

Abstract
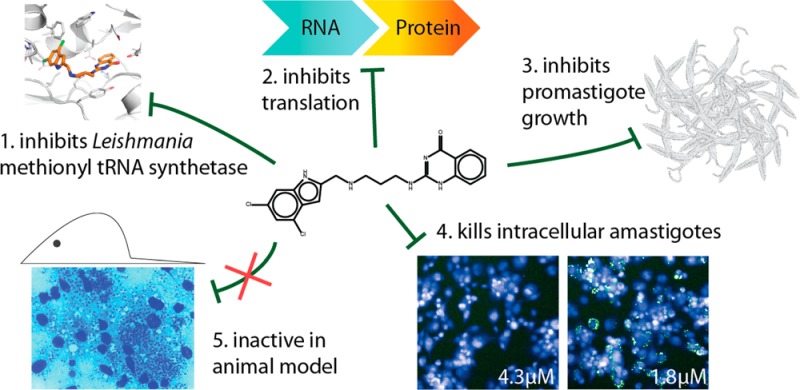
Methionyl-tRNA synthetase (MetRS) has been chemically validated as a drug target in the kinetoplastid parasite Trypanosoma brucei. In the present study, we investigate the validity of this target in the related trypanosomatid Leishmania donovani. Following development of a robust high-throughput compatible biochemical assay, a compound screen identified DDD806905 as a highly potent inhibitor of LdMetRS (Ki of 18 nM). Crystallography revealed this compound binds to the methionine pocket of MetRS with enzymatic studies confirming DDD806905 displays competitive inhibition with respect to methionine and mixed inhibition with respect to ATP binding. DDD806905 showed activity, albeit with different levels of potency, in various Leishmania cell-based viability assays, with on-target activity observed in both Leishmania promastigote cell assays and a Leishmania tarentolae in vitro translation assay. Unfortunately, this compound failed to show efficacy in an animal model of leishmaniasis. We investigated the potential causes for the discrepancies in activity observed in different Leishmania cell assays and the lack of efficacy in the animal model and found that high protein binding as well as sequestration of this dibasic compound into acidic compartments may play a role. Despite medicinal chemistry efforts to address the dibasic nature of DDD806905 and analogues, no progress could be achieved with the current chemical series. Although DDD806905 is not a developable antileishmanial compound, MetRS remains an attractive antileishmanial drug target.
Keywords: drug discovery, kinetoplastids, translation, tRNA synthetase, parasite
Kinetoplastid parasites of the Leishmania species are responsible for leishmaniasis, a neglected tropical disease prevalent in 98 countries, with 350 million people at risk.1 These protozoan parasites are transmitted by the bite of phlebotomine sandflies leading to the development of visceral, cutaneous, or mucocutaneous leishmaniasis; the former of which is fatal if untreated.1 As with most neglected diseases, the treatment options currently available suffer from limitations, including high cost, host toxicity, emerging drug resistance, and suboptimal dosing regimens, leading to a pressing need to discover new therapeutics.2,3
In the search for new antileishmanial therapeutics, we have adopted a balanced approach to drug discovery, with both phenotypic screening campaigns and target-based approaches providing the best opportunity to discover a range of new chemical matter.4,5 A particular challenge associated with target-based approaches is a lack of fully validated drug targets in Leishmania.1 Targets are therefore selected on the basis of those involved in highly essential biological pathways and extrapolation of validation data from related trypanosomatid parasites. Methionyl-tRNA synthetase (MetRS; EC 6.1.1.10) meets both criteria as this enzyme plays a crucial role in protein synthesis,6 a fundamental pathway in all organisms, and has also been validated as a druggable target in the closely related Trypanosoma brucei parasite, with inhibitors of T. brucei MetRS shown to cure T. brucei bloodstream infections in a mouse model of human African trypanosomiasis.7−10 As a family, tRNA synthetases have also been shown to be good targets in the anti-infectives space.11−13 MetRS was therefore prioritized as a target for entry into a Leishmania drug discovery program.
MetRS catalyzes the synthesis of methionyl-tRNA in a two-step reaction; the first step results in the production of a methionyl-AMP intermediate and pyrophosphate, with the second step resulting in the production of methionyl-tRNA and AMP.14 This enzyme is highly conserved among kinetoplastid parasites (both T. brucei and Trypanosoma cruzi MetRS are 76% similar to the Leishmania enzyme at the amino acid level). A key challenge in therapeutically targeting a well conserved enzyme is achieving selectivity over the homologous human protein. As Leishmania MetRS is more closely related to the human mitochondrial MetRS (81% sequence identity in catalytic pocket) compared to the human cytoplasmic enzyme (41% identity in catalytic pocket), assessing selectivity over the human mitochondrial form, as well as human protein synthesis, will be important in a drug discovery program.
In the present study, the validity of MetRS as a target in Leishmania donovani was investigated. Extrapolation of data from T. brucei suggests the L. donovani enzyme will also be an essential and druggable target, with development of a biochemical, high-throughput compatible screening assay possible.7,8,15,42 In addition, the availability of several downstream tools provides an effective route for characterizing and progressing any MetRS inhibitors identified. These include T. brucei and Leishmania major MetRS crystal structures16,17 which provide powerful tools for cocrystallizing any inhibitors identified, thus providing insight into the binding mode with the target. In addition, several phenotypic cell-based screens are available and are routinely used as part of the Leishmania drug discovery pipeline.18 Such assays include the use of free-living promastigote or axenic amastigote parasites (from the insect stage and mammalian stage of the Leishmania life cycle, respectively) and the more complex, but more physiologically relevant, intracellular amastigote assay.19−23 Furthermore, an in vivo model of leishmaniasis allows progression of molecules through to a recognized animal model of this neglected disease.24
Here, we describe the identification of L. donovani MetRS (LdMetRS) inhibitors, characterize the binding mode of our lead molecule using crystallographic and biochemical methods, and confirm on-target activity in cells. Unfortunately, our lead molecule did not show efficacy in our leishmaniasis animal model; we include discussions around potential explanations for this failure to translate.
Results and Discussion
Identification of the LdMetRS Inhibitor DDD806905
Recombinant LdMetRS was produced, and a biochemical enzyme assay, using the BIOMOL Green assay platform, was developed to monitor the first step of the MetRS reaction, namely, the reaction of methionine and ATP to produce methionyl-AMP and pyrophosphate. Assay conditions were refined to ensure optimal screening conditions were employed. This involved assessing buffer conditions and determining the optimal enzyme concentration, assay linearity with respect to time, and Michaelis constants for methionine and ATP (Figure 1A–D). The apparent Km for methionine was determined to be 173 ± 83 μM (with a Hill slope of 0.57 ± 0.06) and 37 ± 3 μM for ATP (with ATP also displaying substrate inhibition with an apparent Ki of 482 ± 44 μM)). The Hill slope of 0.57 observed with methionine can be indicative of negative cooperativity between two subunits of a dimeric protein as observed for MetRS from Bacillus stearothermophilus.25 However, analysis of the oligomeric state of the protein using SEC-MALS (size exclusion chromatography–multi-angle light scattering) suggests the protein is monomeric (Figure S1), supporting previously published crystallography data showing that the L. major crystal structure is monomeric.17 An alternative explanation for the low Hill slope could be conformational selection as previously observed for inhibitors binding to the T. brucei MetRS enzyme.26 In this model, two conformations of the ligand-free enzyme exist, with differing affinities for ligands and different kcat values.27
Figure 1.

LdMetRS assay development summary. (A) Linearity of the assay with respect to enzyme concentration. (B) Methionine Km determination in the presence of a saturating concentration of ATP (200 μM). Solid line is best fit to Hill equation and dotted line to Michaelis–Menten equation. (C) ATP Km determination in the presence of a saturating concentration of methionine (1 mM). (D) Assay linearity with respect to time under the final assay screening conditions of 50 μM methionine and 100 μM ATP either with (closed circles) or without (open circles) 50 nM LdMetRS enzyme. Data are shown as mean ± SD (n = 3 technical replicates).
Using final assay conditions of 50 nM LdMetRS, 50 μM methionine, and 100 μM ATP, a panel of compounds closely related to known inhibitors of bacterial MetRS28 (24 members of a 2-amino benzimidazole series, along with a singleton 2-aminoquinazolin-4-one) was screened. These compounds inhibited the LdMetRS enzyme with a range of potencies (IC50 values of 94 nM to 100 μM) (Table S1), the most active compound being the 2-aminoquinazolin-4-one singleton DDD806905 (IC50 of 94 nM (95% CI, 57–156 nM)) (Figure 2A).
Figure 2.

DDD806905 binding mode. (A) DDD806905 was identified as our lead LdMetRS inhibitor, with an IC50 of 94 nM. (B) Crystal structure of TbMetRS:DDD806905 (PDB 5NFH). DDD806905 bridges the expanded methionine pocket (EMP) and the ligand stabilized auxiliary pocket (AP). The dichloroindole moiety occupies the same site as methionine (C atoms gray). The solvent accessible surface of TbMetRS:DDD806905 is shown in dark green. (C) Comparison of the binding mode of DDD806905 (C atoms gold) compared to TbMetRS:methionyl adenylate (MAMP, C atoms gray, PDB 4EG3). (D) Binding mode of TbMetRS:DDD806905 showing protein side chains that line the binding site. (E) H-bond interactions between quinazolinone moiety of DDD806905 and residues lining the auxiliary pocket. H-bond interactions are shown as dashed lines and water molecules as red spheres, and key residues are labeled. (F) The binding modes of DDD806905 (C atoms gold) compared with aminoquinolone ligand Chem 1312 PDB 4EG5 (C atoms green). (G) Sequence conservation is high between TbMetRS and LdMetRS around the DDD806905 binding site with identical residues colored red and nonidentical colored gray.
The IC50 value for DDD806905 determined in this screening assay was close to the enzyme concentration used in the reaction, meaning DDD806905 was potentially displaying tight binding inhibition under the assay conditions employed. To more accurately define the potency of DDD806905, it is more appropriate to fit the dose response data to the Morrison equation29 (eq 3 in the Supporting Information). Prior to fitting data to the Morrison equation, an accurate determination of the active enzyme concentration is required. This was achieved by titrating the LdMetRS enzyme in the presence of 1 μM DDD806905 (Figure S2A), revealing that 78% of the LdMetRS protein sample is catalytically active. This resulted in an LdMetRS active enzyme concentration of 39 nM. Using this value as a constant and fitting dose response data to the Morrison equation revealed a DDD806905 Kiapp of 41 nM (Figure S2B).
Characterization of DDD806905 Binding and Mode of Inhibition
Determination of the binding mode of the aminoquinazolinone inhibitor DDD806905 was carried out using both crystallographic and biochemical methods. For crystallographic determination of the binding mode, T. brucei MetRS (TbMetRS) protein was used as a structural surrogate (Table S2). Despite an overall sequence identity of only 76% between LdMetRS and the TbMetRS, the residues lining and surrounding the ligand binding site are highly conserved (Figure S3) with a sequence identity of 95%. The only sequence difference within the site is a conservative valine to leucine substitution (TbMetRS Leu 456); therefore, TbMetRS is a valid structural system to understand the mode of action for DDD806905 against LdMetRS.
Previous studies have shown that the binding of aminoquinolone inhibitors stabilize an open conformation of TbMetRS with expansion of the methionine binding pocket and opening of an auxiliary binding pocket.16 The TbMetRS:DDD806905 complex shows the ligand to bridge the auxiliary pocket (AP) and expanded methionine pocket (EMP) of the enzyme (Figure 2B). Overlaying the binding mode of DDD806905 with the intermediate methionyl adenylate (PDB 4EG3)26 shows the linker between the dichloro indole and quinazolinone groups would block the binding of the beta phosphate group of ATP. The ligand stabilized opening of the AP involves a change in rotomer of Tyr250, a consequence of which is the widening of the ATP binding cleft which may reduce the affinity for ATP (Figure 2C). The side chains lining the DDD806905 binding site are largely hydrophobic in nature (Figure 2D) with limited polar interactions. The quinazolinone moiety occupies the AP pocket retaining key bidentate H-bonds between the quinazolinone NH and the amino NH of Asp287 (Figure 2E). The carbonyl moiety forms an H-bond to a water molecule coordinated by the side chain of Tyr250 and the backbone NH of Lys292. The 4,6-dichloro indole moiety occupies the EMP pocket with the 6-chloro atom overlaying with the substrate methionine S atom (Figure 2C). The indole NH does not make any specific interactions with protein. The binding mode of DDD806905 is similar to the published aminoquinolone complex (PDB 4EG5) (Figure 2F) with a root-mean-square deviation of 0.22 Å for the position of all residues within 5 Å of the ligand between TbMetRS:DDD806905 and the aminoquinolone complex 4EG5. The residues that form the ligand binding site and those that facilitate the conformational change in the CP domain are highly conserved between TbMetRS and LdMetRS (Figures 2G and S3), confirming that TbMetRS is a valid model system to understand the binding mode of DDD806905.
Further characterization of the mode of inhibition of DDD806905 was carried out biochemically by varying each substrate concentration at various inhibitor concentrations. Each data set was individually fitted to the Michaelis–Menten equation or a modified high-substrate inhibition Michaelis–Menten equation, and the resulting Lineweaver–Burk plots were examined for diagnostic patterns of competitive, mixed, or uncompetitive inhibition.
Data from these analyses indicate that DDD806905 displays competitive inhibition with respect to methionine binding (Figure 3A), and further fitting to a global competitive inhibition model (eq 4 in the Supporting Information) resulted in a DDD806905 Ki of 18 ± 2 nM (Ki ± SE) being returned. This biochemically determined methionine competitive behavior confirmed the crystallographic data which revealed DDD806905 binds to the methionine pocket of MetRS.
Figure 3.

DDD806905 mode of inhibition. (A) LdMetRS biochemical mode of inhibition studies were carried out with reciprocal plots of A650 and substrate concentration revealing that DDD806905 displays competitive inhibition with respect to methionine with a Ki of 18 nM calculated. (B) Mode of inhibition studies also revealed that DDD806905 displays mixed inhibition with respect to ATP with a Ki of 21 nM calculated. Inhibitor concentrations used were as follows: 2 × IC50 (closed circles), 1 × IC50 (open circles), 0.5 × IC50 (closed triangles), and 0 × IC50 (open triangles).
This DDD806905 mode of inhibition study was repeated using ATP as the variable substrate. Fitting these data sets to the modified high-substrate inhibition Michaelis–Menten equation and plotting a double-reciprocal plot of the data revealed the characteristic substrate inhibition profile shown in Figure 3B (solid lines). To allow global fitting, we excluded the substrate concentrations at which substrate inhibition was observed. The remaining data were fitted to a mixed inhibition model (eq 5 in Supporting Information). It can be seen that DDD806905 displays characteristic mixed inhibition with respect to ATP binding (Figure 3B, dashed lines). This ATP mode of inhibition study returned a DDD806905 Ki of 21 ± 5 nM (Ki ± SE), which is highly comparable to the Ki calculated from the methionine competitive inhibition experiment previously described.
From the ATP study, it should also be noted that the Ki′ value of 227 ± 23 nM indicates DDD806905 binds preferentially to the free enzyme and the inhibitor profile is more closely related to competitive inhibition. This supports kinetic data which reveals there is positive cooperativity between the methionine and ATP pockets (methionine Km shifts from 170 to 1500 μM when either 400 μM ATP (10 × Km) or 80 μM ATP (2 × Km) is used, respectively (Figure S4)), a feature also observed in MetRS from E. coli, B. stearothermophilus, and S. aureus.30−32 Consequently, alterations in ATP concentration can impact both binding of methionine and DDD806905 in the methionine pocket. This interaction between ATP and methionine sites is further confirmed from inhibitor studies using different substrate concentrations. These show that the IC50 of DDD806905 increases when either methionine or ATP concentrations are increased suggesting that both substrates have “competitive like” profiles (Figure S5A–C).
DDD806905 Inhibits in Vitro Translation
Inhibitors of the LdMetRS enzyme would be expected to inhibit protein synthesis. To confirm whether this was the case, DDD806905 was tested for its ability to inhibit protein synthesis in a commercially available Leishmania tarentolae cell extract.33−35 Initial experiments revealed that production of an eGFP protein from a plasmid containing the eGFP gene can be monitored in this system using a 384-well assay format and a standard plate-based reader (Figure 4A).
Figure 4.

DDD806905 inhibits Leishmania in vitro translation. (A) In vitro translation in a Leishmania tarentolae extract was monitored over time by tracking expression of an eGFP construct (closed circles), with a “minus construct” negative control included (open circles). (B) The ability of DDD806905 to inhibit expression of eGFP in the L. tarentolae extract was investigated with this LdMetRS inhibitor inhibiting protein synthesis with an EC50 of 2.2 μM (closed circles). In the presence of an additional 1.5 mM methionine, the EC50 was shifted to 12 μM (open circles), indicative of on-target activity, as DDD806905 is a known methionine competitive inhibitor of LdMetRS. (C) In vitro translation in a HeLa cell extract was also monitored by tracking expression of a GFP construct (closed circles) over time, with “minus construct” (closed triangles) and cycloheximide (protein synthesis inhibitor) controls (open circles) included. When DDD806905 was included at a concentration of 100 μM (open triangles), no inhibition of in vitro translation was observed in this human cell extract. Data are shown as mean ± SD (n = 3 technical replicates (cycloheximide data, n = 2 technical replicates)).
Subsequently, the ability of DDD806905 to inhibit production of this eGFP protein was determined, with DDD806905 shown to inhibit protein synthesis with an EC50 of 2.2 μM (Figure 4B, closed circles). This potency is considerably weaker than that observed in the biochemical enzyme assay, where DDD806905 returned an IC50 of 94 nM. One possible reason for this 23-fold drop off in potency is the difference in substrate concentrations present in the respective assays (the biochemical assay was run using methionine and ATP concentrations of 50 and 100 μM, respectively, whereas the L. tarentolae cell extracts were supplemented with 136 μM methionine and 1200 μM ATP in addition to their endogenous concentrations). As described above, the potency of DDD806905 is seen to shift in the biochemical assay when substrate concentrations are increased, accounting, at least in part, for the different potencies observed in the biochemical assay and L. tarentolae cell extract.
To demonstrate that inhibition of translation in the L. tarentolae cell extract was indeed through inhibition of MetRS, the potency of DDD806905 in the protein synthesis assay was determined in the presence and absence of an additional 1.5 mM methionine. When extra methionine is present in the assay, there is a 5-fold shift in DDD806905 potency (EC50 shift from 2.2 to 12 μM), highly indicative of on-target activity against MetRS (Figure 4B).
It was interesting to note that, when DDD806905 was tested in a similar in vitro translation assay using a human HeLa cell extract, no inhibition of protein synthesis was observed (Figure 4C). Although this observation does not prove selectivity over the more closely related human mitochondrial MetRS enzyme, it does indicate that DDD806905 fails to inhibit the human cytoplasmic MetRS enzyme when tested at a concentration of 100 μM. To investigate potential mitochondrial toxicity, we carried out a mitochondrial protein synthesis assay which revealed some inhibition of mitochondrial protein synthesis with an IC50 of 1.7 μM (95% CI, 1.0–2.8 μM) (Figure S6). In addition, DDD806905 showed toxicity against Leishmania infected, PMA-differentiated THP-1 cells (THP-1 EC50, 10 μM). Reducing mammalian cell toxicity and the level of mitochondrial protein synthesis inhibition would therefore have to be an important goal of a drug development program.
Cellular and in Vivo Efficacy of DDD806905
To investigate whether DDD806905 has antileishmanial activity, it was tested in a L. donovani intracellular amastigote assay, where an EC50 of 2.9 μM (95% CI, 2.2–3.8 μM; n = 8) was determined (Figure S7). Next, we progressed DDD806905 to an in vivo mouse visceral leishmaniasis efficacy model. Unfortunately, no efficacy was observed (Figure 5), with only a 19% knock-down of liver parasite counts observed after 50 mg/kg, twice daily oral dosing for 10 days, compared to vehicle dosed control animals. In contrast, the clinically used control compound miltefosine displayed 99.6% parasite knock-down following 30 mg/kg, once daily oral dosing for 10 days. Blood levels of DDD806905 were measured during the efficacy study and showed that drug total levels were maintained above the MIC (minimum inhibitory concentration = 4.3 μM) measured in the intracellular assay for at least 8 h after dosing (Figure S8A,B). However, due to very high plasma protein binding (fu = 0.006 in mouse plasma and 0.007 in human plasma), blood free levels of DDD806905 were significantly below the in vitro MIC. To gain insight into the importance of protein binding, we performed an in vitro serum shift assay with DDD806905 against Leishmania promastigotes which revealed the expected linear correlation between promastigote EC50 and amount of protein in the media (Figure S9).36 Since in vivo efficacy is usually also driven by the drug free level, the very high plasma protein binding is likely a key reason for the lack of activity for DDD806905 in our visceral leishmaniasis model. Interestingly, a related MetRS inhibitor was also shown not to be curative in an animal model of human African sleeping sickness in spite of having promising in vitro cell activity.7
Figure 5.

DDD806905 mouse visceral leishmaniasis efficacy study with integrated pharmacokinetics. Efficacy of DDD806905 was assessed in a mouse model of visceral leishmaniasis at 50 mg/kg b.i.d. for 10 days, along with vehicle (b.i.d. for 10 days) and miltefosine (30 mg/kg q.d. for 10 days) controls. Mean reduction in liver parasite burden expressed as Leishman Donovan Units (LDU). Miltefosine revealed a 99.6% reduction in liver LDU compared to vehicle control (***p < 0.001), with DDD806905 showing only a 19.4% reduction in liver LDU (integrated pharmacokinetics results shown in Figure S8).
Efficacy of MetRS Inhibition in Promastigotes and Axenic Amastigotes and On-Target Activity
The above experiments showed that DDD806905 was around 10-fold more potent against Leishmania promastigotes compared to intracellular amastigotes (EC50-pro of 0.27 μM (95% CI, 0.25–0.29 μM) (in 10% serum), EC50-intracellular of 2.9 μM (95% CI, 2.2–3.8 μM)). The compound was also found to be 50-fold less active against axenic amastigotes relative to promastigotes (EC50-axam of 13.7 μM (95% CI, 10.5–17.6 μM)). To assess on-target activity and further explore potential life-cycle stage differences in susceptibility, a series of related 2-amino benzimidazoles with varying degrees of LdMetRS potency were tested for their ability to inhibit Leishmania parasite growth, using both promastigote and axenic amastigote Leishmania viability assays (Table S1). As for DDD806905, the compounds in this panel showed a large drop in potency from promastigotes to amastigotes. A reasonable correlation is observed between enzyme data and L. donovani promastigote data (Figure 6A), suggestive of on-target activity, whereas these same compounds showed low, or no, inhibition in the L. donovani axenic amastigote assay (Figure 6B).
Figure 6.

Cellular efficacy of LdMetRS inhibition. (A) A panel of LdMetRS inhibitors show a range of potencies (IC50 94 nM to 100 μM) in the LdMetRS enzymatic assay. Plotting the −log IC50 (pIC50) of this enzymatic data against −log EC50 (pEC50) data from the L. donovani promastigote assay reveals these potencies correlate well. Solid line represents linear regression with a correlation coefficient of 0.76. (B) When the same compounds are tested in the L. donovani axenic amastigote assay, most are inactive. In both (A) and (B), dashed lines represent equipotency in the LdMetRS enzyme assay and the Leishmania phenotypic assay. (C) Confirmation of on-target activity of DDD806905 in L. donovani promastigotes was carried out by testing this compound in the absence (closed circles) and presence (open circles) of excess methionine (2 mM). The EC50 shifts from 0.46 to 1.9 μM in the absence and presence of excess methionine, respectively. Data presented as mean ± SD (n = 3 biological replicates).
While the correlation between enzyme data and promastigote data is indicative of on-target activity, further studies attempted to confirm whether this was the case. Initial efforts to confirm the on-target effect of DDD806905 were explored by attempting to generate MetRS overexpressing and knockout Leishmania cell lines. All efforts to modulate MetRS expression levels in L. donovani proved to be toxic to the parasite, suggesting that tight regulation of this enzyme is important to parasite survival, supporting the validation of MetRS as a highly essential target in Leishmania.
In the absence of MetRS overexpressing or knockout cell lines, alternative methods were used to determine on-target inhibition of MetRS in the parasite. As with the in vitro translation system, DDD806905 was tested in a Leishmania promastigote assay in both the presence and absence of extra methionine. The EC50 of this compound shifted from 0.46 to 1.9 μM after addition of extra methionine, highly indicative of on-target activity (Figure 6C).
Properties of DDD806905 Accounting for Potency Differences between Life-Cycle Stages
To account for the discrepancy in data from the different Leishmania parasite assays, it is noteworthy that these cell assays were performed in media at different pH values (with promastigote and axenic amastigote assays run in media at pH 7.3 and pH 5.6, respectively) and that in the intracellular amastigote assay the parasites reside inside the acidic parasitophorous vacuole. We experimentally determined the pKa’s for DDD806905 (Figure S10A) (pKa of 3.4, 7.5 and 11), and the data shows that, at pH 7.3, 0.01% of the compound is present in an un-ionized state. This falls to essentially 0% at pH 5.6. In addition, the lipophilicity of the compound was significantly reduced at pH 5.6 (measured log D5.6 of 0.3) compared with the lipophilicity at pH 7.3 (measured log D7.3 of 2.6) (Figure S10B). The lower total level of uncharged, membrane permeable, compound in acidic axenic amastigote media compared to neutral promastigote media will result in a lower intracellular compound concentration, providing a potential explanation for the reduced potency in this assay. In the intracellular assay, the parasites also reside in an acidic compartment, but the total acidic volume is much smaller in this setting (volume of acidic organelles only versus volume of media and acidic organelles in the axenic assay), resulting in a higher fraction of uncharged, membrane permeable compound under intracellular conditions.
Attempts were made to increase the fraction of nonionized species by chemically modifying DDD806905 to reduce its basicity. Unfortunately, all attempts to test this hypothesis by modifying the benzylic amine resulted in significant loss of activity against the enzyme (Table 1). In particular, converting the pendant amine to the corresponding amide, i.e., DDD806905 cf. compound 25 (also compound 26 cf. compound 27) or sulphonamide, i.e., compound 26 cf. compound 28 reduced enzyme activity. Similarly, removal of the basic nitrogen by replacement with methylene, i.e., compound 29 cf. compound 26, removed enzyme activity. In addition, substitution of the 2-nitrogen for an oxygen (i.e., DDD806905 cf. compound 31) was not tolerated. Methylating the benzylic amine (compound 30), which is not expected to change basicity, was tolerated, but it did not improve cellular activity.
Table 1. Enzyme Inhibition and Cellular Data for DDD806905 and Analogues.

Therefore, despite medicinal chemistry efforts to reduce the basicity of DDD806905, the SAR suggests that the basic nitrogen is required for activity and DDD806905 remains our lead LdMetRS inhibitor. These results support the hypothesis that the lack of activity seen in the axenic amastigote assay is not due to MetRS being a poor drug target but rather due to poor drug partitioning into axenic amastigotes resulting from the highly protonated state of the active compounds in acidic media.
DDD806905 is Lysosomotropic
Due to its highly protonated state under acidic conditions, it is conceivable that DDD806905 is accumulating in acidic cellular compartments, which could help explain the lower potency in the intracellular assay relative to the promastigote assay and may contribute to the lack of activity in the animal efficacy study. To assess this, we carried out a lysosomal sequestration assay.37,38 The results in Figure 7 reveal that DDD806905 indeed shows the hallmarks of lysosomal accumulation/trapping, whereas, as expected, the nonbasic sulphonamide analogue compound 28 does not. We also confirmed the well-known lysosomal accumulation of the antimalarial drug chloroquine.39 For chloroquine, this is essential as its mode of action depends on accumulation in the acidic food vacuoles of the malaria parasite. In our intracellular Leishmania assay, the host cells present a relatively large lysosomal compartment,40,41 and on the basis of the data presented here, we propose that the THP-1 lysosomes act as a sink for DDD806905, thus reducing the amount of free, membrane permeable compound available to reach the parasites, hence contributing to the potency drop-off seen between promastigotes and intracellular amastigotes. We also measured the volume of distribution (Vdss) for DDD806905 in mice following a single intravenous dose and found it to be high (6 L/kg), indicating accumulation of the compound in tissues, which is in line with the lysosomotropism observed in vitro.
Figure 7.

DDD806905 accumulates in acidic compartments. Ability of compounds to compete with lysotracker red accumulation in THP-1 lysosomes was assessed and revealed that both DDD806905 (open circles) and a positive control compound chloroquine (closed circles) did compete with lysotracker red accumulation in this acidic compartment. Representative data shown (mean ± SD, n = 3 biological replicates). In contrast to DDD806905 and chloroquine, compound 28 (open triangles) does not compete with lysotracker red for accumulation in THP-1 lysosomes. Representative data shown (mean ± SD, n = 2 biological replicates).
Conclusions
We have successfully set up an assay for LdMetRS and established that analogues of bacterial and T. brucei MetRS inhibitors inhibit the Leishmania enzyme. These compounds almost certainly bind in a very similar manner to the Leishmania enzyme as they do to the T. brucei enzyme. We have also demonstrated that these compounds are active against L. donovani promastigotes. Taken together, the correlation between enzyme activity and promastigote activity (Figure 6A), on-target activity in an in vitro protein translation assay (Figure 4B), the cocrystal structure of DDD806905 with MetRS, and the competition experiment with methionine (Figure 6C) provide compelling evidence that the compounds are working on target in promastigotes. However, the compounds showed lower activity against the intramacrophage form of the parasite and no in vivo efficacy. On the basis of the results presented here, the most likely reason for the lack of in vivo efficacy of DDD806905 is its very small free and membrane permeable fraction due to a combination of high protein binding, ionization, and accumulation in acidic compartments. Although this compound has failed to translate into in vivo efficacy, this does not invalidate MetRS as a potential drug target in Leishmania. Indeed, MetRS remains an attractive drug target in this parasite, and further efforts to identify novel starting chemical matter against this enzyme target are underway.
Materials and Methods
Materials and methods can be found in the Supporting Information. These include experimental procedures for LdMetRS and TbMetRS expression and purification; LdMetRS biochemical assays; TbMetRS crystallography; Leishmania and human in vitro translation assays; in vitro Leishmania assays; mouse efficacy study and integrated PK; in vitro physicochemical measurements; chemical synthesis of compounds.
All human biological samples were sourced ethically, and their research use was in accord with the terms of the informed consents. All animal studies were ethically reviewed and carried out in accordance with Animals (Scientific Procedures) Act 1986 and the GSK Policy on the Care, Welfare and Treatment of Animals.
Acknowledgments
We would like to acknowledge the Wellcome Trust for funding (Grants 092340, 105021, 100476). We would like to acknowledge GSK for provision of the TCAMS (Tres Cantos Anti-Malarial Set) for screening against the kinetoplastids and some additional bacterial MetRS inhibitors. We also acknowledge the support of the Protein Production Team in the Division of Biological Chemistry and Drug Discovery (Dundee), the Drug Discovery Unit compound management, and Drug Discovery Unit data management teams. The authors would like to thank Diamond Light Source for beamtime (proposal mx8268) and the staff of beamlines I02 and I04 for assistance with data collection.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsinfecdis.7b00047.
Determination of the oligomeric state of LdMetRS; DDD806905 Kiapp determination; sequence alignment of TbMetRS and LdMetRS; co-operativity between ATP and methionine binding pockets; DDD806905 IC50 under different substrate conditions; DDD806905 inhibiting mitochondrial protein synthesis; DDD806905 Leishmania intracellular amastigote EC50 determination; DDD806905 integrated pharmacokinetics for mouse visceral leishmaniasis efficacy study; DDD806905 EC50 in Leishmania promastigotes using varying serum concentrations; pH effect of ionization and lipophilicity of DDD806905; enzymatic and phenotypic potencies of LdMetRS inhibitors; data measurement and refinement statistics; materials and methods with associated references (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Bilbe G. (2015) Infectious Diseases. Overcoming Neglect of Kinetoplastid Diseases. Science 348, 974–976. 10.1126/science.aaa3683. [DOI] [PubMed] [Google Scholar]
- Croft S. L.; Olliaro P. (2011) Leishmaniasis Chemotherapy--Challenges and Opportunities. Clin. Microbiol. Infect. 17, 1478–1483. 10.1111/j.1469-0691.2011.03630.x. [DOI] [PubMed] [Google Scholar]
- Croft S. L.; Sundar S.; Fairlamb A. H. (2006) Drug Resistance in Leishmaniasis. Clin Microbiol Rev. 19, 111–126. 10.1128/CMR.19.1.111-126.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reguera R. M.; Calvo-Alvarez E.; Alvarez-Velilla R.; Balana-Fouce R. (2014) Target-Based vs. Phenotypic Screenings in Leishmania Drug Discovery: A Marriage of Convenience or a Dialogue of the Deaf?. Int. J. Parasitol.: Drugs Drug Resist. 4, 355–357. 10.1016/j.ijpddr.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert I. H. (2013) Drug discovery for neglected diseases: molecular target-based and phenotypic approaches. J. Med. Chem. 56, 7719–7726. 10.1021/jm400362b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham J. S.; Dawson K. L.; Jackson K. E.; Lim E. E.; Pasaje C. F.; Turner K. E.; Ralph S. A. (2014) Aminoacyl-tRNA synthetases as drug targets in eukaryotic parasites. Int. J. Parasitol.: Drugs Drug Resist. 4, 1–13. 10.1016/j.ijpddr.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata S.; Gillespie J. R.; Kelley A. M.; Napuli A. J.; Zhang Z.; Kovzun K. V.; Pefley R. M.; Lam J.; Zucker F. H.; Van Voorhis W. C.; Merritt E. A.; Hol W. G.; Verlinde C. L.; Fan E.; Buckner F. S. (2011) Selective Inhibitors of Methionyl-tRNA Synthetase have Potent Activity Against Trypanosoma brucei Infection in Mice. Antimicrob. Agents Chemother. 55, 1982–1989. 10.1128/AAC.01796-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata S.; Gillespie J. R.; Ranade R. M.; Koh C. Y.; Kim J. E.; Laydbak J. U.; Zucker F. H.; Hol W. G.; Verlinde C. L.; Buckner F. S.; Fan E. (2012) Urea-Based Inhibitors of Trypanosoma brucei Methionyl-tRNA Synthetase: Selectivity and In Vivo Characterization. J. Med. Chem. 55, 6342–6351. 10.1021/jm300303e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranade R. M.; Gillespie J. R.; Shibata S.; Verlinde C. L.; Fan E.; Hol W. G.; Buckner F. S. (2013) Induced Resistance to Methionyl-tRNA Synthetase Inhibitors in Trypanosoma brucei is Due to Overexpression of the Target. Antimicrob. Agents Chemother. 57, 3021–3028. 10.1128/AAC.02578-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devine W. G.; Diaz-Gonzalez R.; Ceballos-Perez G.; Rojas D.; Satoh T.; Tear W.; Ranade R. M.; Barros-Alvarez X.; Hol W. G.; Buckner F. S.; Navarro M.; Pollastri M. P. (2017) From Cells to Mice to Target: Characterization of NEU-1053 (SB-443342) and Its Analogues for Treatment of Human African Trypanosomiasis. ACS Infect. Dis. 3, 225–236. 10.1021/acsinfecdis.6b00202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.; Lee S. W.; Choi E. C.; Choi S. Y. (2003) Aminoacyl-tRNA Synthetases and their Inhibitors as a Novel Family of Antibiotics. Appl. Microbiol. Biotechnol. 61, 278–288. 10.1007/s00253-003-1243-5. [DOI] [PubMed] [Google Scholar]
- Ochsner U. A.; Sun X.; Jarvis T.; Critchley I.; Janjic N. (2007) Aminoacyl-tRNA Synthetases: Essential and Still Promising Targets for New Anti-Infective Agents. Expert Opin. Invest. Drugs 16, 573–593. 10.1517/13543784.16.5.573. [DOI] [PubMed] [Google Scholar]
- Saint-Leger A.; Sinadinos C.; Ribas de Pouplana L. (2016) The Growing Pipeline of Natural Aminoacyl-tRNA Synthetase Inhibitors for Malaria Treatment. Bioengineered 7, 60–64. 10.1080/21655979.2016.1149270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perona J. J.; Gruic-Sovulj I. (2013) Synthetic and Editing Mechanisms of Aminoacyl-tRNA Synthetases. Top. Curr. Chem. 344, 1–41. 10.1007/128_2013_456. [DOI] [PubMed] [Google Scholar]
- Pedro-Rosa L.; Buckner F. S.; Ranade R. M.; Eberhart C.; Madoux F.; Gillespie J. R.; Koh C. Y.; Brown S.; Lohse J.; Verlinde C. L.; Fan E.; Bannister T.; Scampavia L.; Hol W. G.; Spicer T.; Hodder P. (2015) Identification of Potent Inhibitors of the Trypanosoma brucei Methionyl-tRNA Synthetase Via High-Throughput Orthogonal Screening. J. Biomol. Screening 20, 122–130. 10.1177/1087057114548832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cestari I.; Stuart K. (2013) A spectrophotometric assay for quantitative measurement of aminoacyl-tRNA synthetase activity. J. Biomol. Screening 18 (4), 490–497. 10.1177/1087057112465980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh C. Y.; Kim J. E.; Wetzel A. B.; de van der Schueren W. J.; Shibata S.; Ranade R. M.; Liu J.; Zhang Z.; Gillespie J. R.; Buckner F. S.; Verlinde C. L.; Fan E.; Hol W. G. (2014) Structures of Trypanosoma brucei Methionyl-tRNA Synthetase with Urea-Based Inhibitors Provide Guidance for Drug Design Against Sleeping Sickness. PLoS Neglected Trop. Dis. 8, e2775. 10.1371/journal.pntd.0002775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson E. T.; Kim J. E.; Zucker F. H.; Kelley A.; Mueller N.; Napuli A. J.; Verlinde C. L.; Fan E.; Buckner F. S.; Van Voorhis W. C.; Merritt E. A.; Hol W. G. (2011) Structure of Leishmania major Methionyl-tRNA Synthetase in Complex with Intermediate Products Methionyladenylate and Pyrophosphate. Biochimie 93, 570–582. 10.1016/j.biochi.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Don R.; Ioset J. R. (2014) Screening Strategies to Identify New Chemical Diversity for Drug Development to Treat Kinetoplastid Infections. Parasitology 141, 140–146. 10.1017/S003118201300142X. [DOI] [PubMed] [Google Scholar]
- De Rycker M.; Hallyburton I.; Thomas J.; Campbell L.; Wyllie S.; Joshi D.; Cameron S.; Gilbert I. H.; Wyatt P. G.; Frearson J. A.; Fairlamb A. H.; Gray D. W. (2013) Comparison of a High-Throughput High-Content Intracellular Leishmania donovani Assay with an Axenic Amastigote Assay. Antimicrob. Agents Chemother. 57, 2913–2922. 10.1128/AAC.02398-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuhs A.; De Rycker M.; Manthri S.; Comer E.; Scherer C. A.; Schreiber S. L.; Ioset J. R.; Gray D. W. (2015) Development and Validation of a Novel Leishmania donovani Screening Cascade for High-Throughput Screening Using a Novel Axenic Assay with High Predictivity of Leishmanicidal Intracellular Activity. PLoS Neglected Trop. Dis. 9, e0004094. 10.1371/journal.pntd.0004094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Muylder G.; Ang K. K.; Chen S.; Arkin M. R.; Engel J. C.; McKerrow J. H. (2011) A Screen Against Leishmania Intracellular Amastigotes: Comparison to a Promastigote Screen and Identification of a Host Cell-Specific Hit. PLoS Neglected Trop. Dis. 5, e1253. 10.1371/journal.pntd.0001253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagley M. J.; Saunders E. C.; Simpson K. J.; McConville M. J. (2015) High-Content Assay for Measuring Intracellular Growth of Leishmania in Human Macrophages. Assay Drug Dev. Technol. 13, 389–401. 10.1089/adt.2015.652. [DOI] [PubMed] [Google Scholar]
- Siqueira-Neto J. L.; Moon S.; Jang J.; Yang G.; Lee C.; Moon H. K.; Chatelain E.; Genovesio A.; Cechetto J.; Freitas-Junior L. H. (2012) An Image-Based High-Content Screening Assay for Compounds Targeting Intracellular Leishmania donovani Amastigotes in Human Macrophages. PLoS Neglected Trop. Dis. 6, e1671. 10.1371/journal.pntd.0001671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hommel M.; Jaffe C. L.; Travi B.; Milon G. (1995) Experimental models for leishmaniasis and for testing anti-leishmanial vaccines. Ann. Trop. Med. Parasitol. 89 (Suppl 1), 55–73. 10.1080/00034983.1995.11813015. [DOI] [PubMed] [Google Scholar]
- Mulvey R. S.; Fersht A. R. (1976) Subunit Interactions in the Methionyl-tRNA Synthetase of Bacillus stearothermophilus. Biochemistry 15, 243–249. 10.1021/bi00647a001. [DOI] [PubMed] [Google Scholar]
- Koh C. Y.; Kim J. E.; Shibata S.; Ranade R. M.; Yu M.; Liu J.; Gillespie J. R.; Buckner F. S.; Verlinde C. L.; Fan E.; Hol W. G. (2012) Distinct States of Methionyl-tRNA Synthetase Indicate Inhibitor Binding by Conformational Selection. Structure 20, 1681–1691. 10.1016/j.str.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ainslie G. R. Jr.; Shill J. P.; Neet K. E. (1972) Transients and Cooperativity. A Slow Transition Model for Relating Transients and Cooperative Kinetics of Enzymes. J. Biol. Chem. 247, 7088–7096. [PubMed] [Google Scholar]
- Critchley I. A.; Young C. L.; Stone K. C.; Ochsner U. A.; Guiles J.; Tarasow T.; Janjic N. (2005) Antibacterial Activity of REP8839, a New Antibiotic for Topical Use. Antimicrob. Agents Chemother. 49, 4247–4252. 10.1128/AAC.49.10.4247-4252.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison J. F. (1969) Kinetics of the Reversible Inhibition of Enzyme-Catalysed Reactions by Tight-Binding Inhibitors. Biochim. Biophys. Acta 185, 269–286. 10.1016/0005-2744(69)90420-3. [DOI] [PubMed] [Google Scholar]
- Green L. S.; Bullard J. M.; Ribble W.; Dean F.; Ayers D. F.; Ochsner U. A.; Janjic N.; Jarvis T. C. (2009) Inhibition of Methionyl-tRNA Synthetase by REP8839 and Effects of Resistance Mutations on Enzyme Activity. Antimicrob. Agents Chemother. 53, 86–94. 10.1128/AAC.00275-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanquet S.; Fayat G.; Waller J. P. (1975) The Amino Acid Activation Reaction Catalyzed by Methionyl-Transfer RNA Synthetase: Evidence for Synergistic Coupling Between the Sites for Methionine Adenosine and Pyrophosphate. J. Mol. Biol. 94, 1–15. 10.1016/0022-2836(75)90401-5. [DOI] [PubMed] [Google Scholar]
- Kalogerakos T.; Dessen P.; Fayat G.; Blanquet S. (1980) Proteolytic Cleavage of Methionyl Transfer Ribonucleic Acid Synthetase from Bacillus stearothermophilus: Effects on Activity and Structure. Biochemistry 19, 3712–3723. 10.1021/bi00557a012. [DOI] [PubMed] [Google Scholar]
- Kovtun O.; Mureev S.; Jung W.; Kubala M. H.; Johnston W.; Alexandrov K. (2011) Leishmania cell-free protein expression system. Methods 55, 58–64. 10.1016/j.ymeth.2011.06.006. [DOI] [PubMed] [Google Scholar]
- Kovtun O.; Mureev S.; Johnston W.; Alexandrov K. (2010) Towards the construction of expressed proteomes using a Leishmania tarentolae based cell-free expression system. PLoS One 5, e14388. 10.1371/journal.pone.0014388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mureev S.; Kovtun O.; Nguyen U. T.; Alexandrov K. (2009) Species-independent translational leaders facilitate cell-free expression. Nat. Biotechnol. 27, 747–752. 10.1038/nbt.1556. [DOI] [PubMed] [Google Scholar]
- Copeland R. A. (2000) Determination of Serum Protein Binding Affinity of Inhibitors from Analysis of Concentration-Response Plots in Biochemical Activity Assays. J. Pharm. Sci. 89, 1000–1007. . [DOI] [PubMed] [Google Scholar]
- Nadanaciva S.; Lu S.; Gebhard D. F.; Jessen B. A.; Pennie W. D.; Will Y. (2011) A High Content Screening Assay for Identifying Lysosomotropic Compounds. Toxicol. In Vitro 25, 715–723. 10.1016/j.tiv.2010.12.010. [DOI] [PubMed] [Google Scholar]
- Kazmi F.; Hensley T.; Pope C.; Funk R. S.; Loewen G. J.; Buckley D. B.; Parkinson A. (2013) Lysosomal Sequestration (Trapping) of Lipophilic Amine (Cationic Amphiphilic) Drugs in Immortalized Human Hepatocytes (Fa2N-4 cells). Drug Metab. Dispos. 41, 897–905. 10.1124/dmd.112.050054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley M.; Tilley L. (1998) Quinoline Antimalarials: Mechanisms of Action and Resistance and Prospects for New Agents. Pharmacol. Ther. 79, 55–87. 10.1016/S0163-7258(98)00012-6. [DOI] [PubMed] [Google Scholar]
- Spano A.; Barni S.; Sciola L. (2013) PMA withdrawal in PMA-treated monocytic THP-1 cells and subsequent retinoic acid stimulation, modulate induction of apoptosis and appearance of dendritic cells. Cell Proliferation 46, 328–347. 10.1111/cpr.12030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman R. M.; Brodie S. E.; Cohn Z. A. (1976) Membrane flow during pinocytosis. A stereologic analysis. J. Cell Biol. 68, 665–687. 10.1083/jcb.68.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.