

Abstract
Immunotherapy has been widely explored for applications to both augment and suppress intrinsic host immunity. Clinical achievements have seen a number of immunotherapeutic drugs displace established strategies like chemotherapy in treating immune-associated diseases. However, single drug approaches modulating an individual arm of the immune system are often incompletely effective. Imperfect mechanistic understanding and heterogeneity within disease pathology have seen monotherapies inadequately equipped to mediate complete disease remission. Recent success in applications of combinatorial immunotherapy has suggested that targeting multiple biological pathways simultaneously may be critical in treating complex immune pathologies. Drug delivery approaches through engineered biomaterials offer the potential to augment desired immune responses while mitigating toxic side-effects by localizing immunotherapy. This review discusses recent advances in immunotherapy and highlights newly explored combinatorial drug delivery approaches. Furthermore, prospective future directions for immunomodulatory drug delivery to exploit are provided.
Keywords: Immunotherapy, Combination therapy, Drug delivery, Biomaterials, Immunomodulation, Autoimmunity Cancer
1. Introduction
The complexity of the immune system affords ample opportunities for pathologies to develop, both from aberrant immune activation and misguided immunosuppression. Recent advances delineating immunobiological mechanisms in infectious disease, cancer, and auto-immunity have helped inform novel therapeutic approaches. Immuno-therapy, an increasingly popular methodology, attempts to modulate specific arms of the immune system by interfacing with host biology to augment or suppress natural immune responses. Since Edward Jenner first developed a vaccine for smallpox in the late 18th century, immunotherapy has played a prominent role in improving human health and quality of life. Similarly, allergy immunotherapy has been used clinically for more than a century, yielding positive outcomes for patients with asthma, dietary, and seasonal allergies [1]. Today, immunotherapies are supplanting several well-established clinical treatment paradigms. For example, cancer immunotherapy has emerged in recent decades as an attractive alternative to chemotherapy, as administration of broadly cytotoxic drugs has dangerous and potentially fatal side effects. By contrast, controlled modulation of innate and adaptive immunity can generate robust anti-tumor responses while minimizing systemic toxicity. Recently, a number of innovative immunotherapeutic strategies have garnered attention. Enthusiasm for immunotherapies such as Sipuleucel-T dendritic cell therapy, chimeric antigen receptor (CAR) T cells, and monoclonal antibodies is buoyed by success in clinical trials. Sipuleucel-T immunotherapy, wherein isolated autologous dendritic cells are exogenously activated, loaded with tumor-specific antigen and are re-administered, was the first FDA-approved therapeutic vaccine for cancer of any kind and showed improved survival in men with metastatic prostate cancer [2]. Similarly, adoptive transfer of CD19+ B cell targeting CAR T cells demonstrated sustained remission of acute lymphoblastic leukemia in children and adults [3]. Overall, immune modulating interventions to harness specific features of the immune system are becoming widespread and multipurpose.
The abundance of immunotherapy strategies being explored is in large part due to the expanding number of therapeutic tools. The increased mechanistic understanding and availability of immunomodulatory drugs including recombinant cytokines (e.g., IL-2, TGF-β, IFNγ), small molecule adjuvants (e.g., CpG, MPLA, Pam3CSK4), and monoclonal antibodies (e.g., anti-PD1, anti-CTLA-4, anti-IL-10) have facilitated development of immunotherapy approaches. In cancer immunotherapy alone, there are over 50 immunotherapy agents currently being used in the clinic or in clinical trials [4]. The targets of these single drug approaches are wide ranging, but similar in that they engage an isolated immune pathway. In one clinical trial, for example, immunotherapy with low-dose administration of IL-2 resulted in a dose-dependent increase of FoxP3+ regulatory T cells (Tregs) in patients with type 1 diabetes, a cellular phenotype critically lacking in many autoimmune conditions [5]. On the other hand, recent work using monoclonal antibodies for checkpoint blockade therapy has potentially revolutionized cancer immunotherapy. Nivolumab, a monoclonal antibody that prevents T cell inhibition by impeding programmed death-1 (PD-1) signaling, dramatically increased survival in metastatic melanoma patients, outperforming a standard first-line chemotherapy regimen [6]. In other clinical trials, monoclonal antibody therapy with ipilimumab, a monoclonal antibody that inhibits cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), improved survival in patients with metastatic melanoma by over 10 months and ∼20% exhibited long term survival after five years [7,8]. While clinical achievements from single drug immunotherapies cannot be understated, such approaches are limited by a number of factors. In particular, incomplete understanding of disease pathology and associated immune pathways hinders identification and application of relevant monotherapies. Additionally, heterogeneity within disease pathogenesis and among patient populations can limit the efficacy of drugs that engage individual pathways.
Combinatorial strategies to modulate multiple immune axes in coordination are seen as an attractive strategy to overcome these barriers, the growth of which is well documented [4,9–11]. Combinatorial immunotherapy success is represented by recent clinical trials involving simultaneous administration of nivolumab and ipilimumab [12,13]. This groundbreaking work demonstrated the importance of engaging multiple immune pathways, as metastatic melanoma patients with programmed death ligand 1 (PD-L1) negative tumors displayed significantly reduced survival when administered either agent alone. Conversely, when PD-1 and CTLA-4 monoclonal antibodies were delivered in combination, PD-L1-negative tumor patients had improved survival by over 5 months. While the clinical success of combinatorial immunomodulation has fueled a dramatic increase in such approaches, concerns about toxicity associated with systemic administration have restricted their therapeutic potential. Poorly managed global immunostimulation when using activating adjuvants [14], CAR T cells [15], or monoclonal antibodies [16] can induce life-threatening cytokine storm or autoimmune disease. Similarly, chronic global immunosuppression, often employed for organ transplant and autoimmunity, leaves patients at risk for opportunistic infections and tumor development [17]. To address such problems associated with systemic delivery, controlled-release platforms are increasingly being explored for combination therapies.
Biomaterial-based drug delivery systems have been extensively investigated for immunotherapy applications. Controlled-release systems exhibit a number of advantageous chemical and physical properties that have been exploited to augment immunomodulation compared to soluble drug administration. One frequently employed characteristic, biodegradable materials can deliver sustained release of encapsulated drugs to prolong therapeutic levels, reducing high and/or frequent dose requirements. Similarly, drug degradation and clearance, two critical limitations associated with soluble drug delivery, can be minimized by biomaterial encapsulation. Drug delivery approaches can also enhance the mass and frequency of payload delivered to immune populations, which can facilitate and augment immunotherapy. For example, biomaterial vehicles can be modified to target drug delivery to key cells or tissues. Alternatively, implantable controlled-release platforms have been developed to localize drug delivery at the site of administration, adjacent to relevant immune-associated structures. Such biomaterial approaches are also particularly appealing in order to minimize undesirable systemic immunomodulation. While there is a vast array of bio-materials that have been developed for drug delivery, distinct platforms (e.g., hydrogels, micro- or nanoparticles, liposomes) each offer unique advantages for immunotherapy (Fig. 1). Particle-based systems have been extensively explored to deliver payloads to antigen-presenting cells [18]. Conversely, biomaterial scaffolds have been engineered to actively recruit immune populations in vivo [19]. The various tunable parameters of biomaterial platforms have made them a particularly useful tool for immunotherapy.
Fig. 1.
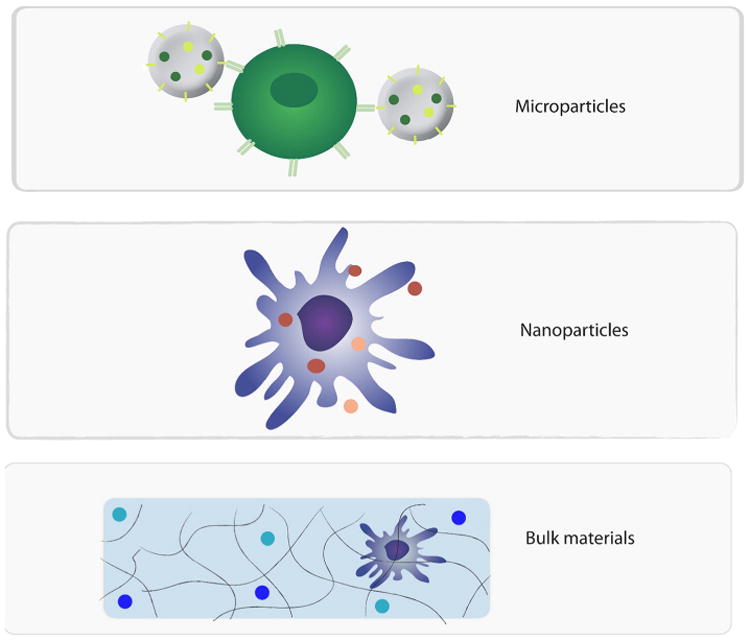
Various biomaterial platforms have been developed for combinatorial drug delivery to modulate immunity. A few of the most frequently applied systems are depicted here. (Top) Micro- and nanoparticle vehicles have been designed for specific targeting/retention to modulate subpopulations of immune cells (e.g., T cells, DCs). (Bottom) Alternatively, bulk materials have been explored to actively recruit immune populations in vivo.
This review highlights recent work on combinatorial drug delivery strategies for immunosuppression and immunostimulation. The primary focus will be on biomaterial platforms (Table 1), although core immunotherapy principles and background results will be discussed for context. Specifically, we emphasize recent experimental work in drug delivery approaches that rely on cellular targeting modalities and controlled-release strategies to recruit immune cells in vivo. While significant progress has been made toward integrating immunomodulatory therapies in the clinic, we also discuss the limitations in current approaches and suggest a number of future directions that show promise.
Table 1.
Biomaterial platforms explored in vivo for combination immunotherapy.
| Reference | Delivery system | Delivery target | Drug(s) delivered | Outcome |
|---|---|---|---|---|
| 46, 47 | PLGA/PROMAXX™ microparticles | DCs | Antisense oligonucleotides against CD40, CD80, and CD86 | Reversed new-onset diabetes in non-obese diabetic (NOD) mice |
| 50 | Polypropylene sulfide nanoparticles | Tumor-draining LNs/DCs | CpG and paclitaxel | Skewed CD4+ T cells to a Th1 phenotype and hindered melanoma growth |
| 54 | PLGA nanoparticles | DCs/B cells | MPLA, imiquimod, and antigen | Produced persistent (N1.5 yr) germinal center formation and plasma-cell responses |
| 62 | PLGA nanoparticles | DCs | Surface conjugated MPLA and encapsulated CpG and antigen | Robust antigen-specific T cell response was dependent on combination delivery |
| 68 | PLGA nanoparticles covalently coupled with α-CD40 | DCs | Surface conjugated α-CD40 and encapsulated Pam3CSK4, poly(I:C), and antigen | α-CD40 coupling improved selective DC uptake and prolonged survival in a melanoma model |
| 69 | PEGylated liposomes covalently coupled with CpG and α-CD40 | DCs | Surface conjugated α-CD40 and CpG | Surface conjugation improved nanoparticle retention at the local tumor site, minimized systemic inflammation, and mitigated melanoma tumor growth in mice |
| 80 | PLGA nanoparticles covalently coupled with α-CD4 | T cells | TGF-β1 and IL-2 | Increased the percentage of regulatory T cells |
| 93 | PEGylated liposomes surrounding PLA-PEG core | Tumor microenvironment/T cells | TGF-β inhibitor and IL-2 | Doubled the number of tumor-infiltrating NK cells and delayed tumor growth in a melanoma model |
| 94 | TLR9-ligand construct | Myeloid/B cells | Stat3 siRNA and CpG | Minimized tumor burden in melanoma and metastatic lung cancer models. Only the covalently linked construct mediated potent anti-tumor effects |
| 19 | PLGA scaffolds | DCs | GM-CSF, CpG, and antigen | Demonstrated a dose-dependent recruitment of DCs and diminished tumor burden in a melanoma model |
| 103 | Silica micro-rods | DCs | GM-CSF, CpG, and antigen | Generated robust humoral immune responses and ameliorated tumor burden |
| 104 | PLGA microparticles | DCs | GM-CSF, TGF-β1, vitamin D3, and antigen | Prevented type 1 diabetes onset in NOD mice |
| 105 | PuraMatrix™ peptide hydrogel containing PLGA microparticles | DCs | GM-CSF, CpG, and antigen | Prevented type 1 diabetes onset in NOD mice |
| 103, 104 | Dextran vinyl sulfone and tetra-functionalized PEG thiol hydrogel & PLGA microparticles | DCs | CCL20 in hydrogel and PLGA MPs encapsulating IL-10 siRNA, CpG and antigen | Induced a robust Th1 response. A later iteration with CpG improved survival in mice immunized with B-cell lymphoma |
| 114 | Alginate scaffold seeded with tumor-specific T cells and silica microparticles | Seeded T cells | GFOGER, α-CD3, α-CD28, α-CD137, and an IL-15 superagonist | Improved T cell migration, viability, and expansion, resulting in augmented tumor suppression |
| 120 | Lipoprotein nanodiscs | DCs | CpG, antigen, and soluble anti-PD-1 and anti-CTLA4 | Nanodiscs alone increased the frequency of antigen-specific T cells. In combination with checkpoint inhibitor therapy, eliminated tumor burden in colon carcinoma and melanoma mouse models |
2. Targeted drug delivery to immune cells
2.1. Targeting antigen-presenting cells
Drug delivery approaches using targeting modalities are seen as particularly appealing for immunotherapy. Targeted drug delivery to disease-relevant organs, tissues, or cells can attenuate the dangerous side effects associated with broad, systemic immunotherapy. Additionally, selective delivery to specific immune compartments can enhance downstream immune responses.
There have been broad efforts developing immunotherapeutics targeting antigen-presenting cells (APCs). Operating at the interface between innate and adaptive immunity, APCs are critical in mediating the balance between immune tolerance and activation. Dendritic cells (DCs) are the most efficient APC in the body [20]. As multifunctional regulators of immunity, various DC subsets exist with differential capacities for immunogenicity [21]. Upon exposure to inflammatory signals, such as engagement of toll-like receptors (TLRs) on DCs by pathogenic ligands (e.g., LPS, CpG), DCs become functionally mature. DC maturation results in phenotypic and functional changes that promote migration to lymphoid organs, secretion of inflammatory cytokines, and T cell activation. However, in a resting state, DCs process and present antigen in an immature fashion, maintaining homeostatic tolerance toward self-antigen [22]. In addition to immature DCs, tolerogenic DC phenotypes can be induced which can promote peripheral tolerance. Tolerogenic DCs employ a variety of modalities to preserve tolerance including induction of T cell anergy or deletion, release of anti-inflammatory cytokines (e.g., TGF-β, IL-10), and expansion of regulatory T cells [23]. Insight into TLRs, DC subpopulations, and the interface between innate and adaptive immunity have driven the continued development of tailored DC-based therapies [24–27]. The robust capacity of APCs to orchestrate immunity, particularly in antigen-specific directions, makes them a frequent target for immunotherapy.
Immunotherapy with exogenously manipulated DCs has demonstrated positive therapeutic outcomes in both preclinical and clinical investigations [28,29]. Since the first clinical trial using DC-based vaccination in 1996 [30], numerous studies have demonstrated induction of antigen-specific T and B cell responses against malignant cancers using DCs primed with tumor-associated antigens [31,32]. Not until 2010, however, was the first DC-based vaccine for cancer immunotherapy, Sipuleucel-T, approved by the FDA. Sipuleucel-T immunotherapy demonstrated success in patients with castration-resistant prostate cancer, improving mean survival time by ∼4 months and increasing tumor-specific antibody titers and antigen-specific T cell proliferation [2]. While the anti-tumor effects demonstrated were modest, as tumor burden was not significantly diminished, the first-of-its-kind regulatory approval generated enthusiasm for DC-based immunotherapy. Alternatively, “negative vaccination” using DCs to induce tolerance has been investigated in models of graft survival, multiple sclerosis, and type 1 diabetes [33-36]. One approach that employed DC adoptive transfer for type 1 diabetes was recently explored in a phase I clinical trial, attempting to modulate autologously isolated DCs toward an immunosuppressive state via a combination of antisense oligonucleotides against co-stimulatory molecules CD40, CD80, and CD86 before being re-administered [37]. While the study was not powered to determine therapeutic benefits, there was no measurable change in C-peptide levels or in regulatory T cell frequency. Notably, however, the approach demonstrated tolerable safety profiles. Similarly, combinatorial drug strategies to modulate DCs ex-vivo have also been an active area of investigation [38,39]. While DC-based immunotherapy has demonstrated clinical efficacy, administration of exogenously-treated DCs exhibits numerous barriers for widespread implementation including high cost, poor yield, and inefficient DC homing to regional lymph nodes [40,41].
A promising strategy to overcome these limitations is the targeted delivery of immunomodulatory factors to DCs in vivo. Nano- and micro-particles of various shapes, sizes, and compositions (e.g., lipids, polymers, metals) have been extensively studied as vehicles for in vivo drug delivery [42]. Particle-based approaches are valuable for drug delivery because they are often highly tunable, provide a sustained release depot for encapsulated agents, reduce systemic toxicity and dosing requirements, and simplify manufacturing, storage and shipping concerns [43,44]. Delivery to endosomes is facilitated when targeting APCs, as particles less than ∼5 μm in diameter are endocytosed by APCs without the need for specific receptor-targeting strategies [45]. This strategy of “passive targeting”, relying on the intrinsic phagocytic activity of APCs for particle-based drug delivery, has drawn significant interest for immunomodulation. Recent work by the Giannoukakis laboratory established microparticles containing antisense oligonucleotides against CD40, CD80, and CD86 co-stimulatory molecule transcripts can combinatorially downregulate DC maturation and ameliorate disease in a type 1 diabetes mouse model, laying the foundation for the DC-based clinical trial mentioned above. Upon subcutaneous injection, microparticles were shown to be taken up by DCs, traffic to draining lymph nodes, augment antigen-specific regulatory T cell proliferation, and reverse new-onset diabetes [46,47].
In contrast to micron-sized particles, which exclusively rely on uptake by APCs in order to traffic to secondary lymphoid organs, nanoparticles can be fabricated small enough to migrate to draining lymph nodes through lymphatic vessels [45]. Reddy et al. demonstrated this effect in a passive targeting strategy to activate lymph node-residing DCs using ultra-small polypropylene sulfide nanoparticles (Fig. 2) [48]. Initially, the authors established the importance of nanoparticle size in lymphatic drainage, as 100 nm nanoparticles were only 10% as efficient in trafficking to draining lymph nodes compared to 25 nm particles. When 25 nm particles were surface-coated with pluronic, previously shown to activate the complement cascade [49], intradermal injection resulted in upregulation of co-stimulatory molecules CD80, CD86, and CD40 on lymph node DCs similar to levels seen by LPS activation. Furthermore, nanoparticles conjugated with ovalbumin (OVA) antigen generated significant antigen-specific humoral and cellular responses. In a follow-up manuscript, Thomas and colleagues took advantage of this lymphatic transport phenomenon using a combinatorial drug cocktail to activate DCs in tumor-draining lymph nodes [50]. Building on their original work, they hypothesized that delivery of adjuvants to tumor-draining lymph nodes, which contain high concentrations of tumor-associated antigens, would induce activated DC phenotypes and generate a potent anti-tumor response. They confirmed their hypothesis in a murine model of melanoma. Intradermal injection of 30 nm nanoparticles encapsulating CpG oligodeoxynucleotide (CpG), a TLR9 agonist, and paclitaxel, a well-established anti-proliferative drug and TLR4 ligand [51], hindered tumor growth and skewed the CD4+ T cell distribution toward a Th1 phenotype. Results also demonstrated the requirement of drainage to tumor-draining lymph nodes for effective therapy, as contralateral injections did not generate as robust antigen-specific CD8+ T cell responses compared to ipsilateral injections upstream of tumor-draining lymph nodes. This work also highlights a recurring theme of controlled-release drug delivery platforms: encapsulation of the combinatorial drug cocktail produced a more efficacious immune response than soluble administration of adjuvants.
Fig. 2.
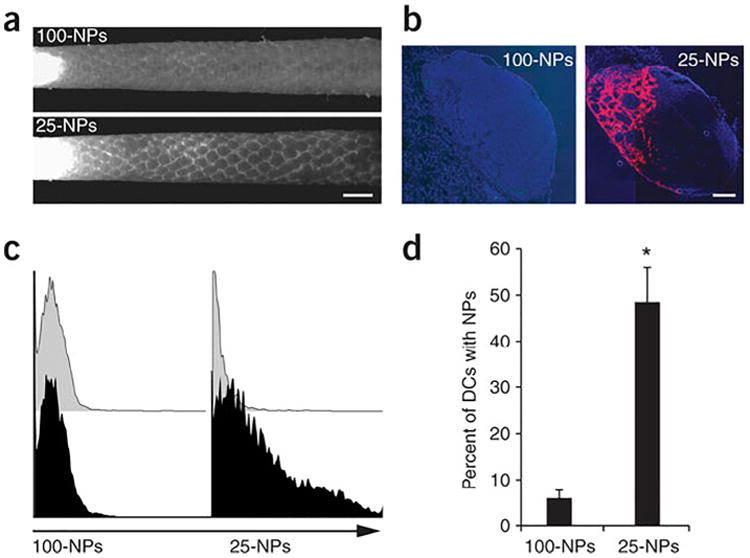
Ultra-small nanoparticles (25 nm) more efficiently drain to lymph nodes and are taken up by DCs than large nanoparticles (100 nm) upon intradermal injection. Fluorescently-labeled nanoparticles are seen draining through lymphatic capillaries (a; scale bar, 1 mm) and from isolated lymph nodes (b; scale bar, 200 μm; blue:cell nuclei, red:nanoparticles), with 25 nm particles trafficking more adeptly. Flow cytometry histograms (c) examined DC uptake of nanoparticles (black) or phosphate-buffered saline (grey) isolated from draining lymph nodes and was quantified (d). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.).
Reprinted by permission from Macmillan Publishers Ltd.: Nature Biotechnology [103], copyright 2014.
As important intermediaries between innate and adaptive immunity, DCs express high concentrations and heterogeneity of TLRs to direct diverse immune responses. Improved immunogenicity through combinatorial TLR agonist delivery is being explored. For example, mice immunized with bone marrow-derived DCs activated with TLR7 and TLR3 agonists improved cytotoxic lymphocyte responses in vivo compared to individual agonists [52]. Similarly, engagement of distinct stimulatory pathways through TLR4 and TLR8 on human DCs in vitro induced cytokine IL-12 and IL-23 levels 50–100 fold higher than those induced by single TLR agonists [53]. Biomaterial drug delivery systems have sought to take advantage of combinatorial TLR approaches. A breakthrough study in 2011 demonstrated the benefits of controlled-release vehicles to deliver two TLR agonists in vivo [54]. Mice subcutaneously injected with poly(lactide-co-glycolide) (PLGA) nanoparticles bearing monophosphoryl lipid A (MPLA; TLR4 agonist), imiquimod (TLR7 agonist), and OVA antigen generated persistent germinal center formation and plasma-cell responses, which were present in lymph nodes for >1.5 years following immunization (Fig. 3). Notably, they found immunization with the combinatorial formulation increased the antigen-specific humoral response compared to nanoparticles with individual TLR agonists. Results also demonstrated that TLR engagement on B cells, in addition to DCs, was critical in producing antibody responses. As cells differentially express TLRs [55], this work suggests that TLR-based vaccines can be optimally designed by rational inclusion of TLR agonists to generate tailored immune responses. Higher order TLR combinations are also being investigated. Recent work described high-throughput methods for combinatorial loading of factors in microparticles [56], as well as a high-throughput microarray to assess DC activation in response to combinations of three TLR-encapsulating microparticles [57,58]. The adjuvant-screening microarray demonstrated microparticle combinations delivering poly(inosinic:cytidylic acid) (poly(I:C)), MPLA, or CpG elicited differential expression of DC activation biomarkers CD86, MHC-II, CCR7, IL-12, and IL-10.
Fig. 3.
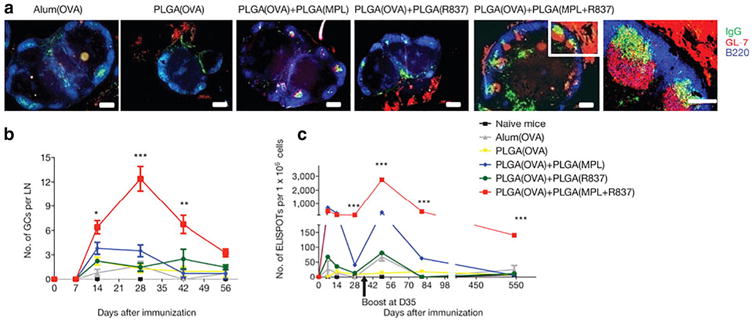
Immunization with combinatorial TLR agonists (MPLA:TLR4 and R837:TLR7) encapsulated in polymeric PLGA nanoparticles induces robust germinal center formation. (a) Draining lymph nodes were isolated four weeks post-immunization and stained for germinal center markers, with combinatorial agonist delivery producing the most persistent formation (scale bar, 200 μm). (b) The number of germinal centers in draining lymph nodes over time was stratified by treatment group. (c) Combinatorial TLR agonists promote longevity (∼1.5 years) of antibody-secreting cells compared to single TLR agonists as determined by an ELISPOT assay. Reprinted by permission from Macmillan Publishers Ltd.: Nature [54], copyright 2011.
Classically, antigen that is endocytosed by DCs is presented by MHC class II molecules and restricted to CD4+ T cell presentation [59]. Cross-presentation is the process by which antigen is released from the endosome into the cytosol and, through alternative pathways, presented on MHC class I molecules to CD8+ cytotoxic T cells [60]. Cross-presentation is vital to vaccines dependent on cytotoxic T cell responses. Controlled-release particles have been shown to be advantageous in generating CD8+ T cell responses. In addition to promoting prolonged antigen presentation, sustained release of antigen from biodegradable microparticles has demonstrated a roughly 1000-fold increase in cross-presentation compared to soluble protein [61]. In a similar vein, a recent nanoparticle-based approach pioneered by the Fahmy laboratory employed combinatorial TLR activation to generate robust CD8+ T cell immunity [62]. The platform utilized PLGA nanoparticles surface-conjugated with MPLA and encapsulated CpG and OVA. An important finding from their research demonstrated the significance of the mode of presentation when designing TLR-based vaccines. CpG efficacy as an immunostimulant, characterized by cytokine profiles, antibody titers, and antigen-specific T cell responses, was maximized when encapsulated in nanoparticles compared to when surface bound. As the receptor for CpG, TLR9, is localized endosomally [63], the authors suggest that physiologically relevant presentation of CpG is important in generating a robust immune response. This work additionally showed the importance of co-delivery, as single TLR agonists failed to replicate the potent CD8+ T cell response generated with the combinatorial formulation.
Strategies for APC targeting have also been developed that bind surface moieties to increase retention and uptake. This approach seeks to minimize off-target drug interactions and can produce more potent immune responses due to the efficiency with which drug cargo is more selectively taken up by APCs. Particulate formulations functionalized with surface moieties including α-CD40, α-DEC205, and α-CD11c antibodies have been explored for DC targeting [64]. As noted earlier, in contrast to microparticles, nano-sized particulates injected intradermally or subcutaneously can migrate and drain through lymphatic vessels. While advantageous in certain applications, nanoparticles are prone to non-specific cellular uptake and this competition for uptake is compounded by hepatic filtration and renal clearance [65]. Thus, targeting strategies to improve DC uptake of nanoparticles is particularly valuable. Recent work characterized the impact of targeting moieties based on particulate size, demonstrating that particles surface conjugated with the human C-type lectin receptor DC-SIGN dramatically improved DC uptake for polymeric nanoparticles, but only produced a modest increase in uptake for micron-sized particles [66]. The choice of targeting moiety is often dictated by the therapeutic intention. DC maturation is undesirable when delivering immunomodulatory drugs for tolerance-inducing purposes. Along these lines, microparticles modified with antibodies against CD11c or DEC-205, an integrin and C-type lectin respectively that are highly expressed on DCs, or functionalized with peptides P-D2 or RGD, targeting intercellular adhesion molecule-4 and surface integrins respectively, were found to efficiently target DCs, both in vitro and in vivo, in a non-activating manner [67].
Conversely, targeting ligands have been studied to improve uptake by DCs and simultaneously augment immunogenicity. Rosalia and colleagues demonstrated that covalent coupling of α-CD40 to PLGA nano-particles containing antigen, Pam3CSK4 and poly(I:C), a TLR2 and TLR3 agonist respectively, improved selective DC uptake and activated DCs in vivo [68]. In a tumor model of melanoma, subcutaneous administration of α-CD40-functionalized adjuvant-bearing nanoparticles improved the antigen-specific CD8+T cell response and prolonged survival compared to mice treated with nanoparticles conjugated with an isotype control. While the authors confirm that conjugation of α-CD40 to nanoparticles augmented nanoparticle uptake and delivery of TLR agonists to DCs, it was not investigated whether DC maturation was improved by engagement of CD40 inflammatory signaling pathways. In another approach, Kwong and colleagues used a combinatorial drug approach delivered intratumorally to generate an anti-tumor response [69]. Intratumoral injection of PEGylated liposomes surface conjugated with CpG and antibody against CD40 significantly mitigated tumor growth in a mouse model of melanoma. Importantly, retention at the local tumor site was confirmed. Surface conjugation of α-CD40 and CpG, the receptors for which are highly expressed on APCs, aided tumor retention. Liposome uptake by CD11c+ DCs and F4/80+ macrophages was observed at extended time periods, 24 and 48 h post injection. Similarly, their drug delivery approach curtailed systemic inflammation, as intratumoral administration of soluble immunomodulators increased serum levels of TNF-alpha and IL-6 and resulted in weight loss compared to the lipo-some formulation.
2.2. Targeting T cells
While APC-targeting therapies attempt to modulate immunity through critical cellular mediators, drug delivery strategies have also been developed to target other immune compartments more directly. Like DC-based therapies, various strategies to modulate T cell numbers and activity ex-vivo for adoptive transfer therapy have been investigated (reviewed in [70]). However, limitations with adoptive cellular transfer similarly plague T cell approaches. T cell targeting strategies have been conducted to work around these limitations. In one approach, researchers developed a targeting strategy to minimize T cell loss upon adoptive transfer. Three days after adoptive transfer of allogeneic T cells bearing the distinct congenic marker Thy1.1, intravenous injection of PEGylated liposomes conjugated with targeting ligands against Thy1.1 effectively marked>95% of transferred cells [71]. As the authors suggest, this targeting approach could be universally applied for adoptive cellular transfer through introduction of distinct surface markers by genetic engineering of cells pre-transfer. In another approach for targeted drug delivery to discrete T cell populations, Vincent et al. developed a MHC-I tetramer construct to deliver payloads to antigen-specific T cells [72]. Conjugation of MHC-I tetramers loaded with islet-specific peptide antigen to saporin, a ribosomal toxin, enriched ablation of antigen-specific, autoreactive CD8+ T cells in a mouse model of type 1 diabetes. Three intravenous injections of the construct reduced diabetes incidence in 8 week old non-obese diabetic mice by 30% after >50 weeks compared to controls. This approach, employing selective delivery to distinct T cell compartments, is desirable in order to minimize global immunotoxicity.
Regulatory T cells (Tregs) are critical for the maintenance of immune tolerance. Impaired Treg function and reduced numbers have been identified as important factors in cases of autoimmunity, organ transplant rejection, and allergy (reviewed in [73]). Immunotherapy strategies to augment Treg activation, proliferation, and function remain highly desirable, and have been explored through a number of diverse drug delivery platforms. In one example, microparticles delivering sustained release of the Treg chemokine CCL22 were shown to recruit Tregs locally and delay organ rejection in a murine allotransplant model [74]. In another unique strategy, the Hubbell laboratory developed an erythrocyte-targeting platform for induction of antigen-specific tolerance [75]. This system was composed of erythrocyte-binding constructs that were coupled with antigen, built around the premise that apoptotic cells, and erythrocytes are frequently recycled, are cleared through self-tolerance promoting pathways (reviewed in [76]). In a follow-up study by the Hubbell group, researchers explored the mechanisms by which the erythrocyte-targeting platform induced tolerance [77]. Results using antigen-specific T-cell-transgenic OT mouse models demonstrated that blockade of PD-1/PD-L1 signaling, but not CTLA-4/CD28, impaired antigen-specific T cell deletion, anergy, and expression of regulatory biomarkers. Additionally, the platform significantly increased the frequency of Tregs compared to administration of soluble antigen. Depletion of Tregs through α-CD25 demonstrated the critical role Tregs played in mediating CD4+ and CD8+ T cell suppression. The concept of antigen coupled to apoptotic cells recently demonstrated promise in a phase I clinical trial for multiple sclerosis [78]. In the trial, patients that received apoptotic leukocytes covalently coupled with various multiple sclerosis-specific peptide antigens demonstrated favorable safety profiles. Furthermore, patients receiving higher doses of apoptotic coupled cells displayed decreases in antigen-specific T cell responses.
As numerous factors have been identified as important modulators for Treg induction, a combinatorial approach engaging multiple pathways has been suggested as an ideal strategy for robust Treg responses. In one such approach, PLGA microparticles were loaded with three known Treg inducing agents: rapamycin, TGF-β1, and IL-2 [79]. Micro-particles were separately loaded with individual factors and demonstrated sustained release of encapsulated agents over 3–4 weeks. Notably, the system was most effective in skewing murine CD4+ T cells to express FoxP3 in vitro when all three microparticles were included. This work also showed the combinatorial microparticle formulation was also effective for induction of human Tregs in vitro, suggesting the translatability of their approach. Using an in vivo targeting scheme, McHugh and colleagues developed a similar approach by targeting delivery of TGF-β and IL-2 to T cells using PLGA nanoparticles surface-conjugated with α-CD4 antibodies [80]. Their results demonstrated that nanoparticles surface modified with α-CD4 dramatically increased binding of T cells by ∼18-fold in vitro. Furthermore, the increased bio-availability of these pro-tolerogenic factors was shown to skew the CD4+ T cell populations in vivo toward a regulatory phenotype, increasing the percentage of CD4+ T cells expressing FoxP3. The authors suggest this approach can also minimize the pleiotropic effects of these cytokines when delivered solubly, as TGF-β is well known to cause severe adverse responses when administered systemically [81]. However, as they note, a potential shared drawback of particle-based systems is non-specific uptake of nanoparticles by APCs through passive phagocytic activity. As such, it may be advantageous to minimize undesirable phagocytosis when designing vehicles for drug delivery to non-APC cellular compartments. Strategies such as functionalizing peptides from membrane proteins CD47 or CD200, reported ubiquitous self-identifiers, have been employed for drug delivery applications to mitigate macrophage-mediated phagocytic clearance [82,83]. Additionally, surface charge has been explored to modulate phagocytic uptake of particulate delivery systems, albeit with varying results. It has been reported that particles exhibiting high negative or positive charges are more readily taken up by DCs and macrophages compared those displaying a neutral surface charge [84,85]. However, concerns about toxicity may limit drug delivery applications for such approaches. In 2014, for example, Getts et al. described how anionic microparticles were taken up by monocytes via the scavenger receptor, MARCO, which resulted in sequestration of monocytes in the spleen and subsequent apoptotic clearance [86]. Similarly, it has been demonstrated that cationic particles can result in the generation of reactive oxygen species or mitochondrial injury pathways [87,88]. Lastly, particle shape has been described as an influential design consideration to avoid phagocytosis. Unlike spherical particles which are phagocytosed within minutes, rat alveolar macrophages had greatly diminished uptake capacity when incubated with ellipsoid polystyrene particles [89]. The initial point of contact between macrophages and particles was shown to be pivotal in internalization kinetics. Macrophages that attached to the spheroid end of an ellipsoid particle internalized it within minutes, however, those that encountered the flat portion of the ellipsoid did not internalize the particle for over 12 h. These approaches have been suggested to increase payload delivery to non-APC targets. Understanding the parameters that effect phagocytosis are critical in designing particle-based drug delivery systems and need to be further explored.
2.3. Alternative targeting strategies
Alternative immunotherapy targeting strategies have also been pursued. A recent innovative targeting approach modulates immunity by “hitchhiking” onto albumin protein draining to peripheral lymphoid organs [90]. Instead of targeting cell surface receptors, Liu et al. sought to localize an immunostimulatory vaccine intranodally by conjugating a lipophilic albumin-binding moiety to CpG. Subcutaneous administration of the albumin-binding construct resulted in an 8-fold increase in accumulation to peripheral lymph nodes compared to soluble CpG. Localization was further amplified as the albumin-binding vaccine showed sustained retention in lymph nodes after seven days. Notably, their approach dramatically increased anti-tumor effects in a murine model, while reducing systemic inflammation by attenuating serum IL-6 and IL-12 levels.
Localization of immunomodulatory agents to sites of concentrated immune activity has also been explored by more direct delivery approaches. In one strategy, researchers highlighted the efficacy of controlled-release particles to elicit robust immunity via intranodal delivery. By mapping the lymphatic system of mice using the tracer dye Evans blue, Jewell et al. intranodally injected PLGA microparticles containing poly(I:C) and OVA as model antigen to generate antigen-specific immunity [91]. Results demonstrated a single intranodal injection of the microparticle vaccine expanded the frequency of OVA-specific CD8+ T cells by over 8-fold compared to intramuscular immunization. Additionally, encapsulation of vaccine components in controlled-release microparticles augmented poly(I:C) intranodal persistence. Fluorescently-conjugated poly(I:C) levels released from microparticles were ∼2.5-fold higher measured after four days compared to an equivalent soluble intranodal dose. This work also displayed a core paradigm of controlled-release drug delivery approaches by reducing the adjuvant dose necessary to achieve similar immunogenicity. Microparticles loaded with a poly(I:C) and antigen generated a stronger OVA-specific CD8+ T cell response than soluble poly(I:C) given at a 10-fold greater dose. Analogous intranodal microparticle immunotherapy has also demonstrated antigen-specific suppression in a mouse model of multiple sclerosis by delivering myelin self-antigen and the immunoregulatory drug rapamycin [92].
Drug delivery approaches have also been explored to localize immuno-therapy agents to the tumor microenvironment. In a strategy to promote T cell proliferation and simultaneously attenuate the immunosuppressive tumor microenvironment, Park and colleagues designed PEGylated liposomes to deliver IL-2 and a TGF-β inhibitor (Fig. 4) [93]. The nanoscale vehicles were composed of a lipid bilayer surrounding a degradable, polymeric core encapsulating the hydrophilic cytokine, IL-2, and the small hydrophobic TGF- β inhibitor, SB505124 (SB). To deliver sustained release of both drugs upon hydrolysis of the internal polylactide-PEG core, SB was solubilized with β-cyclodextrans to enable encapsulation of the small hydrophobic molecule. Solubilization minimized the burst release observed compared to when SB was loaded in the absence of β-cyclodextrans and improved sustained release to over 7 days in vitro. The authors demonstrated the efficacy of this combination therapy as weekly intratumoral injections of nanoscale liposomes significantly improved survival and delayed tumor growth in a mouse model of melanoma, more so than liposomes bearing individual drugs. Strikingly, intratumoral numbers of activated CD8+ T cells and natural killer (NK) cells more than doubled upon liposome administration. Lastly, using NK-depleted mice, they showed liposomal-mediated tumor suppression required NK cells.
Fig. 4.
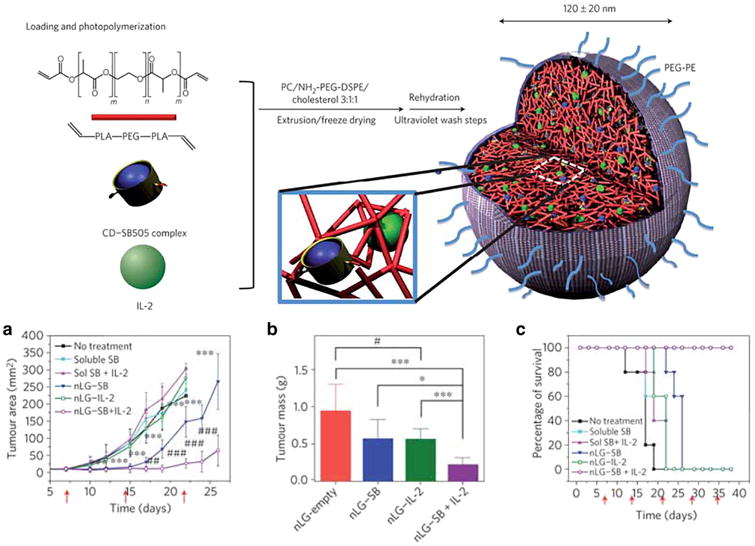
Synthesis scheme of PEGylated liposomes with polymeric cores to deliver the T cell proliferative factor, IL-2, and a TGF-β inhibitor, SB505124. Methacrylated cyclodextrans (CD) were used to solubilize the small, hydrophobic inhibitor (SB505). (Top) CD-SB505 complexes and IL-2 were loaded into a biodegradable polymer core (red) with a PEGylated liposomal exterior (grey). Photoinduced polymerization of the polymer core and the acrylated-CD induces nanoscale liposome gel (nLG) formation, capable of delivering sustained release of both drugs upon hydrolysis of the internal polymeric core. (a–c) B16 melanomas were tracked over time in response to various treatments, with combinatorial nLGs delaying tumor growth most effectively. Red arrows indicate intratumoral treatment administration. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.).
Reprinted by permission from Macmillan Publishers Ltd.: Nature Biotechnology [103], copyright 2014.
Targeting of multiple immune compartments for coordinated activation can also be desirable. In one example, researchers covalently linked CpG to small interfering RNA (siRNA) to preferentially target myeloid and B cells rich in TLR9 [94]. Their combinatorial approach sought to activate the TLR9 immunostimulatory pathway and silence Stat3, an important oncogenic transcription factor that mediates immunosuppressive activity. By covalently linking CpG to Stat3 siRNA, the researchers hypothesized that TLR9 ligation, located endosomally, would facilitate cytosolic escape of the construct and improve siRNA gene silencing. The efficacy of their engineered platform was demonstrated in murine tumor models of melanoma and metastatic lung cancer. Administration of non-conjugated CpG and Stat3 siRNA produced only minor anti-tumor effects compared to the covalently linked combination. Additionally, the frequency of FoxP3+ Tregs within the CD4+ T cell compartment intratumorally diminished from ∼60% to 25% post CpG-Stat3 siRNA peritumoral injections. The authors found that CpG-siRNA gene silencing was dependent on TLR9 expression, though it was unclear whether engagement of TLR9 promoted endosomal release or altered intracellular processing.
3. Controlled-release approaches to recruit immune cells
While targeting systems have been investigated for in vivo drug delivery, controlled-release materials to promote cellular recruitment and localized conditioning have also gained traction for immunotherapy. Traditionally, synthetic biomaterial scaffolds have primarily been investigated in applications for tissue engineering (reviewed in [95]). Scaffolds with high porosity are compatible carriers for cellular engraftment or recruitment and also allow encapsulation of biologics to direct cell function. Many of the same attributes that makes biomaterial structures desirable for tissue engineering can be applied for immunomodulation. Biomaterials can be engrafted with immune cells in adoptive cell therapies to improve leukocyte viability and subsequent immune responses [96]. Similarly, biological signals incorporated into biomaterial scaffolds have been broadly explored for immunomodulation (reviewed in [97]). This section of the review discusses the controlled-release material approaches that have been developed to recruit and modulate immune cells in situ.
Inflammatory responses following biomaterial implantation coincide with recruitment of innate immune cells to the site of injection. This is advantageous for combinatorial therapies that seek to deliver immunomodulatory drugs to APCs. However, the significance of DCs as the most effective APC can be diminished by its low frequency [21]. In response, some drug delivery approaches intend to maximize DC recruitment, differentiation, and proliferation. Granulocyte-macrophage colony-stimulating factor (GM-CSF) is a pleiotropic cytokine produced by a number of myeloid and supporting cells that features prominently in the pursuit of a number of DC-recruiting biologics. GM-CSF has demonstrated capacity for promoting DC proliferation, recruitment of DCs, antigen cross-presentation, and differentiation of monocytes into DCs [98–101]. Incorporation of GM-CSF has been explored both in emerging immunotherapy technologies and clinically. As has been discussed earlier, Sipuleucel-T immunotherapy utilizes GM-CSF to mature and expand DCs upon autologous isolation. GM-CSF has also been used as a supplementary therapeutic for cancer treatment to improve myeloid recovery following chemotherapy (reviewed in [102]).
Recently, a number of groups have combined GM-CSF with immunomodulatory agents in controlled-release materials to activate or suppress DCs. In 2009, researchers in the Mooney laboratory created a synthetic biomaterial scaffold to program DCs in a protective, anti-tumor fashion [19]. The authors fabricated biodegradable PLGA scaffolds that were made macroporous in order to create space for DC recruitment and antigen-presentation. Initial investigations demonstrated scaffolds loaded with GM-CSF were shown to mediate DC recruitment in a dose-dependent manner, improving DC recruitment by up to 12 times that of unloaded scaffolds. Upon the addition of the TLR9 agonist CpG, DC recruitment to the biomaterial was further improved as ∼1.5 million DCs were isolated 7 days following in vivo implantation. Consequently, the highlight of this work was the combinatorial effect CpG and GM-CSF had in mitigating tumor burden in the mouse model of melanoma. PLGA matrices loaded with tumor lysate as antigen, CpG, and GM-CSF increased antigen-specific CD8+ T cell recruitment, DC trafficking to lymph nodes, and resulted in a 90% survival rate by day 90 compared to 0% survival for unloaded scaffolds.
This combinatorial approach of using GM-CSF and CpG to modulate DC maturation has been explored in the same laboratory through other drug delivery vehicles as well. One strategy implemented an injectable material that agglomerated in vivo to form a macroporous scaffold, improving the safety profile by removing the need the surgical implantation [103]. Results from this work show mesoporous silica rods subcutaneously injected can deliver sustained release of GM-CSF, CpG, and OVA as a model antigen to generate a potent OVA-specific cellular and humoral response (Fig. 5). By altering the size of the microrods, they demonstrated that larger aspect-ratio material augmented cellular recruitment, which they attributed to the larger interparticle pores allowing robust cellular infiltration. Notably, they found that three repeated booster injections of the unencapsulated soluble factors were required to produce an OVA IgG2a antibody response comparable to that of the single injection of the microrod technology encapsulating the same factors. This combinatorial drug delivery strategy also significantly minimized tumor growth and survival using an EG.7-OVA tumor mouse model.
Fig. 5.
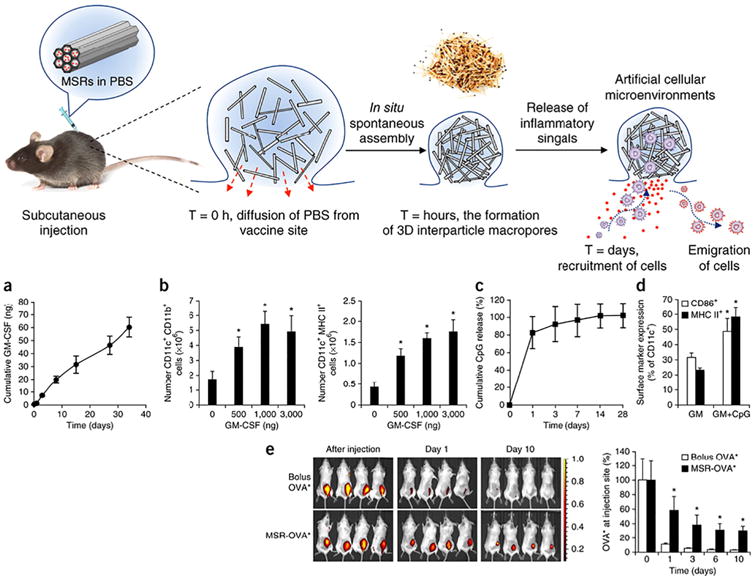
Mesoporous silica rods (MSRs) aggregate upon injection, forming macroporous spaces where host cells can be recruited and educated byencapsulated immunomodulatory drugs, GM-CSF and CpG. (a) In vitro cumulative release of GM-CSF from MSRs (b) CD11c+ CD11b+ (left) and CD11c+ MHC II+ (right) DCs were recruited to MSR scaffolds in a dose-dependent manner. (c) In vitro cumulative release of CpG from MSRs. (d) CpG inclusion in MSRs augmented DC activation, increasing surface expression of CD86 and MHC class II molecules. (e) Fluorescent in vivo imaging demonstrated MSRs loaded with OVA antigen deliver sustained release of protein over an extended period compared to bolus OVA injection.
Reprinted by permission from Macmillan Publishers Ltd.: Nature Biotechnology [103], copyright 2014.
Applications of GM-CSF as a DC chemokine has also been explored in tandem with other immunomodulatory drugs to moderate immune tolerance. Lewis et al. describe a dual-microparticle approach consisting of small (∼1 μm), phagocytosable microparticles and large (∼30 μm), non-phagocytosable microparticles designed to recruit DCs and induce tolerance in type 1 diabetes upon subcutaneous injection [104]. Briefly, large particles encapsulating GM-CSF and the tolerogenic drug TGF-β1 served to create a microparticle depot to recruit and condition DCs, respectively, via extracellular pathways. Simultaneous delivery of small, phagocytosable particles containing insulin B:9–23 peptide, a primary autoantigen for type 1 diabetes, and an additional immunomodulatory factor, vitamin D3, targeted the intracellular delivery of factors to recruited DCs. The premise behind the system was to promote presentation of autoantigen in a tolerogenic context to reeducate aberrant autoreactivity. In vitro characterization demonstrated the efficacy of this system, inducing tolerogenic DC phenotypes with downregulated expression of CD86 and MHC-II molecules, suppression of allogeneic T cell proliferation, and induction of FoxP3+ Tregs. Cumulatively, its effect in vivo resulted in a 40% prevention rate of type 1 diabetes in 4-week old non-obese diabetic mice.
In a similar approach to induce antigen-specific tolerance in a type 1 diabetes model, researchers fabricated a peptide hydrogel that delivered sustained release of GM-CSF to recruit DCs to an immunomodulatory microenvironment [105]. The hydrogel contained PLGA microparticles encapsulating denatured insulin to promote self-tolerance to recruited DCs and, notably, CpG. While CpG is more classically used as a vaccine adjuvant in an inflammatory capacity, the authors note it can elicit anti-inflammatory effects such as inducing IL-10 production [106]. This approach protected disease incidence in 40% of pre-diabetic mice. Additionally, results demonstrated that hydrogel implantation generated a resolvable granulomatous tissue at the site of administration that recruited an array of leukocyte populations and with some characteristics of a tertiary lymphoid organ.
In another DC recruiting strategy pioneered by the Roy group, the DC chemokine MIP3α, also known as CCL20, was loaded into a crosslinkable dextran vinyl sulfone and tetra-functional PEG thiol hydrogel along with PLGA microparticles encapsulating IL-10 targeting siRNA and plasmid DNA antigen [107]. The authors built off their original research which demonstrated microparticles bearing IL-10 siRNA boosted DC maturation, T cell proliferation, and skewed the in vivo T cell response to a Th1 pheno-type [108]. By incorporating a DC chemokine into biodegradable hydrogels, Singh et al. hypothesized that more DCs would encounter siRNA and plasmid antigen and the hydrogel might serve as an artificial DC priming center. The results demonstrated that a sustained chemokine gradient exuded in a controlled-release manner from the hydrogel was pivotal to its success, as the combinatorial system recruited ∼5-fold more DCs over an extended period of time compared to an equivalent bolus dose in vitro. The recruited DCs efficiently infiltrated hydrogels and displayed notably diminished gene expression of IL-10. In vivo results demonstrated that intramuscular injection of the combinatorial DC-recruiting platform induced T-cell class switching toward a Th1 response, inducing CD8+ CTL responses ∼3-fold more robust compared to administration of soluble components alone [109]. An iteration of this work was further explored, when in 2014 Pradhan et al. incorporated the adjuvant CpG into the IL-10 siRNA and plasmid DNA antigen bearing microparticles [110]. Hydrogels formulated with the full combinatorial payload (MIP3α and microparticles containing IL-10 siRNA, CpG, and plasmid DNA) significantly improved prophylactic survival in mice immunized with A20 B-lymphoma cells compared to controls without all four agents.
Incorporation of biological factors into scaffolds can also be accomplished by surface ligation. This may be advantageous when designing systems that benefit from localized effects and to limit systemic toxicity from soluble factors. This approach has been suggested as a potential solution for allograft tolerance, wherein researchers conjugated TGF-β1 to glass beads to generate Tregs [111]. Combinatorial approaches of ligation of biologics has also been explored. Hume and colleagues functionalized IL-10 and TGF-β1 to PEG hydrogels in an effort to minimize DC maturation in vitro [112]. Immature DCs seeded in hydrogels with immobilized pro-tolerogenic cytokines maintained their immature phenotypes following incubation with LPS, displaying reduced expression of MHC-II molecules and IL-12 production. In another example, researchers demonstrated that incorporation of cell-adhesion moieties improved subsequent interaction between recruited T cells and immunomodulatory microenvironment in a hydrogel platform [113].
Specifically, the Anseth laboratory developed a PEG hydrogel functionalized with intercellular adhesion molecule-1 and α-Fas antibodies, where Fas is a well-established death receptor, to modulate localized T cell apoptosis. This platform could be valuable as a complementary immunotherapy on the surface of cell encapsulation materials to reduce allograft rejection. Results show that hydrogels coated with both ligands improve T cell apoptosis by 50% in vitro compared to hydrogel only containing α-Fas antibodies.
Macroporous biomaterial scaffolds contain large surface area which encourages complex biomaterial design and conveys higher order spatiotemporal control of the surrounding microenvironment. In a particularly innovative combinatorial approach, Stephan et al. developed an alginate scaffold implant to improve adoptive transfer of tumor-specific T cells [114]. In the approach, tumor-specific T cells were loaded into the alginate scaffold which was conjugated with the collagen-mimetic peptide (GFOGER) to stimulate T cell migration (Fig. 6). Additionally, the scaffold contained silica microparticles with surface-bound α-CD3, α-CD28, and α-CD137 antibodies to stimulate lymphocyte proliferation and encapsulated an IL-15 superagonist as a T cell survival signal. This multipronged approach minimized numerous steps normally required for adoptive cellular transfer (e.g., T cell expansion and viability, intratumoral delivery), as biological factors included in the alginate scaffold account support these functions in vivo. Notably, upon peritumoral administration of the biopolymer in a murine tumor model, adoptively transferred tumor-specific T cells were isolated from tumor beds and found to suppress tumor growth. The inclusion microparticles bearing survival and proliferative factors was shown to generate robust T cell expansion and improve the viability of encapsulated cells released from the scaffold. Additionally, GFOGER linkage onto the polymer matrix augmented T cell migration and resulted in a significantly higher number of T cells released from the scaffold.
Fig. 6.
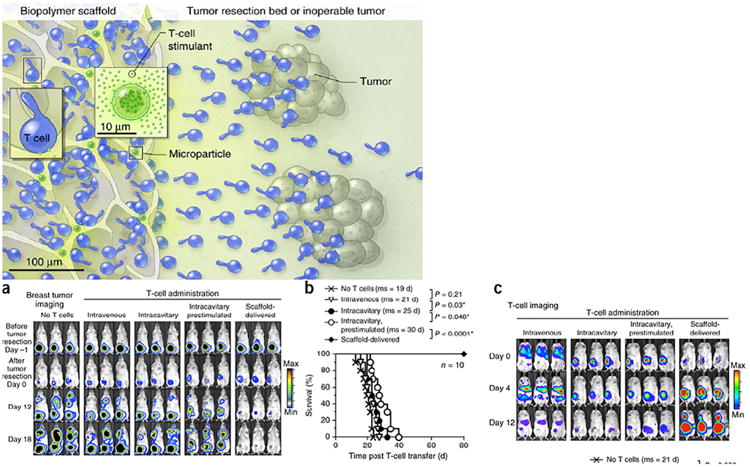
(Top) Illustration of the biomaterial system. Collagen-mimetic peptides conjugated to the polymeric scaffold encourage trafficking of loaded tumor-specific T cells into surrounding tumor beds. Additionally, microparticles in the scaffold encapsulating an IL-15 superagonist and with surface-bound anti-CD3, anti-CD28, and anti-137 stimulate lymphocyte survival and proliferation.(a) Bioluminescent imaging of 4T1 breast tumors demonstrates the potency of the biomaterial system, as minimal tumor presence is observed with the combinatorial technology compared to other treatments. (b) Kaplan-Meier survival curves for treated and control mice (ms:median survival). (c) Bioluminescent imaging of adoptively transferred T cells, showing the scaffold technology encourages robust T cell proliferation and migration.
Reprinted by permission from Macmillan Publishers Ltd.: Nature Biotechnology [114], copyright 2014.
4. Future directions
There is mounting evidence that combinatorial immunotherapy can offer significant advantages in treating complex immune-associated diseases. While we have described a number of approaches that have demonstrated potential as therapeutic strategies, the future of combinatorial drug delivery is not limited to the approaches discussed. The continually expanding body of immunotherapy research provides a strong foundation from which scientists can draw upon. Certain drug delivery opportunities may prove particularly important in establishing successful immunotherapies in the future.
4.1. Combinatorial use of monoclonal antibodies
Checkpoint blockade therapy has buoyed the enthusiasm for monoclonal antibody immunotherapy, as it has produced some of the most successful human clinical trials for cancer in recent years (reviewed in [115]). Drug delivery strategies to exploit the expanding field of monoclonal antibody immunotherapy could be immensely beneficial. For example, controlled-release approaches can aid in mitigating antibody clearance that limits their therapeutic effects [116]. Additionally, localized release adjacent to specific immune-associated structures (e.g., peritumorally, perinodally) could augment immunotherapy and address renewed concerns of systemic toxicity. However, limitations with biomaterial incorporation to deliver sensitive biologics are well established. Functional activity can be lost when attempting to encapsulate or conjugate large proteins to drug delivery vehicles [117]. Novel drug delivery strategies to circumvent these challenges are actively being explored. Recent work, for example, developed a method to reduce antibody oxidation encapsulated in silk biomaterials via methionine functionalization [118]. Strategies that utilize monoclonal antibodies in combination with other immunotherapeutics have also demonstrated promise in pre-clinical investigations. One approach explored this past year saw a vaccine composed of four components (anti-PD-1, tumor-specific antibodies, recombinant IL-2, and albumin-binding CpG-peptide antigen constructs) build upon checkpoint inhibitor blockade therapy to mediate improved, antigen-specific survival in tumor-bearing mice [119]. Another recent approach utilized lipoprotein nanodiscs to deliver multiple tumor-associated epitopes and the adjuvant CpG while intraperitoneally administering checkpoint blockade therapy [120]. The combinatorial technology, when applied in coordination with anti-PD-1 and anti-CTLA4 antibodies, dramatically improved anti-tumoral immunity compared to nanodiscs alone. These results suggest that applications involving monoclonal antibody therapy could benefit from combinatorial and controlled-release approaches.
4.2. Combinatorial use of immunoablation and cellular reprogramming
Often accomplished through monoclonal antibodies, selective depletion of immune compartments has been investigated as an alternative therapy for systemic, cytotoxic chemotherapy. Monoclonal antibodies against CD20+ B cells are frequently employed for Non-Hodgkin Lymphoma [121,122]. Similarly, depletion of CD25+ Tregs has been investigated in a clinical setting for augmenting anti-tumor vaccines [123]. Approaches utilizing immunoablating techniques have also generated interest in autoimmune disease to deplete autoreactive cells in order to reprogram homeostatic immunity. Combination therapy with anti-thymocyte globulin and GM-CSF has been shown to increase Treg frequency, alter DC pheno-types, and improve survival in mouse models of type 1 diabetes [124,125]. This combinatorial approach was validated when explored in clinical trials of type 1 diabetics, improving Treg frequency and preserving β-cell function compared to placebo controls [126,127]. While careful consideration should be given to therapies that completely ablate leukocyte subpopulations for fear of promoting systemic immunosuppression, there is growing evidence that suggests immunoablation in combination with immunomodulation can be a useful approach.
4.3. Combinatorial use with antigen-specific therapy
A recurring theme throughout the research discussed in this review has been the incorporation of disease-specific antigen in combinatorial drug delivery platforms. Antigen-specific immunotherapy is highly desirable to elicit controlled, directional immune responses. Viable antigen-specific therapies are advantageous compared to broadly cytotoxic or immunosuppressive drugs that can result in systemic toxicities. There is a wealth of research on immunotherapy systems that utilize disease-relevant antigen in coordination with immunomodulatory drugs, which indicates the value of this combination (reviewed in [9]). Controlled-release biomaterials have been extensively studied to deliver antigen, as their tunable properties can be beneficial in shaping immune responses. As discussed, sustained release of antigen from biodegradable PLGA nanoparticles has improved both prolonged antigen-presentation and the capacity for cross-presentation in bone-marrow derived DCs [61]. Mimicking long-standing clinical approaches for allergy immuno-therapy, biomaterials have also been studied to deliver self-protein to induce antigen-specific tolerance. In one approach, intravenous administration of polystyrene nanoparticles conjugated to myelin sheath epitopes ameliorated multiple sclerosis in a mouse model [128]. Vaccines that rely on immunomodulatory drugs may be improved upon by the inclusion of antigen. For example, nanoparticles containing the immuno-suppressive drug rapamycin were most effective in generating sustained humoral and cellular responses when antigen was included in the formulation [129,130]. Modulating immunity in antigen-specific immune directions will likely continue to be an important feature of forthcoming combinatorial immunotherapy strategies.
5. Conclusions
There is growing interest in combinatorial drug delivery strategies for immunomodulation. The breadth of new research highlighting the efficacy of multi-drug immunotherapy schemes is perennially expanding. Biomaterial platforms offer numerous advantages to optimize drug delivery for immunotherapy. While there are many challenges associated with successful immunotherapy, we are optimistic that combinatorial drug delivery platforms can help overcome the limitations associated with previous therapeutic interventions.
Acknowledgments
This work was supported by the National Institute of Diabetes and Digestive and Kidney Diseases R01DK091658 and R01DK098589.
Abbreviations
- CAR
chimeric antigen receptor
- Treg
regulatory T cell
- PD-1
programmed death-1
- CTLA-4
cytotoxic T-lymphocyte-associated protein 4
- PD-L1
programmed death ligand 1
- APC
antigen-presenting cell
- DC
dendritic cell
- TLR
tolllike receptor
- OVA
ovalbumin
- PLGA
poly(lactide-co-glycolide)
- MPLA
monophosphoryl lipid A
- poly(I:C)
poly(inosinic:cytidylic acid)
- SB
SB505124
- NK
natural killer
- siRNA
small interfering RNA
- GM-CSF
granulocyte-macrophage colony-stimulating factor
References
- 1.Larche M, Akdis CA, Valenta R. Immunological mechanisms of allergen-specific immunotherapy. Nat Rev Immunol. 2006;6:761–771. doi: 10.1038/nri1934. [DOI] [PubMed] [Google Scholar]
- 2.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB, Xu Y, Frohlich MW, Schellhammer PF, Investigators IS. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363:411–422. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 3.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, Mahnke YD, Melenhorst JJ, Rheingold SR, Shen A, Teachey DT, Levine BL, June CH, Porter DL, Grupp SA. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smyth MJ, Ngiow SF, Ribas A, Teng MW. Combination cancer immunotherapies tailored to the tumour microenvironment. Nat Rev Clin Oncol. 2016;13:143–158. doi: 10.1038/nrclinonc.2015.209. [DOI] [PubMed] [Google Scholar]
- 5.Rosenzwajg M, Churlaud G, Mallone R, Six A, Derian N, Chaara W, Lorenzon R, Long SA, Buckner JH, Afonso G, Pham HP, Hartemann A, Yu A, Pugliese A, Malek TR, Klatzmann D. Low-dose interleukin-2 fosters a dose-dependent regulatory T cell tuned milieu in T1D patients. J Autoimmun. 2015;58:48–58. doi: 10.1016/j.jaut.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, Hassel JC, Rutkowski P, McNeil C, Kalinka-Warzocha E, Savage KJ, Hernberg MM, Lebbe C, Charles J, Mihalcioiu C, Chiarion-Sileni V, Mauch C, Cognetti F, Arance A, Schmidt H, Schadendorf D, Gogas H, Lundgren-Eriksson L, Horak C, Sharkey B, Waxman IM, Atkinson V, Ascierto PA. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320–330. doi: 10.1056/NEJMoa1412082. [DOI] [PubMed] [Google Scholar]
- 7.Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbe C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maio M, Grob JJ, Aamdal S, Bondarenko I, Robert C, Thomas L, Garbe C, Chiarion-Sileni V, Testori A, Chen TT, Tschaika M, Wolchok JD. Five-year survival rates for treatment-naive patients with advanced melanoma who received ipilimumab plus dacarbazine in a phase III trial. J Clin Oncol. 2015;33:1191–1196. doi: 10.1200/JCO.2014.56.6018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Northrup L, Christopher MA, Sullivan BP, Berkland C. Combining antigen and immunomodulators: Emerging trends in antigen-specific immunotherapy for autoimmunity. Adv Drug Deliv Rev. 2015;98:86–98. doi: 10.1016/j.addr.2015.10.020. [DOI] [PubMed] [Google Scholar]
- 10.Pozzilli P, Maddaloni E, Buzzetti R. Combination immunotherapies for type 1 diabetes mellitus. Nat Rev Endocrinol. 2015;11:289–297. doi: 10.1038/nrendo.2015.8. [DOI] [PubMed] [Google Scholar]
- 11.Carstens MR, Fisher RC, Acharya AP, Butterworth EA, Scott E, Huang EH, Keselowsky BG. Drug-eluting microarrays to identify effective chemotherapeutic combinations targeting patient-derived cancer stem cells. Proc Natl Acad Sci U S A. 2015;112:8732–8737. doi: 10.1073/pnas.1505374112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, Ferrucci PF, Hill A, Wagstaff J, Carlino MS, Haanen JB, Maio M, Marquez-Rodas I, McArthur GA, Ascierto PA, Long GV, Callahan MK, Postow MA, Grossmann K, Sznol M, Dreno B, Bastholt L, Yang A, Rollin LM, Horak C, Hodi FS, Wolchok JD. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, Burke MM, Caldwell A, Kronenberg SA, Agunwamba BU, Zhang X, Lowy I, Inzunza HD, Feely W, Horak CE, Hong Q, Korman AJ, Wigginton JM, Gupta A, Sznol M. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122–133. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reed SG, Orr MT, Fox CB. Key roles of adjuvants in modern vaccines. Nat Med. 2013;19:1597–1608. doi: 10.1038/nm.3409. [DOI] [PubMed] [Google Scholar]
- 15.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Voskens CJ, Goldinger SM, Loquai C, Robert C, Kaehler KC, Berking C, Bergmann T, Bockmeyer CL, Eigentler T, Fluck M, Garbe C, Gutzmer R, Grabbe S, Hauschild A, Hein R, Hundorfean G, Justich A, Keller U, Klein C, Mateus C, Mohr P, Paetzold S, Satzger I, Schadendorf D, Schlaeppi M, Schuler G, Schuler-Thurner B, Trefzer U, Ulrich J, Vaubel J, von Moos R, Weder P, Wilhelm T, Goppner D, Dummer R, Heinzerling LM. The price of tumor control: an analysis of rare side effects of anti-CTLA-4 therapy in metastatic melanoma from the ipilimumab network. PLoS One. 2013;8:e53745. doi: 10.1371/journal.pone.0053745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Denton MD, Magee CC, Sayegh MH. Immunosuppressive strategies in transplantation. Lancet. 1999;353:1083–1091. doi: 10.1016/S0140-6736(98)07493-5. [DOI] [PubMed] [Google Scholar]
- 18.Reddy ST, Swartz MA, Hubbell JA. Targeting dendritic cells with biomaterials: developing the next generation of vaccines. Trends Immunol. 2006;27:573–579. doi: 10.1016/j.it.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 19.Ali OA, Huebsch N, Cao L, Dranoff G, Mooney DJ. Infection-mimicking materials to program dendritic cells in situ. Nat Mater. 2009;8:151–158. doi: 10.1038/nmat2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 21.MacDonald KP, Munster DJ, Clark GJ, Dzionek A, Schmitz J, Hart DN. Characterization of human blood dendritic cell subsets. Blood. 2002;100:4512–4520. doi: 10.1182/blood-2001-11-0097. [DOI] [PubMed] [Google Scholar]
- 22.Probst HC, McCoy K, Okazaki T, Honjo T, van den Broek M. Resting dendritic cells induce peripheral CD8+ T cell tolerance through PD-1 and CTLA-4. Nat Immunol. 2005;6:280–286. doi: 10.1038/ni1165. [DOI] [PubMed] [Google Scholar]
- 23.Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol. 2003;21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040. [DOI] [PubMed] [Google Scholar]
- 24.van Duin D, Medzhitov R, Shaw AC. Triggering TLR signaling in vaccination. Trends Immunol. 2006;27:49–55. doi: 10.1016/j.it.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 25.Pulendran B, Smith JL, Caspary G, Brasel K, Pettit D, Maraskovsky E, Maliszewski CR. Distinct dendritic cell subsets differentially regulate the class of immune response in vivo. Proc Natl Acad Sci U S A. 1999;96:1036–1041. doi: 10.1073/pnas.96.3.1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maldonado RA, von Andrian UH. How tolerogenic dendritic cells induce regulatory T cells. Adv Immunol. 2010;108:111–165. doi: 10.1016/B978-0-12-380995-7.00004-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. 2013;31:563–604. doi: 10.1146/annurev-immunol-020711-074950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kandalaft LE, Powell DJ, Jr, Chiang CL, Tanyi J, Kim S, Bosch M, Montone K, Mick R, Levine BL, Torigian DA, June CH, Coukos G. Autologous lysate-pulsed dendritic cell vaccination followed by adoptive transfer of vaccine-primed ex vivo co-stimulated T cells in recurrent ovarian cancer. Oncoimmunology. 2013;2:e22664. doi: 10.4161/onci.22664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clare-Salzler MJ, Brooks J, Chai A, Van Herle K, Anderson C. Prevention of diabetes in nonobese diabetic mice by dendritic cell transfer. J Clin Invest. 1992;90:741–748. doi: 10.1172/JCI115946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hsu FJ, Benike C, Fagnoni F, Liles TM, Czerwinski D, Taidi B, Engleman EG, Levy R. Vaccination of patients with B-cell lymphoma using autologous antigen-pulsed dendritic cells. Nat Med. 1996;2:52–58. doi: 10.1038/nm0196-52. [DOI] [PubMed] [Google Scholar]
- 31.Holtl L, Zelle-Rieser C, Gander H, Papesh C, Ramoner R, Bartsch G, Rogatsch H, Barsoum AL, Coggin JH, Jr, Thurnher M. Immunotherapy of metastatic renal cell carcinoma with tumor lysate-pulsed autologous dendritic cells. Clin Cancer Res. 2002;8:3369–3376. [PubMed] [Google Scholar]
- 32.Yu JS, Wheeler CJ, Zeltzer PM, Ying H, Finger DN, Lee PK, Yong WH, Incardona F, Thompson RC, Riedinger MS, Zhang W, Prins RM, Black KL. Vaccination of malignant glioma patients with peptide-pulsed dendritic cells elicits systemic cytotoxicity and intracranial T-cell infiltration. Cancer Res. 2001;61:842–847. [PubMed] [Google Scholar]
- 33.Taner T, Hackstein H, Wang Z, Morelli AE, Thomson AW. Rapamycin-treated, alloantigen-pulsed host dendritic cells induce ag-specific T cell regulation and prolong graft survival. Am J Transplant. 2005;5:228–236. doi: 10.1046/j.1600-6143.2004.00673.x. [DOI] [PubMed] [Google Scholar]
- 34.Mansilla MJ, Selles-Moreno C, Fabregas-Puig S, Amoedo J, Navarro-Barriuso J, Teniente-Serra A, Grau-Lopez L, Ramo-Tello C, Martinez-Caceres EM. Beneficial effect of tolerogenic dendritic cells pulsed with MOG autoantigen in experimental autoimmune encephalomyelitis. CNS Neurosci Ther. 2015;21:222–230. doi: 10.1111/cns.12342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harrison LC. Vaccination against self to prevent autoimmune disease: the type 1 diabetes model. Immunol Cell Biol. 2008;86:139–145. doi: 10.1038/sj.icb.7100151. [DOI] [PubMed] [Google Scholar]
- 36.Keselowsky BG, Xia CQ, Clare-Salzler M. Multifunctional dendritic cell-targeting polymeric microparticles: engineering new vaccines for type 1 diabetes. Hum Vaccin. 2011;7:37–44. doi: 10.4161/hv.7.1.12916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Giannoukakis N, Phillips B, Finegold D, Harnaha J, Trucco M. Phase I (safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients. Diabetes Care. 2011;34:2026–2032. doi: 10.2337/dc11-0472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anderson AE, Swan DJ, Wong OY, Buck M, Eltherington O, Harry RA, Patterson AM, Pratt AG, Reynolds G, Doran JP, Kirby JA, Isaacs JD, Hilkens CM. Tolerogenic dendritic cells generated with dexamethasone and vitamin D3 regulate rheumatoid arthritis CD4+ T cells partly via transforming growth factor-beta1. Clin Exp Immunol. 2017;187:113–123. doi: 10.1111/cei.12870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lewis JS, Roche C, Zhang Y, Brusko TM, Wasserfall CH, Atkinson M, Clare-Salzler MJ, Keselowsky BG. Combinatorial delivery of immunosuppressive factors to dendritic cells using dual-sized microspheres. J Mater Chem B Mater Biol Med. 2014;2:2562–2574. doi: 10.1039/C3TB21460E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mart In-Fontecha A, Sebastiani S, Hopken UE, Uguccioni M, Lipp M, Lanzavecchia A, Sallusto F. Regulation of dendritic cell migration to the draining lymph node: impact on T lymphocyte traffic and priming. J Exp Med. 2003;198:615–621. doi: 10.1084/jem.20030448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tacken PJ, de Vries IJ, Torensma R, Figdor CG. Dendritic-cell immunotherapy: from ex vivo loading to in vivo targeting. Nat Rev Immunol. 2007;7:790–802. doi: 10.1038/nri2173. [DOI] [PubMed] [Google Scholar]
- 42.Moon JJ, Huang B, Irvine DJ. Engineering nano- and microparticles to tune immunity. Adv Mater. 2012;24:3724–3746. doi: 10.1002/adma.201200446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Uhrich KE, Cannizzaro SM, Langer RS, Shakesheff KM. Polymeric systems for controlled drug release. Chem Rev. 1999;99:3181–3198. doi: 10.1021/cr940351u. [DOI] [PubMed] [Google Scholar]
- 44.Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Preat V. PLGA-based nano-particles: an overview of biomedical applications. J Control Release. 2012;161:505–522. doi: 10.1016/j.jconrel.2012.01.043. [DOI] [PubMed] [Google Scholar]
- 45.Champion JA, Walker A, Mitragotri S. Role of particle size in phagocytosis of polymeric microspheres. Pharm Res. 2008;25:1815–1821. doi: 10.1007/s11095-008-9562-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Engman C, Wen Y, Meng WS, Bottino R, Trucco M, Giannoukakis N. Generation of antigen-specific Foxp3+ regulatory T-cells in vivo following administration of diabetes-reversing tolerogenic microspheres does not require provision of antigen in the formulation. Clin Immunol. 2015;160:103–123. doi: 10.1016/j.clim.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 47.Phillips B, Nylander K, Harnaha J, Machen J, Lakomy R, Styche A, Gillis K, Brown L, Lafreniere D, Gallo M, Knox J, Hogeland K, Trucco M, Giannoukakis N. A microsphere-based vaccine prevents and reverses new-onset autoimmune diabetes. Diabetes. 2008;57:1544–1555. doi: 10.2337/db07-0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reddy ST, van der Vlies AJ, Simeoni E, Angeli V, Randolph GJ, O'Neil CP, Lee LK, Swartz MA, Hubbell JA. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat Biotechnol. 2007;25:1159–1164. doi: 10.1038/nbt1332. [DOI] [PubMed] [Google Scholar]
- 49.Moghimi SM, Hunter AC, Dadswell CM, Savay S, Alving CR, Szebeni J. Causative factors behind poloxamer 188 (Pluronic F68, Flocor)-induced complement activation in human sera. A protective role against poloxamer-mediated complement activation by elevated serum lipoprotein levels. Biochim Biophys Acta. 2004;1689:103–113. doi: 10.1016/j.bbadis.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 50.Thomas SN, Vokali E, Lund AW, Hubbell JA, Swartz MA. Targeting the tumor-draining lymph node with adjuvanted nanoparticles reshapes the anti-tumor immune response. Biomaterials. 2014;35:814–824. doi: 10.1016/j.biomaterials.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 51.Szajnik M, Szczepanski MJ, Czystowska M, Elishaev E, Mandapathil M, Nowak-Markwitz E, Spaczynski M, Whiteside TL. TLR4 signaling induced by lipo-polysaccharide or paclitaxel regulates tumor survival and chemoresistance in ovarian cancer. Oncogene. 2009;28:4353–4363. doi: 10.1038/onc.2009.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Warger T, Osterloh P, Rechtsteiner G, Fassbender M, Heib V, Schmid B, Schmitt E, Schild H, Radsak MP. Synergistic activation of dendritic cells by combined Toll-like receptor ligation induces superior CTL responses in vivo. Blood. 2006;108:544–550. doi: 10.1182/blood-2005-10-4015. [DOI] [PubMed] [Google Scholar]
- 53.Napolitani G, Rinaldi A, Bertoni F, Sallusto F, Lanzavecchia A. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat Immunol. 2005;6:769–776. doi: 10.1038/ni1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kasturi SP, Skountzou I, Albrecht RA, Koutsonanos D, Hua T, Nakaya HI, Ravindran R, Stewart S, Alam M, Kwissa M, Villinger F, Murthy N, Steel J, Jacob J, Hogan RJ, Garcia-Sastre A, Compans R, Pulendran B. Programming the magnitude and persistence of antibody responses with innate immunity. Nature. 2011;470:543–547. doi: 10.1038/nature09737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 56.Acharya AP, Lewis JS, Keselowsky BG. Combinatorial co-encapsulation of hydrophobic molecules in poly(lactide-co-glycolide) microparticles. Biomaterials. 2013;34:3422–3430. doi: 10.1016/j.biomaterials.2013.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Acharya AP, Carstens MR, Lewis JS, Dolgova N, Xia CQ, Clare-Salzler MJ, Keselowsky BG. A cell-based microarray to investigate combinatorial effects of micro-particle-encapsulated adjuvants on dendritic cell activation. J Mater Chem B Mater Biol Med. 2016;4:1672–1685. doi: 10.1039/C5TB01754H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Acharya AP, Clare-Salzler MJ, Keselowsky BG. A high-throughput microparticle microarray platform for dendritic cell-targeting vaccines. Biomaterials. 2009;30:4168–4177. doi: 10.1016/j.biomaterials.2009.04.032. [DOI] [PubMed] [Google Scholar]
- 59.Sallusto F, Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J Exp Med. 1994;179:1109–1118. doi: 10.1084/jem.179.4.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat Rev Immunol. 2012;12:557–569. doi: 10.1038/nri3254. [DOI] [PubMed] [Google Scholar]
- 61.Shen H, Ackerman AL, Cody V, Giodini A, Hinson ER, Cresswell P, Edelson RL, Saltzman WM, Hanlon DJ. Enhanced and prolonged cross-presentation following endosomal escape of exogenous antigens encapsulated in biodegradable nanoparticles. Immunology. 2006;117:78–88. doi: 10.1111/j.1365-2567.2005.02268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Siefert AL, Caplan MJ, Fahmy TM. Artificial bacterial biomimetic nanoparticles synergize pathogen-associated molecular patterns for vaccine efficacy. Biomaterials. 2016;97:85–96. doi: 10.1016/j.biomaterials.2016.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vollmer J, Krieg AM. Immunotherapeutic applications of CpG oligodeoxynucleotide TLR9 agonists. Adv Drug Deliv Rev. 2009;61:195–204. doi: 10.1016/j.addr.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 64.Cruz LJ, Rosalia RA, Kleinovink JW, Rueda F, Lowik CW, Ossendorp F. Targeting nanoparticles to CD40, DEC-205 or CD1 1c molecules on dendritic cells for efficient CD8(+) T cell response: a comparative study. J Control Release. 2014;192:209–218. doi: 10.1016/j.jconrel.2014.07.040. [DOI] [PubMed] [Google Scholar]
- 65.Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5:505–515. doi: 10.1021/mp800051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cruz LJ, Tacken PJ, Fokkink R, Joosten B, Stuart MC, Albericio F, Torensma R, Figdor CG. Targeted PLGA nano- but not microparticles specifically deliver antigen to human dendritic cells via DC-SIGN in vitro. J Control Release. 2010;144:118–126. doi: 10.1016/j.jconrel.2010.02.013. [DOI] [PubMed] [Google Scholar]
- 67.Lewis JS, Zaveri TD, Crooks CP, 2nd, Keselowsky BG. Microparticle surface modifications targeting dendritic cells for non-activating applications. Biomaterials. 2012;33:7221–7232. doi: 10.1016/j.biomaterials.2012.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rosalia RA, Cruz LJ, van Duikeren S, Tromp AT, Silva AL, Jiskoot W, de Gruijl T, Lowik C, Oostendorp J, van der Burg SH, Ossendorp F. CD40-targeted dendritic cell delivery of PLGA-nanoparticle vaccines induce potent anti-tumor responses. Biomaterials. 2015;40:88–97. doi: 10.1016/j.biomaterials.2014.10.053. [DOI] [PubMed] [Google Scholar]
- 69.Kwong B, Liu H, Irvine DJ. Induction of potent anti-tumor responses while eliminating systemic side effects via liposome-anchored combinatorial immunotherapy. Biomaterials. 2011;32:5134–5147. doi: 10.1016/j.biomaterials.2011.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol. 2012;12:269–281. doi: 10.1038/nri3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zheng Y, Stephan MT, Gai SA, Abraham W, Shearer A, Irvine DJ. In vivo targeting of adoptively transferred T-cells with antibody- and cytokine-conjugated liposomes. J Control Release. 2013;172:426–435. doi: 10.1016/j.jconrel.2013.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vincent BG, Young EF, Buntzman AS, Stevens R, Kepler TB, Tisch RM, Frelinger JA, Hess PR. Toxin-coupled MHC class I tetramers can specifically ablate autoreactive CD8 + T cells and delay diabetes in nonobese diabetic mice. J Immunol. 2010;184:4196–4204. doi: 10.4049/jimmunol.0903931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3 + regulatory T cells in the human immune system. Nat Rev Immunol. 2010;10:490–500. doi: 10.1038/nri2785. [DOI] [PubMed] [Google Scholar]
- 74.Jhunjhunwala S, Raimondi G, Glowacki AJ, Hall SJ, Maskarinec D, Thorne SH, Thomson AW, Little SR. Bioinspired controlled release of CCL22 recruits regulatory T cells in vivo. Adv Mater. 2012;24:4735–4738. doi: 10.1002/adma.201202513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kontos S, Kourtis IC, Dane KY, Hubbell JA. Engineering antigens for in situ erythrocyte binding induces T-cell deletion. Proc Natl Acad Sci U S A. 2013;110:E60–E68. doi: 10.1073/pnas.1216353110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Getts DR, McCarthy DP, Miller SD. Exploiting apoptosis for therapeutic tolerance induction. J Immunol. 2013;191:5341–5346. doi: 10.4049/jimmunol.1302070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Grimm AJ, Kontos S, Diaceri G, Quaglia-Thermes X, Hubbell JA. Memory of tolerance and induction of regulatory T cells by erythrocyte-targeted antigens. Sci Rep. 2015;5:15907. doi: 10.1038/srep15907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lutterotti A, Yousef S, Sputtek A, Sturner KH, Stellmann JP, Breiden P, Reinhardt S, Schulze C, Bester M, Heesen C, Schippling S, Miller SD, Sospedra M, Martin R. Antigen-specific tolerance by autologous myelin peptide-coupled cells: a phase 1 trial in multiple sclerosis. Sci Transl Med. 2013;5:188ra175. doi: 10.1126/scitranslmed.3006168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Jhunjhunwala S, Balmert SC, Raimondi G, Dons E, Nichols EE, Thomson AW, Little SR. Controlled release formulations of IL-2, TGF-beta1 and rapamycin for the induction of regulatory T cells. J Control Release. 2012;159:78–84. doi: 10.1016/j.jconrel.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.McHugh MD, Park J, Uhrich R, Gao W, Horwitz DA, Fahmy TM. Paracrine co-delivery of TGF-beta and IL-2 using CD4-targeted nanoparticles for induction and maintenance of regulatory T cells. Biomaterials. 2015;59:172–181. doi: 10.1016/j.biomaterials.2015.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Border WA, Ruoslahti E. Transforming growth factor-beta in disease: the dark side of tissue repair. J Clin Invest. 1992;90:1–7. doi: 10.1172/JCI115821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rodriguez PL, Harada T, Christian DA, Pantano DA, Tsai RK, Discher DE. Minimal “self” peptides that inhibit phagocytic clearance and enhance delivery of nanoparticles. Science. 2013;339:971–975. doi: 10.1126/science.1229568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kim YK, Que R, Wang SW, Liu WF. Modification of biomaterials with a self-protein inhibits the macrophage response. Adv Healthc Mater. 2014;3:989–994. doi: 10.1002/adhm.201300532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Foged C, Brodin B, Frokjaer S, Sundblad A. Particle size and surface charge affect particle uptake by human dendritic cells in an in vitro model. Int J Pharm. 2005;298:315–322. doi: 10.1016/j.ijpharm.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 85.He C, Hu Y, Yin L, Tang C, Yin C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials. 2010;31:3657–3666. doi: 10.1016/j.biomaterials.2010.01.065. [DOI] [PubMed] [Google Scholar]
- 86.Getts DR, Terry RL, Getts MT, Deffrasnes C, Muller M, van Vreden C, Ashhurst TM, Chami B, McCarthy D, Wu H, Ma J, Martin A, Shae LD, Witting P, Kansas GS, Kuhn J, Hafezi W, Campbell IL, Reilly D, Say J, Brown L, White MY, Cordwell SJ, Chadban SJ, Thorp EB, Bao S, Miller SD, King NJ. Therapeutic inflammatory monocyte modulation using immune-modifying microparticles. Sci Transl Med. 2014;6:219ra217. doi: 10.1126/scitranslmed.3007563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Iwaoka S, Nakamura T, Takano S, Tsuchiya S, Aramaki Y. Cationic liposomes induce apoptosis through p38 MAP kinase-caspase-8-Bid pathway in macrophage-like RAW264.7 cells. J Leukoc Biol. 2006;79:184–191. doi: 10.1189/jlb.0405181. [DOI] [PubMed] [Google Scholar]
- 88.Xia T, Kovochich M, Liong M, Zink JI, Nel AE. Cationic polystyrene nanosphere toxicity depends on cell-specific endocytic and mitochondrial injury pathways. ACS Nano. 2008;2:85–96. doi: 10.1021/nn700256c. [DOI] [PubMed] [Google Scholar]
- 89.Champion JA, Mitragotri S. Role of target geometry in phagocytosis. Proc Natl Acad Sci U S A. 2006;103:4930–4934. doi: 10.1073/pnas.0600997103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liu H, Moynihan KD, Zheng Y, Szeto GL, Li AV, Huang B, Van Egeren DS, Park C, Irvine DJ. Structure-based programming of lymph-node targeting in molecular vaccines. Nature. 2014;507:519–522. doi: 10.1038/nature12978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jewell CM, Lopez SC, Irvine DJ. In situ engineering of the lymph node microenvironment via intranodal injection of adjuvant-releasing polymer particles. Proc Natl Acad Sci U S A. 2011;108:15745–15750. doi: 10.1073/pnas.1105200108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tostanoski LH, Chiu YC, Gammon JM, Simon T, Andorko JI, Bromberg JS, Jewell CM. Reprogramming the local lymph node microenvironment promotes tolerance that is systemic and antigen specific. Cell Rep. 2016;16:2940–2952. doi: 10.1016/j.celrep.2016.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Park J, Wrzesinski SH, Stern E, Look M, Criscione J, Ragheb R, Jay SM, Demento SL, Agawu A, Licona Limon P, Ferrandino AF, Gonzalez D, Habermann A, Flavell RA, Fahmy TM. Combination delivery of TGF-beta inhibitor and IL-2by nanoscale liposomal polymeric gels enhances tumour immunotherapy. Nat Mater. 2012;11:895–905. doi: 10.1038/nmat3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kortylewski M, Swiderski P, Herrmann A, Wang L, Kowolik C, Kujawski M, Lee H, Scuto A, Liu Y, Yang C, Deng J, Soifer HS, Raubitschek A, Forman S, Rossi JJ, Pardoll DM, Jove R, Yu H. In vivo delivery of siRNA to immune cells by conjugation to a TLR9 agonist enhances antitumor immune responses. Nat Biotechnol. 2009;27:925–932. doi: 10.1038/nbt.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Place ES, Evans ND, Stevens MM. Complexity in biomaterials for tissue engineering. Nat Mater. 2009;8:457–470. doi: 10.1038/nmat2441. [DOI] [PubMed] [Google Scholar]
- 96.Hori Y, Winans AM, Huang CC, Horrigan EM, Irvine DJ. Injectable dendritic cell-carrying alginate gels for immunization and immunotherapy. Biomaterials. 2008;29:3671–3682. doi: 10.1016/j.biomaterials.2008.05.033. [DOI] [PubMed] [Google Scholar]
- 97.Singh A, Peppas NA. Hydrogels and scaffolds for immunomodulation. Adv Mater. 2014;26:6530–6541. doi: 10.1002/adma.201402105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Haddad D, Ramprakash J, Sedegah M, Charoenvit Y, Baumgartner R, Kumar S, Hoffman SL, Weiss WR. Plasmid vaccine expressing granulocyte-macrophage colony-stimulating factor attracts infiltrates including immature dendritic cells into injected muscles. J Immunol. 2000;165:3772–3781. doi: 10.4049/jimmunol.165.7.3772. [DOI] [PubMed] [Google Scholar]
- 99.Lutz MB, Kukutsch N, Ogilvie AL, Rossner S, Koch F, Romani N, Schuler G. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods. 1999;223:77–92. doi: 10.1016/s0022-1759(98)00204-x. [DOI] [PubMed] [Google Scholar]
- 100.Palucka KA, Taquet N, Sanchez-Chapuis F, Gluckman JC. Dendritic cells as the terminal stage of monocyte differentiation. J Immunol. 1998;160:4587–4595. [PubMed] [Google Scholar]
- 101.Sathe P, Pooley J, Vremec D, Mintern J, Jin JO, Wu L, Kwak JY, Villadangos JA, Shortman K. The acquisition of antigen cross-presentation function by newly formed dendritic cells. J Immunol. 2011;186:5184–5192. doi: 10.4049/jimmunol.1002683. [DOI] [PubMed] [Google Scholar]
- 102.Arellano M, Lonial S. Clinical uses of GM-CSF, a critical appraisal and update. Biologics. 2008;2:13–27. doi: 10.2147/btt.s1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kim J, Li WA, Choi Y, Lewin SA, Verbeke CS, Dranoff G, Mooney DJ. Injectable, spontaneously assembling, inorganic scaffolds modulate immune cells in vivo and increase vaccine efficacy. Nat Biotechnol. 2015;33:64–72. doi: 10.1038/nbt.3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lewis JS, Dolgova NV, Zhang Y, Xia CQ, Wasserfall CH, Atkinson MA, Clare-Salzler MJ, Keselowsky BG. A combination dual-sized microparticle system modulates dendritic cells and prevents type 1 diabetes in prediabetic NOD mice. Clin Immunol. 2015;160:90–102. doi: 10.1016/j.clim.2015.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yoon YM, Lewis JS, Carstens MR, Campbell-Thompson M, Wasserfall CH, Atkinson MA, Keselowsky BG. A combination hydrogel microparticle-based vaccine prevents type 1 diabetes in non-obese diabetic mice. Sci Rep. 2015;5:13155. doi: 10.1038/srep13155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zonneveld-Huijssoon E, van Wijk F, Roord S, Delemarre E, Meerding J, de Jager W, Klein M, Raz E, Albani S, Kuis W, Boes M, Prakken BJ. TLR9 agonist CpG enhances protective nasal HSP60 peptide vaccine efficacy in experimental autoimmune arthritis. Ann Rheum Dis. 2012;71:1706–1715. doi: 10.1136/annrheumdis-2011-201131. [DOI] [PubMed] [Google Scholar]
- 107.Singh A, Suri S, Roy K. In-situ crosslinking hydrogels for combinatorial delivery of chemokines and siRNA-DNA carrying microparticles to dendritic cells. Biomaterials. 2009;30:5187–5200. doi: 10.1016/j.biomaterials.2009.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Singh A, Nie H, Ghosn B, Qin H, Kwak LW, Roy K. Efficient modulation of T-cell response by dual-mode, single-carrier delivery of cytokine-targeted siRNA and DNA vaccine to antigen-presenting cells. Mol Ther. 2008;16:2011–2021. doi: 10.1038/mt.2008.206. [DOI] [PubMed] [Google Scholar]
- 109.Singh A, Qin H, Fernandez I, Wei J, Lin J, Kwak LW, Roy K. An injectable synthetic immune-priming center mediates efficient T-cell class switching and T-helper 1 response against B cell lymphoma. J Control Release. 2011;155:184–192. doi: 10.1016/j.jconrel.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 110.Pradhan P, Qin H, Leleux JA, Gwak D, Sakamaki I, Kwak LW, Roy K. The effect of combined IL10 siRNA and CpG ODN as pathogen-mimicking microparticles on Th1/Th2 cytokine balance in dendritic cells and protective immunity against B cell lymphoma. Biomaterials. 2014;35:5491–5504. doi: 10.1016/j.biomaterials.2014.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yang EY, Kronenfeld JP, Gattas-Asfura KM, Bayer AL, Stabler CL. Engineering an “infectious” Treg biomimetic through chemoselective tethering of TGF-beta1 to PEG brush surfaces. Biomaterials. 2015;67:20–31. doi: 10.1016/j.biomaterials.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hume PS, He J, Haskins K, Anseth KS. Strategies to reduce dendritic cell activation through functional biomaterial design. Biomaterials. 2012;33:3615–3625. doi: 10.1016/j.biomaterials.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hume PS, Anseth KS. Inducing local T cell apoptosis with anti-Fas-functionalized polymeric coatings fabricated via surface-initiated photopolymerizations. Biomaterials. 2010;31:3166–3174. doi: 10.1016/j.biomaterials.2010.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Stephan SB, Taber AM, Jileaeva I, Pegues EP, Sentman CL, Stephan MT. Bio-polymer implants enhance the efficacy of adoptive T-cell therapy. Nat Biotechnol. 2015;33:97–101. doi: 10.1038/nbt.3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016;16:275–287. doi: 10.1038/nrc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Weiner GJ. Building better monoclonal antibody-based therapeutics. Nat Rev Cancer. 2015;15:361–370. doi: 10.1038/nrc3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bilati U, Allemann E, Doelker E. Strategic approaches for overcoming peptide and protein instability within biodegradable nano- and microparticles. Eur J Pharm Biopharm. 2005;59:375–388. doi: 10.1016/j.ejpb.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 118.Guziewicz NA, Massetti AJ, Perez-Ramirez BJ, Kaplan DL. Mechanisms of monoclonal antibody stabilization and release from silk biomaterials. Biomaterials. 2013;34:7766–7775. doi: 10.1016/j.biomaterials.2013.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Moynihan KD, Opel CF, Szeto GL, Tzeng A, Zhu EF, Engreitz JM, Williams RT, Rakhra K, Zhang MH, Rothschilds AM, Kumari S, Kelly RL, Kwan BH, Abraham W, Hu K, Mehta NK, Kauke MJ, Suh H, Cochran JR, Lauffenburger DA, Wittrup KD, Irvine DJ. Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses. Nat Med. 2016 doi: 10.1038/nm.4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kuai R, Ochyl LJ, Bahjat KS, Schwendeman A, Moon JJ. Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat Mater. 2017;16(4):489–496. doi: 10.1038/nmat4822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Cheson BD, Leonard JP. Monoclonal antibody therapy for B-cell non-Hodgkin's lymphoma. N Engl J Med. 2008;359:613–626. doi: 10.1056/NEJMra0708875. [DOI] [PubMed] [Google Scholar]
- 122.Pescovitz MD. Rituximab, an anti-cd20 monoclonal antibody: history and mechanism of action. Am J Transplant. 2006;6:859–866. doi: 10.1111/j.1600-6143.2006.01288.x. [DOI] [PubMed] [Google Scholar]
- 123.Dannull J, Su Z, Rizzieri D, Yang BK, Coleman D, Yancey D, Zhang A, Dahm P, Chao N, Gilboa E, Vieweg J. Enhancement of vaccine-mediated antitumor immunity in cancer patients after depletion of regulatory T cells. J Clin Invest. 2005;115:3623–3633. doi: 10.1172/JCI25947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Xue S, Posgai A, Wasserfall C, Myhr C, Campbell-Thompson M, Mathews CE, Brusko T, Rabinovit A. 2015:3873–3884. doi: 10.2337/db15-0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Parker MJ, Xue S, Alexander JJ, Wasserfall CH, Campbell-Thompson ML, Battaglia M, Gregori S, Mathews CE, Song S, Troutt M, Eisenbeis S, Williams J, Schatz DA, Haller MJ, Atkinson MA. Immune depletion with cellular mobilization imparts immunoregulation and reverses autoimmune diabetes in nonobese diabetic mice. Diabetes. 2009;58:2277–2284. doi: 10.2337/db09-0557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Haller MJ, Gitelman SE, Gottlieb PA, Michels AW, Perry DJ, Schultz AR, Hulme MA, Shuster JJ, Zou B, Wasserfall CH, Posgai A, Mathews CE, Brusko TM, Atkinson MA, Schatz DA. Anti-thymocyte globulin + G-CSF combination therapy leads to sustained immunomodulatory and metabolic effects in a subset of responders with established type 1 diabetes. Diabetes. 2016;65(12):3765–3775. doi: 10.2337/db16-0823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Haller MJ, Gitelman SE, Gottlieb PA, Michels AW, Rosenthal SM, Shuster JJ, Zou B, Brusko TM, Hulme MA, Wasserfall CH, Mathews CE, Atkinson MA, Schatz DA. Anti-thymocyte globulin/G-CSF treatment preserves beta cell function in patients with established type 1 diabetes. J Clin Invest. 2015;125:448–455. doi: 10.1172/JCI78492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Getts DR, Martin AJ, McCarthy DP, Terry RL, Hunter ZN, Yap WT, Getts MT, Pleiss M, Luo X, King NJ, Shea LD, Miller SD. Microparticles bearing encephalitogenic peptides induce T-cell tolerance and ameliorate experimental autoimmune encephalomyelitis. Nat Biotechnol. 2012;30:1217–1224. doi: 10.1038/nbt.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kishimoto TK, Ferrari JD, La Mothe RA, Kolte PN, Griset AP, O'Neil C, Chan V, Browning E, Chalishazar A, Kuhlman W, Fu FN, Viseux N, Altreuter DH, Johnston L, AMR Improving the efficacy and safety of biologic drugs with tolerogenic nanoparticles. Nat Nanotechnol. 2016;11:890–899. doi: 10.1038/nnano.2016.135. [DOI] [PubMed] [Google Scholar]
- 130.Maldonado RA, LaMothe RA, Ferrari JD, Zhang AH, Rossi RJ, Kolte PN, Griset AP, O'Neil C, Altreuter DH, Browning E, Johnston L, Farokhzad OC, Langer R, Scott DW, von Andrian UH, Kishimoto TK. Polymeric synthetic nanoparticles for the induction of antigen-specific immunological tolerance. Proc Natl Acad Sci U S A. 2015;112:E156–E165. doi: 10.1073/pnas.1408686111. [DOI] [PMC free article] [PubMed] [Google Scholar]