week treatment period, most notably UCP1 (Figure 2b). Furthermore, the change in UCP1 expression after treatment was inversely proportional to post-treatment body weight (Figure 2c). The gene encoding the β3-adrenergic receptor, ADRB3, was also more highly expressed after treatment (Figure 2d). Reduced expression of ADRB3 is frequently observed in obesity-associated catecholamine resistance (Collins et al., 1999; Lowell and Flier, 1997). We also observed increased expression of FGF21, a gene associated with the development and activation of beige fat (Owen et al., 2015) (Figure 2e). Other markers of beige fat, DIO2, PRDM16, PPARGC1A, PPARGC1B and ELOVL3, increased in a subset of the responsive patients (Figure 2f and g). Although there was no significant change in DIO2 expression in the entire group, the increase in DIO2 was inversely proportional to post-treatment body weight (Figure 2f), as was observed for UCP1. There was also a trend towards increased expression of ADIPOQ and IL4, which are associated with activation of type 2 immunity and metabolic health in adipose tissue (Figure 2h and i).
Figure 2. Weight loss and gene expression changes in the open label study subjects.

a) Body weight (left panel) and body weight represented as percentage of baseline body weight (right panel) during 12-week treatment and 4-week washout observation period. b) Relative expression of UCP family of genes encoding mitochondrial inner membrane protein. c) Correlation of relative UCP1 expression to weight loss after 12 weeks of treatment. d) Relative expression of ADRB family of genes encoding β-adrenergic receptors. Relative expression of beige fat markers: (e) FGF21, (f) DIO2, (g) PPARGC1A, PPARGC1B, PRDM16, and ELOVL3. f) Correlation of DIO2 expression and weight loss (bottom panel). h) Relative expression of IL4. i) Relative expression of ADIPOQ, the gene encoding adiponectin. n = 6. ** indicates p-value ≤ 0.01; * indicates p-value ≤ 0.05; # indicates P-value ≤ 0.10 (two-tailed paired t-test). c, f Pearson’s correlation with 95% CI. AU, arbitrary unit.
Treatment-emergent adverse events in the study are tabulated in Table S1. We observed two cases of a pruritic, erythematous generalized rash more pronounced in the extremities in two of the six patients, in whom the rashes were biopsied and showed perivascular inflammation that could be consistent with a drug reaction. In both cases, the rash started at approximately 4 weeks and abated with local measures and symptomatic management within 2 weeks despite continued treatment. Both of the patients (Patient 1 and 5) were among those who had a favorable metabolic response to the drug.
Amlexanox improves metabolic parameters in a placebo-controlled double blind study
We also conducted a 12-week long, randomized, double blind, placebo-controlled proof of concept trial (NCT01842282). In this second study, we administered 50 mg amlexanox three times daily (Figure 1a). The study was designed to have 80% power to detect a minimal difference of 0.5 % in HbA1c (primary end-point) in 34 patients, 17 in each group (detailed in Methods). We randomized 42 patients receiving the study medication or placebo (see methods for details on randomization and retention). Baseline characteristics of the groups are shown in Table 1, and show no significant differences, including diabetes duration and background concomitant medication usage. In order to test the drug in a population that was easily accessible in our tertiary care practice, we did not limit the study to drug naïve patients. Each group had 21 patients and gender distribution was balanced, owing to the stratification in randomization schema with respect to sex. Duration of diabetes and number of medications did not differ between the two groups (Table 1). 20 from the drug-treated group and 18 from the placebo group completed the 12 weeks of treatment (Figure 3a). Three patients from the placebo group and one from the treatment group discontinued the study early (within two weeks) either due to discovery of an exclusion criteria on laboratory assessments (elevated CK or HbA1c >10.5% that was higher than the screening value) or subject burden (Table S2). One patient in the placebo group had a non-ST elevation myocardial infarction at week 11 of the treatment period, leading to early unblinding, and was seen for the 12 and 16-week assessments, during which only safety assessments were conducted and no other study procedures were completed. Therefore, most of the paired data are from 17 patients from the placebo group and 20 patients from the amlexanox-treated group. There are some study parameters with less data points available due to funding reasons or patient compliance, as explained in Table S2, and mentioned in figure legends or data tables as applicable. All but one of the amlexanox-treated patients also completed an additional 4 weeks of observation off the study drug.
Table 1.
Baseline characteristics of patients in placebo-controlled trial.
| Parameters | Placebo (n = 21) |
Amlexanox (n = 21) |
p-value | |
|---|---|---|---|---|
| Demographics | Gender (Female/Male) | 10/11 | 10/11 | STRATIFIED |
| Age | 59±8 | 56±8 | 0.21 | |
| Race: Caucasian, African, Asian American | 19, 2, 0 | 19, 1, 1 | 0.51 | |
| History of tobacco use | 8 | 12 | 0.35 | |
| Baseline Medications | Antidiabetics: Metformin, Sulfonylurea, DPP4 inhibitor, TZD, SGLT2 inhibitor, Repaglinide | 19, 9, 4, 0, 0, 0 | 17, 4, 3, 2, 1, 1 | 0.41, 0.18, 1.00, 0.49, 1.00, 1.00 |
| Anti-hypertensives: ACE inhibitor, ARB, CCB, β-blocker, Diuretic, Other | 10, 5, 2, 4, 10, 1 | 11, 3, 4, 4, 7, 1 | 1.00, 0.70, 0.66, 1.00, 0.53, 1.00 | |
| Other: Statins, Aspirin, NSAID, Antidepressants, Thyroid hormone | 12, 6, 4. 7, 3 | 18, 10, 2, 6, 4 | 0.09, 0.34, 0.66 1.00, 1.00 | |
| Medical History | Diabetes history (years) | 9±6 | 8±5 | 0.56 |
| Diabetic Retinopathy, Diabetic Neuropathy | 0, 5 | 3,9 | 0.23, 0.33 | |
| Hyperlipidemia, Hypertension, Heart disease/attack, Stroke | 16, 15, 2, 3 | 19, 14, 3, 1 | 0.41, 1.00, 1.00, 0.61 | |
| Thyroid disease, Kidney disease History of cancer, Arthritis | 3, 4, 5, 14 | 5, 1, 2, 9 | 0.70, 0.34, 0.41, 0.21 | |
| Asthma/Emphysema, Sleep Apnea | 3, 6 | 6, 9 | 0.45, 0.52 | |
| Polycystic-ovarian syndrome | 2 | 1 | 1.00 | |
| Psychiatric diagnosis (depression or anxiety) | 8 | 7 | 1.00 | |
| ROS: Muscle weakness, Loss of Sensation, Tingling hands/feet | 0, 3, 6 | 2, 6, 7 | 0.49, 0.45 1.00 | |
| Physical exam | Weight (kg) | 98±20 | 100±15 | 0.90 |
| Height (cm) | 171±12 | 172±8 | 0.71 | |
| Body mass index (kg/m2) | 33.5±4.6 | 33.9±4.3 | 0.94 | |
| Systolic blood pressure (mm, Hg) | 132±16 | 131±14 | 0.94 | |
| Diastolic blood pressure (mm,Hg) | 76±9 | 75±7 | 0.73 | |
| Pulse | 69±9 | 72±11 | 0.35 | |
| Acanthosis nigricans, Arrhythmias, Murmurs | 4, 1, 1 | 2, 2, 2 | 0.66, 1.00, 1.00 | |
| Liver enlarged, Total Liver span (cm) | 14, 10±2 | 13, 10±2 | 1.00, 0.98 | |
| Biochemical parameters | HbA1c (%) | 7.6±0.8 | 7.7±1.0 | 0.76 |
| Fructosamine (µmol/L) | 281±58 | 278±52 | 0.89 | |
| Fasting glucose (mg/dL) | 165±51 | 151±39 | 0.32 | |
| Fasting insulin (µU/mL) | 22.4±14.2 | 20.1±12.6 | 0.58 | |
| HOMA-IR | 8.48±4.82 | 7.67±6.68 | 0.65 | |
| Triglycerides (mg/dL) | 181±122 | 171±81 | 0.76 | |
| Cholesterol (mg/dL) | 159±35 | 161±32 | 0.84 | |
| HDL (mg/dL) | 42±14 | 42±7 | 1.00 | |
| LDL (mg/dL) | 83±28 | 85±29 | 0.88 | |
| WBC count (K/µl) | 7.0±1.5 | 6.9±1.5 | 0.94 | |
| Hemoglobin (d/dL) | 13.2±1.6 | 13.9±1.1 | 0.11 | |
| Platelets (K/µl) | 241±119 | 219±32 | 0.42 | |
| Creatinine kinase (IU/L) | 124±75 | 178±167 | 0.19 | |
| ALT | 38±18 | 37±17 | 0.82 | |
| CRP (µg/mL) | 2.35±1.88 | 2.90±2.04 | 0.40 | |
| 24 Hour urine protein, quantity (g/24 hr)a | 0.09±0.11 | 0.18±0.40 | 0.42 | |
| % Fat | DEXA Total | 41.13±6.38 | 39.94±7.69 | 0.59 |
| DEXA Trunk | 46.97±4.87 | 45.06±6.13 | 0.27 | |
| MRI Quantitativeb | 12.8±7.8 | 18.0±9.7 | 0.20 | |
Data represent either n per group or mean ± SD.
n = 16 in each group, based on patient returned samples
n = 9 in Placebo, and 11 in Amlexanox group
DPP4: Dipeptidyl peptidase-4, TZD: Thiazolidinediones, SGLT2: Sodium-Glucose Transporter 2, ACE: Angiotensin converting enzyme, ARB: Angiotensin receptor blocker, CCB: Calcium channel blocker, NSAID: Non-steroidal anti-inflammatory agents, ROS: Review of Systems
Figure 3. Measures of glycemic control in the double blind, placebo-controlled trial.

a) Consort diagram: Schematic of randomized double blind placebo-controlled trial. For details on evaluable n of secondary and exploratory end point analyses, refer to Table S2. Difference from baseline in b) Hemoglobin A1c and (c) fructosamine in the placebo- and amlexanox-treated patients left panels; right panels display baseline and 12-week values for each patient connected with a line. d) Insulin and (e) glucose during the mixed meal test. f) Insulin ratio (week 12 divided by baseline area under the curve for insulin measured during the mixed meal test). b: n = 18 placebo, n = 20 amlexanox; c–f: n = 17 placebo, n = 20 amlexanox (n = 7 A–R). ** indicates p-value ≤ 0.01 and * indicates p-value ≤ 0.05 (two tailed t-test, b, c, e unpaired, d paired). Data presented as individual data and mean ± s.e.m. (b, c, f) or mean ± s.e.m. only (d, e). See also Figure S2–S4, and Table S3 and S4.
As mentioned above, there was one serious adverse event of a non ST-elevation MI observed in a placebo-treated subject (Table S3). There were a total 7 cases of rash reported, only one of which (noted in a drug-treated patient classified as a responder) reached the scope we had observed in the previous two patients in whom a biopsy was also conducted (Figure S1). This event also occurred around week 4 and abated by the end of week 6. Treatment was continued and local measures were taken for symptoms of itching and heat. The other rash incidents that were reported were mild and not clinically apparent at the time of evaluation. There were no other adverse events attributable to drug use that were notable. The package insert for Amlexanox tablets in Japan indicated a 5% incidence of rash.
The study revealed a statistically significant difference in the change in HbA1c, which was reduced close to 0.5% more on average in the drug-treated group compared to placebo (Figure 3b, Table S4); this was the primary end point for the study. The difference in glucose control was further substantiated by a reduction in fructosamine levels (Figure 3c, Table S4), a marker for shorter term glucose control and an important secondary endpoint. There was a trend towards reduction in fasting blood glucose (p value = 0.06) and improved insulin sensitivity as shown by reduced HOMA-IR (p value = 0.08) in the amlexanox-treated patients compared to placebo (Figure S2a and b). Improved insulin sensitivity in the amlexanox-treated patients was also indicated in the mixed meal tolerance test, in which the amlexanox-treated patients exhibited a reduction in the early hyperinsulinemic (Figure 3d) and hyperglycemic (Figure 3e) peaks when compared to baseline, similar to that observed in the open label trial (Figure 1k). The ratio (12-weeks/baseline) of the area under the curve for insulin response was significantly reduced during the mixed meal tolerance test in the drug-treated cohort compared to the placebo group (Figure 3f). Importantly, discontinuation of the drug at the end of the study caused an acute increase in fasting glucose levels and a worsening of HOMA-IR (Figure S2c and d), demonstrating that the effects of the drug may be reversible.
One third of the patients exhibited a robust response to amlexanox
Analyses of the data revealed that not all patients responded to amlexanox equally. There were 7 patients in the treatment group who achieved an HbA1c reduction of ≥ 0.5 %, a clinically meaningful threshold value used as an important milestone threshold in diabetes trials both in the US and EU (Karyekar et al., 2013, see also additional resources), while only one patient in the placebo group achieved this result (p value = 0.045 using Fisher’s exact test). We classified 7 patients as responders in the treatment group, and re-analyzed the data looking at the drug-treated responders and non-responders separately. As may be predicted, responders exhibited a significant reduction in fructosamine, which was almost double the reduction observed in the entire drug-treated group, as non-responders did not exhibit a significant decrease in fructosamine (Figure S2e). As expected from how responder status was defined, the same pattern was observed for fasting blood glucose (Figure S2f), and HOMA IR (Figure S2g), although these results did not reach statistical significance due to high variability of these parameters.
We did not observe a statistical difference in weight loss between the placebo and amlexanox-treated groups in this study (Figure S3a); even within the amlexanox responders, there was no apparent reduction in body weight, unlike the earlier pilot study. There was a wide range of weight change among the participants; with some patients gaining weight and others losing weight in all groups (Figure S3b). However, we did note a tendency for reduction in both percent total body fat (Figure S3c) and in percent truncal fat (Figure S3d) in the amlexanox responder subgroup compared to the non-responders (p value = 0.06) without a change in lean mass. Due to budgetary constraints, we studied hepatic fat only in 20 patients with paired MRIs at baseline, and 19 of these completed a repeat scan at 12 weeks. These 20 patients turned out to be 9 placebo-treated, and 11 amlexanox-treated, further divided into 4 treatment-responders and 7 non-responders. Interestingly, only the responders exhibited a reduction in hepatic fat (Figure S3e). In addition, the responder group also displayed a significant increase in both baseline and post-meal HDL level in serum during the meal tolerance test at 12 weeks versus baseline (Figure S4a), which suggests that the drug may alter postprandial lipid metabolism. No effect was observed on postprandial triglyceride levels (Figure S4b). However, fasting and 30-minute post-meal FFA levels decreased significantly in the responder group compared to the non-responder group (Figure S4c). The increase in HDL levels during the post-mixed meal reached statistical significance at 120 minutes in the responder group comparing the baseline response to the post-drug treatment response (p value =0.015). Post mixed meal peptide YY (PYY) (p value =0.024), and gastric inhibitory polypeptide (GIP) (p value =0.07) were attenuated in the amlexanox-treated groups versus placebo but not different in the responders versus non-responders (Figure S4d and e). Ghrelin and Glucagon-like peptide-1 (GLP-1) secretion (pre- and post-mixed meal) were not different between placebo- and drug-treated groups (Figure S4f and g).
Baseline characteristics that separate responders from non-responders
The bimodal distribution of drug-treated subjects into responders (HbA1c reduction of greater than or equal to 0.5%) and non-responders in terms of glucose control suggested that there might be factors segregating these populations, and we thus sought to identify metabolic or hormonal characteristics differentiating non-responding and responding patients. At baseline, there were no major differences among these groups with regard to gender, weight, BMI, fasting insulin, and fasting or postprandial glucose (Figure S5); nor did we see any significant differences in other parameters including fasting serum levels of other hormones or metabolites.
We were able to analyze baseline and 12-week serum samples from this trial to study drug metabolism (Table S5). Amlexanox and its metabolites were identifiable in the serum of the treated patients at 12 weeks (Inagaki et al., 1992; Rankov et al., 1990). Importantly, there was no difference in steady state serum levels of amlexanox or its metabolites in the responders versus non-responders (Figure 4a and b). This result indicates that the difference between responders and non-responders is unlikely to be compliance or drug metabolism; although a full pharmacokinetic study is needed to definitively compare drug metabolism in responders versus non-responders.
Figure 4. Baseline differences in responders versus non-responders.

Serum levels of (a) amlexanox and (b) one of its metabolites at baseline and 12 weeks. a, b) n = 18 placebo, 7 amlex-R (Amlexanox-treated, responder) and 13 amlex-NR (amlexanox treated, non-responder). See also Table S5. c) Genes whose expression was significantly different in responders versus non-responders at baseline in RNA-sequencing analysis. Values presented as log (fold change) in responders relative to non-responders; (n = 4 per group). d) Baseline CRP values in the placebo-controlled trial in the placebo- and drug-treated patients. The latter group is further divided into responders and non-responders. Baseline CRP values for one of the placebo treated patients was excluded as it was obviously compromised. n = 17 placebo, 7 amlex-R, 13 amlex-NR. e) Correlation of baseline CRP values with change in HbA1c from baseline to 12 weeks in amlexanox-treated patients (left panel, n = 20) and placebo controls (right panel, n = 17). Note the significant inverse relationship in the drug treated group compared to lack of correlation in the placebo-treated group. f) Correlation between baseline BMI and serum CRP at baseline in all patients (left panel; p = 0.031, Pearson’s r = 0.36, n = 37,) and FMI (fat mass index) vs. serum CRP in all patients (right panel) at baseline (p =0.0003, Pearson’s r = 0.56, n=37). g) Correlation between liver fat and serum CRP at baseline in patients who were evaluated for liver fat (p = 0.035, Pearson’s r = 0.49, n = 19). # indicates p-value ≤ 0.10, * indicates p-value ≤ 0.05 (two tailed t-test). Data presented as individual data and mean ± s.e.m or 95% CI. CRP: C-reactive protein. BMI: body mass index. See also Figure S4.
Animal studies and our open-label pilot study have indicated that the adipose tissue is an important primary target tissue for amlexanox (Reilly et al., 2015; Reilly et al., 2013). Therefore, we performed RNA-sequencing analysis to compare gene expression in subcutaneous adipose tissue of responders and non-responders prior to drug treatment. These analyses revealed 28 genes with higher expression and 26 genes with lower expression in the responders versus nonresponders at baseline (Figure 4c). These differentially regulated genes are highly enriched for those involved in the inflammatory response, indicating a potential difference in the inflammatory state of the subcutaneous adipose tissue in responders prior to treatment. Of the top 10 genes whose baseline expression was highest in the responders versus non-responders, 6 are involved in pro-inflammatory signaling (Figure 4c). The two genes whose expression was highest in the responders versus non-responders, FOSB and FOSL1, encode leucine zipper proteins that form the AP-1 transcription factor complex when dimerized with JUN family proteins. AP-1 is a transcriptional regulator driving M1 classical macrophage activation, and an important contributor to obesity-associated adipose tissue inflammation (Olefsky and Glass, 2010). Expression of SOCS3 was also higher in the responders; this gene is expressed downstream of activation of STAT3 by proinflammatory cytokines, and contributes to the development of insulin resistance (Rui et al., 2002) (Ueki et al., 2004). CXCL2 expression was also higher in the responders versus non-responders; this chemokine is induced in M1-polarized macrophages and has previously been shown to be elevated in obese versus non-obese human subcutaneous adipose tissue (Rouault et al., 2013). The chemokine CCL2, also known as MCP1, was more highly expressed in responders. MCP1 plays an important role in M1 macrophage recruitment to obese adipose tissue and obesity-associated inflammation (Weisberg et al., 2006). S100A8 was also increased in responders versus non-responders; this gene encodes a damage-associated molecular pattern protein (Sekimoto et al., 2015). The heterodimeric complex of S100A8/S100A9 promotes inflammation by acting as a ligand for TLR4 and is linked to activation of the Nlrp3 inflammasome (Nagareddy et al., 2014). Serum levels of S100A8/A9 have been associated with both obesity and diabetes (Sekimoto et al., 2015). Two additional inflammatory genes were elevated in responders: CYR61, a matricellular protein that modifies the activity of cytokines and promotes a genetic program consistent with M1 macrophage polarization (Bai et al., 2010), and the chemokine CCL14 (Pardigol et al., 1998). Further supporting increased inflammation in the responders at baseline, genes that are thought to be involved in M2 polarized macrophage function were expressed at significantly lower levels in the responders versus non-responders at baseline, including TGFB1 and VAV1. Taken together, this gene expression pattern indicates that increased baseline adipose tissue inflammation may differentiate responders from non-responders.
Given the proinflammatory pattern of gene expression differentiating the responders and non-responders at baseline, we sought to find a serum marker of inflammation that might predict responsiveness. To this end, we measured C-reactive protein (CRP) levels in serum from all patients. CRP levels tended to be higher in responders than non-responders at baseline (Figure 4d). Furthermore, baseline CRP levels correlated with the reduction in HbA1c from baseline to the end of the 12-week amlexanox treatment, while there was no association of baseline CRP with change in HbA1c levels in placebo-treated patients (Figure 4e). Interestingly, baseline CRP was positively correlated with BMI, percent fat and percent liver fat (Figure 4f and g). These correlations support the connection between serum CRP and obesity-associated inflammation (Landgraf et al., 2015) (Capaldo et al., 2013).
Amlexanox responders show a transient increase in IL-6 secretion
Because of the important role of adipocyte IL-6 secretion in controlling glucose levels in rodents treated with amlexanox (Reilly et al., 2015), we also examined IL-6 levels in all subjects during the course of the study. Interestingly, there was a sharp but transient increase in IL-6 levels in serum over 2 to 4 weeks of treatment only in responders, but not non-responding drug-treated subjects (Figure 5a). Although we cannot ascertain the source of IL-6 in these subjects, this transient change is reminiscent of what is seen in obese mice treated with amlexanox, in which IL-6 secreted from subcutaneous adipocytes activates the JAK/STAT3 pathway in liver to reduce expression of gluconeogenic genes such as G6PC, in the process, reducing hepatic glucose output (Reilly et al., 2015).
Figure 5. Responder specific changes in serum IL-6 and subcutaneous fat gene expression.

a) Serum IL-6 levels over the course of the trial: 12 weeks of treatment and 4 weeks of observation period post treatment. b–f, i) Relative expression of genes in the subcutaneous fat at baseline and after 12 weeks of treatment. b–e) Relative expression of beige fat associated genes: (b) UCP1 (Ct values in the non-responder (baseline: 35.1, 12-week: 35.9) and placebo (baseline: 34.7 12-week: 34.4) were not changed after treatment, while the Ct value in the responder was reduced (baseline: 35.9 12-week: 33.8). While these high Ct values indicate low abundance of UCP1, good linearity was observed in the standard curve from Ct 30 to Ct 35), (c) DIO2, (d) PRDM16 (left panel), FGF21 (right panel), and (e) COX5B. f) Relative expression of CCL2. g) Heat map illustrating the genes that were significantly changed from baseline to 12 weeks in a paired analysis of the RNA-sequencing data within each of the three groups. h) Heat map illustrating genes from different pathways that were differentially regulated by treatment in the responders (*HSPB1 was also significantly upregulated in the non-responder group). i) Relative expression in the subcutaneous fat at baseline and after 12 weeks of treatment of LIPE (top panel) and FASN (bottom panel). See also Table S6 and S7. (a: n = 17 placebo, 7 amlex-R and 13 amlex-NR; c–f, i: n = 15 placebo, 7 amlex-R and 13 amlex-NR; g,h: n = 4 per group * indicates p-value ≤ 0.05; # indicates P-value ≤ 0.10 (two-tailed t-test a,unpaired, b–i, paired). Data presented as mean ± s.e.m. Amlex-R: amlexanox-treated responder, Amlex-NR: amlexanox-treated, non-responder.
Amlexanox drives a characteristic transcriptional response in subcutaneous white adipose tissue of responders
In mice, amlexanox treatment increases energy expenditure and restores catecholamine sensitivity in subcutaneous adipose tissue, a key event in the beneficial effects of amlexanox on both glucose handling and browning of the adipose tissue (Mowers et al., 2013; Reilly et al., 2015). Therefore, we performed qPCR analysis on mRNA isolated from the subcutaneous adipose tissue biopsies taken before and after drug treatment to investigate gene expression changes in response to amlexanox. Consistent with the browning effect observed in mice, amlexanox treatment caused a significant increase in the expression of UCP1 (Figure 5b) and DIO2 (Figure 5c) specifically in the amlexanox responders, while no change in the expression of these genes was observed in the non-responders or placebo controls. Further supporting a browning effect of amlexanox, the responders also exhibited increased expression of PRDM16 and FGF21 (Figure 5d). Additionally, we observed that the expression of the oxidative metabolism gene COX5B was also increased specifically in the amlexanox responders (Figure 5e). Previous data has also shown that long-term amlexanox treatment of mice reduces inflammation in visceral white adipose tissue, and to a lesser extent in subcutaneous fat. While there was a trend towards reduced expression of CCL2 in the amlexanox responders, it did not reach statistical significance (Figure 5f). Long-term amlexanox treatment of mice significantly reduces visceral, and to a lesser extent subcutaneous adipose tissue inflammation; however, this effect appears to be indirect, as amlexanox treatment in cell culture does not reduce inflammation in a cell autonomous manner (Mowers et al., 2013).
To investigate further gene expression changes in subcutaneous adipose tissue on a genome-wide scale, we performed unbiased RNA-sequencing analysis. For this analysis, we selected 4 patient samples with the highest quality RNA from each of the treatment groups: placebo, amlexanox responder and amlexanox non-responder. In biopsy samples of placebo-treated patients, the expression of only 2 genes changed after the 12-week treatment; one of these genes, CLDN5, exhibited increased expression in all treatment groups at 12 weeks versus baseline. In biopsy samples from the non-responding patients, the expression of 10 genes changed, all but one of which was also changed in the amlexanox responder group. Strikingly, RNA-sequencing analysis of biopsies from amlexanox responders revealed an additional 1038 genes, the expression of which changed in response to the drug (Figure 5g).
We performed pathway analysis on the genes with increased or decreased expression over the course of treatment in the amlexanox responders (Table S6, Figure 5h). There were 968 genes with increased expression only in patients who responded to amlexanox, while only 78 genes exhibited decreased expression. Interestingly, among the upregulated genes, significant enrichment of genes in the oxidative pathways was observed, including cytochrome C oxidase genes, and a number of genes encoding subunits of mitochondrial complex I. Additionally, an increase in genes associated with insulin signaling was observed, consistent with lower blood glucose. Since the RNA-sequencing analysis was performed on a subset of the patients, we verified the expression in the entire cohort by qPCR of two differentially expressed genes involved in lipid metabolism, FASN and LIPE (Wei et al., 2016). Loss of function mutations of the latter gene have been recently identified in patients with partial lipodystrophy syndromes in humans, linking LIPE directly to adipocyte dysfunction (Farhan et al., 2014; Zolotov et al., 2016). As predicted by the RNA-sequencing analysis, FASN and LIPE expression increased in samples from the amlexanox responders but not in the non-responder or placebo-treated groups (Figure 5i).
In addition to the pathways we had anticipated might change in patients treated with amlexanox, other pathways emerged that we had not previously considered, including the VEGF signaling pathway, which has been shown to play an important role in healthy adipose tissue expansion, and was upregulated in responders (Robciuc et al., 2016). VEGF signaling and subsequent angiogenesis have also been shown to promote the browning of subcutaneous adipose tissue (Sun et al., 2012). We further observed that the expression of HIF1A was significantly down-regulated by amlexanox treatment in the responders (Figure 5h), supporting the idea that amlexanox treatment promotes healthy, but not pathological adipose tissue expansion associated with hypoxia (Trayhurn, 2013). Future studies will be needed to determine the precise role of VEGF signaling and adipose remodeling in response to amlexanox and whether or not it contributes to the development of beige fat.
Discussion
Taken together, the human studies presented here indicate that the anti-diabetic effects of the IKKε/TBK1 inhibitor amlexanox observed in mice may be translated into a subset of obese humans with type 2 diabetes. The primary endpoint of our randomized trial was met, which was a significant effect of the drug on HbA1c (accompanied by reductions in fasting blood glucose and fructosamine) across the patient group. In addition, the characteristics of the patients with a clinically significant improvement in HbA1c were also noteworthy. The drug-related response in these patients was characterized by a transient increase in IL-6, and changes in subcutaneous adipose tissue gene expression, the presumed target of the drug (Reilly et al., 2015). Amlexanox-induced changes in gene expression in the adipose tissue were consistent with many of the changes seen in mice, indicating an overall increase in fuel oxidation and insulin sensitivity. Moreover, the responding patients exhibited a gene expression pattern indicating increased inflammation at baseline, suggesting that there may be a threshold of intrinsic adipose tissue inflammation in patients that are more likely to respond to the drug. This hypothesis is supported by the correlation between elevated baseline CRP and the decrease in HbA1c. Early observations also suggest that amlexanox may lower hepatic fat in patients with NAFLD; although, the number of patients studied with hepatic MRI did not have sufficient power to reach a definitive conclusion. Future studies will prospectively address whether preselecting patients based on an elevated CRP or increased adipocyte inflammation would improve the systemic anti-glycemic and potentially anti-steatotic response.
Increased adipose tissue inflammation in the responders is consistent with our understanding of the molecular mechanism by which amlexanox improves metabolic health. In animal models, amlexanox has effects only on obese mice, presumably since the targets of the drug, IKKε and TBK1, are overexpressed as a consequence of adipose tissue inflammation (Mowers et al., 2013; Reilly et al., 2013). Moreover, only obese mice develop catecholamine resistance (Mowers et al., 2013), consistent with what has been observed in obese patients (Arner, 1999; Bougneres et al., 1997; Horowitz and Klein, 2000; Reynisdottir et al., 1994). Therefore, it is logical that amlexanox responders are characterized by higher baseline adipose tissue inflammation. In future studies, it will be interesting to investigate whether adipose tissue inflammation at baseline also correlates with adipose tissue catecholamine resistance in responsive patients, whether catecholamine sensitivity is restored by amlexanox treatment, and whether there are other mechanisms involved in increased energy expenditure and improved glucose homeostasis in patients who respond to amlexanox treatment.
Amlexanox provided amelioration of high glucose levels in a subset of patients in this study, with little evidence of safety concerns outside of a previously noted rash, which so far appears to be self-limited despite drug continuation in the patients in whom this effect was observed. It will be important to establish an optimal safe dose and a dosing regimen for the drug in future studies. Exploring the utility of combinations with other diabetes or obesity drugs is another area of future investigation. Also, the duration of our trial may not have been long enough to uncover all the effects of the drug (positive or negative). While only a first step in understanding the metabolic effects of amlexanox in humans, these data indicate that impacting energy expenditure pathways in subcutaneous adipose tissue in patients with high baseline inflammation may be beneficial for the treatment of obese patients with type 2 diabetes and NAFLD.
Star Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Alan Saltiel (asaltiel@ucsd.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Subjects were enrolled who fulfilled the following inclusion criteria: age 18 to 60 years, physician-confirmed type 2 diabetes mellitus, BMI between 27 and 40 (for the open label study) or 45 (for the double blind, placebo-controlled trial) kg/m2, and a liver ultrasound confirming presence of fatty infiltration. Prior to enrolling, subjects were on a stable diabetes treatment regimen for twelve weeks, with oral medications only, such as metformin, sulfonylurea, and/or DPP IV inhibitors for the open label study. Sodium glucose transporter inhibitors were also allowed for the placebo controlled study as they were prescribed more commonly by the time this study was launched. We specifically did not target just drug naïve patients as we wanted to understand the applicability of this drug in the greater diabetes field, while we recognized that this may impact the effect size in a negative manner. Women of child-bearing potential were required to have a negative pregnancy test and practice appropriate birth control throughout the entire duration of the study period. Exclusion criteria were: consumption of alcohol of more than 40 grams every week, presence of other liver disease, hemoglobin A1c greater than 10.5%, treatment with insulin or other injectable diabetic drugs, or history of significant comorbidities such as advanced liver disease, hematologic abnormalities, HIV, advanced renal disease, proliferative diabetic retinopathy, clinically relevant unstable coronary artery disease, or active infection. The open label trial was performed in 6 patients, their baseline characteristics are presented in Figure 1b. 42 patients were enrolled in the placebo controlled trial, their baseline characteristics can be found in Table 1. Both studies were reviewed and approved by the University of Michigan IRBMED and the US FDA and conducted under IND117543.
METHOD DETAILS
Subjects were recruited with the assistance of local media outlets and the University of Michigan Diabetes Research Registry. Those who were determined to be appropriate candidates based on the above mentioned inclusion and exclusion criteria were enrolled in clinical trial NCT01842282. Once intermittent data analyses were conducted for the first study, and funding for the placebo-controlled study was obtained, a decision was made to proceed with the placebo-controlled study NCT01975935. For both studies, a screening evaluation was conducted through review of medical records. A screening visit was performed after the subjects gave informed consent where an HbA1c was obtained and an abdominal ultrasound was performed if one was not available within 6 months of evaluation. Eligible patients were admitted to the Michigan Clinical Research Unit (MCRU) on the morning of the baseline evaluation.
Overview of open label trial
The primary endpoint of the study was determined to be safety and tolerability with specific attention to hypoglycemia. In addition, efficacy and mechanistic endpoints deemed exploratory were determined as glucose control, body weight, and hepatic fat as determined by MRI. Other exploratory studies included assessment of insulin sensitivity and gene expression profiles from adipose tissue biopsies. The original plan was to enroll 15 patients to assess the safety and tolerability of the drug in the study population at the doses used to treat asthma. The overall goal for this study was learning to use this drug for metabolic endpoints. Six patients completed all procedures of the study from July 2013 to April 2014, until the funding for the open-label study became available from NIDDK. This study is still left open to allow for further mechanistic inquiry in an open label fashion using the same timelines and dosing schema.
In the open label study, baseline assessments listed in Figure 1a. were carried out on two different days, no more than 10 days apart. First day of baseline assessment consisted of the hyperinsulinemic euglycemic insulin clamp study. The adipose tissue biopsy was obtained within the second hour of the glucose tracer infusion. The second assessment day comprised of the remaining studies (fasting biochemistry, the mixed meal test, anthropometric measurements, dietary evaluations, DEXA scan and MRI of the liver, shown in Figure 1a. Patients were provided instructions for a standardized meal (600 kcal, 55% carbohydrates, 30% fat and 15% protein) and then instructed to fast overnight after 22:00 hours prior to the studies, such that the mixed meal test and the hyperinsulinemic euglycemic clamp measurements could be compared without the confounding changes in the diet during the preceding 24 hours.
After all assessments were completed, subjects were instructed to take amlexanox tablets (manufactured by Takeda Pharmaceuticals and imported into US) in an open-label manner at a dosage of 25 mg PO three times daily. After 2 weeks, the dosage was increased to 50 mg tablets three times daily for the remainder of the 12-week treatment period. They were allowed to continue baseline diabetes therapies without dose adjustments. The only diet intervention was reinforcement of ADA guidelines (American Diabetes et al., 2008). Blood collection was performed at baseline and 1, 2, 4, 6 and 12 weeks of treatment, as well as week 16, which was a follow-up 4 weeks after discontinuation of the study drug.
Safety biochemistry, urinalysis, fasting insulin and glucose were measured at each visit. Diet records and symptom sheets were collected and evaluated at each visit so long as patients brought them. In addition, at baseline and 12-week visits, levels of hemoglobin A1c, thyroid function tests, and fasting adipocytokines were measured. Anthropometric measurements were obtained at baseline, 12-week, and 16-week visits. Hyperinsulinemic euglycemic clamp, and adipose tissue biopsy, mixed meal test, DEXA scan for total body composition, liver MRI, were performed at baseline and 12-week visits. The 12-week assessment also was spread over two days, mirroring the order described for baseline assessments with a 10-day window.
Overview of placebo-controlled trial
The clinical protocol for the placebo-controlled study was similar to the open label trial, but the procedures were simplified to allow for increased recruitment as well as completion of the studies in one day (Figure 1a). The amlexanox tablets or inactive placebo material were coated by University of Michigan Investigational Drug Services using Good Manufacturing Principles. Subjects were started on 50 mg three times a day dose (two capsules of drug or placebo, three times daily) without any titration.
Serum factor analysis
Blood samples after overnight fast were analyzed for Hemoglobin A1c, fructosamine, glucose, insulin, fasting lipids and hepatic function tests, electrolytes, BUN, creatine, creatinine kinase (CK), and thyroid hormone levels in the Clinical Pathology Laboratory of University of Michigan using auto-analyzer equipment on the same day of testing, processed as soon as available. The adipokine levels were determined using a custom ordered commercial multiplex assay (EMD Millipore, Billerica, MA). IL-6 and CRP levels were determined by ELISA (R&D systems, Minneapolis, MN). FFA measurements were performed using a colorimetric NEFA Kit (Wako, Richmond, VA). For these latter measurements, samples were stored at −80 °C and measured all at once at the end of the study.
Hyperinsulinemic euglycemic clamp
An intravenous catheter was placed in the antecubital forearm vein for infusion, and the hand of the opposite arm was placed in a hotbox for sampling. At time 0, samples were collected for CBC and baseline glucose, and a primed, constant rate infusion of [6,6 d2] glucose (obtained from Isotec/Sigma, Miamisburg, OH) (25 umol/kg priming dose; 0.41 umol/kg/min continuous infusion) was initiated. Between 0–3 hours, a retrograde IV was inserted into the arm placed in the hotbox. Beginning at 3 hours and 45 minutes, three arterialized blood samples were obtained in 5 minute intervals for determination of basal hepatic glucose production via isotope dilution of the constant rate infusion of [6,6 d2] glucose. At 4 hours, the hyperinsulinemic euglycemic clamp was started to assess insulin sensitivity, as described previously (DeFronzo et al., 1979). The clamp entailed a primary 2 hour insulin infusion at a rate of 40 mU/m2/min. Plasma glucose concentration was monitored every 5 minutes during the clamp study using a glucose autoanalyzer (Yellow Springs Instrument” (YSI) device, Yellow Springs, OH) and glucose (D20 dextrose solution) was infused at a variable rate to maintain plasma glucose concentration at the subject’s baseline glucose concentration. The infused glucose solution was enriched with [6,6 d2]glucose to limit changes in glucose tracer enrichment in plasma. Blood samples were collected every 20 minutes for insulin and plasma enrichment of [6,6 d2]glucose, as well as in 5 minute intervals during the final 20 minutes of the clamp. Plasma concentrations of glucose were measured using the glucose oxidase assay (Thermo Fisher Scientific, Waltham, MO), and plasma insulin concentration was measured using a radioimmunoassay (Human insulin RIA kit; EMD Millipore, Billerica, MA). Subjects received an IV infusion of potassium (KCl) during the clamp to prevent hypokalemia. Following the clamp, subjects were provided a meal and glucose monitored until stable.
Abdominal fat biopsy
Skin of the abdomen was sterilized in standard fashion and anesthetized using 1% lidocaine subcutaneous injection. A 16-gauge needle connected to a 20 ml syringe was inserted subcutaneously at the desensitized area and repeatedly rotated to obtain tissue. Approximately 4–6 ml of subcutaneous fat and fluid were aspirated at each pass and the procedure repeated until about 0.6 grams of tissue was collected which was washed with PBS and aliquoted in 6 tubes, containing approximately 0.1 grams of adipose tissue. The biopsy area was cleaned and dressed.
Mixed meal test
Subjects first completed a validated visual analogue scale (VAS) questionnaires to assess subjective hunger, feeling of fullness and sweet craving. Baseline samples for glucose, insulin, FFAs as well as incretin hormones for Ghrelin, GIP, GLP-1 and PYY were collected. Incretin hormones were measured using a multiplex assay (EMD Millipore, Billerica, MA). The tubes for the incretin hormones contained Pefabloc SC (Sigma-Aldrich, St. Louis, MO) and DPP-IV inhibitor (EMD Millipore, Billerica, MA) to inhibit DPP-IV and other proteases. Subjects then consumed a liquid load of Optifast (Optifast; Noartis, Minneapolis, MN; 474 ml, 320 kcal, 50% carbohydrate, 35% protein, 15% fat) within a 15-minute period. Additional blood samples were collected at 30, 60, 90, 120, and 180 minutes after Optifast consumption. VAS assessments were repeated at 180 minutes (Selimoglu et al., 2009; Simonis-Bik et al., 2011).
Body composition
Body composition was evaluated using anthropometric measurements, including skin thickness and waist and hip circumferences using standardized techniques (Haarbo et al., 1991). Fat and lean body mass were estimated using dual X-ray absorptiometry (DEXA) (GE Lunar Prodigy, model PA +41744, Madison, WI) (Haarbo et al., 1991). Fat mass index (Total fat mass divided by patient’s height squared at baseline, FMI) was calculated.
Hepatic fat quantification
Hepatic fat studies were performed on a 3 Tesla clinical MRI system (Philips Healthcare, Best, The Netherlands) using the vendor-provided torso phased-array surface coil positioned on the abdomen. In addition to breath-hold single-shot turbo-spin-echo sequences for localization, fat was quantified using two image-based and one spectroscopy-based technique as follows. In “MRI method1”, fifteen axial 2D slices were acquired mid-liver in one 16-second breath-hold using a fast-field-echo (FFE) sequence at 6 echo times (TE = n*1.15ms, where n=1,2… 6). Other nominal acquisition settings were: TR = 200ms; flip angle = 20o; acquisition matrix = 280(R/L) by 183(A/P); parallel imaging SENSE = 2; FOV = 450mm; and slice thickness = 5mm. These magnitude-valued images were processed offline using in-house MatLab routines (The MathWorks Inc., Natick, MA) to derive water and fat signal fractions calculated on a pixel-by-pixel basis by least-squared minimization fit of signal versus TE using a water plus six peak lipid model including T2* decay(Yu et al., 2008). In “MRI method2”, a multi-echo Dixon product sequence was performed in a breath-hold lasting at least 16 seconds. Nominal acquisition conditions of this FFE sequence were: 77 axial 3D slices; 6 echos (TE=0.96ms + n*0.7ms, n=0,1,5); TR= 6.2ms; flip angle = 3°; acquisition matrix = 160(R/L) by 160(A/P); parallel imaging SENSE = 2; FOV = 400mm; and slice thickness = 3mm. These complex-valued data were reconstructed online using a multiple lipid peak model with T2* decay to generate quantitative maps of fat-fraction and T2* decay. MRI-based fat fraction estimates by MRI methods 1 and 2 were quantified by pixel mean and standard deviation from freehand volumes-of-interest over at least 5 central slices through the right lobe of the liver avoiding large vessels. Lastly the “MRS method”, was a two-echo, single-voxel Point-RESolved Spectroscopy (PRESS) sequence performed in a single breath-hold. The 30x30x30mm3 PRESS voxel was positioned in the right lobe of the liver avoiding large vessels. MRS acquisition settings were: TR=4000ms; TE=45 and 65ms; number of averages = 2; performed 3 times over 3 separate 16 second breath-holds. Time-domain MRS data were filtered, 1D Fourier transformed, 0th and 1st-order phase-corrected and quantified by integral peak areas using off-line MatLab routines. The T2 of water and lipid peaks were calculated individually so that T2-corrected water and fat fractions at TE=0 could be estimated. MRS-derived fat fraction was quantified by mean and standard deviation over the three breath-hold data. Both of the MRI methods used agreed well with MR Spectroscopy as reported previously, which is considered the gold standard, although MR Spectroscopy (MRS) is rather inefficient since only one small region of the liver is sampled (Ajluni et al., 2017).
Amlexanox and metabolite products measurement
For measure of amlexanox and metabolite products, LC-MS/MS analysis was utilized. Blood biosamples were isolated from patients at baseline and 12 weeks following initiation of amlexanox treatment and plasma extracted using standard protocols. 40 µL plasma was added to 160 µL of ice-cold 80:20 methanol:water to extract metabolites and precipitate protein. Samples were vortexed for 15 minutes followed by being centrifuged at 14000g for 10 minutes at 4°C. The supernatant (170 µL) was transferred a fresh 96-well plate and dried down in vacuo using a vacuum concentrator. Samples were re-suspended in 50 µL of 80:20 methanol:water and put on the UPLC autosampler, which was maintained at 4 °C. The injection volume was 2 µL for each sample. Compounds were separated on a Thermo Vanquish UPLC using a Millipore (Sequant) Zic-pHILIC 2.1×150mm 5um column maintained at 25 °C. Compounds were eluted via a 19-minute linear gradient starting from 90:10 acetonitrile:20mM ammonium bicarbonate to 45:55 acetonitrile:20 mM ammonium bicarbonate. A Thermo Q-Exactive orbitrap mass spectrometer was operated in negative ion mode using a heated electrospray ionization (HESI) source at 35,000 resolution, 100 ms ion trap time for MS1 and 17,500 resolution, 50 ms ion trap time for MS2 collection. Data was collected over a mass range of m/z 200–400, using a sheath gas flow rate of 40 units, auxillary gas flow rate of 20 units, sweep gas flow rate of 2 units, spray voltage of 2.5 kV, capillary inlet temperature of 275 °C, auxillary gas heater temperature of 350 °C and an S-lens RF level of 45. For MS2 collection, MS1 ions were isolated using a 1.0 m/z window and fragmented using a normalized collision energy of 35. Fragmented ions were placed on dynamic exclusion for 30 seconds before being allowed to be fragmented again. Raw profile data was loaded in Progenesis QI (Version 2.2, Nonlinear Dynamics, Waters). Peak peaking was done with a sensitivity set to 3 and a minimum peak width of 0.025 minutes. Amlexanox and breakdown products were identified using comparison of accurate mass, retention time and MS2 features with pure standards, with relative quantitation of levels using MS1 intensity values.
RNA isolation
RNA was extracted from the biopsy samples using the RNeasy lipid tissue kit (Qiagen) for the open label trial. For the placebo controlled trial, RNA was extracted from the biopsy samples using the PureLink RNA mini kit (Life Technology). First, the connective tissue was removed and the samples were cleaned in RNAlater solution (Life Technology); followed by homogenization in Trizol (Life Technology) and isolation using the kit.
Q-PCR analysis
We used the Superscript IV First-Strand Synthesis System for reverse transcription–PCR (Invitrogen) of up to 200 ng mRNA in a 20 µL reaction, with a 3:1 mixture of random hexamers/oligo dT primers for reverse transcription. Real-time PCR amplification was performed on samples in triplicate using 4 µL of 1:20 diluted cDNA in each 20uL reaction and Power SYBR Green PCR Master Mix (Applied Biosystems) on the Applied Biosystems 7900HT for the open label trial and Applied Biosystems QuantStudio5 for the placebo-controlled trial. Fast real-time PCR System and quantified using an internal standard curve and RPLP0 as the control gene. The sequences of all primers used in this study are listed in Table S7.
RNA-sequencing analysis
Libraries were prepared from 100 ng total RNA (TrueSeq Stranded Total RNA, Illumina) and singled-ended sequencing was performed on the Illumina HiSeq 2500, using bar-coded multiplexing and a 100-bp read length, yielding a median of 12.7 M reads per sample. Read alignment, junction finding and read counts per gene was accomplished using STAR v2.5.1b (Dobin et al., 2013). Paired differential gene expression was analyzed with edgeR v3.16.2 (Robinson et al., 2010) while unpaired expression was analyzed with Cuffdiff v2.2.1 (Trapnell et al., 2013), using GRCh37 and UCSC hg19 as the reference assembly and annotation respectively. Pathway analysis was performed with DAVID Bioinformatics Resources 6.7 (http://david.abcc.ncifcrf.gov/) (Huang da et al., 2009a, b).
QUANTIFICATION AND STATISTICAL ANALYSIS
Study data were collected and managed using REDCap electronic data capture tools hosted at the University of Michigan (Harris et al., 2009). Data were exported using automated export procedures of REDcap and the statistical analyses were performed using SAS version 9.2 (Cary, NC), Prism, and R version 3.2.3 (Vienna, Austria). For each parameter, maximum and minimum values and descriptive statistics were generated and inspected by the study team for outliers. All primary analyses on biochemical and clinical outcomes were completed while study team remained blinded. Only responder analyses and molecular investigations were conducted subsequent to unblinding. Figures were drawn on GraphPad Prism version 6.05 and 7.02 (La Jolla, CA). Correlations were investigated by calculating Pearson’s correlation coefficient. Data were analyzed using two tailed t-test or Wilcoxon rank sum test as applicable. Categorical ratios were analyzed by Fisher’s exact tests as applicable. A p-value ≤ 0.05 was considered statistically significant. Missing data were not imputed and a modified intention to treat analysis was used for the placebo-controlled trial. A correction for multiple comparisons was not employed as the study had one primary outcome and all other analyses were either secondary or exploratory (hypothesis generating). Responder versus non-responder statistical analysis was performed only within the amlexanox treated group using a two tailed t-test; since there was only one placebo treated responder statistical analysis within this group would not be meaningful. Statistical details can be found in the figure legends. Sample size n represents the number of patients.
Sample size and power calculations for the double blind, placebo controlled trial
This double blind placebo controlled trial was designed to have 80% power to detect a minimal difference of 0.5% in HbA1c (primary end-point) in 34 patients, 17 in each group. The estimates for mean and SD for the change in HbA1c were taken from a published study of Canagliflozin at the time being developed as a monotherapy for diabetes (Rosenstock et al., 2012). Assuming 20% drop out, we sought to randomize 40 patients. When two patients discontinued very early due to discovery of an exclusion criteria from baseline assessments, (see below) we added two additional patients to recruitment allocation and randomized a total of 42 patients. Recruitment for the study began in April 2014 and completed in January 2016. All study follow-up was completed on May 21, 2016. There were many other study parameters described above which were either secondary or exploratory. Of note, fructosamine and the percent of patients achieving an HbA1c > 0.5% (which is a clinically significant endpoint for a 12-week period) were deemed important secondary parameters.
Randomization and unblinding
Block randomization within two strata defined by gender was used. Randomization numbers, and associated treatment, were generated by the statistician using Excel and given directly to the investigational pharmacist. The investigational pharmacist assigned the randomization numbers sequentially within gender once a study number was given to a screened and eligible subject and allocated the treatment based on the randomization number. Only one member of the investigator team (primary coordinator) had access to randomization numbers assigned to the subjects and entered this information to the database. No investigator had access to the treatments associated with the randomization numbers. The statistician had no access to the patients and received data only when the trial was completed. Unblinding procedure for the study was performed after the primary analyses plan was completed and responder analyses were undertaken. One subject was unblinded prematurely due to an SAE of acute myocardial infarction occurring at 11 weeks of the trial and the previously determined protocol procedure was performed for this subject who was assigned to placebo.
History of the study progression, hardships, protocol amendments and oversight
A draft version of the protocol was created prior to seeking funding for the study and obtaining funding. At that stage, power calculations were completed for change in HbA1c as the primary outcome. The study had a second aim listing hepatic steatosis and an exploratory aim of identifying unique characteristics of drug-treated patients who achieve a clinically significant reduction of HbA1c (accepted as ≥ 0.5% reduction from baseline). The study section required a single endpoint which was revised as HbA1c. After the study commenced, there were two major amendments both of which were approved by the IRB, and reported to the NIDDK and the FDA with no objections raised: due to funding restrictions, we completed the MRI studies only on half of the study population. We also increased the goal recruitment to 42 patients from 40 due to the very early withdrawals of two subjects (these are the two discovered to have abnormal laboratory values that were not noted prior to baseline visit). Other amendments undertaken were related to study personnel changes. The study progression, data collection and subject safety were reviewed by a two person DSMB meeting at least twice a year. The only SAE that could have affected the conduct of the trial was reported to the DSMB on the day of study team notification and the premature unblinding procedure was followed.
Elimination/dropout for the double blind, placebo controlled trial
One patient from the placebo and one patient from the treatment group were terminated within the first two days of the study due to the discovery of an exclusion criteria. The patient from the placebo group was noted to have an elevated HbA1c above 10.5% and had fasting glucose values >300 mg/dL for two days and it was not safe for the patient to stay untreated. He had taken four doses of the assigned treatment. One patient assigned to the treatment group was found to have an elevated CK at baseline and was terminated prior to treatment administration. Two additional patients from the placebo group withdrew from the study within two weeks due to perceived subject burden. One patient in the placebo group had a non-ST elevation myocardial infarction at week 11 of the treatment period, leading to early unblinding, and was seen for the 12 and 16-week assessments, during which only noninvasive assessments were conducted. All but one of the amlexanox-treated patients also completed an additional 4 weeks of observation off the study drug. Lastly, the MRI studies for determination of liver fat were attempted in 20 completed in 19 subjects in a paired fashion due to funding restrictions, one other subject (the placebo treated subject who had the SAE) had only baseline MRI performed. The numbers included in the final study analyses for key study parameters are summarized in Table S2 with reasons for exclusions, there were only very limited occasions where the full data set as described above was not available due to either order entry error or a sample that was obviously compromised.
Supplementary Material
Table S5: Related to Figure 4. Amlexanox and amlexanox metabolites in serum. (excel file)
Highlights.
12 weeks amlexanox treatment significantly reduced HbA1c in obese diabetic subjects
amlexanox treatment did not affect all subjects equally
the amlexanox treated responders had higher baseline inflammation
energy expenditure-associated genes were increased in responders
Acknowledgments
We are indebted to the patients for volunteering in this study and making themselves available for future studies. We also acknowledge the Michigan Clinical Research Unit (MCRU) staff and the dieticians supporting the metabolic research unit (led by Sarah Ball, RD and Theresa Han-Markey, RD). We thank Dr. Morton B. Brown for assistance with statistical analyses. Dr. Corey Powell from University of Michigan C-SCAR group reviewed the statistical analyses for consistency and scientific rigor after the manuscript had been written. Drs. Richard Auchus and Meng Tan provided DSMB support. This study was primarily supported by NIH grant R21DK098776 to EAO and ARS. Other important support included K01DK105075 to SMR, DK057978, DK090962, HL088093, and HL105278, the Glenn Foundation, and the Leona M. and Harry B. Helmsley Charitable Trust (2012-PG-MED-002) to RME and DK100319 and DK060591 to ARS. Infrastructure and data management support has been provided by the NIH Clinical and Translational Science Awards grant UL1TR000433, the Nutrition Obesity Research Centers grant P30 DK089503, NIH institutional grant DK034933 and P30DK0063491.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions
The studies were conceived by EAO and ARS jointly. The regulatory framework was executed by EAO and AHN. The trial team was led by EAO and included LB, EK, NA, AHN, RM, RH, and DR. Biopsy samples were collected by EAO, LB and JH, and cleaned by SMR, AN and RH. TLC performed MRI and MRS acquisition protocol design and analyses. The data were assembled, and quality controlled by RM, DR, LB, AN and RH. EAO is ultimately responsible from generation of clinical data and ensuring GCP. EAO, SMR and RM carried out secondary analyses from trial data. RNA extraction from biopsy samples was performed by SMR, AVG, BP and PZ, Q-PCR analysis was done by SMR, AVG and BP, and the RNAseq by RY, MA, MD, RME and analyzed by CL. SMR and AVG measured serum factors. MJ and KL performed measurements of amlexanox and its metabolites in serum. Further statistical analysis was performed by SMR and CL in regards to molecular data. EAO, SMR, RM, EK, LB, CL and AVG contributed to the construction of data into figures and tables. The manuscript was written primarily by ARS, EAO and SMR and refined by RM and AVG. All authors have read and approved the final manuscript.
DATA AND SOFTWARE AVAILABILITY
RNA-sequencing analysis data reported in this paper have been deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database, Accession # SRP094784.
Database from the placebo-controlled trial will be made available at the time of publication.
ADDITIONAL RESOURCES
Open label trial: https://clinicaltrials.gov/ct2/show/NCT01842282
Open label trial protocol: https://drive.google.com/open?id=0B5xvZGFuNKhgMm9TRURWdFRsOUE
Placebo controlled trial: https://clinicaltrials.gov/ct2/show/NCT01975935
Placebo controlled trial Protocol: https://drive.google.com/open?id=0B5xvZGFuNKhgei1YZUlmYWtFOW8
Guidelines for clinically significant effects: https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM071624.pdf
References
- Ajluni N, Meral R, Neidert AH, Brady GF, Buras E, McKenna B, DiPaola F, Chenevert TL, Horowitz JF, Buggs-Saxton C, et al. Spectrum of Disease Associated with Partial Lipodystrophy (PL): Lessons from a Trial Cohort. Clin Endocrinol (Oxf) 2017 doi: 10.1111/cen.13311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Diabetes, A. Bantle JP, Wylie-Rosett J, Albright AL, Apovian CM, Clark NG, Franz MJ, Hoogwerf BJ, Lichtenstein AH, Mayer-Davis E, et al. Nutrition recommendations and interventions for diabetes: a position statement of the American Diabetes Association. Diabetes Care. 2008;31(Suppl 1):S61–78. doi: 10.2337/dc08-S061. [DOI] [PubMed] [Google Scholar]
- Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM, Wynshaw-Boris A, Poli G, Olefsky J, Karin M. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med. 2005;11:191–198. doi: 10.1038/nm1185. [DOI] [PubMed] [Google Scholar]
- Arner P. Catecholamine-induced lipolysis in obesity. Int J Obes Relat Metab Disord. 1999;23(Suppl 1):10–13. doi: 10.1038/sj.ijo.0800789. [DOI] [PubMed] [Google Scholar]
- Bai T, Chen CC, Lau LF. Matricellular protein CCN1 activates a proinflammatory genetic program in murine macrophages. J Immunol. 2010;184:3223–3232. doi: 10.4049/jimmunol.0902792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bougneres P, Stunff CL, Pecqueur C, Pinglier E, Adnot P, Ricquier D. In vivo resistance of lipolysis to epinephrine. A new feature of childhood onset obesity. J Clin Invest. 1997;99:2568–2573. doi: 10.1172/JCI119444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capaldo B, Di Bonito P, Iaccarino M, Roman MJ, Lee ET, Devereux RB, Riccardi G, Howard BV, de Simone G. Cardiovascular characteristics in subjects with increasing levels of abnormal glucose regulation: the Strong Heart Study. Diabetes Care. 2013;36:992–997. doi: 10.2337/dc12-1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla A, Nguyen KD, Goh YP. Macrophage-mediated inflammation in metabolic disease. Nature reviews. Immunology. 2011;11:738–749. doi: 10.1038/nri3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang SH, Bazuine M, Lumeng CN, Geletka LM, Mowers J, White NM, Ma JT, Zhou J, Qi N, Westcott D, et al. The protein kinase IKKepsilon regulates energy balance in obese mice. Cell. 2009;138:961–975. doi: 10.1016/j.cell.2009.06.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins S, Daniel KW, Rohlfs EM. Depressed expression of adipocyte beta-adrenergic receptors is a common feature of congenital and diet-induced obesity in rodents. Int J Obes Relat Metab Disord. 1999;23:669–677. doi: 10.1038/sj.ijo.0800894. [DOI] [PubMed] [Google Scholar]
- DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol. 1979;237:E214–223. doi: 10.1152/ajpendo.1979.237.3.E214. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farhan SM, Robinson JF, McIntyre AD, Marrosu MG, Ticca AF, Loddo S, Carboni N, Brancati F, Hegele RA. A novel LIPE nonsense mutation found using exome sequencing in siblings with late-onset familial partial lipodystrophy. Can J Cardiol. 2014;30:1649–1654. doi: 10.1016/j.cjca.2014.09.007. [DOI] [PubMed] [Google Scholar]
- Glass CK, Olefsky JM. Inflammation and lipid signaling in the etiology of insulin resistance. Cell Metab. 2012;15:635–645. doi: 10.1016/j.cmet.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haarbo J, Gotfredsen A, Hassager C, Christiansen C. Validation of body composition by dual energy X-ray absorptiometry (DEXA) Clin Physiol. 1991;11:331–341. doi: 10.1111/j.1475-097x.1991.tb00662.x. [DOI] [PubMed] [Google Scholar]
- Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)--a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42:377–381. doi: 10.1016/j.jbi.2008.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horowitz JF, Klein S. Whole body and abdominal lipolytic sensitivity to epinephrine is suppressed in upper body obese women. Am J Physiol Endocrinol Metab. 2000;278:E1144–1152. doi: 10.1152/ajpendo.2000.278.6.E1144. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–917. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009a;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009b;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Inagaki M, Michimata H, Minato K, Sunaga Y, Kobayashi S, Tani G, Nakazawa T. Inhibitory effect of amlexanox on asthmatic attacks in an aspirin sensitive asthmatic. Nihon Kyobu Shikkan Gakkai Zasshi. 1992;30:1180–1185. [PubMed] [Google Scholar]
- Karyekar CS, Frederich R, Ravichandran S. Clinically relevant reductions in HbA1c without hypoglycaemia: results across four studies of saxagliptin. Int J Clin Pract. 2013;67:759–767. doi: 10.1111/ijcp.12212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackey DE, Olefsky JM. Regulation of metabolism by the innate immune system. Nat Rev Endocrinol. 2016;12:15–28. doi: 10.1038/nrendo.2015.189. [DOI] [PubMed] [Google Scholar]
- Landgraf K, Rockstroh D, Wagner IV, Weise S, Tauscher R, Schwartze JT, Loffler D, Buhligen U, Wojan M, Till H, et al. Evidence of early alterations in adipose tissue biology and function and its association with obesity-related inflammation and insulin resistance in children. Diabetes. 2015;64:1249–1261. doi: 10.2337/db14-0744. [DOI] [PubMed] [Google Scholar]
- Lowell BB, Flier JS. Brown adipose tissue, beta 3-adrenergic receptors, and obesity. Annu Rev Med. 1997;48:307–316. doi: 10.1146/annurev.med.48.1.307. [DOI] [PubMed] [Google Scholar]
- Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. The Journal of clinical investigation. 2007;117:175–184. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest. 2011;121:2111–2117. doi: 10.1172/JCI57132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino H, Saijo T, Ashida Y, Kuriki H, Maki Y. Mechanism of action of an antiallergic agent, amlexanox (AA-673), in inhibiting histamine release from mast cells. Acceleration of cAMP generation and inhibition of phosphodiesterase. International archives of allergy and applied immunology. 1987;82:66–71. doi: 10.1159/000234292. [DOI] [PubMed] [Google Scholar]
- Mowers J, Uhm M, Reilly SM, Simon J, Leto D, Chiang SH, Chang L, Saltiel AR. Inflammation produces catecholamine resistance in obesity via activation of PDE3B by the protein kinases IKK{varepsilon} and TBK1. Elife. 2013;2:e01119. doi: 10.7554/eLife.01119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagareddy PR, Kraakman M, Masters SL, Stirzaker RA, Gorman DJ, Grant RW, Dragoljevic D, Hong ES, Abdel-Latif A, Smyth SS, et al. Adipose tissue macrophages promote myelopoiesis and monocytosis in obesity. Cell Metab. 2014;19:821–835. doi: 10.1016/j.cmet.2014.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annual review of physiology. 2010;72:219–246. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- Owen BM, Mangelsdorf DJ, Kliewer SA. Tissue-specific actions of the metabolic hormones FGF15/19 and FGF21. Trends Endocrinol Metab. 2015;26:22–29. doi: 10.1016/j.tem.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardigol A, Forssmann U, Zucht HD, Loetscher P, Schulz-Knappe P, Baggiolini M, Forssmann WG, Magert HJ. HCC-2, a human chemokine: gene structure, expression pattern, and biological activity. Proc Natl Acad Sci U S A. 1998;95:6308–6313. doi: 10.1073/pnas.95.11.6308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankov G, Sasaki K, Fukuda M. Pharmacodynamics of Amlexanox (AA-673) in normal and anaphylactic rat conjunctiva and its effect on histamine concentration. Ophthalmic research. 1990;22:359–364. doi: 10.1159/000267047. [DOI] [PubMed] [Google Scholar]
- Reilly SM, Ahmadian M, Zamarron BF, Chang L, Uhm M, Poirier B, Peng X, Krause DM, Korytnaya E, Neidert A, et al. A subcutaneous adipose tissue-liver signalling axis controls hepatic gluconeogenesis. Nat Commun. 2015;6:6047. doi: 10.1038/ncomms7047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly SM, Chiang SH, Decker SJ, Chang L, Uhm M, Larsen MJ, Rubin JR, Mowers J, White NM, Hochberg I, et al. An inhibitor of the protein kinases TBK1 and IKK-varepsilon improves obesity-related metabolic dysfunctions in mice. Nat Med. 2013;19:313–321. doi: 10.1038/nm.3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynisdottir S, Ellerfeldt K, Wahrenberg H, Lithell H, Arner P. Multiple lipolysis defects in the insulin resistance (metabolic) syndrome. J Clin Invest. 1994;93:2590–2599. doi: 10.1172/JCI117271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robciuc MR, Kivela R, Williams IM, de Boer JF, van Dijk TH, Elamaa H, Tigistu-Sahle F, Molotkov D, Leppanen VM, Kakela R, et al. VEGFB/VEGFR1-Induced Expansion of Adipose Vasculature Counteracts Obesity and Related Metabolic Complications. Cell Metab. 2016;23:712–724. doi: 10.1016/j.cmet.2016.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenstock J, Aggarwal N, Polidori D, Zhao Y, Arbit D, Usiskin K, Capuano G, Canovatchel W, Canagliflozin DIASG. Dose-ranging effects of canagliflozin, a sodium-glucose cotransporter 2 inhibitor, as add-on to metformin in subjects with type 2 diabetes. Diabetes Care. 2012;35:1232–1238. doi: 10.2337/dc11-1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouault C, Pellegrinelli V, Schilch R, Cotillard A, Poitou C, Tordjman J, Sell H, Clement K, Lacasa D. Roles of chemokine ligand-2 (CXCL2) and neutrophils in influencing endothelial cell function and inflammation of human adipose tissue. Endocrinology. 2013;154:1069–1079. doi: 10.1210/en.2012-1415. [DOI] [PubMed] [Google Scholar]
- Rui L, Yuan M, Frantz D, Shoelson S, White MF. SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J Biol Chem. 2002;277:42394–42398. doi: 10.1074/jbc.C200444200. [DOI] [PubMed] [Google Scholar]
- Sekimoto R, Fukuda S, Maeda N, Tsushima Y, Matsuda K, Mori T, Nakatsuji H, Nishizawa H, Kishida K, Kikuta J, et al. Visualized macrophage dynamics and significance of S100A8 in obese fat. Proc Natl Acad Sci U S A. 2015;112:E2058–2066. doi: 10.1073/pnas.1409480112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selimoglu H, Duran C, Kiyici S, Guclu M, Ersoy C, Ozkaya G, Erturk E, Tuncel E, Imamoglu S. Comparison of composite whole body insulin sensitivity index derived from mixed meal test and oral glucose tolerance test in insulin resistant obese subjects. Endocrine. 2009;36:299–304. doi: 10.1007/s12020-009-9213-z. [DOI] [PubMed] [Google Scholar]
- Simonis-Bik AM, Boomsma DI, Dekker JM, Diamant M, de Geus EJ, t Hart LM, Heine RJ, Kramer MH, Maassen JA, Mari A, et al. The heritability of beta cell function parameters in a mixed meal test design. Diabetologia. 2011;54:1043–1051. doi: 10.1007/s00125-011-2060-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K, Wernstedt Asterholm I, Kusminski CM, Bueno AC, Wang ZV, Pollard JW, Brekken RA, Scherer PE. Dichotomous effects of VEGF-A on adipose tissue dysfunction. Proc Natl Acad Sci U S A. 2012;109:5874–5879. doi: 10.1073/pnas.1200447109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trayhurn P. Hypoxia and adipose tissue function and dysfunction in obesity. Physiol Rev. 2013;93:1–21. doi: 10.1152/physrev.00017.2012. [DOI] [PubMed] [Google Scholar]
- Ueki K, Kondo T, Tseng YH, Kahn CR. Central role of suppressors of cytokine signaling proteins in hepatic steatosis, insulin resistance, and the metabolic syndrome in the mouse. Proc Natl Acad Sci U S A. 2004;101:10422–10427. doi: 10.1073/pnas.0402511101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, Dixit VD. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nature medicine. 2011;17:179–188. doi: 10.1038/nm.2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X, Song H, Yin L, Rizzo MG, Sidhu R, Covey DF, Ory DS, Semenkovich CF. Fatty acid synthesis configures the plasma membrane for inflammation in diabetes. Nature. 2016;539:294–298. doi: 10.1038/nature20117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, Charo I, Leibel RL, Ferrante AW., Jr CCR2 modulates inflammatory and metabolic effects of high-fat feeding. The Journal of clinical investigation. 2006;116:115–124. doi: 10.1172/JCI24335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW., Jr Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Shimakawa A, McKenzie CA, Brodsky E, Brittain JH, Reeder SB. Multiecho water-fat separation and simultaneous R2* estimation with multifrequency fat spectrum modeling. Magn Reson Med. 2008;60:1122–1134. doi: 10.1002/mrm.21737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolotov S, Xing C, Mahamid R, Shalata A, Sheikh-Ahmad M, Garg A. Homozygous LIPE mutation in siblings with multiple symmetric lipomatosis, partial lipodystrophy, and myopathy. Am J Med Genet A. 2016 doi: 10.1002/ajmg.a.37880. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S5: Related to Figure 4. Amlexanox and amlexanox metabolites in serum. (excel file)