

Abstract
Staphylococcus aureus is a pathogenic bacterium that causes a variety of mild to lethal human diseases. The rapid spread of multidrug-resistant strains makes the discovery of new antimicrobial agents critical. Dihydroorotase (PyrC), the third enzyme in the bacterial pyrimidine biosynthesis pathway, is structurally and mechanistically distinct from its mammalian counterpart. It has been confirmed to be essential in S. aureus making it an attractive antibacterial drug target. No protocol to express and purify S. aureus PyrC (SaPyrC) has been reported. To obtain the SaPyrC enzyme and overcome anticipated solubility problems, the SaPyrC gene was cloned into the pET-SUMO vector. The N-terminal His-SUMO fused SaPyrC was expressed in E. coli BL21 (DE3) with an HRV 3C protease recognition site inserted between the SUMO tag and SaPyrC to allow for improved cleavage by HRV protease. Purification of cleaved protein using HisTrap affinity and gel filtration columns resulted in native SaPyrC with estimated 95 % purity and 40 % yield. Both His-SUMO tagged and native SaPyrC form dimers, and enzyme characterization studies have shown that the His-SUMO tag affects enzyme activity slightly. Forward and reverse kinetic rate constants for both tagged and native SaPyrC were determined, and pH profiling studies revealed the optimal pH values for forward and reverse reactions.
Keywords: Staphylococcus aureus, dihydroorotase, purification, characterization, SUMO, pH profiling
Introduction
The problem of bacterial drug resistance has grown to epidemic proportions. The resulting high global cost of illness and increased morbidity and mortality demand the immediate attention of the drug discovery community [1, 2]. One particularly troublesome organism is Staphylococcus aureus, a Gram-positive pathogenic bacterium that causes a wide array of disease. A large number of multiple-drug resistant (MDR) S. aureus strains have been reported within recent years, including hospital- and community-acquired strains of methicillin-resistant S. aureus (MRSA), vancomycin-intermediate and vancomycin-resistant S. aureus (VISA and VRSA), daptomycin-non-susceptible S. aureus (DNSSA), and linezolid-non-susceptible S. aureus (LNSSA) [3–6]. The emergence of S. aureus drug resistance is so rapid that there have even been reports of the organism developing resistance to new antibacterial agents only one year after approval [7]. One solution to this problem is the identification and characterization of new antibacterial drug targets, inhibition of which by novel agents could be expected to bypass current S. aureus resistance mechanisms.
An attractive and novel target for antibacterial drug design is the pyrimidine biosynthesis pathway. Pyrimidines are essential building blocks in RNA and DNA, and their de novo biosynthesis has previously been shown to be critical for the growth of pathogenic bacteria [8]. Although a de novo pyrimidine biosynthetic pathway is also present in mammals, the reactions are carried out by a multifunctional enzyme complex (Class I); whereas in most of the bacteria the pathway consists of distinct and separate enzymes for each step (Class II) [9]. This lends validity to the potential of designing selective inhibitors for enzymes in the bacterial pathway. The dihydroorotase enzyme, PyrC, is the third enzyme in the bacterial pyrimidine biosynthesis pathway and catalyzes an important step in the overall synthesis, as shown in Figure 1 [10]. Importantly, the S. aureus PyrC enzyme (SaPyrC) has been identified as essential for the survival of S. aureus [11]. Although there is strong biological evidence for the design of PyrC inhibitors for use as antibacterial agents, there have been relatively few reports in the literature and none specifically for S. aureus [12–14].
Figure 1. The Bacterial de novo Pyrimidine Biosythetic Pathway.

The dihydroorotase enzyme step is highlighted.
Although the pyrimidine biosynthetic pathway is an active area of antimicrobial research, purification and characterization of the PyrC enzyme has only been reported for a small number of clinically relevant bacteria. Washabaugh, et al. reported an early purification of PyrC from E. coli using a multi-step protocol involving two steps of precipitation, heating, anion-exchange and high-performance liquid chromatography [15]. Porter, et al. reported a simplified purification of Escherichia coli PyrC using three steps: precipitation, gel filtration and anion-exchange chromatography [9]. Another multi-step purification of PyrC from Pseudomonas putida was reported by Ogawa and coworkers which involved multiple steps of precipitation and ion-exchange chromatography [16]. A simpler purification protocol was reported by Wang and coworkers for Klebsiella pneumoniae PyrC that used Ni2+-affinity chromatography to obtain the enzyme at approximately 95 % purity [17]. Recently, Kim, et al. reported the first purification of E. coli PyrC enzyme using a fusion tag [18]. In this study, they were able to achieve high expression of the PyrC enzyme in the soluble fraction using end-to-end fusion with the maltose-binding protein (MBP) and a one-step purification using amylose resin affinity chromatography. This study was significant not only because it was the first report of PyrC purification using a fusion protein tag, but also because it achieved the purification using a low number of steps, which differed significantly from previously reported PyrC purifications. There have been no reports to date of the cloning, expression, or purification by any means, of the PyrC enzyme from S. aureus.
Herein, we report the cloning, expression and purification of the dihydroorotase (PyrC) enzyme from Staphylococcus aureus using a His-SUMO fusion tag. SUMO, small ubiquitin-related modifier, is an 11-kDa protein that is posttranslationally attached and removed from eukaryotic proteins and has been shown to be involved in cellular processes such as apoptosis, stress response, cell cycle progression, protein activation, and protein stability [19]. Because of the absence of the SUMO pathway in prokaryotes, eukaryotic SUMO (typically of yeast origin) is an ideal candidate for recombinant fusion to target proteins for prokaryotic expression and purification, particularly when combined with a SUMO protease, such as yeast Ulp1 [20]. The principal advantages for using the SUMO fusion protein include enhanced expression and solubility, potentially greater yield over larger fusion proteins such as MBP and GST, and decreased erroneous cleavage within the target protein [21–23]. Additionally, the use of a His purification tag in combination with the SUMO fusion protein allowed for the development of a more efficient purification protocol when compared to the early E. coli PyrC purifications discussed above. In these studies we tested both SUMO protease and HRV protease to identify optimal cleavage conditions and characterized the purified, native PyrC enzyme using both enzyme kinetic analysis and pH profiling.
Materials and methods
Reagents, chemicals, biologicals, and equipment
The SaPyrC gene was obtained in a pMCSG19C vector from the Center for Structural Genomics of Infectious Diseases at Northwestern University. The Taq Polymerase and the Champion pET SUMO Protein Expression System (catalog #K300–01) were ordered from Invitrogen Life Technologies (Grand Island, NY). The kit contained the pET SUMO vector, PCR and transformation reagents, Mach1-T1 competent E. coli cells, and BL21 (DE3) ‘One Shot’ chemically competent E. coli cells. The SUMO protease (catalog #4010) was obtained from LifeSensors (Malvern, PA). The QuikChange Lightning Site-Directed Mutagenesis Kit (catalog #210518) was ordered from Stratagene (La Jolla, CA). QIAquick PCR Purification Kit and QIAprep Spin Miniprep Kit were obtained from QIAGEN (Valencia, CA). Luria-Bertani (LB) media was obtained from Fisher Scientific (Pittsburgh, PA). The AKTAxpress and AKTApurifier FPLC systems, the HisTrap HP column (5 ml, 16 × 25 mm), and HiLoad 16/60 Superdex 200 PG (120 ml, 16 × 600 mm) and HiLoad 16/60 Superdex 75 PG (120 ml, 16 × 600 mm) gel filtration columns were obtained from GE Healthcare (Piscataway, NJ). Kanamycin, isopropyl β-D-thiogalactopyranoside (IPTG), N-carbamoyl-L-aspartic acid (CA-asp) and L-dihydroorotase (DHO) acid were ordered from Sigma-Aldrich Chemicals (St. Louis, MO). Recombinant type 14 3C protease from human rhinovirus (HRV 3C, catalog # 71493) was purchased from Novagen (San Diego, CA).
SaPyrC cloning and plasmid construction
Instructions from the manual provided with the Champion pET SUMO Protein Expression System were followed for cloning and transformation protocols unless otherwise noted below (Invitrogen, MAN0000440). The gene encoding SaPyrC contained in the pMCSG19C vector was the template for PCR amplification using forward primer 5′-ATGAAATTAATTAAAAACGGTAAAGTATTACAAAATGGCGAATTACAACAAGC-3′ and reverse primer 5′-TTATTTATCCCCCTCAAATTTAACTTCGCCTTCAACC-3′. The 50-μL PCR mixture consisted of 1 μL of DNA template, 5 μL of 10× PCR buffer, 0.5 μL of 50 mM dNTP mix, 1 μL each of forward primer and reverse primers (150 ng/μL stock concentrations), 1 μL of Taq polymerase, and 40.5 μL of molecular biology grade H2O. The reaction conditions were as follows: initial denaturation (94 °C, 120 seconds); 40 cycles of denaturation (94 °C, 30 seconds), annealing (54 °C, 30 seconds), and elongation (72 °C, 105 seconds); and final extension (72 °C, 20 minutes). The PCR products were analyzed by electrophoresis on a 0.8 % agarose gel stained with ethidium bromide and cleaned with a QIAquick PCR Purification Kit. The PCR product, containing 3′ deoxyadenosine (A) residues, was ligated by T4 DNA Ligase into linearized pET SUMO vector, containing deoxythymidine (T) overhangs. The 10-μL ligation reaction was carried out at 25 °C for 30 minutes and consisted of 4 μL of PCR product (1:1 vector/insert molar ratio), 1 μL of 10× Ligation Buffer, 2 μL of pET SUMO vector (25 ng/μL), 1 μL of T4 DNA Ligase, and 2 μL of H2O. Ligation products were transformed into ‘One Shot’ Mach1–1T chemically competent E. coli cells for plasmid propagation by heat shock at 42 °C for 30 seconds, and transformants were incubated overnight at 37 °C on LB agar plates with kanamycin (50 μg/ml). Individual colonies were selected and grown overnight in 5 ml of LB media (50 μg/ml kanamycin) and plasmid DNA was isolated by QIAprep Spin Miniprep Kit for sequencing. Clones containing the correct SaPyrC sequence were transformed into BL21 (DE3) ‘One Shot’ E. coli expression cells by heat shock (42 °C for 30 seconds) and were incubated overnight at 37 °C on LB agar plates (50 μg/ml kanamycin). Resulting colonies were inoculated in LB media with 2 % glucose and were stored at −80 °C with 20 % glycerol concentration as a cryoprotectant.
The QuikChange Lightning Site-Directed Mutagenesis Kit manual guidelines were followed for the PCR mutagenesis protocol to insert an HRV protease recognition site between the SUMO tag and the SaPyrC gene sequence. The sequences of the mutagenic oligonucleotide primers were 5′-GAGAACAGATTGGTGGTCTGGAAGTTCTGTTCCAGGGCCCGATGAAATTAATTAAAAACGGTAAAGTATTAC-3′ for forward and 5′-GTAATACTTTACCGTTTTTAATTAATTTCATCGGGCCCTGGAACAGAACTTCCAGACCACCAATCTGTTCTC-3′ for reverse. They were designed based on the desired eight amino acid HRV protease sequence (CTGGAAGTTCTGTTCCAGGGCCCG). Separate forward and reverse PCR reactions were created to circumvent annealing competition between complementary primers and the vector template. The two 25-μL reactions consisted of 0.5 μL of template (pET SUMO vector containing SaPyrC insert), 1 μL of forward or reverse primer (150 ng/μL stock), and 22.5 μL of PCR mix. The PCR mixture consisted of 1 μL of dNTP mix, 5 μL of 10X reaction buffer, 1.5 μL of QuikSolution reagent, 1 μL of QuikChange Lightning Enzyme, and 39 μL of H2O. The reaction conditions were as follows: initial denaturation (95 °C, 120 seconds); 8 cycles of denaturation (95 °C, 20 seconds), annealing (60 °C, 10 seconds), and elongation (68 °C, 4 minutes); and final extension (68 °C, 5 minutes). The forward and reverse PCR reaction products were then mixed and the PCR reaction was repeated at the same conditions as the separate reactions. Following this, 2 μL of Dpn I enzyme was gently mixed into the amplification products by pipetting up and down several times. The reaction mixture was then briefly centrifuged and incubated at 37 °C for 15 minutes. The mixture was transformed into XL10-Gold ultra-competent cells by heat shock at 42 °C for 30 seconds, and the sequencing verification and transformation of verified plasmids into BL21 (DE3) cells was performed as described above.
Expression and purification of recombinant His-SUMO-SaPyrC
The stored BL21 (DE3) cells containing the plasmid construct were grown in 1 ml LB media with kanamycin (50 μg/ml) shaking for 8 hours at 37 °C. The culture was diluted 1:50 with fresh LB media (50 μg/ml kanamycin), grown 16 hours shaking at 37 °C, and used to inoculate a 1-L LB culture (50 μg/ml kanamycin) with starting OD600 of approximately 0.1. The culture was shaken at 37 °C until OD600 reached 0.6, and were then induced with 0.5 mM IPTG and shaken at 25°C for 16 hours. Induced cells were harvested by centrifugation at 8000 rpm for 20 minutes (Beckman, JA-10 rotor), weighed, and frozen at −80°C. The thawed pellet (5.77 g wet weight) was dissolved in 60 ml of lysis buffer (50 mM Tris, pH 8.0, 500 mM NaCl, 20 mM imidazole, 1 % Triton X-100, 0.025 mg/ml DNase I, 1 mg/ml lysozyme) and sonicated (Sonic Dismembrator Model 500, Fisher Scientific) at 65 % amplitude by 100 × 3 seconds bursts separated by 6 seconds off time. Crude extract was separated by centrifugation at 18000 rpm for 30 minutes (Beckman, JA-20 rotor). The crude extract was filtered through a 0.22-μm membrane filter and loaded onto a 5-ml HisTrap HP affinity column equilibrated with binding buffer A (50 mM Tris, pH 8.0, 500 mM NaCl, 20 mM imidazole). Ten column volumes of binding buffer were applied to wash unbound impurities followed by elution with buffer B (50 mM Tris, pH 8.0, 500 mM NaCl, 500 mM imidazole) at a 3 ml/min flow rate and step gradients of 10 %, 50 %, and 100 % buffer B. Samples were collected in 2-ml fractions. The SaPyrC content in the eluted fractions was analyzed by SDS-PAGE gel electrophoresis and the absorbance assay described in further detail below. Protein concentrations were determined by Bradford assay using an albumin standard [24].
Removal of His-SUMO tag and gel filtration purification
The buffer conditions and amount of HRV 3C protease used in the following methods were based on guidelines from Novagen with slight modification to extend the digestion time and increase the buffer salt concentration (see Results and Discussion). Purified His-SUMO-SaPyrC was dialyzed in 4 L of cleavage buffer (50 mM Tris, pH 7.5, 500 mM NaCl, 1 mM TCEP) for 16 hours and collected into a 50-mL conical tube. HRV 3C protease (1000 U, 500 μL) was added to the collected protein sample and the digestion mixture was rocked slowly at 4 °C for 72 hours. At various time points 10-μL samples of the digestion mixture were taken for cleavage efficiency analysis with SDS-PAGE gel electrophoresis (see Figure S3, Supplementary Content). The mixture was then loaded onto a 5-ml HisTrap HP column equilibrated with binding buffer without imidazole (50 mM Tris, pH 8.0, 500 mM NaCl) and eluted as described above. The flow-through containing cleaved SaPyrC was collected as well as fractions containing any uncleaved fusion product (for kinetic analysis, discussed below). Both pooled cleaved and uncleaved SaPyrC were concentrated to under 5 ml using an Amicon ultra centrifugal filter (Millipore) and loaded onto an equilibrated (50 mM Tris, pH 8.0, 200 mM NaCl, 1 mM TCEP) HiLoad 16/60 Superdex 200 PG gel filtration column. The protein was loaded at 1 ml/minute and the length of elution was 1.5 column volumes. The eluate was collected in 3-ml fractions and samples were analyzed by SDS-PAGE gel electrophoresis and absorbance assay for purity and activity, respectively.
Prior to the HRV protease cleavage site insertion, SUMO protease was tested following guidelines provided by LifeSensors. Nickel column purified His-SUMO-SaPyrC was dialyzed (20 mM Tris, pH 7.5, 150 mM NaCl, 2 mM DTT) at 4 °C overnight. The cleavage reaction was prepared by adding 1 unit of SUMO protease per 100 μg of tagged protein. The mixture was then incubated for 1 h at 30 °C with slow rocking and then incubated over night at 4 °C. SDS-PAGE gel samples were taken before the reaction, after the 1-hr reaction, and after the overnight incubation to evaluate cleavage efficiency (see Figure S4, Supplementary Content).
Enzyme activity assay
The SaPyrC enzyme assay was optimized by adjusting the assay buffer pH and final enzyme dilution to obtain a linear reaction rate of at least ten minutes with a maximum slope. Dihydroorotate (DHO), the product of the forward reaction, has a peak UV absorption at 230 nm, and the enzyme reaction can be monitored by following a decrease in absorbance at 230 nm as the enzymatic reaction is run in reverse [17]. The SaPyrC enzyme activity was measured by monitoring the conversion of DHO to CA-asp using a SpectraMax Plus384 Absorbance Microplate Reader (Molecular Devices) at 25 °C. The absorbance assays were run in 96-well UV plates (Corning Life Sciences, catalog #3635) using a 200 μL total volume. The assays performed during the purification used 2 μL of enzyme samples added to 148 μL assay buffer (50 mM Tris, pH 8.3), and the reaction was initiated by adding 50 μL of DHO substrate (100 μM final concentration). Absorbance was measured at 10-second intervals after 30 seconds of mixing. Slopes were calculated from the linear portion of the monitored reaction. Assays were performed in quadruplicate using two controls of 100 μM DHO in assay buffer with no enzyme. Enzyme rates were calculated using the measured slopes, path length of 0.44 cm, and the extinction coefficient (ε) of DHO at 230 nm, 1.17 mM−1 cm−1 [25].
pH-rate profiling
The enzyme activity of native SaPyrC at varying pH levels was tested to form a pH-rate profile for the forward and reverse reactions at 25 °C. Reactions were performed using the 96-well UV plate assay described above and varying pH levels from 4 to 11 in 0.5 pH intervals. The final enzyme concentration used in the pH profile assays was 0.1 μM while the DHO and CA-asp concentrations used were 0.1 mM and 0.3 mM, respectively. Three buffers were used in the experiments, depending on the pH being tested. MES buffer was used for pH 4.0 to 5.5, HEPES was used for pH 6.0 to 8.0, and Tris was used for pH 8.5 to 11.0. The buffer concentration was 50 mM for all experiments. Substrate stocks in the varying pH assay buffers were mixed and their corresponding pH enzyme stock was added to start the reaction. Absorbance at 230 nm was read at 10-second intervals after an initial 30 seconds of mixing. Slopes from linear ranges were calculated to determine enzyme reaction rates.
Enzyme kinetic characterization
All enzyme kinetic experiments were performed using both the cleaved enzyme and the His-SUMO tagged enzyme for comparison. The enzyme was characterized by determining the Michaelis constant (Km) and Kcat/Km for both the forward and reverse reactions of CA-asp cyclization to form DHO, and the reverse ring cleavage. The direction of the reaction is dependent on both the added substrate and the buffer pH, as has been previously published for both mammalian and bacterial dihydroorotase [26, 27]. Buffer, pH (see above), and enzyme concentrations were optimized for the forward and reverse reactions, respectively, to maximize the slope and obtain an extended linear range for slope calculations. A modified version of the 200-μL assay described above was used in these studies. For the forward reaction, 50 μL of protein (0.1 μM final concentration) was added to 150 μL of varying CA-asp concentrations (final concentrations of 0, 10, 15, 25, 50, 150, 300, 600, 900, 1000, 1500, 2000, 2500, 3000, 4000, 5000 μM) in 50 mM MES buffer, pH 5.8. For the reverse reaction, 50 μL of protein (0.1 μM final concentration) was added to 150 μL of varying DHO concentrations (final concentrations of 0, 50, 75, 125, 250, 500, 750 and 1000 μM) in 50 mM Tris buffer, pH 8.3. Absorbance was recorded every 10 seconds at 230 nm after 30 seconds of mixing. Initial slopes were determined from the linear portion of the monitored enzyme reaction. Reactions were performed in triplicate at 25 °C for both native and His-SUMO-tagged SaPyrC. Kinetic constants were estimated by fitting the assay data (slopes calculated from linear portion of absorbance vs. time plots) to equation (1) using the program SigmaPlot v11.0 (Systat Software, Inc., San Jose California USA):
| (1) |
Kcat values were calculated by dividing VMax by the enzyme concentration (obtained by Bradford assay as discussed above).
Results and discussion
Cloning, expression and purification of His-SUMO tagged SaPyrC
The pyrC gene from Staphylococcus aureus, subspecies aureus, strain MW2 (NCBI Gene ID: 1003196, Locus tag: MW1084) contains a total length of 1,275 base pairs and encodes a 424 residue protein product with a mass of 46.37 kDa [28]. Figure 2 shows a structure-based sequence alignment of S. aureus, E. coli, and B. anthracis PyrC, with the active site residues indicated. SaPyrC has a low overall (16 %) and active site (50 %) sequence identity to E. coli, and a high overall (60 %) and active site (100 %) sequence identity to Bacillus anthracis; both of which are clinically relevant organisms for which PyrC work has been reported [15, 29, 30]. The S. aureus MW2 strain is methicillin-resistant and has been typically associated with community-acquired infection [31]. To overexpress this target protein, the pyrC gene, which was obtained in a pMCSG19C vector, was PCR amplified and cloned into the pET SUMO vector, a T7lac promoted vector carrying an N-terminal His6 -SUMO tag with SUMO protease cleavage site and kanamycin resistance determinant (see Methods above). After the cloned pyrC gene was confirmed by sequencing, the plasmid vector was transformed into E. coli strain BL21 (DE3) cells for overexpression of the PyrC target protein. The use of the pET SUMO expression system allowed for expedient plasmid construction, transformation, and expression of large amounts of soluble His6-SUMO-SaPyrC with minimal optimization. The insertion of the PCR amplified pyrC gene into the pET SUMO vector required only a single ligation step due to the 3′ nontemplate-dependent addition of a single deoxyadenosine (A) by Taq polymerase and the 5′ single deoxythymidine (T) residues on the linear vector.
Figure 2. Dihydroorotase (PyrC) Structure-Based Sequence Alignment.

The PyrC sequence alignment from three clinically relevant bacterial species, S. aureus (SA), E. coli (EC), and B. anthracis (BA), is shown. Black boxes indicate the active site residues, defined as all residues within a five angstrom radius surrounding the active site zinc metals in the respective apo structures.
Figure 3 shows the SaPyrC expression and purification gel analysis. The gel (lanes 2 and 3) shows that overexpression of His-SUMO-tagged SaPyrC proceeded with no evidence of leaky expression prior to induction at the desired cell density (see Methods). SDS-PAGE analysis further shows that crude extract from the disrupted cells (lane 4) had a very high expression of target protein. In addition to this high expression, the target protein was almost entirely present in the soluble fraction (lanes 5 and 6), which we attribute to our use of the SUMO fusion tag. A typical SaPyrC purification run (from a 1-L LB media culture) results in a total wet cell weight of roughly 6 grams, on average. Table 1 shows the results of a typical SaPyrC purification, including total and specific activity, yield and purity. Parenthetical values in Table 1 under ‘Total Protein’ reflect the difference in sizes of the His-SUMO tagged and native SaPyrC enzymes [32]. The parenthetical values shown under ‘Total Enzyme Activity’ reflect a difference in the enzyme activity noted between the purified native and purified His-SUMO-tagged SaPyrC enzyme. These values were used in the calculation of the yield. The His-SUMO tagged SaPyrC enzyme possesses approximately 85 % the activity of the native enzyme (further discussed below). The values shown in parentheses have been weighted accordingly and were used in the calculation of ‘Specific Activity’ and ‘Purity’.
Figure 3. SaPyrC Expression and Purification, Gel Documentation and Analysis.
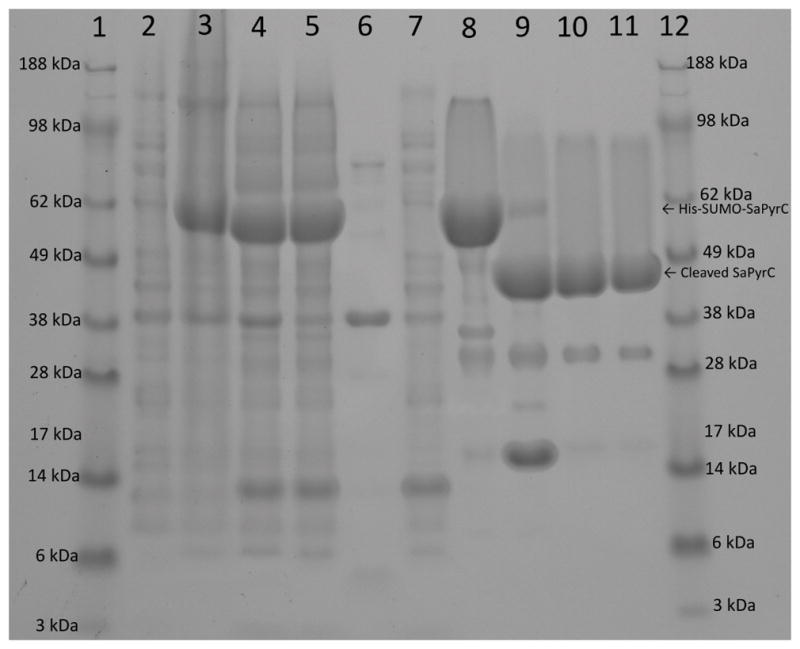
Samples were loaded in 10 μL amounts in 1× SDS loading dye, and unless noted, samples were taken unaltered from resulting steps described in the methods and diluted 2-fold with 2× SDS loading dye. Lane 1 ladder, lane 2 culture before IPTGA, lane 3 culture after IPTGA, lane 4 crude lysateB, lane 5 supernatantB, lane 6 pelletB, lane 7 flow through (nickel column 1), lane 8 pooled peak (nickel column 1), lane 9 final digestion after 72 hours, lane 10 pooled peak cleaved proteinC (nickel column 2), lane 11 pooled peak cleaved protein gel filtrationC, lane 12 ladder. A, B adjusted to same concentrations C concentrated prior to taking gel sample
Table 1.
Purification of Recombinant SaPyrC
| Step | Volume (ml) | Concentration (mg/ml) d, e | Total Protein (mg)e | Total Enzyme Activity (Units)f, g | Specific Activity (Units/mg) h | Yield (%)i | Purity (%) i |
|---|---|---|---|---|---|---|---|
| Crude Lysatea | 60 | 7.7 | 462 (354) | 61.8 (72.7) | 0.21 | 100 | 29 |
| Clarified Extract | 57 | 7.5 | 427 (327) | 60.9 (71.6) | 0.22 | 98 | 31 |
| Tagged SaPyrC HisTrapb | 17.6 | 6.6 | 116.2 (89) | 37.6 (44.2) | 0.50 | 61 | 70 |
| Native SaPyrC HisTrapc | 18.4 | 3.7 | 68.1 | 39.3 | 0.58 | 54 | 81 |
| Native SaPyrC size exclusion (pooled peak) | 17.0 | 2.5 | 42.5 | 28.7 | 0.68 | 39 | 95 |
From 5.77 g of wet weight E. coli cell pellet (from 1 L culture).
First Nickel HisTrap Column FPLC purification step (pooled fractions).
Second Nickel HisTrap Column FPLC purification step, after tag cleavage (pooled fractions).
The protein concentration was determined by Bradford assay using BSA as the standard.
Parenthetical values reflect calculated mass of native protein.
Enzyme activity measured by UV assays as described in methods (200 μL volume, 2 μL protein, 0.44 cm pathlength; 1.17 mM−1 cm−1 extinction coefficient).
Parenthetical values reflect estimated activity based upon lower enzyme efficiency of tagged SaPyrC (see Discussion).
Specific Activity calculated from parenthetical estimates of Total Protein and Total Activity for tagged protein.
Yield is calculated from Total Enzyme Activity; Purity is calculated from Specific Activity, with 95% final purity estimated the gel.
The purification of the cleaved SaPyrC enzyme consisted of three chromatographic steps: immobilized metal ion affinity (IMAC) column chromatography of the His-SUMO tagged enzyme, IMAC column chromatography of the cleaved enzyme (collected in flow through), and size exclusion chromatography. A step gradient elution was used to collect His-SUMO-tagged SaPyrC from the Ni IMAC column. Elution of the target enzyme occurred at 50 % buffer B (or 260 mM imidazole), resulting in a large, distinct peak on the chromatograph (not shown). A smaller, yet still prominent peak was also seen during washing steps with low buffer B concentration on initial purification runs. SDS-PAGE analysis of the corresponding fractions revealed that a small amount of target enzyme was being washed from the column along with impurities. We attributed this to the column capacity being exceeded as a result of the very high yield from the target expression. Our belief was verified in subsequent purification runs when only half the crude extract obtained from 1-L cultures was loaded onto a 5-ml HisTrap HP affinity column at a time. This resulted in elimination of any detectable target protein loss in the flow through during washing steps (Figure 3, lane 7). Thus reduction in loss of protein can be achieved at the IMAC purification step by reducing the amount of extract being loaded during purification, though yield is still very high without taking such a step. The purity of the His-SUMO-tagged SaPyrC after this first step was estimated to be 70 %, with a yield of 60 % (Table 1). We note the appearance of a large band at approximately 14 kDa in lanes 4, 5, and 7 of Figure 3, which is likely the lysozyme enzyme used in the lysis buffer (see Methods).
Bradford assays were used to determine protein concentration at various stages of purification; however A280 values showed similar protein concentration values when tested on corresponding fractions. Thus either method appears to be reliable for protein concentration measurement. The SaPyrC enzyme is stable at high concentrations, under refrigeration and after freezing. Both tagged and native SaPyrC were stable when concentrated above 15 mg/ml without precipitation. Neither tagged nor cleaved enzyme showed any reduction in activity or precipitation after one month of refrigeration at 4 °C in gel filtration buffer. Additionally, we noted no appreciable loss of enzyme activity after a cycle of liquid nitrogen freezing and thawing.
One interesting feature noted during the initial purification of SaPyrC was a marked loss of enzyme activity after the first step of purification, in which the His-SUMO tagged SaPyrC enzyme was collected by Ni-IMAC column chromatography. The enzyme activity in the crude extract was significantly higher than that of the Ni column purified sample, though the gel analysis indicated a definite improvement in purity. This can be explained as interference of activity by the high imidazole concentration in the pooled fraction, which likely coordinates to the active site zinc atoms in the SaPyrC enzyme and thereby inhibits the enzyme. This effect on metalloenzymes by high concentration imidazole, and imidazole derivatives, has been reported previously [33, 34]. We were able to confirm this by retesting the fraction after dialysis to remove the imidazole. The activity obtained (reflected in Table 1) was significantly improved and was more closely aligned to the level of purity seen in the gel analysis (Figure 3).
SUMO tag removal and purification of native SaPyrC
Ideally, SUMO protease produces native protein by cleaving the SUMO tag at the protease site immediately before the target protein, which is then removed using Ni IMAC. In these studies, the cleavage reaction was prepared following the optimal buffer components, pH, and SUMO protease to tagged protein ratio. One hour incubation at 30 °C or overnight incubation at 4 °C is recommended for 90 % efficiency, and both were performed in succession here to ensure maximal cleavage. However, despite the extra time given at optimal conditions, SUMO protease cleavage was ineffective as visualized by SDS-PAGE analysis (see Figure S4, Supplementary Content). We hypothesized that the structured N-terminus of SaPyrC may have blocked SUMO protease access to the cleavage site. To overcome this issue, PCR mutagenesis was used to insert an 8 amino acid HRV 3C protease cleavage site between the SUMO protease cleavage site and the SaPyrC gene. We chose this approach over the addition of extra, random amino acids to extend the SUMO cleavage site away from the native enzyme because the resulting cleavage product (after HRV cleavage) would differ less from the native enzyme.
After mutagenesis, DNA sequencing, and transformation into BL21 (DE3) cells (see Methods), the expression and purification protocol of His-SUMO-tagged SaPyrC was repeated with no observed difference in effect on expression level or solubility. The nickel column purified tagged protein was dialyzed and mixed with HRV 3C protease for 72 hours in accordance with the Novagen recommended buffer conditions with minor modifications for improved efficiency. First, 500 mM NaCl was used instead of 150 mM, for increased efficiency as recommended in the manual. Although we used 1 unit of HRV 3C protease per 116 μg of fusion protein, which was slightly less than the recommended ratio (1 U/100 μg), we increased the cleavage time from 16 hours to 72 hours (see Methods). The gel analysis of the cleavage reaction showed that cleavage was still occurring even at 72 hours (Figure S3, Supplementary Content). These modifications allowed for efficient cleavage, estimated at approximately 80 %, which surpassed the SUMO protease efficiency, approximately 30 % (Figure S3 & S4, Supplementary Content). The results of HRV cleavage of the His-SUMO-tagged SaPyrC enzyme at the 72 hour time point can be seen in Figure 3, lane 9. The band visible at approximately 14 kDa in this lane is the cleaved His-SUMO tag.
Isolation of the native SaPyrC protein from the cleavage reaction was accomplished by a second step of Ni IMAC chromatography (Figure 3, lane 10). The native protein was collected in the flow through, concentrated to 5 ml, and then gel filtrated as the final purification step (Figure 3, lanes 11). The remaining His-SUMO-tagged SaPyrC was collected in fractions at the 50 % Buffer B UV-Vis peak, concentrated to below 5 ml, and gel filtrated for use in comparison characterization studies, discussed below. The final SaPyrC product contained only two extra amino acid residues, glycine and proline at the N-terminus. The purification results in an average yield of 40 mg of native protein per 1-L culture (6.5 – 7 mg of purified protein per gram of wet weight cells) with an estimated purity of 95 %.
It is known that E. coli PyrC and B. anthracis PyrC are functional dimers. The dimerization of SaPyrC was tested and confirmed by analysis of the gel filtration using proteins purified under the same conditions previously as standards (Figure 4). The 16/60 SD 75 (see Methods) gel filtration column was used to run His-SUMO SaPyrC (60.7 kDa) and cleaved SaPyrC (46.5 kDa) along with three other proteins with three different molecular weights, 138 kDa, 94 kDa, 67 kDa, respectively. As shown in Figure 4, both the His-SUMO-tagged SaPyrC and cleaved SaPyrC eluted from the gel filtration column as dimers, which indicates that the His-SUMO tag does not interfere with SaPyrC dimerization.
Figure 4. SaPyrC Gel Filtration Analysis.

A. UV profile of gel filtration of His-sumo SaPyrC (Dimer, 121.4 kDa). B. UV profile of gel filtration of cleaved SaPyrC (Dimer, 93 kDa). Column volume is 120 ml, and void volume is approximately 45 ml. The void volume was determined from two large proteins, IgG (158 kDa) and Ferritin (440 kDa), that eluted out together between 40 ml and 48 ml elution volume.
The mechanism of the dihydroorotase enzyme (PyrC) has been experimentally and theoretically investigated [9, 35, 36]. PyrC belongs to the aminohydrolase (AHS) enzyme superfamily and catalyzes the reversible interconversion of carbamoyl aspartate to dihydroorotate. The PyrC active site contains two zinc metals coordinated by four histidine residues and bridged by a hydroxide molecule that functions as the nucleophile during the hydrolysis of dihydroorotate during the reverse reaction [37]. In E. coli, the catalytic efficiency of the reaction for both directions has been demonstrated to be influenced by pH, with the forward reaction favored at lower pH (below pH 6.0) and the reverse reaction favored at higher pH (kinetic pKa = 6.1 for reverse reaction) [9]. It was postulated that the protonation state of the bridging hydroxide group is the key determinant; the deprotonated hydroxide being required for dihydroorotate hydrolysis. Because no pH-rate analysis has been performed using the SaPyrC enzyme, we have performed a detailed pH-rate analysis to identify the optimal pH for both the forward and reverse SaPyrC reactions. This data, in combination with the enzyme kinetic parameters discussed below, will facilitate the development of a balanced assay for high throughput screening purposes.
The results of the pH-rate profile analyses are shown in Figure 5. SaPyrC enzyme velocities were calculated at varying pH levels for both the forward and reverse reactions (see Methods). In keeping with previous observations made with other bacterial species, the SaPyrC enzyme efficiency was influenced by the assay pH. For the cyclization reaction (forward), the maximum enzyme velocity was seen at pH 6 (92.8 μmol s−1 mg−1) and for the hydrolysis reaction (reverse) the maximum velocity was seen at pH 9 (123.7 μmol s−1 mg−1). This data confirms that previous observations related to PyrC pH-rate dependence are applicable to SaPyrC as well. We note that the maximum enzyme velocity that we observed for the reverse reaction is greater than the maximum velocity observed for the forward reaction. This is in keeping with previously reported rates for the E. coli PyrC enzyme, which showed approximately 0.5 log higher kcat/Km values for the reverse reaction over the forward reaction [9].
Figure 5. pH Effect of the Ring Cyclization and Cleavage Activities of Recombinant SaPyrC.
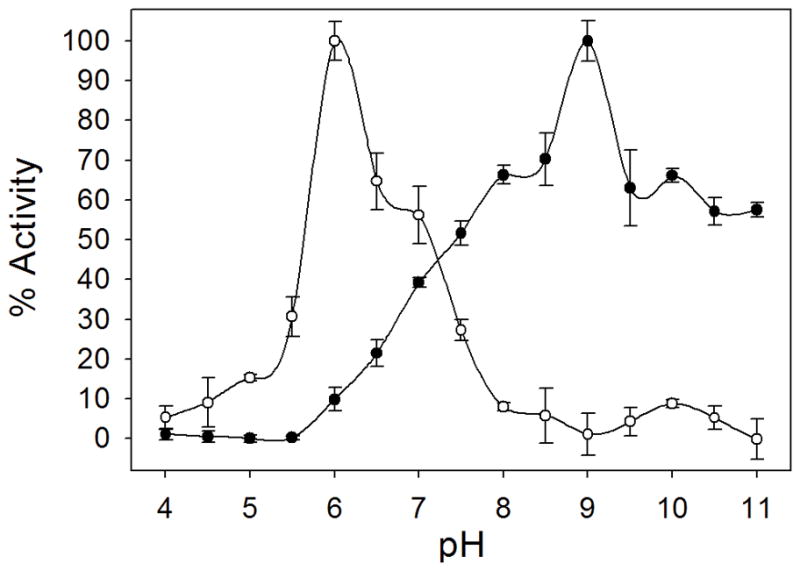
The maximal activities of ring cyclization (open circles) and cleavage (closed circles) were 92.8 and 123.7 μmol seconds−1 mg−1, respectively. SaPyrC enzyme % activities at each pH were calculated with the maximal activities as 100 %.
SaPyrC kinetic characterization
The S. aureus dihydroorotase enzyme is a di-metalloenzyme that is dependent upon two zinc atoms in the active site for the reaction to take place. The activity of the enzyme can be monitored by following an increase or decrease in the UV absorption of the dihydroorotate product at 230 nm as the enzyme reaction proceeds in the forward or reverse direction, respectively. UV absorption in 200-μL, 96-well plates was used to check the activity of the fractions collected during purification as well as for all kinetics studies discussed below (see Methods). In order to optimize screening assay conditions, the Km values for both the carbamoyl aspartate substrate and the dihydroorotate product were determined. This was accomplished by performing a detailed kinetic analysis of SaPyrC by varying the concentrations of each as the reaction was run in both the forward and reverse directions. All kinetic studies were completed using both the purified His-SUMO-SaPyrC and purified native SaPyrC enzyme obtained after the second step of nickel column purification and gel filtration and at the optimal pH levels identified above (see Methods).
Table 2 and Figure 6 show the kinetic characterization results for both tagged and cleaved SaPyrC in both directions. The calculated Km values of His-SUMO tagged SaPyrC and native SaPyrC for conversion of Ca-Asp to DHO (forward reaction) were 334 μM and 348 μM, respectively; which fall within reasonable error to be considered equivalent. For the reverse reaction (hydrolysis of DHO to Ca-Asp), the Km values for the His-SUMO tagged SaPyrC and native SaPyrC were 167 μM and 118 μM, which were significantly different. This may indicate either a decreased affinity of the DHO ligand for the His-SUMO tagged enzyme or an effect of the tag on the catalytic efficiency of the reverse reaction (depending on the rate of conversion compared to the substrate dissociation). Km values for the substrates of E. coli PyrC have been previously reported [9]. In these studies, the DHO substrate also showed a lower Km value over Ca-Asp, in this case 80 μM and 1.7 mM, respectively.
Table 2.
Enzyme kinetic characterization
| Enzyme | Reaction | Km (μM) | Vmax (μM s−1) | Kcat (s−1) | Kcat/Km (mM−1s−1) |
|---|---|---|---|---|---|
| Native SaPyrC | Forward (CA-asp substrate) | 348 ± 62 | 0.248± 0.011 | 2.48 | 7.13 |
| His-SUMO-SaPyrC | Forward (CA-asp substrate) | 334 ± 27 | 0.190 ± 0.004 | 1.90 | 5.69 |
| Native SaPyrC | Reverse (DHO substrate) | 118 ± 15 | 0.155 ± 0.006 | 1.55 | 13.1 |
| His-SUMO-SaPyrC | Reverse (DHO substrate) | 167 ± 20 | 0.187 ± 0.007 | 1.87 | 11.2 |
Figure 6. Kinetic Parameter Determination of the Ring Cyclization and Ring Cleavage Reactions.
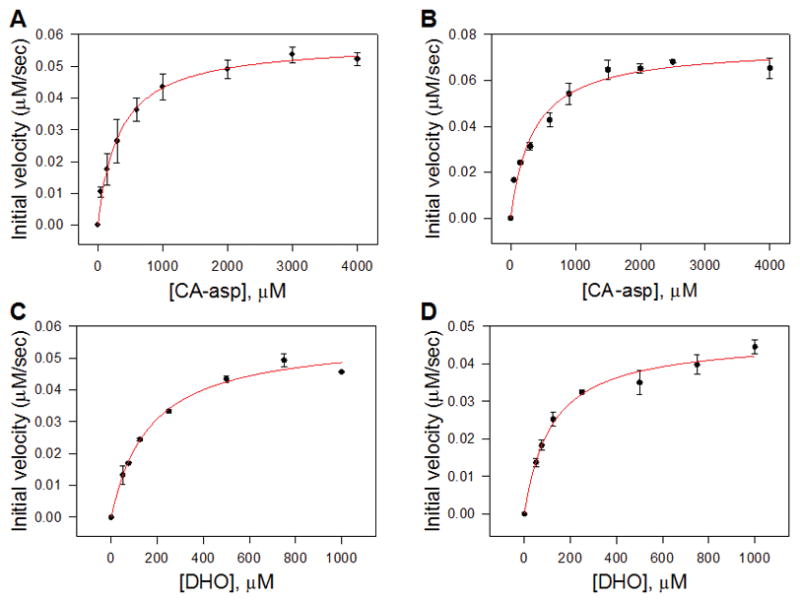
Shown are the fitted curves used to determine Michaelis-Menten constants (Km) of the reverse reaction (ring cleavage) of His-SUMO tagged SaPyrC (A), and cleaved SaPyrC (B); and the fitted curves of the forward reaction (ring cyclization) of His-SUMO tagged SaPyrC (C) and cleaved SaPyrC (D).
Interestingly, for SaPyrC, the Vmax of the native enzyme was increased over the tagged enzyme for the forward reaction, but decreased over the tagged enzyme for the reverse reaction, which indicates that the expression tag affects the maximal rate and thus the overall enzyme efficiency of SaPyrC (Kcat/Km), as shown in Table 2. For example, for the reverse reaction, tagged enzyme showed an efficiency of 85 % of that observed from the native enzyme. This was taken into account when preparing the purification table (Table 1) as we mentioned above. Because the tagged enzyme does retain significant activity, a single-step nickel column purification is able to produce active, SUMO tagged enzyme that is of sufficient purity and activity for high-throughput enzyme assays (after dialysis to remove imidazole). For studies requiring pure, native enzyme such as crystallography and advanced inhibitor testing, cleavage followed by size exclusion chromatography is appropriate.
Conclusions
Drug resistant strains of Staphylococcus aureus cause serious infections in healthcare and community settings and are epidemic in many areas. SaPyrC, a crucial enzyme in its pyrimidine biosynthesis pathway, is necessary for the survival of the organism and a very attractive, and unexploited, antibacterial drug target. In these studies, we have cloned the SaPyrC gene into a pET SUMO vector and optimized a high expression and three-step purification protocol to obtain active tagged and native enzyme. The SUMO tag allowed for very high, inducible expression with negligible insoluble target protein, and a purification scheme significantly less complex than many previously reported. Cleavage of the SUMO tag with SUMO protease was noted to be very inefficient, and was therefore accomplished by cloning the HRV cleavage site into the construct and using HRV 3C protease to cleave the SUMO tag. We have further kinetically optimized the SaPyrC enzymatic reaction and performed a pH-rate profile analysis to identify optimal enzyme assay conditions for high-throughput inhibitor screening. The tagged product of the single-step Ni-IMAC purification is of sufficient purity and yield to be useful in such studies, while the native enzyme product of the three-step purification will be useful for pending crystallography trials and secondary assays once inhibitors are identified.
Supplementary Material
Highlights.
The purification of the S. aureus PyrC enzyme using a His-SUMO tag is described.
Protein solubility and yield were high with the use of the His-SUMO tag.
Improved tag cleavage was achieved with HRV 3C protease over SUMO protease.
Forward and reverse kinetic rate constants for SaPyrC were calculated experimentally.
Optimal pH values for forward and reverse reactions were determined.
Acknowledgments
KEH was supported during a portion of this work by NIDCR 5T32-DE018381, UIC College of Dentistry, MOST program. The authors thank the Elisabetta Sabini of the Center for Structural Genomics of Infectious Diseases at Northwestern University for providing the SaPyrC gene. The authors thank ChemAxon, Ltd. for an academic research license of their Marvin suite which was used in the production of Figure 1.
Abbreviations
- CA-asp
N-carbamoyl-L-aspartic acid
- DHO
L-dihydroorotase
- DNSSA
daptomycin-non-susceptible S. aureus
- HRV 3C protease
Recombinant type 14 3C protease from human rhinovirus
- IPTG
isopropyl β-D-thiogalactopyranoside
- LNSSA
linezolid-non-susceptible S. aureus
- MBP
maltose-binding protein
- MDR
multiple-drug resistant
- MRSA
methicillin-resistant S. aureus
- PyrC
dihydroorotase
- SaPyrC
S. aureus dihydroorotase
- SUMO
small ubiquitin-related modifier
- TCEP
tris (2-carboxyethyl) phosphine
- VISA
vancomycin-intermediate S. aureus
- VRSA
vancomycin-resistant S. aureus
Footnotes
Supplementary content includes the S. aureus PyrC full nucleotide sequence (with and without His-SUMO tag), the full protein sequence (with and without His-SUMO tag), gel documentation of collected Ni column fractions and gel filtrated fractions of His-SUMO SaPyrC (Figure S1), gel documentation of collected gel filtrated fractions of cleaved SaPyrC (Figure S2), gel documentation of the HRV protease cleavage reaction (Figure S3), and gel documentation of the SUMO protease cleavage reaction (Figure S4).
References
- 1.Furuya EY, Lowy FD. Antimicrobial-resistant bacteria in the community setting. Nature reviews. 2006;4:36–45. doi: 10.1038/nrmicro1325. [DOI] [PubMed] [Google Scholar]
- 2.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis. 2009;48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 3.Gould IM, David MZ, Esposito S, Garau J, Lina G, Mazzei T, Peters G. New insights into meticillin-resistant Staphylococcus aureus (MRSA) pathogenesis, treatment and resistance. Int J Antimicrob Agents. 2012;39:96–104. doi: 10.1016/j.ijantimicag.2011.09.028. [DOI] [PubMed] [Google Scholar]
- 4.Moellering RC., Jr MRSA: the first half century. The Journal of antimicrobial chemotherapy. 2012;67:4–11. doi: 10.1093/jac/dkr437. [DOI] [PubMed] [Google Scholar]
- 5.Nannini E, Murray BE, Arias CA. Resistance or decreased susceptibility to glycopeptides, daptomycin, and linezolid in methicillin-resistant Staphylococcus aureus. Current opinion in pharmacology. 2010;10:516–521. doi: 10.1016/j.coph.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 6.Hentschke M, Saager B, Horstkotte MA, Scherpe S, Wolters M, Kabisch H, Grosse R, Heisig P, Aepfelbacher M, Rohde H. Emergence of linezolid resistance in a methicillin resistant Staphylococcus aureus strain. Infection. 2008;36:85–87. doi: 10.1007/s15010-007-7220-7. [DOI] [PubMed] [Google Scholar]
- 7.Tsiodras S, Gold HS, Sakoulas G, Eliopoulos GM, Wennersten C, Venkataraman L, Moellering RC, Ferraro MJ. Linezolid resistance in a clinical isolate of Staphylococcus aureus. Lancet. 2001;358:207–208. doi: 10.1016/S0140-6736(01)05410-1. [DOI] [PubMed] [Google Scholar]
- 8.Samant S, Lee H, Ghassemi M, Chen J, Cook JL, Mankin AS, Neyfakh AA. Nucleotide biosynthesis is critical for growth of bacteria in human blood. PLoS Pathog. 2008;4:e37. doi: 10.1371/journal.ppat.0040037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Porter TN, Li Y, Raushel FM. Mechanism of the dihydroorotase reaction. Biochemistry. 2004;43:16285–16292. doi: 10.1021/bi048308g. [DOI] [PubMed] [Google Scholar]
- 10.Schenk-Groninger R, Becker J, Brendel M. Cloning, sequencing, and characterizing the Lactobacillus leichmannii pyrC gene encoding dihydroorotase. Biochimie. 1995;77:265–272. doi: 10.1016/0300-9084(96)88135-1. [DOI] [PubMed] [Google Scholar]
- 11.Forsyth RA, Haselbeck RJ, Ohlsen KL, Yamamoto RT, Xu H, Trawick JD, Wall D, Wang L, Brown-Driver V, Froelich JM, KGC, King P, McCarthy M, Malone C, Misiner B, Robbins D, Tan Z, Zhu ZyZY, Carr G, Mosca DA, Zamudio C, Foulkes JG, Zyskind JW. A genome-wide strategy for the identification of essential genes in Staphylococcus aureus. Mol Microbiol. 2002;43:1387–1400. doi: 10.1046/j.1365-2958.2002.02832.x. [DOI] [PubMed] [Google Scholar]
- 12.Li Y, Raushel FM. Inhibitors designed for the active site of dihydroorotase. Bioorganic chemistry. 2005;33:470–483. doi: 10.1016/j.bioorg.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 13.Lee M, Chan CW, Graham SC, Christopherson RI, Guss JM, Maher MJ. Structures of ligand-free and inhibitor complexes of dihydroorotase from Escherichia coli: implications for loop movement in inhibitor design. J Mol Biol. 2007;370:812–825. doi: 10.1016/j.jmb.2007.05.019. [DOI] [PubMed] [Google Scholar]
- 14.Lee M, Maher MJ, Guss JM. Structure of the T109S mutant of Escherichia coli dihydroorotase complexed with the inhibitor 5-fluoroorotate: catalytic activity is reflected by the crystal form. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2007;63:154–161. doi: 10.1107/S1744309107004009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Washabaugh MW, Collins KD. Dihydroorotase from Escherichia coli. Purification and characterization. J Biol Chem. 1984;259:3293–3298. [PubMed] [Google Scholar]
- 16.Ogawa J, Shimizu S. Purification and characterization of dihydroorotase from Pseudomonas putida. Arch Microbiol. 1995;164:353–357. doi: 10.1007/BF02529982. [DOI] [PubMed] [Google Scholar]
- 17.Wang CC, Tsau HW, Chen WT, Huang CY. Identification and characterization of a putative dihydroorotase, KPN01074, from Klebsiella pneumoniae. The protein journal. 2010;29:445–452. doi: 10.1007/s10930-010-9272-2. [DOI] [PubMed] [Google Scholar]
- 18.Kim GJ, Lee DE, Kim HS. High-level expression and one-step purification of cyclic amidohydrolase family enzymes. Protein Expr Purif. 2001;23:128–133. doi: 10.1006/prep.2001.1484. [DOI] [PubMed] [Google Scholar]
- 19.Peroutka RJ, III, Orcutt SJ, Strickler JE, Butt TR. SUMO fusion technology for enhanced protein expression and purification in prokaryotes and eukaryotes. Methods Mol Biol. 2011;705:15–30. doi: 10.1007/978-1-61737-967-3_2. [DOI] [PubMed] [Google Scholar]
- 20.Walls D, Loughran ST. Tagging recombinant proteins to enhance solubility and aid purification. Methods Mol Biol. 2011;681:151–175. doi: 10.1007/978-1-60761-913-0_9. [DOI] [PubMed] [Google Scholar]
- 21.Malakhov MP, Mattern MR, Malakhova OA, Drinker M, Weeks SD, Butt TR. SUMO fusions and SUMO-specific protease for efficient expression and purification of proteins. Journal of structural and functional genomics. 2004;5:75–86. doi: 10.1023/B:JSFG.0000029237.70316.52. [DOI] [PubMed] [Google Scholar]
- 22.Marblestone JG, Edavettal SC, Lim Y, Lim P, Zuo X, Butt TR. Comparison of SUMO fusion technology with traditional gene fusion systems: enhanced expression and solubility with SUMO. Protein Sci. 2006;15:182–189. doi: 10.1110/ps.051812706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Catanzariti AM, Soboleva TA, Jans DA, Board PG, Baker RT. An efficient system for high-level expression and easy purification of authentic recombinant proteins. Protein Sci. 2004;13:1331–1339. doi: 10.1110/ps.04618904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 25.Sander EG, Wright LD, McCormick DB. Evidence for function of a metal ion in the activity of dihydroorotase from Zymobacterium oroticum. J Biol Chem. 1965;240:3628–3630. [PubMed] [Google Scholar]
- 26.Christopherson RI, Jones ME. The effects of pH and inhibitors upon the catalytic activity of the dihydroorotase of multienzymatic protein pyr1–3 from mouse Ehrlich ascites carcinoma. J Biol Chem. 1980;255:3358–3370. [PubMed] [Google Scholar]
- 27.Taylor WH, Taylor ML, Balch WE, Gilchrist PS. Purification of properties of dihydroorotase, a zinc-containing metalloenzyme in Clostridium oroticum. Journal of bacteriology. 1976;127:863–873. doi: 10.1128/jb.127.2.863-873.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Voyich JM, Vuong C, DeWald M, Nygaard TK, Kocianova S, Griffith S, Jones J, Iverson C, Sturdevant DE, Braughton KR, Whitney AR, Otto M, DeLeo FR. The SaeR/S gene regulatory system is essential for innate immune evasion by Staphylococcus aureus. The Journal of infectious diseases. 2009;199:1698–1706. doi: 10.1086/598967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mehboob S, Mulhearn DC, Truong K, Johnson ME, Santarsiero BD. Structure of dihydroorotase from Bacillus anthracis at 2.6 A resolution. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2010;66:1432–1435. doi: 10.1107/S1744309110037085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Washabaugh MW, Collins KD. Dihydroorotase from Escherichia coli. Sulfhydryl group-metal ion interactions. J Biol Chem. 1986;261:5920–5929. [PubMed] [Google Scholar]
- 31.Baba T, Takeuchi F, Kuroda M, Yuzawa H, Aoki K, Oguchi A, Nagai Y, Iwama N, Asano K, Naimi T, Kuroda H, Cui L, Yamamoto K, Hiramatsu K. Genome and virulence determinants of high virulence community-acquired MRSA. Lancet. 2002;359:1819–1827. doi: 10.1016/s0140-6736(02)08713-5. [DOI] [PubMed] [Google Scholar]
- 32.Burgess RR. Preparing a purification summary table. Methods Enzymol. 2009;463:29–34. doi: 10.1016/S0076-6879(09)63004-4. [DOI] [PubMed] [Google Scholar]
- 33.Schilling S, Niestroj AJ, Rahfeld JU, Hoffmann T, Wermann M, Zunkel K, Wasternack C, Demuth HU. Identification of human glutaminyl cyclase as a metalloenzyme. Potent inhibition by imidazole derivatives and heterocyclic chelators. J Biol Chem. 2003;278:49773–49779. doi: 10.1074/jbc.M309077200. [DOI] [PubMed] [Google Scholar]
- 34.Huang KF, Liu YL, Wang AH. Cloning, expression, characterization, and crystallization of a glutaminyl cyclase from human bone marrow: a single zinc metalloenzyme. Protein Expr Purif. 2005;43:65–72. doi: 10.1016/j.pep.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 35.Liao RZ, Yu JG, Raushel FM, Himo F. Theoretical investigation of the reaction mechanism of the dinuclear zinc enzyme dihydroorotase. Chemistry. 2008;14:4287–4292. doi: 10.1002/chem.200701948. [DOI] [PubMed] [Google Scholar]
- 36.Lee M, Maher MJ, Christopherson RI, Guss JM. Kinetic and structural analysis of mutant Escherichia coli dihydroorotases: a flexible loop stabilizes the transition state. Biochemistry. 2007;46:10538–10550. doi: 10.1021/bi701098e. [DOI] [PubMed] [Google Scholar]
- 37.Thoden JB, Phillips GN, Jr, Neal TM, Raushel FM, Holden HM. Molecular structure of dihydroorotase: a paradigm for catalysis through the use of a binuclear metal center. Biochemistry. 2001;40:6989–6997. doi: 10.1021/bi010682i. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.