

Abstract
Vascular disease and heart failure impart an enormous burden in terms of global morbidity and mortality. Although there are many different causes of cardiac and vascular disease, most share an important pathological mechanism: oxidative stress. In the failing heart, oxidative stress occurs in the myocardium and correlates with left ventricular dysfunction. Reactive oxygen species (ROS) negatively affect myocardial calcium handling, cause arrhythmias, and contribute to cardiac remodeling by inducing hypertrophic signaling, apoptosis, and necrosis. Similarly, oxidative balance in the vasculature is tightly regulated by a wealth of pro- and anti-oxidant systems that orchestrate region-specific ROS production and removal. ROS also regulate multiple vascular cell functions, including: endothelial and smooth muscle cell growth, proliferation, and migration; angiogenesis; apoptosis; vascular tone; host defenses; and genomic stability. However, excessive levels of ROS promote vascular disease via direct and irreversible oxidative damage to macromolecules, as well as disruption of redox-dependent vascular wall signaling processes.
Keywords: cardiac, reactive oxygen species, vascular
With the basic biology of oxidative stress and telomeres already reviewed in Part 1 of this series, here in Part 2 we turn our attention to the impact of oxidative stress on the heart and vasculature, and, in particular, to the pathophysiological role of oxidative stress in heart failure and vascular disease (Central Illustration).
Central Illustration. Mechanisms, Sources, and Implications of Oxidative Stress in Cardiovascular Disease and Heart Failure.
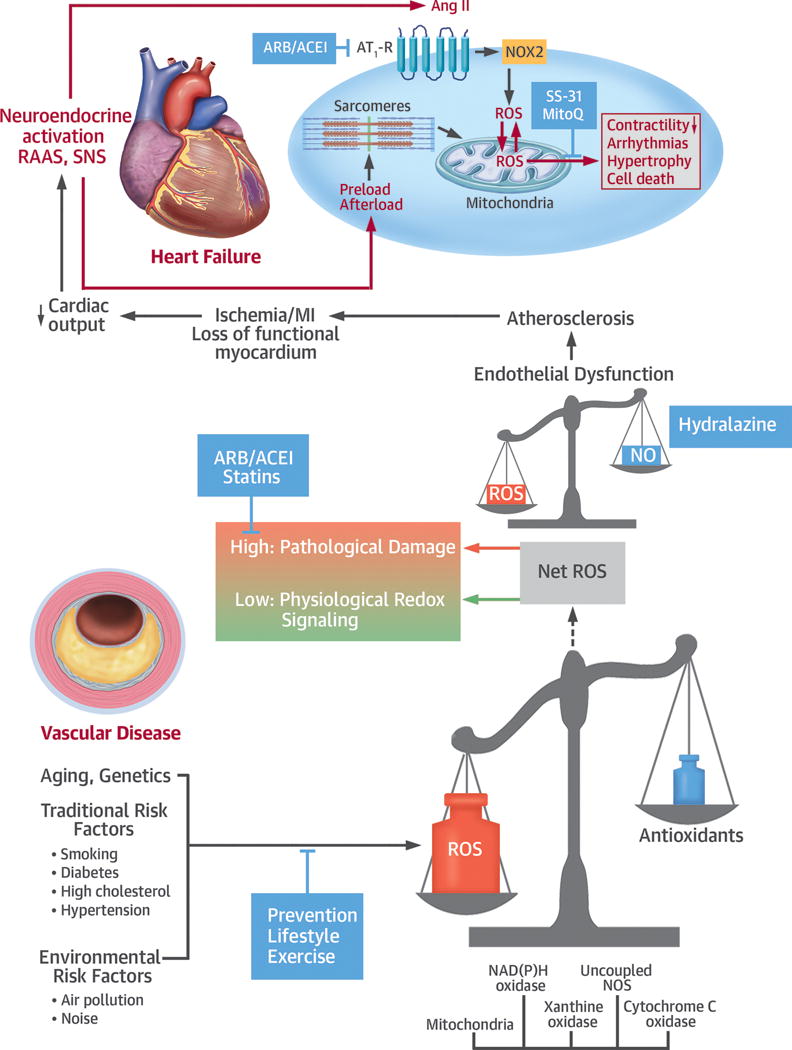
Aging, genetic predisposition, traditional risk factors, and environmental factors can induce oxidative stress, particularly in vessels, where nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) and uncoupled nitric oxide synthase (NOS) are dominant sources. At physiological levels, a low increase in net reactive oxygen species (ROS) can induce protective effects through redox signaling mediating, for example, improved antioxidative capacity. If the generation of ROS outweighs antioxidative capacity, then at higher ROS levels cell damage and endothelial dysfunction arise, which contribute to the development of atherosclerosis. Through ischemia in the context of myocardial infarction (MI), this can induce the loss of functional myocardium and, eventually, heart failure. Although heart failure also arises via other mechanisms, including diabetes and primary cardiomyopathic processes, ultimately neuroendocrine activation via the renin-angiotensin-aldosterone system (RAAS) and angiotensin II receptor type 1 (AT1-R), and the sympathetic nervous system (SNS), combined with increased pre- and after-load, impose additional oxidative stress on the heart. Specific mechanisms leading to increased cardiac oxidative stress then include receptor-induced activation of NOX2 and mitochondrial redox mismatch. As a consequence, oxidation of mitochondrial NADPH gives rise to hydrogen peroxide (H2O2), which plays a causal role in contractile dysfunction, arrhythmia, and ultimately maladaptive cardiac remodeling through hypertrophy and cell death. Potential points of intervention are through lifestyle change, exercise, and medication to reduce risk factors and environmental stressors. Medical options include hydralazine to improve endothelial function, angiotensin-converting enzyme inhibitors (ACEI)/angiotensin receptor blockers (ARBs), and statins. More recently, drugs directly targeted to mitochondria have been developed, such as SS-31 or MitoQ, whose clinical efficacy is currently under evaluation. Ang II = angiotensin II; NO = nitric oxide.
PATHOPHYSIOLOGICAL ROLE OF OXIDATIVE STRESS IN HEART FAILURE
INTRODUCTION
Heart failure (HF) is characterized by activation of the sympathetic nervous and renin-angiotensin-aldosterone systems. This neuroendocrine activation is associated with oxidative stress in the myocardium and vasculature. As outlined in Part 1 of this review, oxidative stress is an imbalance between the generation and detoxification of reactive oxygen species (ROS) (1,2). In patients with HF, oxidative stress occurs in the myocardium (3,4) and plasma, and correlates with left ventricular (LV) dysfunction (5). ROS negatively affect myocardial calcium (Ca2+) handling, cause arrhythmias, and contribute to cardiac remodeling by inducing hypertrophic signaling, apoptosis, and necrosis (for review see (6,7)). Enzymatic sources for ROS, such as the nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOXs), uncoupled nitric oxide (NO) synthase, and mitochondria are all considered relevant sources of ROS in HF, causing vascular and myocardial dysfunction (2). Importantly, mitochondria amplify ROS derived from NOXs and may thereby function as “redox hubs” in cardiac physiology and disease (8,9).
In the initial part of this review, we focus on the crucial role of ROS in HF causing vascular and myocardial dysfunction. We also address the vitamin paradox by exploring why attempts to reduce oxidative stress in patients at cardiovascular risk (e.g., with vitamin E) caused, rather than prevented, the development of HF (2).
ENZYMATIC SUPEROXIDE SOURCES IN HF AND FUNCTIONAL CONSEQUENCES FOR THE VASCULATURE AND MYOCARDIUM
NADPH OXIDASE
In experimental models and patients with HF, myocardial superoxide (O2−)-generating NOX activity is increased (3,4). Specific NOX isoforms exist in endothelial cells (ECs), smooth muscle cells (SMCs), adventitial cells, and cardiac myocytes (10). In the latter, physiological stretch activates the sarcolemmal and transverse (T)-tubule–localized NOX2 (X-ROS signaling), and these ROS sensitize nearby ryanodine receptors to trigger Ca2+ release from the sarcoplasmic reticulum (11). This mechano-chemotransduction also involves NO synthases and calmodulin-dependent protein kinase II (12), raising the possibility that peroxynitrite (ONOO−) and methionine oxidation are involved.
Canonical activation of NOX2 is through Gq-coupled angiotensin II (ATII) receptors. Subpressor doses of ATII induce cardiac hypertrophy that is abolished by NOX2 deletion, although NOX2 deletion does not prevent HF development in response to severe pressure overload induced by aortic constriction (6). In animal models of myocardial infarction, NOX2 inactivation reduces infarct size and ameliorates HF development, but it is unclear whether this is related to vascular NOX or phagocytic NOX located in inflammatory cells. At higher ATII doses that induce hypertension, depletion of inflammatory cells attenuates some features of oxidative stress, such as endothelial dysfunction, vascular ROS formation, and also arterial hypertension (13).
In contrast to NOX2, NOX4 does not require any regulatory subunits, and it constitutively produces hydrogen peroxide (H2O2), rather than O2− (6). In the heart, NOX4 is located in the endoplasmic reticulum (ER), nucleus, and possibly also in mitochondria (6). Stimuli that activate NOX4 include ischemia, hypoxia, and adrenergic stimuli, which are all present and enhanced in HF. However, the role of NOX4 in HF development is controversial, as mice lacking cardiac NOX4 have been shown to exhibit both reduced and aggravated maladaptive remodeling in pressure overload-induced HF using differing model systems (6,14). Furthermore, NOX4-deficient mice exhibit partial protection against ischemic myocardial injury (15), whereas double knockout of NOX2 and NOX4 increases susceptibility to ischemic myocardial injury, perhaps through altered ROS production and down-regulation of stress-response pathways, such as hypoxia-inducible factor (HIF)-1α (15). As both myocardial and endothelial NOX4 promote angiogenesis, their absence from cardiac myocytes or vessels could also lead to capillary rarefaction and sensitivity to ischemic injury (16,17).
MITOCHONDRIA
In mitochondria, O2− is generated by the electron transfer chain (ETC), but is rapidly dismutated to H2O2 by manganese-dependent superoxide dismutase (Mn-SOD). H2O2 is then eliminated by antioxidative enzymes (i.e., glutathione peroxidase and peroxiredoxin), which require NADPH for regeneration (Figure 1). In HF, a functional block of complex I provokes excessive production of O2−, which is transformed to H2O2 and hydroxyl radicals (OH•; via the Fenton reaction) (18). Besides increased production, the elimination of ROS in the mitochondrial matrix is compromised by dynamic oxidation of the antioxidative capacity, resulting in increased net emission of ROS from mitochondria (Figure 1) (concept of “redox-optimized ROS balance”; for reviews see (19,20)).
Figure 1. Mitochondrial ROS Generation in HF.
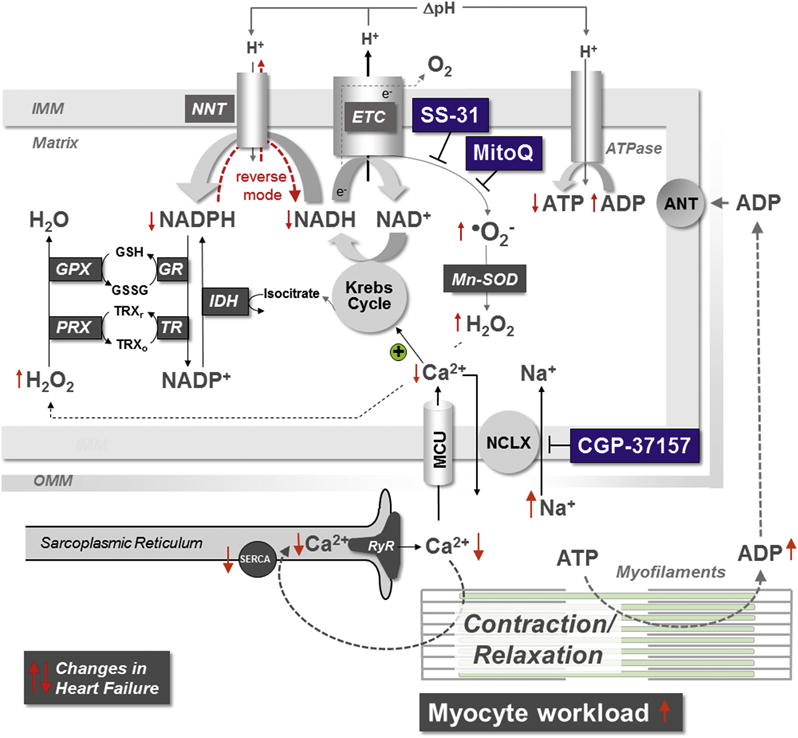
In HF, the regulation of mitochondrial ROS emission is controlled by ion handling and mechanical workload. Increased myocyte workload and dysregulation of calcium (Ca2+) and sodium (Na+) handling reduce mitochondrial Ca2+ accumulation, causing NADH and NADPH oxidation. NADPH depletion provokes H2O2 emission from mitochondria. ANT = adenine nucleotide translocator; ADP = adenosine diphosphate; ATP = adenosine triphosphate; ATPase = adenosine triphosphatase; GPX = glutathione peroxidase; GR = glutathione reductase; GSH/GSSG = reduced/oxidized glutathione; HF = heart failure; H2O2 = hydrogen peroxide; IDH = isocitrate dehydrogenase; IMM = inner mitochondrial membrane; MCU = mitochondrial calcium uniporter; NADH = nicotinamide adenine dinucleotide; NADPH = nicotinamide adenine dinucleotide phosphate; NCLX = mitochondrial sodium/calcium (and lithium) exchanger; NNT = nicotinamide nucleotide transhydrogenase; O2− = superoxide; OMM = outer mitochondrial membrane; PRX = peroxiredoxin; ROS = reactive oxygen species; RyR = ryanodine receptor; SERCA = sarcoplasmic reticulum calcium adenosine triphosphatase; TR = thioredoxin reductase; TRXr/o = reduced/oxidized thioredoxin.
In mitochondria, the Krebs cycle generates nicotinamide adenine dinucleotide (NADH), which donates electrons to the ETC to produce adenosine triphosphate (ATP). However, the Krebs cycle also produces substrates that regenerate NADPH, which, in turn, regenerates antioxidative enzymes (Figure 1) (20,21). In HF, defects in cytosolic Ca2+ and sodium (Na+) handling in cardiac myocytes (e.g., reduced sarcoplasmic reticulum Ca2+ release, elevated Na+) reduce mitochondrial Ca2+ accumulation and thereby hinder the regeneration of NADH and NADPH, which, on the one hand, compromises ATP production, while, on the other hand, provoking the emission of ROS from mitochondria (20).
In HF, limited substrates for ATP production (i.e., NADH) and ROS elimination (i.e., NADPH) face an increased energetic demand imposed by elevated cardiac preload, afterload, and heart rate. Workload increases adenosine diphosphate (ADP) production, and ADP accelerates respiration, which consumes NADH (Figure 1). In this scenario, the mitochondrial transhydrogenase (nicotinamide nucleotide transhydrogenase [NNT]), which under physiological conditions regenerates NADPH, then paradoxically consumes NADPH towards NADH and ATP production, but at the cost of antioxidative capacity (21). The resulting increase in mitochondrial H2O2 emission induces necrosis, LV dysfunction, and death (21). In fact, targeting the H2O2-eliminating enzyme catalase to mitochondria prevents the development of HF in response to pressure overload and/or neurohormonal activation (22,23). Taken together, these data indicate that in HF, increased production and decreased elimination of ROS in cardiac myocytes conspire to elevate net mitochondrial ROS emission, which plays a causal role in the pathobiology of HF.
UNCOUPLED ENDOTHELIAL NITRIC OXIDE SYNTHASE IN HF
In patients with HF, concomitant endothelial dysfunction can be improved with acute administration of oral or intra-arterial vitamin C, suggesting that oxidative stress contributes to endothelial dysfunction in this condition (24); an observation that is also supported by preclinical studies. In fact, uncoupling of endothelial NO synthase (eNOS) in both the vasculature and the myocardium contribute to HF development in animal models, a condition where NO production by the synthase is converted to O2− generation (25). Similarly, several other enzymes downstream in the NO cascade are also inhibited (e.g., soluble guanylyl cyclase or cyclic guanosine monophosphate [cGMP]-dependent kinase I), leading to an impairment of NO signaling in vascular SMCs (4).
XANTHINE/XANTHINE OXIDASE
In chronic HF, serum uric acid levels rise with increased purine catabolism resulting from tissue hypoxia, apoptosis, and/or enhanced or up-regulated xanthine oxidoreductase activity (reviewed in (26)). Xanthine oxidase (XO) activity and expression are also increased, and its inhibition improves coronary and peripheral endothelial dysfunction, as well as contractile efficacy in patients with HF due to coronary artery disease (27). However, despite these promising effects in smaller trials (27), larger multicenter trials of XO inhibitors in patients with HF were neutral (28).
CROSSTALK BETWEEN DIFFERENT ROS SOURCES
ROS biology likely includes self-supporting mechanisms in which ROS trigger further ROS formation from various sources through the activation of “redox switches”. For instance, oxidation of thiol residues of xanthine dehydrogenase converts this enzyme to a O2− producing oxidase, and uncoupling of eNOS also involves redox-sensitive kinases, tetrahydrobiopterin (BH4) depletion, and disruption of the enzyme’s zinc-sulfur complex (8). These mechanisms may be initiated by the “escape” of mitochondrial ROS, which may occur in a burst-like fashion as a consequence of increased membrane permeability under de-energized conditions in the failing heart. Conversely, ROS derived from NOX2 require further mitochondrial ROS release to trigger maladaptive remodeling and diastolic dysfunction in response to ATII infusion (9), a process termed ROS-induced ROS release (Figure 1). In these studies, ROS scavenging in the cytosol and/or peroxisomes (by N-acetyl cysteine or targeting catalase to these compartments) was ineffective, whereas reducing mitochondrial ROS (by targeting catalase to mitochondria or by the tetrapeptide SS-31, see later discussion) prevented pathology (9,23,29). These data indicate that mitochondria could resemble a final “afterburner” that integrates ROS signals from various cellular sources and accounts for detrimental ROS-induced remodeling and HF development (Figure 1) (for a more mechanistic review, see also (9,20,29)).
THERAPEUTIC STRATEGIES IN HF
ANTIOXIDANTS/VITAMINS
Despite a predominant role of ROS in the pathophysiology of HF, large clinical trials have failed to show any benefits of antioxidants such as vitamins E and C (1,2). A possible explanation is that these vitamins are not specifically targeted to the sites of ROS generation (e.g., mitochondria) that are most important in pathological conditions. Furthermore, vitamins react much more slowly with ROS than ROS can interact with their direct counterpart NO to induce damage, and therefore exceptionally high concentrations of vitamins would be required to override these kinetic constraints (1). However, when administered in high doses, vitamin E worsens, rather than improves vascular function (30), which may be related to the formation of the pro-oxidative vitamin E radical (31). This may also explain why long-term treatment with vitamin E does not prevent, but rather induces HF and acute left heart decompensation, as shown in the HOPE (32) and HOPE-TOO trials (33).
TARGETING MITOCHONDRIAL REDOX AND ROS
On the basis of this experience with vitamins C and E, a more promising approach may be to target more specifically the most relevant compartments for ROS production (Figure 1). As reviewed previously (34), mitochondrial ROS can be targeted in at least 3 different ways. First, due to the negative mitochondrial membrane potential (ΔΨm ≈ −180 mV), antioxidants can be coupled as “cargo” to lipophilic cations, which accumulate several hundred-fold in the matrix. This is discussed in greater detail in a subsequent section.
Second, some drugs, such as the tetrapeptide SS-31, which binds to cardiolipin (35), accumulate in mitochondria because they bind to mitochondria-specific structures. Cardiolipin is an important phospholipid of the inner mitochondrial membrane that controls the assembly of ETC complexes (35). Oxidation of cardiolipin in HF (36) hampers ETC assembly, and thereby provokes deviation of electrons to oxygen, which increases O2− formation. SS-31 is not a direct antioxidant (37), but binds specifically to cardiolipin and prevents its oxidation, thereby improving electron transfer along the ETC and reducing aberrant production of O2− (35). In various preclinical HF models, SS-31 reduced oxidative stress, preserved mitochondrial function, and prevented necrosis, maladaptive remodeling, LV dysfunction, and death (21,35). Although, in a first clinical trial involving 297 patients with acute myocardial infarction, a single application of SS-31 did not reduce infarct size, it was, however, safe (38), and is currently undergoing evaluation for longer-term treatment in patients with HF (NCT02814097, NCT02788747 and NCT02914665) (35).
A third option to target mitochondrial ROS is to interfere with cellular ion handling. Because an elevated cytosolic Na+ concentration accelerates mitochondrial Ca2+ extrusion, inhibition of the mitochondrial Na+/Ca2+ exchanger with CGP-37157 (Figure 1) prevents oxidation of NADH and NADPH, emission of ROS, maladaptive cardiac remodeling, and arrhythmias in a guinea pig model of systolic HF (20,39). This compound, however, is yet to be evaluated in humans.
Although the listed interventions target oxidative stress, there is also some evidence that in HF, reductive stress may be relevant to disease development and a potential target for treatment. Down-regulation of complex I in HF may reduce the redox state of NADH/NAD+, thereby limiting the availability of NAD+ to the mitochondrial NAD+-dependent deacetylase sirtuin 3 (SIRT3) (40). The ensuing hyperacetylation of mitochondrial proteins induces an energetic deficit and contractile dysfunction, whereas elevating NAD+ synthesis via the NAD+ salvage pathway by supplementing with the NAD+ precursor, nicotinamide mononucleotide (NMN), prevented these changes (40). Due to its deacetylase activity, SIRT3 is therefore protective in various cardiac pathologies (40,41). Although hyperacetylation of mitochondrial proteins can occur in common forms of HF (40), it has also been identified as an important downstream consequence of mutations in the human αB-crystallin gene that are responsible for the development of desmin-related cardiomyopathy (42).
ANGIOTENSIN-CONVERTING ENZYME INHIBITORS AND ANGIOTENSIN-1-RECEPTOR BLOCKERS
As ATII is a key upstream trigger of ROS formation from NADPH oxidase and, in turn, mitochondria (9), it is reasonable that antiotensin 1 (AT1) antagonists and angiotensin-converting enzyme (ACE) inhibitors may exert their beneficial clinical effects in part through antioxidative mechanisms. In fact, ACE inhibitors ameliorate inflammatory processes in the vessel wall (4), prevent SMC proliferation, and, in particular, prevent activation of the vascular and phagocytic NADPH oxidase (4,43). Thus, although vitamin E therapy can be considered as a secondary therapy that scavenges already formed ROS, ACE inhibitors (and also AT-1 receptor blockers) can be thought of as a primary therapy that blocks ROS production at the enzymatic source (43).
HYDRALAZINE
Another drug with often underestimated beneficial effects on the balance between O2− and NO (the “nitroso-redox balance”), which is impaired in HF (44), is hydralazine. Clinical treatment with nitrates (e.g., nitroglycerin or isosorbide dinitrate [ISDN]), induces tolerance to its vascular and hemodynamic effects, mediated largely by endothelial dysfunction (for review see (45)). When combined with hydralazine, however, nitrate tolerance is prevented because hydralazine inhibits nitroglycerin-induced vascular O2− and ONOO− formation both in vitro (46) and in vivo (47) (Figure 2). Also, in failing cardiac myocytes, hydralazine improves cytosolic Ca2+ handling, in part by scavenging O2− formation (48). Thus, the antioxidant action of hydralazine and the prevention of tolerance development in response to ISDN (49) may explain, at least in part, why this combination improves morbidity and mortality in patients with chronic congestive HF (50).
Figure 2. Efficacy of Hydralazine and Nitrates in HF.
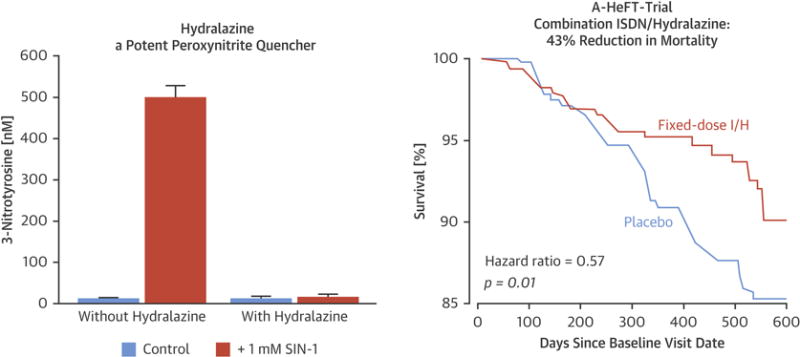
The combination of hydralazine and ISDN therapy is efficacious in the treatment of chronic HF with reduced ejection fraction. (Left) The powerful inhibitory effect of hydralazine on protein tyrosine nitration in rat smooth muscle cells by in situ generated ONOO− (caused by 3-morpholino sydnonimine [SIN-1] administration) is illustrated. Reprinted, with permission, from Daiber et al. (46). (Right) The marked effect of combined ISDN + hydralazine (I/H) on survival in patients with HF and reduced left ventricular ejection fraction is illustrated, as demonstrated in the African-American Heart Failure (A-HeFT) Trial. Reprinted, with permission, from Taylor et al. (50). ISDN = isosorbide dinitrate;
CONCLUSIONS REGARDING OXIDATIVE STRESS IN HF
Although at lower concentrations, ROS serve physiological (protective) functions (6), higher levels of oxidative stress triggered by neurohormones and hemodynamic alterations play a causal role in the initiation and progression of HF. In recent years, the importance of ROS compartmentalization has been progressively recognized, and instead of global, nonspecific antioxidative approaches (e.g., with vitamins), specific treatments targeted toward these compartments (in particular, mitochondria) may hold promise for ongoing and future drug development. These treatments may add to the established benefit provided by the upstream therapy of neuroendocrine antagonism.
OXIDATIVE STRESS IN THE VASCULATURE
PATHOPHYSIOLOGY OF ROS IN THE VASCULATURE
Vascular dysfunction or disease refers to different pathological states affecting the venous and/or arterial system that may give rise to adverse cardiovascular events. Among these pathologies are atherosclerosis, arterial remodeling, restenosis, and thrombosis. Different risk factors predisposing to cardiovascular disease (CVD), such as hypercholesterolemia, diabetes, obesity, hypertension, and aging, lead to vascular dysfunction and disease, partially through oxidative stress, making the latter a common denominator to risk factor–induced vascular disease, as well as an attractive therapeutic and preventive target (51,52). Under physiological conditions, oxidative balance is sustained by a tightly regulated wealth of pro- and anti-oxidant systems distributed within different cellular compartments to orchestrate region-specific ROS production (Figure 3). In fact, ROS have been shown to regulate multiple cellular functions, including: EC and SMC growth, proliferation, and migration; angiogenesis; EC and SMC apoptosis; vascular tone; host defense; and genomic stability (53). Excessive levels of oxidants over antioxidants, however, cause a deviation in the redox state, defined as oxidative stress. This mediates vascular disease through direct and irreversible oxidative damage to macromolecules, as well as disruption of redox-dependent signaling processes in the vascular wall (54).
Figure 3. Enzymes Determining Redox Balance Through Production or Scavenging of ROS.
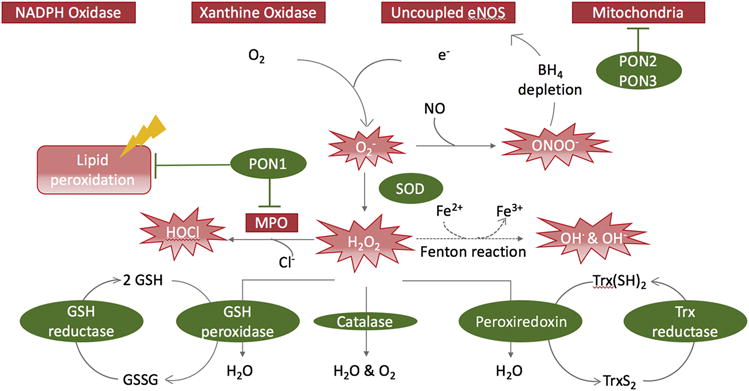
In the vasculature, O2− is generated by NADPH oxidase, xanthine oxidase, mitochondria, and uncoupled eNOS. Superoxide dismutase (SOD) converts O2− to H2O2. Through the Fenton reaction, H2O2 can spontaneously convert to the hydroxyl radical OH•. Due to its extreme reactivity, OH• can damage most cellular compartments. Immune cell–secreted myeloperoxidase (MPO) can mediate oxidation of chloride to hypochlorous acid (HOCl) using H2O2, and inactivate NO via oxidation to nitrite. By chlorination, HOCl inactivates multiple biomolecules, such as the eNOS substrate L-arginine and lipoproteins. The antioxidant enzymes glutathione (GSH) peroxidase, catalase, and peroxiredoxin convert H2O2 to oxygen and water. Paraoxonases (PON) 2 and 3 inhibit mitochondrial ROS production, whereas PON1 inhibits myeloperoxidase (MPO) and protects from oxidative stress mediated lipid-peroxidation. BH4 = tetrahydrobiopterin; eNOS = endothelial nitric oxide synthase; NO = nitric oxide; Other abbreviations as in Figure 1.
Among the signal transduction pathways and targets modulated by ROS are proximal tyrosine kinases, such as c-Src, platelet-derived growth factor receptor, epidermal growth factor receptor, the low molecular weight guanosine-5′-triphosphate (GTP)-binding protein Ras, mitogen-activated protein kinases (MAPKs), such as extracellular signal-regulated kinases (ERK1/2), p38, and c-Jun N-terminal kinases, as well as the serine/threonine kinase Akt/protein kinase B (55). Furthermore, ROS directly regulate several classes of genes that are crucial for vascular function (e.g., adhesion molecules, chemotactic factors, vasoactive substances, and antioxidant enzymes), whereas ROS also indirectly modulate genomic activity through transcription factors such as activator protein 1, nuclear transcription factor kappa-light-chain-enhancer of activated B cells (NF-κB), and HIF-1.
In conditions of oxidative stress, however, ROS mediate pathological vascular changes, such as endothelial dysfunction, by disrupting the vasoprotective NO signaling cascade (56). NO is a key antiatherogenic player, which inhibits platelet adhesion and aggregation, powerfully augments vasodilation through stimulation of cGMP production by SMCs, decreases leukocyte-endothelial interactions, and reduces SMC proliferation (25). It is generated from L-arginine by eNOS in the presence of its essential cofactor BH4. Superoxide (O2−) rapidly scavenges NO to produce ONOO− (57), a potent oxidant in its own right. This rapid reaction between O2− and NO, yielding ONOO−, is about 3 to 4 times faster than the dismutation of O2− by the superoxide dismutases (SODs). Therefore, depending on the rates of tissue O2− production, this pathway can be of major importance, particularly as ONOO− is cytotoxic and causes damage to proteins, lipids, and DNA (58,59). Furthermore, in these settings, NO is not only depleted, but peroxynitrite-induced oxidation of BH4 leads to a state of eNOS uncoupling (60), where oxygen reduction takes place independently from NO production, turning eNOS into a pro-oxidant enzyme (61).
ROS also play a key role in the inflammatory component of atherosclerosis, where they mediate inflammasome formation, which, in turn, supports secretion and processing of the proinflammatory cytokines interleukin (IL)-1β) and IL-8 via caspase 1 activation (62).
IMBALANCE IN VASCULAR ROS PRODUCTION AND SCAVENGING: ROS-PRODUCING ENZYMES
Oxidative balance is tightly regulated by several key enzymes that either produce or dispose of free radicals (Figure 3). Although many enzymes are known to produce ROS in the vascular wall, 4 enzymes in particular seem to play an especially prominent role, namely NADPH oxidase, enzymes of the mitochondrial respiratory chain, XO, and uncoupled eNOS. Further sources of vascular ROS include lipoxygenase, cyclooxygenase, and cytochrome P 450 monooxygenase.
NADPH oxidases
NADPH oxidases are major sources of vascular ROS and are unique in their sole commitment to ROS-production. They produce O2− from molecular oxygen using NADPH as an electron donor, and are found in various vascular cell types, including ECs (NOX2 and 4), SMCs (NOX1 and 4), and fibroblasts (NOX2 and 4) (62,63). The multisubunit enzyme complexes rely on up to 5 regulatory subunits and, in some cases, on a small GTPase (Rac1 or 2) for correct assembly and activation. Evidence for NADPH oxidase activation in vascular disease stems from animal models of hypertension (genetic and ATII-induced), diabetes, and hypercholesterolemia.
Genetic disruption or overexpression of NOX1 in mice is associated with reduced or increased blood pressure responses to ATII, respectively (64,65). The systemic deletion of NOX2 or the p47phox subunit in apolipoprotein E-deficient (ApoE−/−) mice was shown to decrease atherosclerotic burden (66,67) and diminish blood pressure response to ATII (67). In humans, atherosclerosis correlates with increased coronary expression of NADPH oxidase subunits (p22phox, NOX2, p47phox, and p67phox) (68).
Xanthine oxidase
Xanthine oxidase (XO) produces O2− and H2O2, and is a source of O2− in diseased human coronary arteries (68). It is expressed by ECs in response to several stimuli, including ATII (69). It is also produced in a circulating form by the liver, a mechanism of production that is promoted by hypercholesterolemia, and can adhere to ECs through interaction with glycosaminoglycans (70). The activity of both endothelial and plasma XO is elevated in experimental models of atherosclerosis and in human atherosclerotic plaques. Consistent with this, XO inhibitors improve endothelial function in aortic ring explant assays from hyperlipidemic animals (70), attenuate endothelial dysfunction in heavy smokers (71), and reduce the development of atherosclerosis in ApoE−/− mice (72). However, clinical data concerning XO inhibitors are controversial (56).
Mitochondrial dysfunction and contribution to ROS
Under physiological conditions, mitochondria represent a key source of O2− by reducing approximately 1% of consumed oxygen by only a single electron to O2−. In this respect, the ETC enzyme complexes NADH dehydrogenase (complex I) and ubiquinone-cytochrome b-c1 (complex III) play an integral role, whereas the amount of mitochondrial O2− released also depends on Mn-SOD activity. In pathophysiological conditions, evidence suggests that mitochondrial dysfunction (73) and mitochondrial ROS generation could be linked to atherogenesis through promotion of other ROS sources and the release of mitochondrial apoptotic signals (74,75). In ECs, mitochondrial dysfunction due to Mn-SOD deficiency was indeed correlated to accelerated atherogenesis in ApoE−/− mice (76). Accordingly, specific targeting of antioxidants to mitochondria has raised hopes in preclinical studies, but has yet to be translated to treatment of human vascular disease.
Dysfunctional eNOS and inducible NO synthase
As mentioned, oxidative stress causes endothelial dysfunction through O2− scavenging of NO to produce ONOO−, followed by uncoupling of oxygen reduction from NO synthesis by eNOS, causing it to produce not NO, but O2−. This pathological process, termed eNOS uncoupling, is evident in several animal models and in humans with endothelial dysfunction. The peroxynitrite-mediated depletion of the essential cofactor BH4 is considered a significant (but not the only) cause of eNOS uncoupling. In addition, the inducible form of NO synthase (iNOS) is expressed in the vasculature in pathological states such as inflammation, oxidative stress, or sepsis. iNOS produces excessive amounts of NO and mediates impaired vasoconstriction and endothelium-dependent vasodilation. Proposed mechanisms for impaired contractile responses include continuous guanylyl cyclase activation (77), disturbed vascular calcium regulation (78), and oxidative alteration of catecholamines (79). Concurrently, impaired vasodilation may be further worsened by decreased eNOS activity, resulting from competition with iNOS for BH4 and NO scavenging by O2− (80). Additionally, continuous NO exposure can impair the link between endothelial receptors and calcium-calmodulin–dependent activation of eNOS (81) and the sensitivity of SMCs to NO (82,83). In vivo, genetic deficiency of iNOS was seen to reduce atherosclerosis in ApoE−/− mice (84). In humans, iNOS expression in plaques from patients with transplant coronary artery disease correlates with nitrotyrosine residues and protein nitrosylation, both markers of ONOO− formation, which is an important mediator of atherogenesis (85).
IMBALANCE IN VASCULAR ROS PRODUCTION AND SCAVENGING: ANTIOXIDANT ENZYMES
Counterbalancing the ROS-producing factors discussed in the previous section, the following act as the major defenses against vascular ROS.
Superoxide dismutase
Three SOD isoforms exist in humans. SOD1 (copper, zinc [Cu-Zn]-SOD) is a soluble enzyme of the cytoplasm and mitochondrial intermembrane space. SOD2 (Mn-SOD) exists in the mitochondrial matrix, whereas SOD3 (extracellular [EC]-SOD) is secreted into the extracellular space. All isozymes catalyze the dismutation of O2− to H2O2 and oxygen, thus blunting superoxide-mediated NO inactivation. High levels of superoxide dismutation products, however, can cross cellular membranes to form proatherogenic molecules, thus rendering the SOD effect dose-dependent. Accordingly, SOD1-overexpressing mice develop more pronounced fatty streaks on an atherogenic diet (86), whereas combined overexpression of SOD and catalase retards atherogenesis in ApoE−/− mice (87).
SOD2 is the frontline defense against mitochondrial O2−. The role of this enzyme is underlined by the fact that SOD2 knockout mice die within days of birth, whereas heterozygous SOD2 deficiency mediates mitochondrial dysfunction and DNA damage, as well as atherosclerosis in ApoE−/− mice (74). Vascular SOD3 is produced by macrophages and SMCs, the same cell types implicated in iNOS induction (88). Thus, SOD3 may also prevent formation of ONOO−, as well as O2−. The functional role of SOD3 in atherogenesis, however, remains poorly understood (89).
Catalase
Catalase, a tetramer of 4 polypeptides, catalyzes the decomposition of H2O2 to oxygen and water by virtue of 4 porphyrin heme iron groups. The enzyme’s overexpression blunts atherosclerosis (87) and inhibits ATII-mediated aortic wall hypertrophy (90). The differing contributions of various ROS to vascular disease are underlined by the observation that some atherosclerosis models, like ApoE−/− mice or a high fat diet, are attenuated by catalase, but not SOD1 overexpression alone (87,91).
Paraoxonases
Three members make up the paraoxonase (PON) family: PON1, 2, and 3. Despite their varying functions, localizations, and activities, all exhibit antiatherogenic properties, most likely through inhibition of oxidative stress and lipid peroxidation (92). PON1, mainly secreted by the liver, associates with high-density lipoprotein (HDL) in the plasma, where it protects HDL and low-density lipoprotein (LDL) against peroxidation and degrades cholesteryl esters and lipoproteins found in oxidized lipoproteins. It inhibits ROS production by myeloperoxidase (MPO), an enzyme secreted by immune cells upon activation and an accepted biomarker in acute coronary syndrome (93), thereby reducing oxidative stress, inflammation, and monocyte attraction.
PON2 is localized intracellularly in membranes of the ER and mitochondria, and exerts antioxidative effects in vascular cells (94,95). It has been proposed that PON2 interacts with coenzyme Q10 (96,97) and thus inhibits mitochondrial O2− generation, which is of relevance both to atherosclerosis and cancer. In line with this, PON2 has antiatherogenic effects in mice (92) and possibly humans, where its expression is lost in vessels with advanced atherosclerotic disease (98). PON3 is found both in serum and cells, and appears to exert similar effects to PON2 (99). Like the other members of the family, PON3 attenuates atherogenesis in mice (100) and is lost in vascular cells from patients with atherosclerosis (101).
Glutathione peroxidase
Glutathione peroxidase (GPx) exists in 8 isoforms, and reduces H2O2 to water and lipid peroxides to their respective alcohols. GPx1, among the most abundant GPx enzymes, is expressed in many cell types. GPx1 deficiency in mice raises sensitivity to myocardial ischemia/reperfusion injury and atherosclerotic burden in ApoE−/− and diabetic mice (102–104). In humans, GPx1 activity in erythrocytes correlates inversely to the risk of cardiovascular events in patients with coronary artery disease (105).
GPx4 is unique in function and structure. It is dispersed among various subcellular locations, such as the ER, cytoplasm, mitochondria, and plasma membrane. Apart from H2O2, it can detoxify a broad selection of lipid and cholesterol hydroperoxides. GPx4 overexpression in ApoE−/− mice blunts atherogenesis by inhibiting lipid peroxidation and vascular cell sensitivity to oxidized lipids (106).
Heme oxygenase
Heme oxygenase (HO) catalyzes the first part of the disintegration of heme to carbon monoxide (CO), biliverdin, and free ferrous iron. It exists in 3 isoforms, with HO-1 representing the inducible, HO-2 the constitutive, and HO-3 the enzymatically-inactive variant. HO-1 expression is induced by oxidative stress, hypoxia, and certain cytokines. It is implicated in protection from adverse vascular remodeling and atherosclerosis (107). Mechanistically, the breakdown of pro-oxidant heme to the ROS scavenger bilirubin is thought to be of importance. Apart from radical scavenging, bilirubin also inhibits the assembly and activity of NADPH oxidase (108). Reduced heme availability can additionally interfere with the formation of NOX2, and thereby NADPH oxidase (109). Although toxic at very high levels, CO produced in small amounts by HO exhibits anti-inflammatory, antiproliferative, and vasodilatory functions, with each of these being implicated in adverse vascular remodeling (109). Different genetic models of HO deficiency and overexpression support its role in atherosclerosis (110).
Thioredoxin
Thioredoxin (Trx) consists of 2 systems, Trx1/thioredoxin-reductase (TrxR1) and mitochondrial Trx2/TrxR2, and is found in ECs, SMCs, and fibroblasts. It has direct and indirect antioxidant actions, and plays an accepted vasoprotective role. Recently, Trx was shown to reverse age-related hypertension and arterial stiffness by improving vascular redox and restoring eNOS function (111).
Small nonprotein antioxidants
Nonenzymatic, low molecular weight compounds, including metabolic products like uric acid, bilirubin, and glutathione, as well as exogenous composites, such as antioxidant vitamins (mainly C and E) and polyphenols, add to cellular antioxidant systems. Glutathione determines the intracellular redox state, whereas bilirubin and uric acid scavenge extracellular ROS. Evidence for a protective role of these small nonprotein antioxidants in vivo, however, is still limited. Vitamin C (L-ascorbate) scavenges several reactive oxygen and nitrogen species, and restores α-tocopherol from its radical state (112). Furthermore, vitamin C stabilizes BH4 (113) and, in turn, eNOS. Among the 8 tocopherols and tocotrienols constituting vitamin E, α-tocopherol is the most active and its major antioxidant action is thought to be scavenging of lipid peroxides. Nevertheless, upon radical-scavenging, α-tocopherol itself is converted into a pro-oxidant radical (tocopheroxyl radical), potentially limiting its in vivo efficacy. Finally, some of the polyphenolic antioxidants in food (e.g., vegetables, cocoa) and beverages (e.g., green tea, red wine) have been demonstrated to inhibit the activity of NADPH oxidases or support the activation of antioxidant enzymes, possibly explaining positive data from epidemiological studies (114,115).
CARDIOVASCULAR RISK FACTORS, ATHEROSCLEROSIS, AND ROS
Although dealt with at the level of the entire cardiovascular system in Part 3 of this review series, it is important to note from the vascular perspective that classical cardiovascular risk factors associate with increased oxidative stress and specifically with endothelial dysfunction. Indeed, oxidative stress is likely to be the common pathological mechanism through which different risk factors contribute to the development of vascular disease, rendering it an attractive therapeutic target (Figure 4).
Figure 4. ROS Mediate CVD in Response to Several Different Risk Factors.
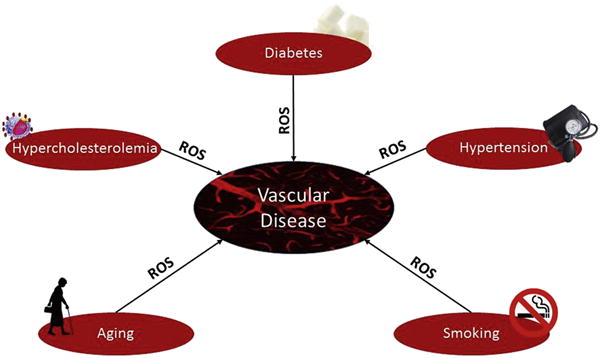
Vascular disease relates to a variety of pathological states, and is almost invariably prompted by classical and nonclassical risk factors. ROS play a central role in mediating the noxious effects of classical risk factors, and thus represent attractive therapeutic and preventive targets. CVD = cardiovascular disease; ROS = reactive oxygen species.
Atherosclerosis, as a chronic arterial inflammatory state, is strongly linked to oxidative stress (116). Indeed, increased ROS favor proinflammatory gene expression, including vascular cell adhesion molecule-1, intercellular adhesion molecule-1, monocyte chemotactic protein 1, and E-selectin, which in turn, are transcribed by redox-sensitive transcription factors like NF-κB, activator protein-1, HIF-1β, and early growth response protein 1. These genes are crucially involved in the early phases of atherogenesis, such as endothelial-leukocyte interactions (117). The same transcription factors also play a role in the proliferative signaling associated with SMC growth and vascular remodeling (118). MAPKs, as well as receptor and nonreceptor tyrosine kinases, are known to be activated through ROS. For instance, p38 MAPK is activated by ATII-mediated O2− production through NADPH oxidase, which, in turn, stimulates the fibroblast migration and SMC proliferation that contribute to the atherogenic process (Figure 5).
Figure 5. Mechanisms Through Which Cardiovascular Risk Factors Mediate Atherogenesis and Available Pharmacological Tools for Targeting Crucial Pathogenic Factors.
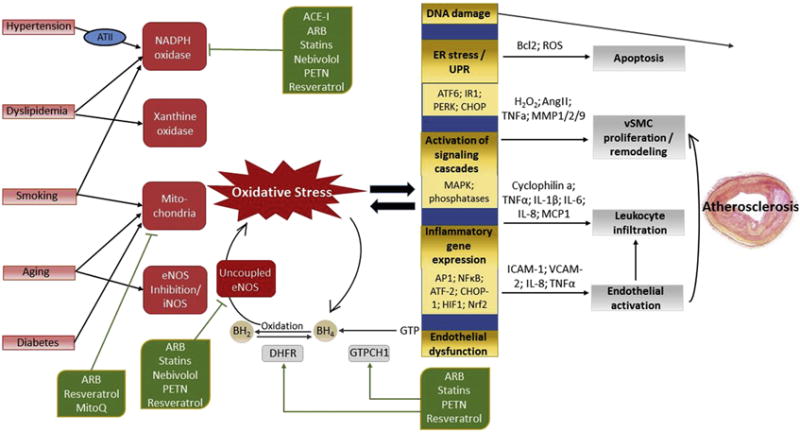
Cardiovascular risk factors promote oxidative stress by stimulation of various pro-oxidant enzyme systems, tipping the redox balance in favor of oxidation, with O2− as the key mediator. Hypertension activates NADPH oxidase via angiotensin II (ATII) and angiotensin 1 receptor (AT1R) signaling. The main ROS sources in states of hypercholesterolemia are NADPH oxidase and xanthine oxidase (activated by oxidized low-density lipoprotein), and the hypercholesterolemia-mediated up-regulation of AT1R. Hyperglycemia, as seen in diabetes, associates primarily with mitochondrial ROS production, which can secondarily activate NADPH oxidase. Compounds contained in cigarette smoke activate NADPH oxidase, which mediates mitochondrial dysfunction and thus ROS production. Aging is associated with increased mitochondrial dysfunction and reduced eNOS activity. Superoxide from all sources can inactivate vasoprotective NO by its reaction to ONOO−, which, in turn, mediates uncoupling of eNOS-mediated oxygen reduction and NO− production by oxidation of the essential cofactor tetrahydrobiopterin (BH4).
The enzymes dihydrofolate reductase (DHFR) and GTPCH1 counteract eNOS uncoupling by replenishing BH4 levels via regeneration and de novo synthesis, respectively. ATII decreases BH4 levels, not only by activating NADPH oxidase, but also by down-regulation of DHFR. NADPH oxidase has been seen to be inhibited by angiotensin-converting enzyme inhibitors (ACEIs), ATII receptor type 1 blockers (ARBs), 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors (statins), the β-blocker nebivolol, the plant-derived polyphenol resveratrol, and the organic nitrate pentaerithrityl tetranitrate (PETN). The same compounds inhibit eNOS uncoupling by activating GTPCH1 and DHFR. Mitochondrial O2− generation is decreased by ARBs, MitoQ and resveratrol. Other abbreviations as in Figures 1 and 3.
We have recently shown that oxidative stress can directly promote endothelial-to-mesenchymal transition (EndMT) (116). Although indispensable for the normal development of the cardiovascular system (119), EndMT in the adult is generally considered to be pathological and may arise in in the setting of chronic inflammatory diseases, such as atherosclerosis. We identified that high concentrations of H2O2 cause EndMT, with the extent of EndMT-derived mesenchymal cells (fibroblasts, SMCs) in atherosclerotic plaques related to an advanced plaque phenotype and human plaque instability (116). We are currently engaged in further unraveling the relevant signaling pathways, and are actively investigating possible ways to manipulate EndMT for therapeutic gain.
Oxidative stress is also a critical component of hypertension, where NADPH oxidase represents the primary source of ROS. Mechanistically, ATII, through angiotensin 1 receptor (AT1R) signaling, mediates the vascular up-regulation and activation of NADPH oxidase (120,121) promoted by rapid translocation of the small GTPase Rac 1 (Ras-related C3 botulinum toxin substrate 1) to the cellular membrane (122), or by phosphorylation and membrane translocation of the p47phox subunit (123). Mechanical stretch, as observed in hypertension, has also been directly linked to p47phox translocation and NADPH oxidase activation (124,125). The role of oxidative stress in hypertension is further supported by the blood pressure-lowering effects of genetic deletion or pharmacological inhibition of NADPH oxidase (67,69,126).
Hyperglycemia promotes cellular oxidative stress (127) with the mitochondrial respiratory chain as the primary source of O2− (128). Mitochondrial O2− mediates stimulation of protein kinase C and generation of advanced glycation end-products (128). Protein kinase C and advanced glycation end-products in turn activate NADPH oxidase and inhibit eNOS via post-translational modification (129), whereas NADPH oxidase-mediated eNOS uncoupling further promotes oxidative stress (130). Insulin resistance, as found in type 2 diabetes, may inhibit GTP cyclohydrolase 1 (GTPCH1) activity and thus de novo BH4 synthesis. Furthermore, the insulin-mediated activation of eNOS via the phosphoinositide 3 kinase (PI3K)/Akt pathway is disrupted in a state of insulin resistance. Increased levels of ATII found in patients with diabetes could contribute to NADPH oxidase activation and reduce BH4 recycling from BH2 by dihydrofolate reductase (131).
In hypercholesterolemia, NADPH oxidase and XO appear to be key sources of O2− (68); in fact, uncoupling of eNOS is possibly secondary to oxidative stress induced by these enzymes (132,133), as observed in LDL cholesterol–treated ECs, ApoE−/− mice, and hypercholesterolemic humans (134,135). LDL cholesterol and, especially, oxidized LDL (oxLDL) cholesterol are known to promote generation of O2− and ONOO−, and thereby regulate monocyte adhesion, inflammatory vascular gene expression, and redox-sensitive transcription factors, such as NF-κB and activator protein-1. Cellular lipoxygenases (LOs) have been proposed as a possible enzymatic source of oxLDL cholesterol. Accordingly, elevated levels of 15-LO have been observed in atherosclerotic lesions of the human and rabbit aorta (136). Adding to the direct effects of LDL cholesterol on vascular ROS production, dyslipidemia may potentiate the effects of ATII via up-regulation of the AT1R (137).
In vascular aging, 2 characteristic ROS-related features have been defined, namely endothelial dysfunction and central arterial stiffness (138). Endothelial dysfunction is mediated by reduced NO bioavailability, either through decreased synthesis or increased degradation. Studies provide conflicting evidence on age-related changes in eNOS protein expression (139–141). However, more recent data are suggestive of an age-dependent eNOS uncoupling (142), favoring oxidative stress through decreased availability of NO and increased O2− and ONOO− production. The perturbed redox balance promotes inflammation, which drives increased NADPH oxidase expression and O2− production through mediators, such as blood-borne tumor necrosis factor α. In parallel, age-dependent activation of the renin-angiotensin-aldosterone system may further stimulate NADPH oxidases through ATII (143). However, age-dependent central arterial stiffness involves a loss of elastic fibers partly mediated by proteases with elastinolytic activity, including matrix metalloproteinases. These, in turn, have been linked to ROS through a ROS-induced activation of cyclophilin A, which mediates matrix metalloproteinase 9, tumor necrosis factor α, and IL-6 expression via the NF-κB and ERK pathways.
Components of cigarette smoke are also known to stimulate endothelial NADPH oxidase (144) and diminish mitochondrial function, thus inducing mitochondrial oxidative stress (145). Elevated O2− and ONOO− formation mediate vascular DNA damage and inflammation, thereby accelerating vascular aging. Via oxidation of LDL cholesterol, cigarette smoke enhances its pro-oxidative potential (146). Through oxidative eNOS uncoupling, smokers suffer from endothelial dysfunction that may be attenuated by BH4 supplementation (147).
THE ANTIOXIDANT PARADOX: CLINICAL TRIALS AND THEIR SO FAR DISAPPOINTING OUTCOMES
The term antioxidant paradox refers to the fact that despite the crucial role ROS play in the pathogenesis of CVDs, supplementation of dietary antioxidants has shown little to no therapeutic effects (148).
A large meta-analysis showed that antioxidant supplements to exert no beneficial effects (vitamin C and selenium), or that they even potentially raise overall mortality (vitamin E, β-carotene) (149), although this inference was later repudiated because re-examination of data showed that the analyzed studies did not include mortality as a primary endpoint (150). A number of mechanisms could explain these unexpected clinical results. First, the antioxidants used may have been inappropriate. Indeed, at the time of initiation of clinical trials with natural antioxidants, no data from animal trials with these compounds were available. In fact, animal trials of that era had been carried out with more potent antioxidants, such as probucol or probucol analogues (151). Also, accessibility of the administered natural compounds to the exact site where ROS are produced (especially within cellular organelles such as mitochondria) may be limited. Additionally, H2O2 and hypochlorous acid (HOCl), which are strongly implicated in vascular damage, are not scavenged by antioxidant vitamins (152). Furthermore, any antioxidant would likely be oxidized rapidly to its inactive form, with little hope of reversal. Apart from the depletion of the active form, this could mediate an accumulation of the oxidized and potentially pro-oxidant form of the parent molecule (153,154). Moreover, the reaction of one type of ROS with a certain antioxidant could potentially generate another ROS, which the original antioxidant cannot scavenge. Due to the nature of ROS as signaling molecules, chronic antioxidant exposure might lead to compensatory up-regulation of ROS producing systems.
Regarding the dosages used, there may have been inconsistencies between different studies, as no information was made available on vascular antioxidant concentrations or efficacy (e.g., isoprostane levels as biomarkers for lipid peroxidation). Such information is critical, given the high rate constants of the reactions between endogenous molecules such as NO and ROS; indeed, for an antioxidant to work effectively, its reaction with ROS must outcompete that of the ROS with its target molecule. In line with this, vitamin E would have to exceed the concentration of NO 1 million-fold, which is in the millimolar range, which is unlikely to be achieved, even with the highest clinically administered doses (63).
Additionally, several clinical trials were performed on high-risk patients, in which vascular disease was advanced (151) and thus difficult to reverse. Also, several of these patients were already treated with acetylsalicylic acid (aspirin), making the treatment with an additional antioxidant less likely to be effective. Perhaps longer-term prospective studies with regular efficacy monitoring that are carried out in healthy individuals treated with a single antioxidant would achieve different results.
POTENTIAL THERAPEUTIC TARGETS
The so far disappointing clinical results suggest a limited efficacy of conventional antioxidant treatments. Thus, novel strategies aimed at preventing ROS production or stimulating endogenous antioxidant systems should be considered. In this context, enzymes of the NADPH oxidase family, especially those containing NOX1 or NOX2 catalytic subunits, are attractive targets, as they represent key sources of ROS in vascular disease states (63). The organizer subunit p47phox is a requisite for full activation of these isoforms. Without it, however, the enzymes can still be activated, albeit less efficiently. Interestingly, given the importance of NADPH oxidase activity for immune cell function, it potentially makes partial inhibition clinically more attractive than full blockade. Several animal studies on atherosclerosis, hypertension, and stroke have demonstrated the efficacy of p47phox inhibitors, such as Nox2ds-tat and apocynin (155–159). However, improvements are still needed in effectiveness, pharmacokinetics, and specificity before clinical testing of these compounds may be commenced.
Considerable advances have been made in delivering antioxidants to mitochondria. A well-established approach is conjugation of an antioxidant to a lipophilic cation such as triphenylphosphonium (TTP), which, due to its positive charge and the large negative potential gradient across the inner mitochondrial membrane, concentrates 100- to 500-fold higher within mitochondria than in the cytoplasm. The compound MitoQ follows this principle, harboring a quinone moiety, and diminishing radical production and mitochondrial damage without interfering with respiratory function. It was seen to blunt myocardial ischemia/reperfusion injury, nitrate-related endothelial damage, blood pressure, doxorubicin-induced heart damage, and sepsis-associated heart and liver injury (160). Two phase II trials on Parkinson’s disease and hepatitis C have not reported any adverse effects, although benefits could only be shown against hepatitis C–induced liver damage. Further clinical studies focusing on cardiovascular endpoints will be needed. Other compounds relying on the same principle, such as MitoE (tocopherol), MitoTEMPOL (4-hydroxy-2,2,6,6-tetramethyl-piperidine-1-oxyl), SkQ (plastoquinone fused to TTP), Mito-Apocynin, Mito-Peroxidase, or MitoSOD have raised hopes in preclinical trials, but their clinical efficacy is yet to be demonstrated.
Experimental evidence has also highlighted gene therapy as a potential approach to attenuate vascular oxidative stress. Endothelial GTPCH1 overexpression increases BH4 levels, and inhibits eNOS uncoupling and O2− production in ApoE−/− mice (161). Gene therapy with SOD3 was shown to reduce ROS levels, stimulate endothelial regeneration, and inhibit in-stent restenosis in hyperlipidemic rabbits (162). Furthermore, overexpression of catalase in ApoE−/− mice was shown to decrease atherosclerosis, an effect further potentiated by SOD1 overexpression (87).
Novel therapeutic targets could also include the activator protein-1 transcription factor JunD, the mitochondrial adaptor p66Shc, and SIRT1, a family-member of nicotinamide adenine dinucleotide (NAD)-dependent proteins. They are intimately involved in controlling ROS production/scavenging in diverse disease conditions. Whereas JunD and SIRT1 preserve endothelial function by attenuating oxidative stress, p66Shc promotes mitochondrial ROS production and apoptosis (138). All of these genes have been implicated in age-dependent vascular dysfunction; however, p66Shc has also been implicated in the vascular pathobiology triggered by various cardiovascular risk factors, including smoking, diabetes (127,163), and hypertension (125). Ongoing chemical library screening studies are aiming to reveal compounds capable of enhancing JunD activity in the vasculature, whereas the SIRT1 activator SRT2104 has already been investigated in clinical trials (164).
The ER is intrinsically linked to free radicals, whereby oxidative stress mediates ER stress and vice versa. A major responsive signaling pathway in this respect is the unfolded protein response, mediating endothelial inflammation and dysfunction through regulation of eNOS expression/activity, as well as being implicated in resolving ER stress. When ER stress prevails, cells induce apoptosis via the unfolded protein response with the C/EBP homologous protein (CHOP) as a central proapoptotic factor. Apoptosis and CHOP are specifically implicated in plaque stability (165). Consequently, the ER folding capacity and mediators of the unfolded protein response are compelling therapeutic targets, with CHOP and its role in atherosclerosis being especially appealing (166,167).
CONCLUSIONS REGARDING OXIDATIVE STRESS IN THE VASCULATURE
The vasculature is an important site of ROS and oxidative stress in human biology and disease. Although, as in cardiac myocytes, certain ROS can modulate physiological responses at low concentrations ROS become pathological, at higher levels. The failure of clinical antioxidants across several clinical trials has been disappointing, but there is realistic hope that better-targeted, and more specific and concentrated therapies may demonstrate efficacy.
CONDENSED ABSTRACT.
Although many different factors promote cardiac and vascular disease, most share an important pathological mechanism: oxidative stress. In the failing heart, oxidative stress occurs in the myocardium and correlates with left ventricular dysfunction. Reactive oxygen species (ROS) negatively affect myocardial calcium handling, cause arrhythmias, and contribute to adverse cardiac remodeling. Similarly, oxidative balance in the vasculature is tightly regulated by a wealth of pro- and anti-oxidant systems that orchestrate region-specific ROS production and removal. Although at low levels ROS regulate multiple physiological vascular cell functions, excessive ROS levels promote vascular disease by disruption of redox-dependent vascular signaling and oxidative damage.
Acknowledgments
The authors thank Margot Neuser for graphical support on Figures 1 and 2. This study was partly supported by the Center for Translational Vascular Biology (CTVB) and funded by the “Stiftung Mainzer Herz”. Dr. Münzel is PI of the DZHK (German Center for Cardiovascular Research), Partner Site Rhein-Main, Mainz, Germany. Dr. Maack’s research is supported by the Deutsche Forschungsgemeinschaft (Heisenberg Programm, SFB 894, Ma2528/7-1) and Corona Stiftung. Dr. Camici is the recipient of a Sheikh Khalifa’s Foundation Assistant Professorship at the Faculty of Medicine, University of Zurich and he acknowledges research support from “Alfred and Annemarie von Sick Grants for Translational and Clinical Research Cardiology and Oncology” and the Foundation for Cardiovascular Research–Zurich Heart House. Dr. Kovacic acknowledges research support from the National Institutes of Health (R01HL130423), the American Heart Association (14SFRN20490315; 14SFRN20840000), and The Leducq Foundation (Transatlantic Network of Excellence Award).
ABBREVIATIONS AND ACRONYMs
- ATII
angiotensin II
- eNOS
endothelial nitric oxide synthase
- H2O2
hydrogen peroxide
- HF
heart failure
- NADPH
nicotinamide adenine dinucleotide phosphate
- NO
nitric oxide
- NOX
nicotinamide adenine dinucleotide phosphate oxidase (NADPH oxidase)
- O2−
superoxide
- ROS
reactive oxygen species
- SOD
superoxide dismutase
Footnotes
Disclosures: The authors have reported that they have no relationships relevant to the contents of this paper to disclose.
References
- 1.Gori T, Münzel T. Oxidative stress and endothelial dysfunction: therapeutic implications. Ann Med. 2011;43:259–72. doi: 10.3109/07853890.2010.543920. [DOI] [PubMed] [Google Scholar]
- 2.Münzel T, Gori T, Bruno RM, Taddei S. Is oxidative stress a therapeutic target in cardiovascular disease? Eur Heart J. 2010;31:2741–8. doi: 10.1093/eurheartj/ehq396. [DOI] [PubMed] [Google Scholar]
- 3.Maack C, Kartes T, Kilter H, et al. Oxygen free radical release in human failing myocardium is associated with increased activity of rac1-GTPase and represents a target for statin treatment. Circulation. 2003;108:1567–74. doi: 10.1161/01.CIR.0000091084.46500.BB. [DOI] [PubMed] [Google Scholar]
- 4.Mollnau H, Oelze M, August M, et al. Mechanisms of increased vascular superoxide production in an experimental model of idiopathic dilated cardiomyopathy. Arterioscler Thromb Vasc Biol. 2005;25:2554–9. doi: 10.1161/01.ATV.0000190673.41925.9B. [DOI] [PubMed] [Google Scholar]
- 5.Belch JJ, Bridges AB, Scott N, Chopra M. Oxygen free radicals and congestive heart failure. Br Heart J. 1991;65:245–8. doi: 10.1136/hrt.65.5.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burgoyne JR, Mongue-Din H, Eaton P, Shah AM. Redox signaling in cardiac physiology and pathology. Circ Res. 2012;111:1091–106. doi: 10.1161/CIRCRESAHA.111.255216. [DOI] [PubMed] [Google Scholar]
- 7.Wagner S, Rokita AG, Anderson ME, Maier LS. Redox regulation of sodium and calcium handling. Antioxid Redox Signal. 2013;18:1063–77. doi: 10.1089/ars.2012.4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kröller-Schön S, Steven S, Kossmann S, et al. Molecular mechanisms of the crosstalk between mitochondria and NADPH oxidase through reactive oxygen species-studies in white blood cells and in animal models. Antioxid Redox Signal. 2014;20:247–66. doi: 10.1089/ars.2012.4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maack C, Böhm M. Targeting mitochondrial oxidative stress in heart failure throttling the afterburner. J Am Coll Cardiol. 2011;58:83–6. doi: 10.1016/j.jacc.2011.01.032. [DOI] [PubMed] [Google Scholar]
- 10.Lassègue B, San Martín A, Griendling KK. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ Res. 2012;110:1364–90. doi: 10.1161/CIRCRESAHA.111.243972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prosser BL, Ward CW, Lederer WJ. X-ROS signaling: rapid mechano-chemo transduction in heart. Science. 2011;333:1440–5. doi: 10.1126/science.1202768. [DOI] [PubMed] [Google Scholar]
- 12.Jian Z, Han H, Zhang T, et al. Mechanochemotransduction during cardiomyocyte contraction is mediated by localized nitric oxide signaling. Sci Signal. 2014;7:ra27. doi: 10.1126/scisignal.2005046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wenzel P, Knorr M, Kossmann S, et al. Lysozyme M-positive monocytes mediate angiotensin II-induced arterial hypertension and vascular dysfunction. Circulation. 2011;124:1370–81. doi: 10.1161/CIRCULATIONAHA.111.034470. [DOI] [PubMed] [Google Scholar]
- 14.Kuroda J, Ago T, Matsushima S, Zhai P, Schneider MD, Sadoshima J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc Natl Acad Sci U S A. 2010;107:15565–70. doi: 10.1073/pnas.1002178107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsushima S, Kuroda J, Ago T, et al. Broad suppression of NADPH oxidase activity exacerbates ischemia/reperfusion injury through inadvertent downregulation of hypoxia-inducible factor-1α and upregulation of peroxisome proliferator-activated receptor-α. Circ Res. 2013;112:1135–49. doi: 10.1161/CIRCRESAHA.111.300171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Craige SM, Chen K, Pei Y, et al. NADPH oxidase 4 promotes endothelial angiogenesis through endothelial nitric oxide synthase activation. Circulation. 2011;124:731–40. doi: 10.1161/CIRCULATIONAHA.111.030775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang M, Brewer AC, Schröder K, et al. NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proc Natl Acad Sci U S A. 2010;107:18121–6. doi: 10.1073/pnas.1009700107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ide T, Tsutsui H, Kinugawa S, et al. Direct evidence for increased hydroxyl radicals originating from superoxide in the failing myocardium. Circ Res. 2000;86:152–7. doi: 10.1161/01.res.86.2.152. [DOI] [PubMed] [Google Scholar]
- 19.Aon MA, Cortassa S, O’Rourke B. Redox-optimized ROS balance: a unifying hypothesis. Biochim Biophys Acta. 2010;1797:865–77. doi: 10.1016/j.bbabio.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nickel A, Kohlhaas M, Maack C. Mitochondrial reactive oxygen species production and elimination. J Mol Cell Cardiol. 2014;73:26–33. doi: 10.1016/j.yjmcc.2014.03.011. [DOI] [PubMed] [Google Scholar]
- 21.Nickel AG, von Hardenberg A, Hohl M, et al. Reversal of mitochondrial transhydrogenase causes oxidative stress in heart failure. Cell Metab. 2015;22:472–84. doi: 10.1016/j.cmet.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 22.Dai DF, Hsieh EJ, Liu Y, et al. Mitochondrial proteome remodelling in pressure overload-induced heart failure: the role of mitochondrial oxidative stress. Cardiovasc Res. 2012;93:79–88. doi: 10.1093/cvr/cvr274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dai DF, Johnson SC, Villarin JJ, et al. Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Gαq overexpression-induced heart failure. Circ Res. 2011;108:837–46. doi: 10.1161/CIRCRESAHA.110.232306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hornig B, Arakawa N, Kohler C, Drexler H. Vitamin C improves endothelial function of conduit arteries in patients with chronic heart failure. Circulation. 1998;97:363–8. doi: 10.1161/01.cir.97.4.363. [DOI] [PubMed] [Google Scholar]
- 25.Förstermann U, Münzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation. 2006;113:1708–14. doi: 10.1161/CIRCULATIONAHA.105.602532. [DOI] [PubMed] [Google Scholar]
- 26.Lee BE, Toledo AH, Anaya-Prado R, Roach RR, Toledo-Pereyra LH. Allopurinol, xanthine oxidase, and cardiac ischemia. J Investig Med. 2009;57:902–9. doi: 10.2310/JIM.0b013e3181bca50c. [DOI] [PubMed] [Google Scholar]
- 27.Baldus S, Müllerleile K, Chumley P, et al. Inhibition of xanthine oxidase improves myocardial contractility in patients with ischemic cardiomyopathy. Free Radic Biol Med. 2006;41:1282–8. doi: 10.1016/j.freeradbiomed.2006.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hare JM, Mangal B, Brown J, et al. OPT-CHF Investigators Impact of oxypurinol in patients with symptomatic heart failure. Results of the OPT-CHF study. J Am Coll Cardiol. 2008;51:2301–9. doi: 10.1016/j.jacc.2008.01.068. [DOI] [PubMed] [Google Scholar]
- 29.Dai DF, Chen T, Szeto H, et al. Mitochondrial targeted antioxidant peptide ameliorates hypertensive cardiomyopathy. J Am Coll Cardiol. 2011;58:73–82. doi: 10.1016/j.jacc.2010.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Keaney JF, Jr, Gaziano JM, Xu A, et al. Low-dose alpha-tocopherol improves and high-dose alpha-tocopherol worsens endothelial vasodilator function in cholesterol-fed rabbits. J Clin Invest. 1994;93:844–51. doi: 10.1172/JCI117039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stocker R. The ambivalence of vitamin E in atherogenesis. Trends Biochem Sci. 1999;24:219–23. doi: 10.1016/s0968-0004(99)01404-8. [DOI] [PubMed] [Google Scholar]
- 32.Heart Outcomes Prevention Evaluation Study Investigators. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. N Engl J Med. 2000;342:145–53. doi: 10.1056/NEJM200001203420301. [DOI] [PubMed] [Google Scholar]
- 33.Lonn E, Bosch J, Yusuf S, et al. HOPE and HOPE-TOO Trial Investigators Effects of long-term vitamin E supplementation on cardiovascular events and cancer: a randomized controlled trial. JAMA. 2005;293:1338–47. doi: 10.1001/jama.293.11.1338. [DOI] [PubMed] [Google Scholar]
- 34.Smith RA, Hartley RC, Cochemé HM, Murphy MP. Mitochondrial pharmacology. Trends Pharmacol Sci. 2012;33:341–52. doi: 10.1016/j.tips.2012.03.010. [DOI] [PubMed] [Google Scholar]
- 35.Szeto HH. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br J Pharmacol. 2014;171:2029–50. doi: 10.1111/bph.12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sparagna GC, Lesnefsky EJ. Cardiolipin remodeling in the heart. J Cardiovasc Pharmacol. 2009;53:290–301. doi: 10.1097/FJC.0b013e31819b5461. [DOI] [PubMed] [Google Scholar]
- 37.Brown DA, Hale SL, Baines CP, et al. Reduction of early reperfusion injury with the mitochondria-targeting peptide bendavia. J Cardiovasc Pharmacol Ther. 2014;19:121–32. doi: 10.1177/1074248413508003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gibson CM, Giugliano RP, Kloner RA, et al. EMBRACE STEMI study: a Phase 2a trial to evaluate the safety, tolerability, and efficacy of intravenous MTP-131 on reperfusion injury in patients undergoing primary percutaneous coronary intervention. Eur Heart J. 2016;37:1296–303. doi: 10.1093/eurheartj/ehv597. [DOI] [PubMed] [Google Scholar]
- 39.Liu T, Takimoto E, Dimaano VL, et al. Inhibiting mitochondrial Na+/Ca2+ exchange prevents sudden death in a guinea pig model of heart failure. Circ Res. 2014;115:44–54. doi: 10.1161/CIRCRESAHA.115.303062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee CF, Chavez JD, Garcia-Menendez L, et al. Normalization of NAD+ redox balance as a therapy for heart failure. Circulation. 2016;134:883–94. doi: 10.1161/CIRCULATIONAHA.116.022495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koentges C, Bode C, Bugger H. SIRT3 in cardiac physiology and disease. Front Cardiovasc Med. 2016;3:38. doi: 10.3389/fcvm.2016.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rajasekaran NS, Connell P, Christians ES, et al. Human αB-crystallin mutation causes oxido-reductive stress and protein aggregation cardiomyopathy in mice. Cell. 2007;130:427–39. doi: 10.1016/j.cell.2007.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Münzel T, Keaney JF., Jr Are ACE inhibitors a “magic bullet” against oxidative stress? Circulation. 2001;104:1571–4. doi: 10.1161/hc3801.095585. [DOI] [PubMed] [Google Scholar]
- 44.Hare JM. Nitroso-redox balance in the cardiovascular system. N Engl J Med. 2004;351:2112–4. doi: 10.1056/NEJMe048269. [DOI] [PubMed] [Google Scholar]
- 45.Münzel T, Daiber A, Gori T. More answers to the still unresolved question of nitrate tolerance. Eur Heart J. 2013;34:2666–73. doi: 10.1093/eurheartj/eht249. [DOI] [PubMed] [Google Scholar]
- 46.Daiber A, Oelze M, Coldewey M, et al. Hydralazine is a powerful inhibitor of peroxynitrite formation as a possible explanation for its beneficial effects on prognosis in patients with congestive heart failure. Biochem Biophys Res Commu. 2005;338:1865–74. doi: 10.1016/j.bbrc.2005.10.106. [DOI] [PubMed] [Google Scholar]
- 47.Münzel T, Kurz S, Rajagopalan S, et al. Hydralazine prevents nitroglycerin tolerance by inhibiting activation of a membrane-bound NADH oxidase. A new action for an old drug. J Clin Invest. 1996;98:1465–70. doi: 10.1172/JCI118935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dulce RA, Yiginer O, Gonzalez DR, et al. Hydralazine and organic nitrates restore impaired excitation-contraction coupling by reducing calcium leak associated with nitroso-redox imbalance. J Biol Chem. 2013;288:6522–33. doi: 10.1074/jbc.M112.412130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Münzel T, Steven S, Daiber A. Organic nitrates: update on mechanisms underlying vasodilation, tolerance and endothelial dysfunction. Vascul Pharmacol. 2014;63:105–13. doi: 10.1016/j.vph.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 50.Taylor AL, Ziesche S, Yancy C, et al. African-American Heart Failure Trial Investigators Combination of isosorbide dinitrate and hydralazine in blacks with heart failure. N Engl J Med. 2004;351:2049–57. doi: 10.1056/NEJMoa042934. [DOI] [PubMed] [Google Scholar]
- 51.Hulsmans M, Holvoet P. The vicious circle between oxidative stress and inflammation in atherosclerosis. J Cell Mol Med. 2010;14:70–8. doi: 10.1111/j.1582-4934.2009.00978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Madamanchi NR, Hakim ZS, Runge MS. Oxidative stress in atherogenesis and arterial thrombosis: the disconnect between cellular studies and clinical outcomes. J Thromb Haemost. 2005;3:254–67. doi: 10.1111/j.1538-7836.2004.01085.x. [DOI] [PubMed] [Google Scholar]
- 53.Freed JK, Gutterman DD. Mitochondrial reactive oxygen species and vascular function: less is more. Arterioscler Thromb Vasc Biol. 2013;33:673–5. doi: 10.1161/ATVBAHA.13.301039. [DOI] [PubMed] [Google Scholar]
- 54.Stocker R, Keaney JF., Jr Role of oxidative modifications in atherosclerosis. Physiol Rev. 2004;84:1381–478. doi: 10.1152/physrev.00047.2003. [DOI] [PubMed] [Google Scholar]
- 55.Griendling KK, Sorescu D, Lassègue B, Ushio-Fukai M. Modulation of protein kinase activity and gene expression by reactive oxygen species and their role in vascular physiology and pathophysiology. Arterioscler Thromb Vasc Biol. 2000;20:2175–83. doi: 10.1161/01.atv.20.10.2175. [DOI] [PubMed] [Google Scholar]
- 56.Förstermann U. Nitric oxide and oxidative stress in vascular disease. Pflugers Archiv. 2010;459:923–39. doi: 10.1007/s00424-010-0808-2. [DOI] [PubMed] [Google Scholar]
- 57.Gryglewski RJ, Palmer RM, Moncada S. Superoxide anion is involved in the breakdown of endothelium-derived vascular relaxing factor. Nature. 1986;320:454–6. doi: 10.1038/320454a0. [DOI] [PubMed] [Google Scholar]
- 58.Beckman JS. Oxidative damage and tyrosine nitration from peroxynitrite. Chem Res Toxicol. 1996;9:836–44. doi: 10.1021/tx9501445. [DOI] [PubMed] [Google Scholar]
- 59.Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol. 1996;271:C1424–37. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 60.Laursen JB, Somers M, Kurz S, et al. Endothelial regulation of vasomotion in apoE-deficient mice: implications for interactions between peroxynitrite and tetrahydrobiopterin. Circulation. 2001;103:1282–8. doi: 10.1161/01.cir.103.9.1282. [DOI] [PubMed] [Google Scholar]
- 61.Harrison DG, Chen W, Dikalov S, Li L. Regulation of endothelial cell tetrahydrobiopterin pathophysiological and therapeutic implications. Adv Pharmacol. 2010;60:107–32. doi: 10.1016/B978-0-12-385061-4.00005-2. [DOI] [PubMed] [Google Scholar]
- 62.Li H, Horke S, Förstermann U. Vascular oxidative stress, nitric oxide and atherosclerosis. Atherosclerosis. 2014;237:208–19. doi: 10.1016/j.atherosclerosis.2014.09.001. [DOI] [PubMed] [Google Scholar]
- 63.Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov. 2011;10:453–71. doi: 10.1038/nrd3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dikalova A, Clempus R, Lassègue B, et al. Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation. 2005;112:2668–76. doi: 10.1161/CIRCULATIONAHA.105.538934. [DOI] [PubMed] [Google Scholar]
- 65.Matsuno K, Yamada H, Iwata K, et al. Nox1 is involved in angiotensin II-mediated hypertension: a study in Nox1-deficient mice. Circulation. 2005;112:2677–85. doi: 10.1161/CIRCULATIONAHA.105.573709. [DOI] [PubMed] [Google Scholar]
- 66.Barry-Lane PA, Patterson C, van der Merwe M, et al. p47phox is required for atherosclerotic lesion progression in ApoE−/− mice. J Clin Invest. 2001;108:1513–22. doi: 10.1172/JCI11927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Landmesser U, Cai H, Dikalov S, et al. Role of p47phox in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension. 2002;40:511–5. doi: 10.1161/01.hyp.0000032100.23772.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Guzik TJ, Sadowski J, Guzik B, et al. Coronary artery superoxide production and nox isoform expression in human coronary artery disease. Arterioscler Thromb Vasc Biol. 2006;26:333–9. doi: 10.1161/01.ATV.0000196651.64776.51. [DOI] [PubMed] [Google Scholar]
- 69.Landmesser U, Spiekermann S, Preuss C, et al. Angiotensin II induces endothelial xanthine oxidase activation: role for endothelial dysfunction in patients with coronary disease. Arterioscler Thromb Vasc Biol. 2007;27:943–8. doi: 10.1161/01.ATV.0000258415.32883.bf. [DOI] [PubMed] [Google Scholar]
- 70.White CR, Darley-Usmar V, Berrington WR, et al. Circulating plasma xanthine oxidase contributes to vascular dysfunction in hypercholesterolemic rabbits. Proc Natl Acad Sci U S A. 1996;93:8745–9. doi: 10.1073/pnas.93.16.8745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Guthikonda S, Sinkey C, Barenz T, Haynes WG. Xanthine oxidase inhibition reverses endothelial dysfunction in heavy smokers. Circulation. 2003;107:416–21. doi: 10.1161/01.cir.0000046448.26751.58. [DOI] [PubMed] [Google Scholar]
- 72.Schröder K, Vecchione C, Jung O, et al. Xanthine oxidase inhibitor tungsten prevents the development of atherosclerosis in ApoE knockout mice fed a Western-type diet. Free Radic Biol Med. 2006;41:1353–60. doi: 10.1016/j.freeradbiomed.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 73.Ramachandran A, Levonen AL, Brookes PS, et al. Mitochondria, nitric oxide, and cardiovascular dysfunction. Free Radic Biol Med. 2002;33:1465–74. doi: 10.1016/s0891-5849(02)01142-5. [DOI] [PubMed] [Google Scholar]
- 74.Ballinger SW, Patterson C, Knight-Lozano CA, et al. Mitochondrial integrity and function in atherogenesis. Circulation. 2002;106:544–9. doi: 10.1161/01.cir.0000023921.93743.89. [DOI] [PubMed] [Google Scholar]
- 75.Zeini M, López-Fontal R, Través PG, Benito G, Hortelano S. Differential sensitivity to apoptosis among the cells that contribute to the atherosclerotic disease. Biochem Biophys Res Comm. 2007;363:444–50. doi: 10.1016/j.bbrc.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 76.Ohashi M, Runge MS, Faraci FM, Heistad DD. MnSOD deficiency increases endothelial dysfunction in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. 2006;26:2331–6. doi: 10.1161/01.ATV.0000238347.77590.c9. [DOI] [PubMed] [Google Scholar]
- 77.Gunnett CA, Lund DD, McDowell AK, Faraci FM, Heistad DD. Mechanisms of inducible nitric oxide synthase-mediated vascular dysfunction. Arterioscler Thromb Vasc Biol. 2005;25:1617–22. doi: 10.1161/01.ATV.0000172626.00296.ba. [DOI] [PubMed] [Google Scholar]
- 78.Chen SJ, Li SY, Shih CC, Liao MH, Wu CC. NO contributes to abnormal vascular calcium regulation and reactivity induced by peritonitis-associated septic shock in rats. Shock. 2010;33:473–8. doi: 10.1097/SHK.0b013e3181bea334. [DOI] [PubMed] [Google Scholar]
- 79.Shelkovnikov S, Gonick HC. Peroxynitrite but not nitric oxide donors destroys epinephrine: HPLC measurement and rat aorta contractility. Life Sci. 2004;75:2765–73. doi: 10.1016/j.lfs.2004.04.044. [DOI] [PubMed] [Google Scholar]
- 80.Virdis A, Colucci R, Fornai M, et al. Cyclooxygenase-2 inhibition improves vascular endothelial dysfunction in a rat model of endotoxic shock: role of inducible nitric-oxide synthase and oxidative stress. J Pharmacol Exp Ther. 2005;312:945–53. doi: 10.1124/jpet.104.077644. [DOI] [PubMed] [Google Scholar]
- 81.Kessler P, Bauersachs J, Busse R, Schini-Kerth VB. Inhibition of inducible nitric oxide synthase restores endothelium-dependent relaxations in proinflammatory mediator-induced blood vessels. Arterioscler Thromb Vasc Biol. 1997;17:1746–55. doi: 10.1161/01.atv.17.9.1746. [DOI] [PubMed] [Google Scholar]
- 82.Eguchi D, D’Uscio LV, Wambi C, et al. Inhibitory effect of recombinant iNOS gene expression on vasomotor function of canine basilar artery. Am J Physiol Heart Circ Physiol. 2002;283:H2560–6. doi: 10.1152/ajpheart.00415.2002. [DOI] [PubMed] [Google Scholar]
- 83.Marfella R, Di Filippo C, Esposito K, et al. Absence of inducible nitric oxide synthase reduces myocardial damage during ischemia reperfusion in streptozotocin-induced hyperglycemic mice. Diabetes. 2004;53:454–62. doi: 10.2337/diabetes.53.2.454. [DOI] [PubMed] [Google Scholar]
- 84.Kuhlencordt PJ, Chen J, Han F, Astern J, Huang PL. Genetic deficiency of inducible nitric oxide synthase reduces atherosclerosis and lowers plasma lipid peroxides in apolipoprotein E-knockout mice. Circulation. 2001;103:3099–104. doi: 10.1161/01.cir.103.25.3099. [DOI] [PubMed] [Google Scholar]
- 85.Ravalli S, Albala A, Ming M, et al. Inducible nitric oxide synthase expression in smooth muscle cells and macrophages of human transplant coronary artery disease. Circulation. 1998;97:2338–45. doi: 10.1161/01.cir.97.23.2338. [DOI] [PubMed] [Google Scholar]
- 86.Tribble DL, Gong EL, Leeuwenburgh C, et al. Fatty streak formation in fat-fed mice expressing human copper-zinc superoxide dismutase. Arterioscler Thromb Vasc Biol. 1997;17:1734–40. doi: 10.1161/01.atv.17.9.1734. [DOI] [PubMed] [Google Scholar]
- 87.Yang H, Roberts LJ, Shi MJ, et al. Retardation of atherosclerosis by overexpression of catalase or both Cu/Zn-superoxide dismutase and catalase in mice lacking apolipoprotein E. Circ Res. 2004;95:1075–81. doi: 10.1161/01.RES.0000149564.49410.0d. [DOI] [PubMed] [Google Scholar]
- 88.Fukai T, Ushio-Fukai M. Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxid Redox Signal. 2011;15:1583–606. doi: 10.1089/ars.2011.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sentman ML, Brännstrom T, Westerlund S, et al. Extracellular superoxide dismutase deficiency and atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 2001;21:1477–82. doi: 10.1161/hq0901.094248. [DOI] [PubMed] [Google Scholar]
- 90.Zhang Y, Griendling KK, Dikalova A, Owens GK, Taylor WR. Vascular hypertrophy in angiotensin II-induced hypertension is mediated by vascular smooth muscle cell-derived H2O2. Hypertension. 2005;46:732–7. doi: 10.1161/01.HYP.0000182660.74266.6d. [DOI] [PubMed] [Google Scholar]
- 91.Yang H, Zhou L, Wang Z, et al. Overexpression of antioxidant enzymes in ApoE-deficient mice suppresses benzo(a)pyrene-accelerated atherosclerosis. Atherosclerosis. 2009;207:51–8. doi: 10.1016/j.atherosclerosis.2009.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Witte I, Foerstermann U, Devarajan A, Reddy ST, Horke S. Protectors or traitors: the roles of PON2 and PON3 in atherosclerosis and cancer. J Lipids. 2012;2012:342806. doi: 10.1155/2012/342806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Huang Y, Wu Z, Riwanto M, et al. Myeloperoxidase, paraoxonase-1, and HDL form a functional ternary complex. J Clin Invest. 2013;123:3815–28. doi: 10.1172/JCI67478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Horke S, Witte I, Wilgenbus P, Krüger M, Strand D, Förstermann U. Paraoxonase-2 reduces oxidative stress in vascular cells and decreases endoplasmic reticulum stress-induced caspase activation. Circulation. 2007;115:2055–64. doi: 10.1161/CIRCULATIONAHA.106.681700. [DOI] [PubMed] [Google Scholar]
- 95.Ng CJ, Bourquard N, Grijalva V, et al. Paraoxonase-2 deficiency aggravates atherosclerosis in mice despite lower apolipoprotein-B-containing lipoproteins: anti-atherogenic role for paraoxonase-2. J Biol Chem. 2006;281:29491–500. doi: 10.1074/jbc.M605379200. [DOI] [PubMed] [Google Scholar]
- 96.Altenhöfer S, Witte I, Teiber JF, et al. One enzyme, two functions: PON2 prevents mitochondrial superoxide formation and apoptosis independent from its lactonase activity. J Biol Chem. 2010;285:24398–403. doi: 10.1074/jbc.M110.118604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Devarajan A, Bourquard N, Hama S, et al. Paraoxonase 2 deficiency alters mitochondrial function and exacerbates the development of atherosclerosis. Antioxid Redox Signal. 2011;14:341–51. doi: 10.1089/ars.2010.3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fortunato G, Di Taranto MD, Bracale UM, et al. Decreased paraoxonase-2 expression in human carotids during the progression of atherosclerosis. Arterioscler Thromb Vasc Biol. 2008;28:594–600. doi: 10.1161/ATVBAHA.107.154658. [DOI] [PubMed] [Google Scholar]
- 99.Schweikert EM, Devarajan A, Witte I, et al. PON3 is upregulated in cancer tissues and protects against mitochondrial superoxide-mediated cell death. Cell Death Differ. 2012;19:1549–60. doi: 10.1038/cdd.2012.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Shih DM, Xia YR, Wang XP, et al. Decreased obesity and atherosclerosis in human paraoxonase 3 transgenic mice. Circ Res. 2007;100:1200–7. doi: 10.1161/01.RES.0000264499.48737.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Marsillach J, Camps J, Beltran-Debón R, et al. Immunohistochemical analysis of paraoxonases-1 and 3 in human atheromatous plaques. Eur J Clin Invest. 2011;41:308–14. doi: 10.1111/j.1365-2362.2010.02411.x. [DOI] [PubMed] [Google Scholar]
- 102.Lewis P, Stefanovic N, Pete J, et al. Lack of the antioxidant enzyme glutathione peroxidase-1 accelerates atherosclerosis in diabetic apolipoprotein E-deficient mice. Circulation. 2007;115:2178–87. doi: 10.1161/CIRCULATIONAHA.106.664250. [DOI] [PubMed] [Google Scholar]
- 103.Torzewski M, Ochsenhirt V, Kleschyov AL, et al. Deficiency of glutathione peroxidase-1 accelerates the progression of atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2007;27:850–7. doi: 10.1161/01.ATV.0000258809.47285.07. [DOI] [PubMed] [Google Scholar]
- 104.Yoshida T, Maulik N, Engelman RM, et al. Glutathione peroxidase knockout mice are susceptible to myocardial ischemia reperfusion injury. Circulation. 1997;96 II-216-20. [PubMed] [Google Scholar]
- 105.Blankenberg S, Rupprecht HJ, Bickel C, et al. AtheroGene Investigators Glutathione peroxidase 1 activity and cardiovascular events in patients with coronary artery disease. N Engl J Med. 2003;349:1605–13. doi: 10.1056/NEJMoa030535. [DOI] [PubMed] [Google Scholar]
- 106.Guo Z, Ran Q, Roberts LJ, II, et al. Suppression of atherogenesis by overexpression of glutathione peroxidase-4 in apolipoprotein E-deficient mice. Free Radic Biol Med. 2008;44:343–52. doi: 10.1016/j.freeradbiomed.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Stocker R, Perrella MA. Heme oxygenase-1: a novel drug target for atherosclerotic diseases? Circulation. 2006;114:2178–89. doi: 10.1161/CIRCULATIONAHA.105.598698. [DOI] [PubMed] [Google Scholar]
- 108.Jiang F, Roberts SJ, Datla, Dusting GJ. NO modulates NADPH oxidase function via heme oxygenase-1 in human endothelial cells. Hypertension. 2006;48:950–7. doi: 10.1161/01.HYP.0000242336.58387.1f. [DOI] [PubMed] [Google Scholar]
- 109.Taille C, El-Benna J, Lanone S, et al. Induction of heme oxygenase-1 inhibits NAD(P)H oxidase activity by down-regulating cytochrome b558 expression via the reduction of heme availability. J Biol Chem. 2004;279:28681–8. doi: 10.1074/jbc.M310661200. [DOI] [PubMed] [Google Scholar]
- 110.Hoekstra KA, Godin DV, Cheng KM. Protective role of heme oxygenase in the blood vessel wall during atherogenesis. Biochem Cell Biol. 2004;82:351–9. doi: 10.1139/o04-006. [DOI] [PubMed] [Google Scholar]
- 111.Hilgers RH, Kundumani-Sridharan V, Subramani J, et al. Thioredoxin reverses age-related hypertension by chronically improving vascular redox and restoring eNOS function. Sci Transl Med. 2017;9:eaaf6094. doi: 10.1126/scitranslmed.aaf6094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.May JM. How does ascorbic acid prevent endothelial dysfunction? Free Radic Biol Med. 2000;28:1421–9. doi: 10.1016/s0891-5849(00)00269-0. [DOI] [PubMed] [Google Scholar]
- 113.Heller R, Unbehaun A, Schellenberg B, Mayer B, Werner-Felmayer G, Werner ER. L-Ascorbic acid potentiates endothelial nitric oxide synthesis via a chemical stabilization of tetrahydrobiopterin. J Biol Chem. 2001;276:40–7. doi: 10.1074/jbc.M004392200. [DOI] [PubMed] [Google Scholar]
- 114.Wallerath T, Deckert G, Ternes T, et al. Resveratrol, a polyphenolic phytoalexin present in red wine, enhances expression and activity of endothelial nitric oxide synthase. Circulation. 2002;106:1652–8. doi: 10.1161/01.cir.0000029925.18593.5c. [DOI] [PubMed] [Google Scholar]
- 115.Wallerath T, Li H, Gödtel-Ambrust U, Schwarz PM, Förstermann U. A blend of polyphenolic compounds explains the stimulatory effect of red wine on human endothelial NO synthase. Nitric Oxide. 2005;12:97–104. doi: 10.1016/j.niox.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 116.Evrard SM, Lecce L, Michelis KC, et al. Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nat Commun. 2016;7:11853. doi: 10.1038/ncomms11853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kunsch C, Medford RM. Oxidative stress as a regulator of gene expression in the vasculature. Circ Res. 1999;85:753–66. doi: 10.1161/01.res.85.8.753. [DOI] [PubMed] [Google Scholar]
- 118.Stocker R, Keaney JF., Jr New insights on oxidative stress in the artery wall. J Thromb Haemost. 2005;3:1825–34. doi: 10.1111/j.1538-7836.2005.01370.x. [DOI] [PubMed] [Google Scholar]
- 119.Kovacic JC, Mercader N, Torres M, Boehm M, Fuster V. Epithelial-to-mesenchymal and endothelial-to-mesenchymal transition: from cardiovascular development to disease. Circulation. 2012;125:1795–808. doi: 10.1161/CIRCULATIONAHA.111.040352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Nickenig G, Harrison DG. The AT1-type angiotensin receptor in oxidative stress and atherogenesis: part I: oxidative stress and atherogenesis. Circulation. 2002;105:393–6. doi: 10.1161/hc0302.102618. [DOI] [PubMed] [Google Scholar]
- 121.Zalba G, San José G, Moreno MU, et al. Oxidative stress in arterial hypertension: role of NAD(P)H oxidase. Hypertension. 2001;38:1395–9. doi: 10.1161/hy1201.099611. [DOI] [PubMed] [Google Scholar]
- 122.Laufs U, Kilter H, Konkol C, Wassmann S, Böhm M, Nickenig G. Impact of HMG CoA reductase inhibition on small GTPases in the heart. Cardiovasc Res. 2002;53:911–20. doi: 10.1016/s0008-6363(01)00540-5. [DOI] [PubMed] [Google Scholar]
- 123.Touyz RM, Yao G, Schiffrin EL. c-Src induces phosphorylation and translocation of p47phox: role in superoxide generation by angiotensin II in human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2003;23:981–7. doi: 10.1161/01.ATV.0000069236.27911.68. [DOI] [PubMed] [Google Scholar]
- 124.Grote K, Flach I, Luchtefeld M, et al. Mechanical stretch enhances mRNA expression and proenzyme release of matrix metalloproteinase-2 (MMP-2) via NAD(P)H oxidase-derived reactive oxygen species. Circ Res. 2003;92:e80–6. doi: 10.1161/01.RES.0000077044.60138.7C. [DOI] [PubMed] [Google Scholar]
- 125.Spescha RD, Glanzmann M, Simic B, et al. Adaptor protein p66Shc mediates hypertension-associated, cyclic stretch-dependent, endothelial damage. Hypertension. 2014;64:347–53. doi: 10.1161/HYPERTENSIONAHA.113.02129. [DOI] [PubMed] [Google Scholar]
- 126.Li H, Witte K, August M, et al. Reversal of endothelial nitric oxide synthase uncoupling and up-regulation of endothelial nitric oxide synthase expression lowers blood pressure in hypertensive rats. J Am Coll Cardiol. 2006;47:2536–44. doi: 10.1016/j.jacc.2006.01.071. [DOI] [PubMed] [Google Scholar]
- 127.Camici GG, Schiavoni M, Francia P, et al. Genetic deletion of p66Shc adaptor protein prevents hyperglycemia-induced endothelial dysfunction and oxidative stress. Proc Natl Acad Sci U S A. 2007;104:5217–22. doi: 10.1073/pnas.0609656104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107:1058–70. doi: 10.1161/CIRCRESAHA.110.223545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Funk SD, Yurdagul A, Jr, Orr AW. Hyperglycemia and endothelial dysfunction in atherosclerosis: lessons from type 1 diabetes. Int J Vasc Med. 2012;2012:569654. doi: 10.1155/2012/569654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Hink U, Li H, Mollnau H, et al. Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ Res. 2001;88:E14–22. doi: 10.1161/01.res.88.2.e14. [DOI] [PubMed] [Google Scholar]
- 131.Chalupsky K, Cai H. Endothelial dihydrofolate reductase: critical for nitric oxide bioavailability and role in angiotensin II uncoupling of endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 2005;102:9056–61. doi: 10.1073/pnas.0409594102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Pritchard KA, Jr, Groszek L, Smalley DM, et al. Native low-density lipoprotein increases endothelial cell nitric oxide synthase generation of superoxide anion. Circ Res. 1995;77:510–8. doi: 10.1161/01.res.77.3.510. [DOI] [PubMed] [Google Scholar]
- 133.Stepp DW, Ou J, Ackerman AW, Welak S, Klick D, Pritchard KA., Jr Native LDL and minimally oxidized LDL differentially regulate superoxide anion in vascular endothelium in situ. Am J Physiol Heart Circ Physiol. 2002;283:H750–9. doi: 10.1152/ajpheart.00029.2002. [DOI] [PubMed] [Google Scholar]
- 134.Stroes E, Kastelein J, Cosentino F, et al. Tetrahydrobiopterin restores endothelial function in hypercholesterolemia. J Clin Invest. 1997;99:41–6. doi: 10.1172/JCI119131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Xia N, Daiber A, Habermeier A, et al. Resveratrol reverses endothelial nitric-oxide synthase uncoupling in apolipoprotein E knockout mice. J Pharmacol Exp Ther. 2010;335:149–54. doi: 10.1124/jpet.110.168724. [DOI] [PubMed] [Google Scholar]
- 136.Ylä-Herttuala S, Rosenfeld ME, Parthasarathy S, et al. Gene expression in macrophage-rich human atherosclerotic lesions. 15-Lipoxygenase and acetyl low density lipoprotein receptor messenger RNA colocalize with oxidation specific lipid-protein adducts. J Clin Invest. 1991;87:1146–52. doi: 10.1172/JCI115111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Nickenig G, Bäumer AT, Temur Y, Kebben D, Jockenhövel F, Böhm M. Statin-sensitive dysregulated AT1 receptor function and density in hypercholesterolemic men. Circulation. 1999;100:2131–4. doi: 10.1161/01.cir.100.21.2131. [DOI] [PubMed] [Google Scholar]
- 138.Camici GG, Savarese G, Akhmedov A, Lüscher TF. Molecular mechanism of endothelial and vascular aging: implications for cardiovascular disease. Eur Heart J. 2015;36:3392–403. doi: 10.1093/eurheartj/ehv587. [DOI] [PubMed] [Google Scholar]
- 139.Donato AJ, Gano LB, Eskurza I, et al. Vascular endothelial dysfunction with aging: endothelin-1 and endothelial nitric oxide synthase. Am J Physiol Heart Circ Physiol. 2009;297:H425–32. doi: 10.1152/ajpheart.00689.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lüscher TF, Yang ZH, Diederich D, Bühler FR. Endothelium-derived vasoactive substances: potential role in hypertension, atherosclerosis, and vascular occlusion. J Cardiovasc Pharmacol. 1989;14(Suppl 6):S63–9. [PubMed] [Google Scholar]
- 141.van der Loo B, Labugger R, Skepper JN, et al. Enhanced peroxynitrite formation is associated with vascular aging. J Exp Med. 2000;192:1731–44. doi: 10.1084/jem.192.12.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yang YM, Huang A, Kaley G, Sun D. eNOS uncoupling and endothelial dysfunction in aged vessels. Am J Physiol Heart Circ Physiol. 2009;297:H1829–36. doi: 10.1152/ajpheart.00230.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Dikalov SI, Nazarewicz RR. Angiotensin II-induced production of mitochondrial reactive oxygen species: potential mechanisms and relevance for cardiovascular disease. Antioxid Redox Signal. 2013;19:1085–94. doi: 10.1089/ars.2012.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Jaimes EA, DeMaster EG, Tian RX, Raij L. Stable compounds of cigarette smoke induce endothelial superoxide anion production via NADPH oxidase activation. Arterioscler Thromb Vasc Biol. 2004;24:1031–6. doi: 10.1161/01.ATV.0000127083.88549.58. [DOI] [PubMed] [Google Scholar]
- 145.Csiszar A, Podlutsky A, Wolin MS, Losonczy G, Pacher P, Ungvari Z. Oxidative stress and accelerated vascular aging: implications for cigarette smoking. Front Biosci (Landmark Ed) 2009;14:3128–44. doi: 10.2741/3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Steffen Y, Vuillaume G, Stolle K, et al. Cigarette smoke and LDL cooperate in reducing nitric oxide bioavailability in endothelial cells via effects on both eNOS and NADPH oxidase. Nitric Oxide. 2012;27:176–84. doi: 10.1016/j.niox.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 147.Heitzer T, Brockhoff C, Mayer B, et al. Tetrahydrobiopterin improves endothelium-dependent vasodilation in chronic smokers : evidence for a dysfunctional nitric oxide synthase. Circ Res. 2000;86:E36–41. doi: 10.1161/01.res.86.2.e36. [DOI] [PubMed] [Google Scholar]
- 148.Halliwell B. The antioxidant paradox: less paradoxical now? Br J Clin Pharmacol. 2013;75:637–44. doi: 10.1111/j.1365-2125.2012.04272.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: systematic review and meta-analysis. JAMA. 2007;297:842–57. doi: 10.1001/jama.297.8.842. [DOI] [PubMed] [Google Scholar]
- 150.Biesalski HK, Grune T, Tinz J, Zöllner I, Blumberg JB. Reexamination of a meta-analysis of the effect of antioxidant supplementation on mortality and health in randomized trials. Nutrients. 2010;2:929–49. doi: 10.3390/nu2090929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Steinberg D, Witztum JL. Is the oxidative modification hypothesis relevant to human atherosclerosis? Do the antioxidant trials conducted to date refute the hypothesis? Circulation. 2002;105:2107–11. doi: 10.1161/01.cir.0000014762.06201.06. [DOI] [PubMed] [Google Scholar]
- 152.Touyz RM. Reactive oxygen species, vascular oxidative stress, and redox signaling in hypertension: what is the clinical significance? Hypertension. 2004;44:248–52. doi: 10.1161/01.HYP.0000138070.47616.9d. [DOI] [PubMed] [Google Scholar]
- 153.Witting PK, Upston JM, Stocker R. Role of α-tocopheroxyl radical in the initiation of lipid peroxidation in human low-density lipoprotein exposed to horse radish peroxidase. Biochemistry. 1997;36:1251–8. doi: 10.1021/bi962493j. [DOI] [PubMed] [Google Scholar]
- 154.Witting PK, Willhite CA, Davies MJ, Stocker R. Lipid oxidation in human low-density lipoprotein induced by metmyoglobin/H2O2: involvement of α-tocopheroxyl and phosphatidylcholine alkoxyl radicals. Chem Res Toxicol. 1999;12:1173–81. doi: 10.1021/tx9900472. [DOI] [PubMed] [Google Scholar]
- 155.Chen H, Song YS, Chan PH. Inhibition of NADPH oxidase is neuroprotective after ischemia-reperfusion. J Cereb Blood Flow Metab. 2009;29:1262–72. doi: 10.1038/jcbfm.2009.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Jackman KA, Miller AA, De Silva TM, Crack PJ, Drummond GR, Sobey CG. Reduction of cerebral infarct volume by apocynin requires pretreatment and is absent in Nox2-deficient mice. Br J Pharmacol. 2009;156:680–8. doi: 10.1111/j.1476-5381.2008.00073.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Jung O, Schreiber JG, Geiger H, Pedrazzini T, Busse R, Brandes RP. gp91phox-containing NADPH oxidase mediates endothelial dysfunction in renovascular hypertension. Circulation. 2004;109:1795–801. doi: 10.1161/01.CIR.0000124223.00113.A4. [DOI] [PubMed] [Google Scholar]
- 158.Kahles T, Luedike P, Endres M, et al. NADPH oxidase plays a central role in blood-brain barrier damage in experimental stroke. Stroke. 2007;38:3000–6. doi: 10.1161/STROKEAHA.107.489765. [DOI] [PubMed] [Google Scholar]
- 159.Selemidis S, Sobey CG, Wingler K, Schmidt HH, Drummond GR. NADPH oxidases in the vasculature: molecular features, roles in disease and pharmacological inhibition. Pharmacol Ther. 2008;120:254–91. doi: 10.1016/j.pharmthera.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 160.Smith RA, Murphy MP. Animal and human studies with the mitochondria-targeted antioxidant MitoQ. Ann N Y Acad Sci. 2010;1201:96–103. doi: 10.1111/j.1749-6632.2010.05627.x. [DOI] [PubMed] [Google Scholar]
- 161.Alp NJ, McAteer MA, Khoo J, Choudhury RP, Channon KM. Increased endothelial tetrahydrobiopterin synthesis by targeted transgenic GTP-cyclohydrolase I overexpression reduces endothelial dysfunction and atherosclerosis in ApoE-knockout mice. Arterioscler Thromb Vasc Biol. 2004;24:445–50. doi: 10.1161/01.ATV.0000115637.48689.77. [DOI] [PubMed] [Google Scholar]
- 162.Brasen JH, Leppänen O, Inkala M, et al. Extracellular superoxide dismutase accelerates endothelial recovery and inhibits in-stent restenosis in stented atherosclerotic Watanabe heritable hyperlipidemic rabbit aorta. J Am Coll Cardiol. 2007;50:2249–53. doi: 10.1016/j.jacc.2007.08.038. [DOI] [PubMed] [Google Scholar]
- 163.Pagnin E, Fadini G, de Toni R, Tiengo A, Calò L, Avogaro A. Diabetes induces p66shc gene expression in human peripheral blood mononuclear cells: relationship to oxidative stress. J Clin Endocrinol Metab. 2005;90:1130–6. doi: 10.1210/jc.2004-1283. [DOI] [PubMed] [Google Scholar]
- 164.Venkatasubramanian S, Noh RM, Daga S, et al. Cardiovascular effects of a novel SIRT1 activator, SRT2104, in otherwise healthy cigarette smokers. J Am Heart Assoc. 2013;2:e000042. doi: 10.1161/JAHA.113.000042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001;21:1249–59. doi: 10.1128/MCB.21.4.1249-1259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Thorp E, Li G, Seimon TA, Kuriakose G, Ron D, Tabas I. Reduced apoptosis and plaque necrosis in advanced atherosclerotic lesions of Apoe−/− and Ldlr−/− mice lacking CHOP. Cell Metab. 2009;9:474–81. doi: 10.1016/j.cmet.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Tsukano H, Gotoh T, Endo M, et al. The endoplasmic reticulum stress-C/EBP homologous protein pathway-mediated apoptosis in macrophages contributes to the instability of atherosclerotic plaques. Arterioscler Thromb Vasc Biol. 2010;30:1925–32. doi: 10.1161/ATVBAHA.110.206094. [DOI] [PubMed] [Google Scholar]