

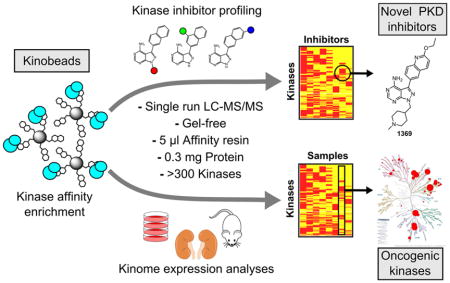
Abstract
ATP-competitive protein kinase inhibitors are important research tools and therapeutic agents. Because there are >500 human kinases that contain highly conserved active sites, the development of selective inhibitors is extremely challenging. Methods to rapidly and efficiently profile kinase inhibitor targets in cell lysates are urgently needed to discover selective compounds and to elucidate the mechanisms of action for polypharmacological inhibitors. Here, we describe a protocol for microgram-scale chemoproteomic profiling of ATP-competitive kinase inhibitors using kinobeads. We employed a gel-free in situ digestion protocol coupled to nanoflow liquid chromatography-mass spectrometry to profile ~200 kinases in single analytical runs using as little as 5 μl of kinobeads and 300 μg of protein. With our kinobead reagents, we obtained broad coverage of the kinome, monitoring the relative expression levels of 312 kinases in a diverse panel of 11 cancer cell lines. Further, we profiled a set of pyrrolopyrimidine- and pyrazolopyrimidine-based kinase inhibitors in competition-binding experiments with label-free quantification, leading to the discovery of a novel selective and potent inhibitor of protein kinase D (PKD) 1, 2 and 3. Our protocol is useful for rapid and sensitive profiling of kinase expression levels and ATP-competitive kinase inhibitor selectivity in native proteomes.
Keywords: Kinobeads, SILAC, kinase inhibitor, protein kinase D, target identification, proteomics
Graphical Abstract
Introduction
Protein kinases regulate cellular signaling by modulating the activity, localization and interactions of their substrates.1 Kinase-dependent signaling pathways are commonly dysregulated in cancer and a number of targeted small molecule kinase inhibitor therapeutics have been developed over the last two decades.2, 3 Numerous kinase inhibitors are in clinical and preclinical trials as anti-cancer drugs and are widely used as chemical genetic probes to study cell signaling. Kinase inhibitors that are used in the clinic can vary significantly in their target selectivity. For example, lapatinib only potently inhibits EGFR and ERBB2 while dasatinib inhibits over 30 tyrosine and serine/threonine kinases.4–7 Because the selectivities of many kinase inhibitors are not known and can be expected to impact diverse aspects of cellular signaling, their mechanisms of action as therapeutic agents are often unclear which can lead to undesirable off-target effects and toxicity.8 Identifying mechanisms of action and targets of kinase inhibitors greatly facilitates the discovery of novel small molecule therapeutics and basic research tool compounds.9, 10
Several approaches have been developed to profile kinase inhibitors against large panels of recombinant or native cellular kinases.6, 11–15 Affinity chromatography-based methods that use immobilized kinase inhibitors and quantitative mass spectrometry (MS) to probe full length kinases from whole cellular proteomes –commonly referred to as kinobead-based methods– are of particular value because they can simultaneously assay multiple proteins in a cell or tissue sample instead of recombinant kinases.11–13, 16–19 Such approaches can profile over 200 kinases in single experiments and the use of stable isotope labeling methods, like SILAC, isobaric chemical labeling, or reductive dimethylation, can increase analytical throughput.20–23
Kinobead-based proteomic assays have greatly enhanced our ability to study the kinome and the method has been progressively improved to achieve higher throughput and sensitivity.11, 24–29 Here, we prepared a set of kinobead reagents using seven non-selective kinase inhibitors that provide broad kinome coverage and applied an improved chemoproteomics protocol requiring reduced amounts of starting protein material and single nanoLC-MS/MS analytical runs. With in-situ digestion of enriched kinases from as little as 5 μl of kinase affinity resin and 300 μg of cellular protein per pulldown experiment, we were able to quantify ~200 protein and lipid kinases from a human cell line in single analytical runs. We demonstrate the strength of the approach by i) profiling relative kinase expression levels in a diverse panel of 11 cancer cell lines and ii) identifying a highly selective inhibitor of the mammalian protein kinase D (PKD) family of serine/threonine kinases among compounds that were originally designed to target Toxoplasma gondii (T. gondii) calcium-dependent protein kinase 1 (TgCDPK1). This novel PKD inhibitor should be a useful chemical genetic tool compound to examine the role of mammalian PKDs in cell signaling and disease.
Experimental Section
The kinobead affinity reagents (Fig. 1a) as well as the inhibitors 1553, 1561, 1649 and 1369 (Fig. 4a) were synthesized in-house. The kinobead affinity reagents were immobilized on ECH Sepharose 4B (GE Healthcare Bio-Sciences, Pittsburgh, PA) according to the previously published protocol.24
Figure 1. Non-selective kinase affinity reagents and experimental protocol for enrichment/soluble competition and LC-MS/MS analysis of cellular kinases.

a) Non-selective type I (1–3) and type II (4–7) kinase inhibitor analogs used to generate the kinobead affinity matrices in these studies. b) Our kinase inhibitor profiling workflow features microgram-scale affinity enrichment of cellular kinases with kinobeads in SILAC cell lysates, soluble competition, on-bead digest of protein samples, and single-shot nanoLC-MS/MS analysis.
Figure 4. Kinobead chemoproteomics profiling of kinase inhibitors.

a) Structures of the four pyrrolopyrimidine and pyrazolopyrimidine compounds developed in our laboratory as well as the parent compounds NA-PP1, NA-PP2 and NM-PP1. b) Volcano plot from a moderated two-tailed, two sample t-test applied to the log2 transformed MaxQuant LFQ intensities of the label-free 1649 competition (50 μM) and DMSO control experiments (five replicates each). Significantly competed kinases are shown in red and non-competed kinases in black. Other proteins are in gray. Added soluble competitor leads to a depletion of the target kinases from the kinobead affinity matrix. Hit kinases appear to the left of the cut-off line indicating an FDR<0.05. c) Box and whisker plots of all log2 LFQ ratios calculated by a moderated two sample t-test for soluble competition experiments with 50 μM of 1553, 1561, 1649 or 1369 over DMSO control. The plot was overlaid with the protein and lipid kinase LFQ ratios (in red).
Cell culture and harvest
U-2 OS, HCT 116, K-562, BJ-5ta, DU4475, HL-60, HS 604.T, Jurkat, NCI-H211, P3HR-1, SH-SY5Y, and HeLa cells were from the American Type Culture Collection (ATCC, Manassas, VA) and cultured in ATCC recommended media supplemented with FBS (Seradigm, Radnor, PA) and Penicillin-Streptomycin-Glutamine (100x, Thermo Scientific, Rockford, IL) in an incubator at 37°C and 5% CO2. For SILAC labeling, HeLa, U-2 OS and HCT-116 cells were grown in custom -Lys/-Arg DMEM (Caisson Labs, North Logan, UT) supplemented with 10% dialyzed FBS (Sigma, St Louis, MO), Penicillin-Streptomycin-Glutamine (100x), 200 μg/ml proline and SILAC amino acids (0.2 mM natural isotope abundance Lys/Arg for light label, 0.2 mM Lys-D4/Arg-13C6 for medium label, 0.2 mM Lys-13C6,15N2/Arg-13C6,15N4 for heavy label, Cambridge Isotope Labs, Andover, MA). Cells were grown for at least 5 cell doublings in SILAC medium and harvested when reaching 90% confluency or a density of 1×106 cells/ml. For harvesting, cells were rinsed twice with ice cold PBS and lysed in 750 μl/20 million cells of mod. RIPA buffer (50 mM Tris, 150 mM NaCl, 10 mM NaF, 0.25% Na-deoxycholate, 1% NP-40 and 5% glycerol) containing Halt™ Protease Inhibitor Cocktail (100x, Thermo Scientific, Rockford, IL). The ice-cold cell lysate was then vortexed five times intermittently for 3 seconds and clarified by centrifugation at 21,000 rcf and 4°C for 20 min. The protein content of the samples was determined using the Pierce 660 nm Assay Reagent (Thermo Scientific, Rockford, IL). U-2 OS and HCT 116 cell lysates encoded with the same SILAC amino acids and label-free cell lysates were mixed in a protein ratio of 1:1 and snap frozen in liquid nitrogen and stored at −80°C until used.
Kinase affinity enrichment/soluble competition experiments and sample digestion
For kinase enrichment, 10 μl of a 50% kinobead slurry in 20% aq. ethanol (complete kinobeads, equal amounts of affinity reagents 1–7) was pipetted into a 1.5 ml microtube and washed twice with 100 μl of mod. RIPA buffer. 300 μg of cell extract (2 mg/ml protein concentration, 150 μl) were pipetted into the tubes and the slurry was agitated on an end-over-end rotator for 3h at 4°C. For soluble competition experiments, SILAC and LFQ lysates were pre-incubated for 20 min at 4°C with either DMSO (Ctrl, 1% final concentration) or the compound at the desired concentration (DMSO solution, 1% final). After the 3 h pulldown, the supernatant was aspirated and the beads were washed twice with 100 μl mod. RIPA buffer and three times with 100 μl of 50 mM Tris, 150 mM NaCl (pH = 7.8, TBS) and the beads were re-suspended in 25 μl freshly prepared tris-buffered urea (8 M urea, 50 mM tris, pH = 7.8) containing 5 mM tris(2-carboxyethyl)phosphine (TCEP) and 10 mM 2-chloroacetamide (CAM) and agitated on a Thermomixer (Eppendorf, Hauppauge, NY) for 30 min at 1400 rpm and 37°C. For duplex and triplex SILAC labeled samples, the beads from the heavy, medium and light channels were mixed after the mod. RIPA wash step and further treated as described but with 2 to 3-times the amount of buffers/reagents. For digestion, the slurry was diluted two-fold with 100 mM aq. triethylammonium bicarbonate solution (TEAB, urea concentration ≤4 M), the pH was adjusted to 8.5 with 1 N NaOH and 0.4 μg of LysC (Wako Chemicals, Richmond, VA) for every 5 μl of settled affinity resin was added. The slurry was shaken for 2 h at 1400 rpm and 37°C. Then the slurry was further diluted 2-fold with 100 mM TEAB (urea concentration ≤2 M) and 0.4 μg of MS grade trypsin (Thermo Scientific, Rockford, IL) for every 5 μl of settled affinity resin was added. Samples were digested overnight on a Thermomixer at 37°C, diluted two-fold with 5% aq. acetonitrile (ACN) containing 0.1% trifluoroacetic acid (TFA) and acidified with formic acid (FA, 1% final). Peptides were extracted using StageTips30 and then analyzed in single nanoLC-MS/MS runs. For kinase abundance profiling using kinobead fractionation, lysates were incubated for 1 h at 4°C with 10 μl of a 50% slurry of affinity matrix 1. The supernatant was transferred into a tube containing 10 μl of a 50% slurry of the affinity matrices 2, 4, 5 and 6 (1:1:1:1 mixture) and incubated for another 1h at 4°C. Then the supernatant was transferred into a tube containing 10 μl of a 50% slurry of the affinity matrices 3 and 7 (1:1 mixture) and incubated for another 1h at 4°C. The kinobead fractions were then processed separately as described above and analyzed in single nanoLC-MS/MS runs.
LC-MS/MS and data analysis
Peptides were separated on a Thermo-Dionex RSLCNano UHPLC instrument (Sunnyvale, CA) with 10 cm long fused silica capillary columns made in-house with a laser puller (Sutter, Novato, CA) and packed with 3 μm 120 Å reversed phase C18 beads (Dr. Maisch, Ammerbuch, DE). The LC gradient was 90 min long with 10–35% B at 200 nL/min. LC solvent A was 0.1% acetic acid and LC solvent B was 0.1% acetic acid, 99.9% acetonitrile. MS data was collected with a Thermo Orbitrap Elite spectrometer. Data-dependent analysis was applied using Top15 selection with CID fragmentation. Raw files were analyzed by MaxQuant/Andromeda31 version 1.5.2.8 using protein, peptide and site FDRs of 0.01 and a score minimum of 40 for modified peptides, 0 for unmodified peptides; delta score minimum of 17 for modified peptides, 0 for unmodified peptides. MS/MS spectra were searched against the UniProt human database (updated July 22nd, 2015). MaxQuant search parameters: Variable modifications included Oxidation (M). Carbamidomethyl (C) was a fixed modification. Max. labeled amino acids was 3, max. missed cleavages was 2, enzyme was Trypsin/P, max. charge was 7, SILAC labels were Arg0/Lys0 (light), Arg6/Lys4 (medium), Arg10/Lys8 (heavy). The MaxQuant re-quantification feature was enabled. The initial search tolerance for FTMS scans was 20 ppm and 0.5 Da for ITMS MS/MS scans. Data was further processed using the Perseus software package (version 1.5.2.6), the R environment, Origin Pro 8.0 and Microsoft Excel. Because our goal is to quantify kinases for secondary analyses (for e.g. inhibitor profiling), we only consider proteins “identified” if MaxQuant is able to compute a protein intensity value; this is a more stringent criterion of protein identification as identifying peptides need to have good MS/MS spectrums and quantifiable ion chromatograms. Further, missing protein intensity values in LFQ analyses were imputed using Perseus by sampling from a distribution downshifted by 1.3 and having a width of 0.2.32 Imputed values are reported in the SI-Excel file 3. Significantly competed kinases in the 50 μM competitor experiment were determined by applying a moderated two-tailed, two-sample t-test in Perseus with an FDR of 0.05 and a small constant s0 = 0.3. Further, a fold-change cut off for the log2 LFQ ration of 1 was applied. The MS raw files were uploaded to the MassIVE proteomics repository (https://massive.ucsd.edu) under the accession number MSV000080122.
Kinobead competitor titration assay
The inhibitors 1369 and 1649 were prepared in DMSO at 12 different concentrations ranging from 50 μM to 0.01 μM (i.e 50, 25, 10, 5, 2, 1, 0.4, 0.2, 0.08, 0.04, 0.02, 0.01 μM). Kinobead soluble competition experiments were performed as described with the complete kinobead mixture in label-free lysate (1:1 mixture of HCT116 and U-2 OS cell lysates) in triplicates and peptide samples were analyzed in single analytical runs. Kinase LFQ ratios were calculated as the difference of the log2 intensity of a soluble competition experiment and the average intensity of the DMSO control experiment (five replicates) to yield three LFQ ratio values for each protein at each concentration. For plotting of the titration curve and for calculation of the IC50s the mean and the standard deviation (SD) was calculated from these three values. Graphs were plotted and analyzed using GraphPad Prism software package (V5.0a) using a least squares nonlinear regression model for curve fitting (One site - Fit logIC50 function).
In-vitro kinase activity assay
The kinase activities of PKD1 (CarnaBio USA, Natick, MA), PKD2 (EMD Millipore, Billerica, MA) and PKD3 (Enzo Life Sciences, Farmingdale, NY) were profiled in presence of myelin basic protein (MBP) as the substrate and the inhibitors in 3-fold dilutions at 10 different concentration starting at 10 μM as the highest concentration. The total reaction volume was 30.2 μl. For each reaction, 15 μl 2X KRB2-buffer (50 mM HEPES pH 7.5, 20 mM MgCl2, 1.2 mM EGTA), 0.8 μl H2O, 0.15 μl 10 mg/ml BSA, 0.04 μl 1.43 M β-mercaptoethanol (BME), 0.06 μl 1 M Na3VO4 and 6 μl of a 1 mg/ml MBP solution were mixed. 2 μl of 15X kinase stock solutions (75 nM for PKD1, 75 nM for PKD2 and 112.5 for PKD3) along with 1.2 μl of the 25X inhibitor solution in DMSO were added and the reaction was incubated for 30 min at RT. Then 5 μl of ATP-[γ32P] (0.04 μCi/μl in H2O) were added, the reactions mixed by pipetting and incubated for 8 h at RT. 4.6 μl aliquots of the reaction mixture were spotted onto nitrocellulose, the membranes washed 3 times with 0.5% phosphoric acid and dried with acetone. Membranes were wrapped in plastic wrap and placed in a phosphor screen for autoradiography and exposed overnight. Phospho imaging was performed using a Typhoon FL 9000 instrument (GE Healthcare, Pittsburgh, PA) and the raw data was processed with the GraphPad Prism software package (V5.0a) using the One site - Fit logIC50 function for curve fitting.
Correlation analysis of mRNA and protein expression data
For the correlation of kinase mRNA and protein expression, we only included kinases quantified with MaxQuant protein intensity values in at least 3 out of 11 cell lines in the kinobead kinase enrichment experiments (true for 269 out of 312 kinases, see SI-Excel file 1 and 2); extracting log10 mRNA intensity values from the CCLE mRNA dataset and the log10 MaxQuant protein intensity values, we then computed the Pearson’s product-moment correlation coefficient r for each of these 269 genes (see Fig. 2c and SI-Excel file 2).
Figure 2. Analysis of kinases enriched from a diverse panel of cell lines using kinobeads.

a) Hierarchical selection criteria for 11 diverse human cancer cell lines for kinome profiling. b) Human kinome dendrogram showing the 312 protein and lipid kinases enriched from the panel of 11 cancer cell lines. Red circles represent the means of the log10 MaxQuant protein intensities of kinases calculated from triplicate pulldowns of each cell line. c) Ranked plot of the Pearson’s r for the correlation between kinase mRNA expression data derived from the CCLE database and the protein expression levels based on MaxQuant protein intensity. For correlation analysis, only kinases quantified in at least 3 out of 11 cell lines were considered. Kinases previously identified as cancer drivers are highlighted in gray and marked with their gene names. Correlation between mRNA and protein expression data ranged widely. A table containing all 269 correlated kinases with the corresponding Pearson’s r values can be found in SI-Excel file 2.
Results and discussion
Synthesis and evaluation of individual kinobead affinity reagents
In developing our kinobead reagents, we wanted to identify a set of immobilized kinase inhibitors that could efficiently and broadly enrich the human kinome. Based on previous profiling studies, we selected seven affinity reagents derived from non-selective type I and type II kinase inhibitor scaffolds as synthetic targets (Compounds 1–7 Fig. 1a).5, 24, 28, 33–35 The inhibitor analogs 1 and 3–5 were described previously and the affinity reagents 2, 6 and 7 were newly developed in our laboratory. Compounds 1–7 were immobilized on carboxy-derivatized sepharose using amide coupling chemistry to yield the corresponding affinity matrices. To assess each affinity reagent’s ability to specifically enrich kinases from cell lysates, we performed a single series of affinity pulldown-soluble competition experiments using duplex SILAC-labeled HeLa cell lysate as previously described (Fig. 1b).5, 18 Experiments were done in quadruplicate, with two SILAC label swap experiments in which SILAC labels were switched between control and inhibitor competition experiments. The label swap experiment helps to distinguish true hits from non-selective hits as the former show an inversion of the sign of the log2 SILAC ratios, while contaminants and false positives do not.18 Soluble competitors were tested at 50 μM of each non-immobilized probe 1–3 and 6 and 50 μM of foretinib (EXEL-2880) for probe 7 (Fig. 1a. From the resulting dataset we can simultaneously determine both the relative enrichment and specificity of kinase interactions with each probe. The affinity reagents 4 and 5 have been characterized in 5 and the corresponding data were extracted from the published dataset.
To estimate the kinase enrichment efficiency of each probe matrix, the protein MS signal intensity from the DMSO control experiments was extracted from the dataset. These intensity values were used as an approximate measure of kinase abundance in the pulldown samples. We analyzed how many protein and lipid kinases could be identified with each probe, i.e. that can be assigned a protein intensity value in at least one out of four replicates. We found that the majority of kinases were captured by the type I probes 1 (n = 115) and 2 (n = 107), representing 148 (73%) out of the 203 total kinases identified in HeLa cell lysate. Probes 3 (n = 80), 4 (n = 82), 5 (n = 57), 6 (n = 38) and 7 (n = 66) contributed to the capture of an additional 55 kinases from all major kinase families (SI-Fig. 1). Probes 4–7 were very efficient in the enrichment of members of the tyrosine kinase (TK) and tyrosine kinase-like (TKL) families of protein kinases and are highly complementary to the type I probes 1–3 in terms of kinome coverage (for a heat map of kinase intensities for each probe see SI-Fig. 1). This evaluation directly assessed the value of each probe in the kinobead mixtures for capturing complementary components of the kinome.
Quantifying changes in kinase abundance due to the presence of soluble competitor identified kinases that interact strongly and specifically with probes 1–7 (SI-Fig. 2). For each probe, ca. 75% of those kinases that were identified with a protein intensity value in the DMSO control experiments could also be assigned a quantitative SILAC ratio between control and competition. In turn, of these quantified kinases 45% to 67% were competed off the individual kinobead reagents in presence of 50 μM of the corresponding free probe. This was interpreted as an evidence for a specific-probe kinase interaction. These findings are important because only kinases that bind to the immobilized kinobead reagent through specific interactions (e.g. interaction with the ATP-binding pocket of kinases) are accessible to kinome-scale profiling of kinase inhibitors.
The soluble competition experiments between the novel affinity reagent 7 and its parent compound, the clinical, multi-tyrosine kinase inhibitor foretinib (EXEL-2880), demonstrate the utility of our kinase inhibitor profiling workflow (SI-Fig. 3).36 Our profiling of foretinib targets from cellular lysates identified its specific interaction with 29 protein and lipid kinases. Most of these kinases have been previously found to interact with recombinant kinase domains in a phage display binding assay (SI-Fig. 3),37 supporting the accuracy of our approach. Further, a number of non-kinase proteins were found to be specifically competed off the affinity matrices with the competitor probes 1–7. These could be either proteins interacting with enriched kinases or non-kinase targets of the tested affinity reagents (SI-Table 1).
Kinome expression analysis of 11 diverse cancer cell lines with kinobeads
Close to 20 % of all protein and lipid kinases are classified as oncogenic drivers, and measurement of the expression levels of these kinases are thought to be informative for the state of the disease.38 Proteomics methods are increasingly recognized as important, orthogonal tools that can measure aberrant protein expression other than quantification of mRNA transcripts.39 Current proteomics technologies cannot sample a whole proteome; selective biochemical enrichment of subsets of proteins is a powerful approach to improve analytical coverage. Because kinase affinity reagents facilitate broad and efficient enrichment of the kinome, they can be used to quantify kinase expression in cells and tissue samples.11, 40, 41 For this reason, we wished to examine our affinity reagents’ potential to broadly enrich and quantify kinases from extracts of a diverse set of human cancer cell lines.
To select a small but representative panel of cancer cell lines with high and diverse expression of kinases, we took advantage of available mRNA expression data from the Cancer Cell Line Encyclopedia (CCLE).42 Using unsupervised clustering of kinase mRNA intensities from 1037 CCLE cell lines, we first chose cell lines from different clusters (primarily classified by distinct tissue of origin) that also contained the most kinases ranked in the top 10th percentile of expression. Because this selection filter generated many possible combinations with high kinase diversity and mRNA abundance from different cancer etiologies, we then chose cell lines based on practical considerations such as availability (ATCC); high proliferation rates; amenability to cell culture (see Fig. 2a for a hierarchical order of selection criteria). This procedure led to the selection of a panel of 11 cell lines; these were BJ-5ta (human foreskin fibroblast), DU4475 (skin carcinoma), HL-60 (acute promyelocytic leukemia), Hs 604.T (Hodgkin’s lymphoma), Jurkat (acute T-cell leukemia), K-562 (chronic myelogenous leukemia), NCI-H211 (small cell lung cancer), P3HR-1 (Burkitt’s lymphoma), SH-SY5Y (neuroblastoma), HCT 116 (colorectal carcinoma) and U-2 OS (osteosarcoma).
To increase our ability to sample the kinome, we combined affinity reagents 1–7 (Fig. 1a) into three different kinobead affinity matrices with overlapping but distinct kinase target profiles. These were matrix 1 (compound 1), matrix 2 (compounds 2, 4, 5 and 6) and matrix 3 (compounds 3, 7). We applied our previously published pulldown and on-bead digestion protocol to obtain kinase-enriched samples for single-shot nanoLC-MS/MS analysis in unlabeled cell lysates.5 Each lysate was incubated with kinobead matrices 1, 2 and 3 for 1 h at 4°C, consecutively, and MS data combined for protein identification. We identified an average of 220 protein and lipid kinases for each cell line in three analytical runs (SI-Fig. 4). From three replicate analyses per cell line, we identified a total of 312 kinases across the 11 cancer cell line panel (SI-Fig. 5). The enriched kinases derive from all major branches of the kinome,1 have high average sequence coverage (~32 %), with protein MS intensities spanning five orders of magnitude (Fig. 2b and heat map in SI-Fig. 5). Both the protein intensity and sequence coverage of kinases were found to be substantially higher compared to non-kinases (SI-Fig. 6). Two-thirds of the total protein MS intensity in MS analyses were derived from protein and lipid kinases, further demonstrating that our kinobead reagents provide specific and efficient kinome enrichment (SI-Excel file 1).
Although mRNA quantification is widely used to measure gene expression, it is well known that the correlation between protein and mRNA expression levels can vary greatly.43–45 We used our kinome analyses to examine the correlation between protein and mRNA expression of 312 kinases in our panel of 11 CCLE lines. As a measure of linear correlation between mRNA and protein expression (as determined by kinobead-LC-MS/MS analysis), we computed the Pearson moment-correlation coefficient, r, for the log10 intensities of mRNA and protein MS signal across all cell lines for each individual kinase (Fig. 2c, see also Experimental Section). This analysis showed the trend that the correlation of relative mRNA and protein expression levels in our dataset varied considerably between individual kinases ranging from very strong positive correlation (>0.9) to very strong negative correlation (<-0.9). Our correlation analysis contained 58 of the 91 recently proposed “cancer driver kinases” 38. These genes were evenly distributed across the range of calculated r values, highlighting the relevance of the kinobead approach in the interpretation of kinome expression data from human cancer cell lines/tumor samples (“Cancer driver kinases” highlighted in gray Fig. 2c); we observe, for example, no correlation between mRNA expression and protein abundance for important oncogenic kinases like AKT1, BRAF and MTOR. We noticed a subtle but significant correlation between the average MaxQuant protein intensity and the Pearson’s r for mRNA-protein intensity correlation (lower kinase intensity with lower r). The average correlation of mRNA and protein intensity improved when missing values in the proteomics dataset were replaced by imputed values representing low MS signal; this led to a poorer correlation between average protein intensity and Pearson’s r (for a detailed discussion of these observations, see SI-Fig. 7). Because kinases are important functional nodes in cellular signaling pathways, our analyses underscore the importance of measuring both kinase mRNA and protein levels in cancer profiling. Our kinobead-based enrichment workflow, which can measure the abundance of >300 kinases from ≤1 mg of input protein, would enable rapid and targeted analyses of cancer kinomes.
Impact of Label-free vs. SILAC quantification on coverage in kinobead analyses
Stable isotope labeling of proteins/peptides can facilitate accurate quantification of proteins and multiplexing of experiments in single analytical MS runs. Precursor-MS stable isotope labeling approaches like SILAC or reductive dimethylation are robust, convenient, and provide high quantitative precision.23 Conversely, each labeled state increases sample complexity and may lead to decreased analytical depth, particularly in unfractionated LC-MS analyses of highly complex peptide mixtures where sample complexity exceeds the sampling speeds of the mass spectrometer.46 Although kinome affinity enrichment should mitigate the problem of sample complexity, we wanted to examine the effect of sample multiplexing on the depth of kinome coverage.
To directly assess the impact of SILAC labeling on the performance of our kinobead analysis, we compared triplicate kinobead pulldowns using non-labeled (label-free, LF), duplex SILAC-, or triplex SILAC-labeled cell extracts derived from HCT 116 and U-2 OS cells (1:1 mixture). Together, the two cell lines yielded 276 of the 312 kinases identified from the 11 cancer cell line panel and provide sufficient breadth of kinome expression for our purposes (SI-Fig. 4b).
To increase analytical throughput in these experiments, we used a single kinobead reagent mixture obtained by combining the individual affinity reagents 1–7 in equal amounts. Our previous kinase affinity pulldowns with 12.5 μl of affinity resin (settled volume) and 1 mg of protein yielded enough peptide sample for ca. 3 analytical LC-MS/MS runs (10–15 μg). Based on these observations, we scaled down our protocol to use 5 μl of affinity resin (settled volume) and 300 μg of protein for each LF sample and for every SILAC channel (light, medium and heavy, 1:1 mixtures). Peptides were then analyzed in single analytical LC-MS/MS runs. SILAC labeling resulted in a two- or three-fold increase in the numbers of peptide precursors, for duplex- and triplex-SILAC, respectively. We evaluated each experiment by standard metrics, including total number of proteins and the number of lipid and protein kinases identified and quantified per LC-MS run as well as the distribution of protein intensities and sequence coverage for kinases and non-kinases (Fig. 3). The bar plot (Fig. 3a) compares the number of peptides, proteins and kinases identified in single-shot LC-MS runs and the Venn diagram (Fig. 3b) compares the identity of kinases identified for LF and SILAC-labeled HCT116/U-2 OS samples; SILAC labeling led to a ~30% decrease in the number of identified peptides and proteins as well as the number of quantified proteins and kinases compared to LF samples. Accordingly, the total number of identified protein and lipid kinases dropped from 211 for LF samples to 157 and 160 for duplex and triplex SILAC labeled samples, respectively. Unsurprisingly, 37 of the 39 kinases not detected in duplex or triplex labeled samples that could be detected in the LF samples were found in the lowest MS intensity quartile (Venn diagram Fig. 3b). In this set of experiments, no further drop in identified/quantified proteins was observed upon increase of the multiplexing from duplex to triplex SILAC.
Figure 3. Comparison of label free (LF), duplex SILAC, and triplex SILAC performance.

a) Bar plots comparing the number of identified and quantified peptides, proteins and kinases between label-free and multiplexed (duplex and triplex) SILAC experiments performed in a 1:1 mixture of HCT 116 and U-2 OS cell lysate (see Experimental Section). We consider a peptide or protein “identified” only if an intensity value was computed by MaxQuant. A protein was considered “quantified” if a SILAC ratio was computed by MaxQuant. The values are the mean numbers from three LC-MS experiments and the error bars are the standard deviation (S.D.). Identified kinases are the total from all three runs (intensity value for a kinase could be computed for at least one out of three experiments). *: A protein was considered quantified in LF experiments if a LFQ intensity was computed by MaxQuant. b) Venn diagram comparing the number of identified kinases between LF and SILAC samples.
We found that use of just 5 μl of a combined kinobead reagent (1–7) in 300 μg of cellular protein was sufficient to identify >200 kinases in single analytical LC-MS/MS runs; this is a significant reduction in reagents, protein input and LC-MS analysis time. Comparable with the experiments done in the 11 cancer cell lines with three kinobead reagents, the use of a single kinobead reagent yielded peptide samples in which 64% of the protein intensity signal was derived from protein and lipid kinases. This was independent of LF or SILAC quantification. Further, the distribution of protein intensities and sequence coverage between protein kinases and background proteins was found to be highly similar to the previous experiments (for reference see SI-Fig. 6). For these reasons we decided to use this protocol in subsequent kinase inhibitor profiling experiments.
Kinome chemoproteomics profiling of T. gondii CDPK1 inhibitors
Compounds based on pyrazolopyrimidine and pyrrolopyrimidine scaffolds, like 1553, 1561, 1649 and 1369 (Fig. 4a), have been developed as selective T. gondii and Cryptosporidium parvum (C. parvum) CDPK1 (Tg/CpCDPK1) inhibitors. These compounds contain substituents that exploit the rare and small glycine gatekeeper residues of Tg/CpCDPK1, which allows selectivity for these parasitic kinases over mammalian kinases like Src and Abl.47–54 However, it has been shown previously that the related compounds NA-PP1 and NM-PP1, which have been designed to target mutant kinases that contain a glycine gatekeeper residue, can also inhibit several other kinases, including the protein kinase D (PKD) isoenzymes (Fig. 4a).55, 56 Off-target inhibition of human kinases by compounds targeting parasite kinases may lead to unwanted side effects when applied in animal models or humans. For this reason, we used our kinobead-based inhibitor profiling method to determine if any of our Tg/CpCDPK1 inhibitors would also inhibit mammalian kinases.
The four compounds, 1553, 1561, 1649 and 1369 were selected from a larger pool of CDPK inhibitors for kinome-wide profiling based on structural diversity (Fig. 4a). For the profiling of the inhibitors, we carried out kinobead soluble competition experiments in the LF lysate mixture of the human U-2 OS and HCT 116 cancer cell lines (hereafter referred to as LFQ master mix); we adapted the single kinobead reagent workflow (equal amounts of 1–7, Fig. 1a) used to compare SILAC and LF quantification of kinase enriched samples in the LF master mix to increase kinome coverage.
In a first round of experiments all four inhibitors were profiled at a single (high) concentration of 50 μM (five replicates) to determine all kinases that potentially interact with the competitors. To determine specific kinase interactors from the LFQ competition experiments, putative hits had to pass two criteria: (1) achieve an FDR of <0.05 in a moderated two sample t-test; and (2) show a log2 LFQ intensity difference value ≤1.0 (hereafter LFQ ratio). Testing the panel of compounds, including DMSO control experiments, resulted in 25 analytical LC-MS/MS runs. In this dataset, 31000 values for the MaxQuant LFQ protein intensity were determined and 214 protein and lipid kinases were identified (see SI-Excel file 3). Figure 4b shows a representative volcano plot obtained by comparing the five replicate 1649 soluble competition experiments (50 μM) with five DMSO ctrl experiments by means of the moderated t-test in the Perseus software package (see Experimental section for details).32 Hit kinases appear on the left side of the plot with negative log2 LFQ ratios. To illustrate the data distribution, we plotted LFQ ratios from the 50 μM soluble competition experiments with 1553, 1561, 1649 and 1369 (box and whisker plots Fig. 4c).
MS-based label-free quantification can fail if a protein species is detected in one sample, e.g. the DMSO control of a pulldown experiment but not in the corresponding soluble competition experiment. This problem was observed when profiling the Tg/CpCDPK1 inhibitors due to exhaustive depletion of high affinity kinase targets from the kinobeads. In order to assign LFQ ratios to these high affinity kinase targets, we replaced missing values in the dataset by data imputation using the Perseus software (see Experimental section, SI-Excel file 3 and Fig. 5).
Figure 5. Initial kinome target profiling of T. gondii CDPK1 inhibitors.

The heat map shows all putative drug-kinase interactions detected in the kinobead-competition assay at 50 μM 1553, 1561, 1649, or 1369 in the LFQ master mix. The panel’s color scale indicates the mean log2 LFQ ratios from 5 replicate LFQ pulldown experiments. For 1553, one replicate LC-MS run failed, so only four replicates were used for the analyses. Hits had to fulfill two criteria: (1) a log2 LFQ ratio ≥1 (i.e. 2-fold) and (2) a FDR <0.05 in a moderated two-tailed, two sample t-test of log2 LFQ protein intensity values from DMSO ctrl against competition experiments. a: one or more LFQ ratio values were obtained by data imputation.
The heat map in Figure 5 shows the mean log2 LFQ ratios (quintuplicate experiments) for the significant kinase hits identified (for exact log2 ratio values and −log10 P-values see SI-Table 2). We found that in these experiments at 50 μM competitor concentration, the compounds 1561, 1553 and 1649 interact with 28, 27, and 41 protein kinases, respectively, whereas inhibitor 1369 only interacts with 19 kinases. The majority (36 out of 46) of potential targets identified were serine/threonine kinases and almost exclusively members of the CAMK, TKL and STE families of protein kinases (Fig. 5, also see kinome dendrograms in SI-Fig. 8). Strikingly, all inhibitors competed PKD1, PKD2 and PKD3 very efficiently. The data also suggest that, besides the PKD isoforms, 13 other kinases are common targets of all four compounds (Fig. 5). Among these, kinases that were competed with high LFQ ratios (log2 ratio >5) were the activin receptor typ-1B (ACVR1B), aurora kinase A (AURKA), LIM domain kinase 1 (LIMK1), STE20-like serine/threonine-protein kinase (SLK), serine/threonine-protein kinase 10 (STK10, also known as LOK), receptor-interacting serine/threonine-protein kinase 2 (RIPK2) and the TGF-beta receptor type 1 (TGFBR1). RIPK2 was also identified as target of the parent compounds NA-PP1 and NM-PP1 in a previous study.55 Except for the 2-ethyloxynaphtyl-substituted 1369, the compounds also interacted with a number of tyrosine kinases, most notably the Src-family kinase YES1, the ephrin receptor A1 (EPHA1), proto-oncogene protein-tyrosine kinase receptor Ret (RET) and protein-tyrosine kinase 6 (PTK6).
To gain further insights into the relative affinities of the compounds to their putative human target kinases, we performed an inhibitor titration experiment in the LFQ master mix cell lysate with the most unselective compound, i.e. 1649, and the most selective compound, that is 1369. Soluble competitors were applied at 12 different concentrations ranging from 50 μM to 10 nM and pulldowns were performed in triplicate. This resulted in 72 soluble competition experiments and five DMSO control pulldowns that were analyzed in 77 LC-MS/MS runs. For plotting of titration curves, LFQ ratios were calculated from the log2 protein intensity values of the DMSO control and competition for all kinases that were found to be putative interactors of 1649 and 1369 in the 50 μM single-dose experiments (see Experimental section, SI-Excel file 3, Fig. 6 and SI-Fig. 9). The 1369 and 1649 titration curves for PKD1, PKD2 and PKD3 (the compounds’ putative main off-targets, Fig. 6a and b) suggest that 1369 has a higher relative affinity for all three isoforms, as at the lowest tested competitor concentration all PKDs were still competed off the kinobeads. This is reflected by the consistently ~2 to 4-fold lower IC50s of 1369 for the PKDs compared to 1649, i.e. <10 nM, 11 nM and 9.3 nM for 1369 vs. 26 nM, 23 nM and 39 nM for 1649 against PKD1, PKD2 and PKD3, respectively (Table 1a). In case of 1369 the major common off-target RIPK2 was calculated to have an IC50 of 3.6 μM which is ~100-fold higher than for the PKD isoforms (Fig. 6c and Table 1a). Conversely, 1649 shows an IC50(RIPK2) of 301 nM which is only about 10-fold higher than for the PKD isoforms (Fig. 6d and Table 1a). Strikingly, 1649 also shows 5–10-fold lower IC50 values for all other common off-targets as compared to 1369 (ranging from 4.4 μM to 500 nM). Further, dose response curves could be drawn for a number of kinases that were found to interact with 1649 but weakly or not at all with 1369 (SI-Fig. 9 and Table 1a). Two of these targets that showed an IC50 lower than 20 μM were LIMK1 (600 nM) and RIPK1 (13 μM).
Figure 6. Competitor titration curves for 1649 and 1369.

Panels a) and b) show the decrease of log2 LFQ intensity ratios for the PKD isoforms (main targets) with decreasing competitor concentration. Panels c) and d) show the decrease of log2 LFQ ratios for the six other shared major off-targets (competed by all inhibitors tested at 50 μM) with decreasing competitor concentration. Log2 LFQ intensity ratios are the mean of three ratios calculated from the protein intensity of a single replicate and the mean of the DMSO control intensity and the error bars are the standard deviation (S.D.). The competitors were applied at 12 different concentrations ranging from 50 μM to 10 nM. IC50 values for competition binding of the target kinase between the kinobeads resin and the soluble competitor were calculated from the curves using a least-squares non-linear regression model in GraphPad Prism (see Table 1a for computed IC50 values).
Table 1.
IC50 values (nM) of 1369 and 1649 for their kinase targets as determined by chemoproteomics profiling and a recombinant kinase activity assay.a
| Chemoproteomics titration experiments | ||
|---|---|---|
| Target | 1369 | 1649 |
| ACVR1B | 7000 ± 5700 | 1700 ± 570 |
| AURKA | 16000 ± 12000 | 1900 ± 580 |
| AURKB | n.a. | >20000 |
| LIMK1 | n.a. | 600 ± 200 |
| PRKD1 | <10 | 26 ± 14 |
| PRKD2 | 11 ± 9.5 | 23 ± 12 |
| PRKD3 | 9.3 ± 11 | 39 ± 24 |
| PTK6 | n.a. | >20000 |
| RIPK1 | n.a. | 13000 ± 3700 |
| RIPK2 | 3600 ± 1100 | 300 ± 72 |
| SIK2 | n.a. | >20000 |
| SLK | >20000 | 4400 ± 1400 |
| STK10 | >20000 | 1100 ± 350 |
| TGFBR1 | 7100 ± 3600 | 500 ± 220 |
| Recombinant kinase activity assay | ||
| Target | 1369 | 1649 |
| PRKD1 | 140 ± 23 | 67 ± 2.8 |
| PRKD2 | 43 ± 9.0 | 31 ± 5.0 |
| PRKD3 | 54 ± 9.0 | 31 ± 4.7 |
IC50 values are given in nM ± S.D. IC50s that were calculated to be higher than 20 μM are indicated by the placeholder “>20000”, IC50s estimated to be lower than 10 nM are indicated by the placeholder “<10” and for these values no S.D. is given.
Conclusively, chemoproteomics profiling of our CDPK inhibitors revealed that the three PKD isoforms are the major kinase off-targets in the human kinome. In an in vitro kinase activity assay using recombinant PKD1, 2 and 3, both 1649 and 1369 were found to inhibit kinase activity at nanomolar concentrations (30–140 nM IC50s, see Table 1b). Our results indicate that compound 1369 is a highly selective (~100-fold over the most relevant off-target RIPK2) and potent inhibitor against the PKD isoforms and is a useful tool compound for dissecting the roles of these kinases in cells and in vivo.
Conclusion
The wide dynamic range of protein abundance and the current limits of analytical instrumentation in proteomics analyses can be mitigated by approaches that enrich specific sub-proteomes, like the kinome, for study. This is especially important when studying disease in animal models or clinical samples where protein amounts may be a limiting factor.
In this study, we describe a novel, streamlined MS-based kinobead chemoproteomics protocol that can broadly enrich the kinome for downstream analyses. Our workflow employs microgram-scale kinase affinity enrichment (as little as 5 μl of kinobead resin, and 300 μg of input cell lysate), in-situ proteolytic digestion, and single-shot nanoLC-MS/MS analysis to achieve increased analytical throughput and sensitivity. We can apply this protocol to measure the relative abundance of the kinome across different samples, quantifying proteins with either SILAC multiplexing or label-free quantification. Likewise, this protocol is well suited for profiling of small to medium scale libraries of ATP-competitive kinase inhibitors in a competition binding assay. We anticipate that the workflow can be adapted to automated sample handling to facilitate the screening of large compound libraries.
To test the analytical depth of the method, we profiled a diverse panel of 11 cancer cell lines which resulted in the quantification of 312 protein and lipid kinases. When analyzing the linear relationship between these protein expression data and mRNA expression data obtained by the CCLE project using Pearson’s product-moment correlation analysis, we observed a poor average correlation for individual kinases across the cell lines panel (mean r = 0.34, Fig. 2c). Imputation of missing protein intensity values improved the correlation substantially (mean r = 0.47, no strong negative correlations observed) which suggests that the imputed (low) intensity values correctly estimated low protein expression (for a detailed discussion see SI-Fig. 7). This supports the assumption that the majority of detected kinases are quantified accurately. We anticipate that with our protocol similar results can be obtained from tissue samples.
We also used our optimized kinobead workflow in soluble competition experiments to profile four T. gondii CDPK1 inhibitors to identify possible targets in the human kinome. We observed that all four inhibitors interacted strongly with nine serine/threonine kinases, most prominently PKD1, 2, and 3; subsequent competitor titration experiments using 1649 and 1369 showed that the latter is highly selective for PKDs amongst the 214 human kinases profiled (Fig. 6 and Table 1). Gratifyingly, an in vitro kinase inhibition assay faithfully reproduced the results from the chemoproteomics inhibitor titration experiments, confirming that 1649 and 1369 have nanomolar IC50s (i.e. 30–140 nM) for the PKD isoforms. While our data suggest that 1369 and related compounds inhibit the human PKD isoforms, they still have >100-fold lower IC50s for TgCDPK1.53 Accordingly, off-target effects related to the inhibition of mammalian kinases may not present a significant issue in a physiological setting. More importantly, our findings indicate that 1369 is an ideal chemical genetic tool for probing PKD function in cell signaling.
Supplementary Material
Acknowledgments
We wish to thank members of the Ong lab, in particular Dr. Ho-Tak Lau, and the Maly lab for fruitful discussions and revision of the manuscript. Research reported in this publication was supported by the National Institutes of Health under award numbers R01GM083926, 5R01AI111341, R21EB018384, and R21CA177402. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. M.G. was supported by a post-doctoral fellowship of the DFG (German Research Foundation, GO 2358/1-1).
Footnotes
The authors declare that there are no competing financial interests.
Associated Content
- SI_Excel-1. Protein intensity values of kinome profiling of 11 cancer cell lines.
- SI_Excel-2. mRNA and protein intensity values for expression correlation analysis.
- SI_Excel-3. Protein intensity values of inhibitor profiling.
References
- 1.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298(5600):1912–34. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 2.Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9(1):28–39. doi: 10.1038/nrc2559. [DOI] [PubMed] [Google Scholar]
- 3.Wu P, Nielsen TE, Clausen MH. FDA-approved small-molecule kinase inhibitors. Trends in Pharmacological Sciences. 2015;36(7):422–39. doi: 10.1016/j.tips.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 4.Rix U, Hantschel O, Durnberger G, Remsing Rix LL, Planyavsky M, Fernbach NV, Kaupe I, Bennett KL, Valent P, Colinge J, Kocher T, Superti-Furga G. Chemical proteomic profiles of the BCR-ABL inhibitors imatinib, nilotinib, and dasatinib reveal novel kinase and nonkinase targets. Blood. 2007;110(12):4055–63. doi: 10.1182/blood-2007-07-102061. [DOI] [PubMed] [Google Scholar]
- 5.Golkowski M, Brigham JL, Perera GK, Romano GE, Maly DJ, Ong SE. Rapid profiling of protein kinase inhibitors by quantitative proteomics. Medchemcomm. 2014;5(3):363–369. doi: 10.1039/C3MD00315A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, Hocker M, Treiber DK, Zarrinkar PP. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011;29(11):1046–51. doi: 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- 7.Rusnak DW, Lackey K, Affleck K, Wood ER, Alligood KJ, Rhodes N, Keith BR, Murray DM, Knight WB, Mullin RJ, Gilmer TM. The effects of the novel, reversible epidermal growth factor receptor/ErbB-2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Mol Cancer Ther. 2001;1(2):85–94. [PubMed] [Google Scholar]
- 8.Hopkins AL. Network pharmacology: the next paradigm in drug discovery. Nat Chem Biol. 2008;4(11):682–90. doi: 10.1038/nchembio.118. [DOI] [PubMed] [Google Scholar]
- 9.Bamborough P. System-based drug discovery within the human kinome. Expert Opin Drug Discov. 2012;7(11):1053–70. doi: 10.1517/17460441.2012.724056. [DOI] [PubMed] [Google Scholar]
- 10.Miduturu CV, Deng X, Kwiatkowski N, Yang W, Brault L, Filippakopoulos P, Chung E, Yang Q, Schwaller J, Knapp S, King RW, Lee JD, Herrgard S, Zarrinkar P, Gray NS. High-throughput kinase profiling: a more efficient approach toward the discovery of new kinase inhibitors. Chemistry and Biology. 2011;18(7):868–79. doi: 10.1016/j.chembiol.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bantscheff M, Eberhard D, Abraham Y, Bastuck S, Boesche M, Hobson S, Mathieson T, Perrin J, Raida M, Rau C, Reader V, Sweetman G, Bauer A, Bouwmeester T, Hopf C, Kruse U, Neubauer G, Ramsden N, Rick J, Kuster B, Drewes G. Quantitative chemical proteomics reveals mechanisms of action of clinical ABL kinase inhibitors. Nat Biotechnol. 2007;25(9):1035–44. doi: 10.1038/nbt1328. [DOI] [PubMed] [Google Scholar]
- 12.McAllister FE, Niepel M, Haas W, Huttlin E, Sorger PK, Gygi SP. Mass spectrometry based method to increase throughput for kinome analyses using ATP probes. Analytical Chemistry. 2013;85(9):4666–74. doi: 10.1021/ac303478g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patricelli MP, Nomanbhoy TK, Wu J, Brown H, Zhou D, Zhang J, Jagannathan S, Aban A, Okerberg E, Herring C, Nordin B, Weissig H, Yang Q, Lee JD, Gray NS, Kozarich JW. In situ kinase profiling reveals functionally relevant properties of native kinases. Chem Biol. 2011;18(6):699–710. doi: 10.1016/j.chembiol.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Anastassiadis T, Deacon SW, Devarajan K, Ma H, Peterson JR. Comprehensive assay of kinase catalytic activity reveals features of kinase inhibitor selectivity. Nature Biotechnology. 2011;29(11):1039–45. doi: 10.1038/nbt.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fedorov O, Marsden B, Pogacic V, Rellos P, Muller S, Bullock AN, Schwaller J, Sundstrom M, Knapp S. A systematic interaction map of validated kinase inhibitors with Ser/Thr kinases. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(51):20523–8. doi: 10.1073/pnas.0708800104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oda Y, Owa T, Sato T, Boucher B, Daniels S, Yamanaka H, Shinohara Y, Yokoi A, Kuromitsu J, Nagasu T. Quantitative chemical proteomics for identifying candidate drug targets. Analytical Chemistry. 2003;75(9):2159–65. doi: 10.1021/ac026196y. [DOI] [PubMed] [Google Scholar]
- 17.Cuatrecasas P, Wilchek M, Anfinsen CB. Selective enzyme purification by affinity chromatography. Proc Natl Acad Sci U S A. 1968;61(2):636–43. doi: 10.1073/pnas.61.2.636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ong SE, Schenone M, Margolin AA, Li X, Do K, Doud MK, Mani DR, Kuai L, Wang X, Wood JL, Tolliday NJ, Koehler AN, Marcaurelle LA, Golub TR, Gould RJ, Schreiber SL, Carr SA. Identifying the proteins to which small-molecule probes and drugs bind in cells. Proc Natl Acad Sci U S A. 2009;106(12):4617–22. doi: 10.1073/pnas.0900191106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rix U, Superti-Furga G. Target profiling of small molecules by chemical proteomics. Nat Chem Biol. 2009;5(9):616–24. doi: 10.1038/nchembio.216. [DOI] [PubMed] [Google Scholar]
- 20.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1(5):376–86. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 21.Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin DJ. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004;3(12):1154–69. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 22.Hsu JL, Huang SY, Chow NH, Chen SH. Stable-isotope dimethyl labeling for quantitative proteomics. Analytical Chemistry. 2003;75(24):6843–52. doi: 10.1021/ac0348625. [DOI] [PubMed] [Google Scholar]
- 23.Lau HT, Suh HW, Golkowski M, Ong SE. Comparing SILAC- and stable isotope dimethyl-labeling approaches for quantitative proteomics. J Proteome Res. 2014;13(9):4164–74. doi: 10.1021/pr500630a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wissing J, Jansch L, Nimtz M, Dieterich G, Hornberger R, Keri G, Wehland J, Daub H. Proteomics analysis of protein kinases by target class-selective prefractionation and tandem mass spectrometry. Mol Cell Proteomics. 2007;6(3):537–47. doi: 10.1074/mcp.T600062-MCP200. [DOI] [PubMed] [Google Scholar]
- 25.Sharma K, Weber C, Bairlein M, Greff Z, Keri G, Cox J, Olsen JV, Daub H. Proteomics strategy for quantitative protein interaction profiling in cell extracts. Nat Methods. 2009;6(10):741–4. doi: 10.1038/nmeth.1373. [DOI] [PubMed] [Google Scholar]
- 26.Lemeer S, Zorgiebel C, Ruprecht B, Kohl K, Kuster B. Comparing immobilized kinase inhibitors and covalent ATP probes for proteomic profiling of kinase expression and drug selectivity. J Proteome Res. 2013;12(4):1723–31. doi: 10.1021/pr301073j. [DOI] [PubMed] [Google Scholar]
- 27.Ruprecht B, Zecha J, Heinzlmeir S, Medard G, Lemeer S, Kuster B. Evaluation of Kinase Activity Profiling Using Chemical Proteomics. ACS Chem Biol. 2015;10(12):2743–52. doi: 10.1021/acschembio.5b00616. [DOI] [PubMed] [Google Scholar]
- 28.Medard G, Pachl F, Ruprecht B, Klaeger S, Heinzlmeir S, Helm D, Qiao H, Ku X, Wilhelm M, Kuehne T, Wu Z, Dittmann A, Hopf C, Kramer K, Kuster B. Optimized chemical proteomics assay for kinase inhibitor profiling. J Proteome Res. 2015;14(3):1574–86. doi: 10.1021/pr5012608. [DOI] [PubMed] [Google Scholar]
- 29.Heinzlmeir S, Kudlinzki D, Sreeramulu S, Klaeger S, Gande SL, Linhard V, Wilhelm M, Qiao H, Helm D, Ruprecht B, Saxena K, Medard G, Schwalbe H, Kuster B. Chemical Proteomics and Structural Biology Define EPHA2 Inhibition by Clinical Kinase Drugs. ACS Chem Biol. 2016;11(12):3400–3411. doi: 10.1021/acschembio.6b00709. [DOI] [PubMed] [Google Scholar]
- 30.Rappsilber J, Mann M, Ishihama Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc. 2007;2(8):1896–906. doi: 10.1038/nprot.2007.261. [DOI] [PubMed] [Google Scholar]
- 31.Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M. Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res. 2011;10(4):1794–805. doi: 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- 32.Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods. 2016 doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- 33.Zhang L, Holmes IP, Hochgrafe F, Walker SR, Ali NA, Humphrey ES, Wu J, de Silva M, Kersten WJ, Connor T, Falk H, Allan L, Street IP, Bentley JD, Pilling PA, Monahan BJ, Peat TS, Daly RJ. Characterization of the novel broad-spectrum kinase inhibitor CTx-0294885 as an affinity reagent for mass spectrometry-based kinome profiling. J Proteome Res. 2013;12(7):3104–16. doi: 10.1021/pr3008495. [DOI] [PubMed] [Google Scholar]
- 34.Statsuk AV, Maly DJ, Seeliger MA, Fabian MA, Biggs WH, 3rd, Lockhart DJ, Zarrinkar PP, Kuriyan J, Shokat KM. Tuning a three-component reaction for trapping kinase substrate complexes. Journal of the American Chemical Society. 2008;130(51):17568–74. doi: 10.1021/ja807066f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ranjitkar P, Brock AM, Maly DJ. Affinity reagents that target a specific inactive form of protein kinases. Chem Biol. 2010;17(2):195–206. doi: 10.1016/j.chembiol.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qian F, Engst S, Yamaguchi K, Yu P, Won KA, Mock L, Lou T, Tan J, Li C, Tam D, Lougheed J, Yakes FM, Bentzien F, Xu W, Zaks T, Wooster R, Greshock J, Joly AH. Inhibition of tumor cell growth, invasion, and metastasis by EXEL-2880 (XL880, GSK1363089), a novel inhibitor of HGF and VEGF receptor tyrosine kinases. Cancer Research. 2009;69(20):8009–16. doi: 10.1158/0008-5472.CAN-08-4889. [DOI] [PubMed] [Google Scholar]
- 37.Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, Hocker M, Treiber DK, Zarrinkar PP. Comprehensive analysis of kinase inhibitor selectivity. Nature Biotechnology. 2011;29(11):1046–51. doi: 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- 38.Fleuren ED, Zhang L, Wu J, Daly RJ. The kinome ‘at large’ in cancer. Nat Rev Cancer. 2016;16(2):83–98. doi: 10.1038/nrc.2015.18. [DOI] [PubMed] [Google Scholar]
- 39.Mertins P, Mani DR, Ruggles KV, Gillette MA, Clauser KR, Wang P, Wang X, Qiao JW, Cao S, Petralia F, Kawaler E, Mundt F, Krug K, Tu Z, Lei JT, Gatza ML, Wilkerson M, Perou CM, Yellapantula V, Huang KL, Lin C, McLellan MD, Yan P, Davies SR, Townsend RR, Skates SJ, Wang J, Zhang B, Kinsinger CR, Mesri M, Rodriguez H, Ding L, Paulovich AG, Fenyo D, Ellis MJ, Carr SA. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature. 2016;534(7605):55–62. doi: 10.1038/nature18003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu Z, Doondeea JB, Gholami AM, Janning MC, Lemeer S, Kramer K, Eccles SA, Gollin SM, Grenman R, Walch A, Feller SM, Kuster B. Quantitative chemical proteomics reveals new potential drug targets in head and neck cancer. Mol Cell Proteomics. 2011;10(12):M111 011635. doi: 10.1074/mcp.M111.011635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gholami AM, Hahne H, Wu Z, Auer FJ, Meng C, Wilhelm M, Kuster B. Global proteome analysis of the NCI-60 cell line panel. Cell Rep. 2013;4(3):609–20. doi: 10.1016/j.celrep.2013.07.018. [DOI] [PubMed] [Google Scholar]
- 42.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, Reddy A, Liu M, Murray L, Berger MF, Monahan JE, Morais P, Meltzer J, Korejwa A, Jane-Valbuena J, Mapa FA, Thibault J, Bric-Furlong E, Raman P, Shipway A, Engels IH, Cheng J, Yu GK, Yu J, Aspesi P, Jr, de Silva M, Jagtap K, Jones MD, Wang L, Hatton C, Palescandolo E, Gupta S, Mahan S, Sougnez C, Onofrio RC, Liefeld T, MacConaill L, Winckler W, Reich M, Li N, Mesirov JP, Gabriel SB, Getz G, Ardlie K, Chan V, Myer VE, Weber BL, Porter J, Warmuth M, Finan P, Harris JL, Meyerson M, Golub TR, Morrissey MP, Sellers WR, Schlegel R, Garraway LA. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603–7. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Greenbaum D, Colangelo C, Williams K, Gerstein M. Comparing protein abundance and mRNA expression levels on a genomic scale. Genome Biol. 2003;4(9):117. doi: 10.1186/gb-2003-4-9-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maier T, Guell M, Serrano L. Correlation of mRNA and protein in complex biological samples. FEBS Letters. 2009;583(24):3966–73. doi: 10.1016/j.febslet.2009.10.036. [DOI] [PubMed] [Google Scholar]
- 45.Nagaraj N, Wisniewski JR, Geiger T, Cox J, Kircher M, Kelso J, Paabo S, Mann M. Deep proteome and transcriptome mapping of a human cancer cell line. Mol Syst Biol. 2011;7:548. doi: 10.1038/msb.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tebbe A, Klammer M, Sighart S, Schaab C, Daub H. Systematic evaluation of label-free and super-SILAC quantification for proteome expression analysis. Rapid Communications in Mass Spectrometry. 2015;29(9):795–801. doi: 10.1002/rcm.7160. [DOI] [PubMed] [Google Scholar]
- 47.Ojo KK, Larson ET, Keyloun KR, Castaneda LJ, Derocher AE, Inampudi KK, Kim JE, Arakaki TL, Murphy RC, Zhang L, Napuli AJ, Maly DJ, Verlinde CL, Buckner FS, Parsons M, Hol WG, Merritt EA, Van Voorhis WC. Toxoplasma gondii calcium-dependent protein kinase 1 is a target for selective kinase inhibitors. Nat Struct Mol Biol. 2010;17(5):602–7. doi: 10.1038/nsmb.1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Johnson SM, Murphy RC, Geiger JA, DeRocher AE, Zhang Z, Ojo KK, Larson ET, Perera BG, Dale EJ, He P, Reid MC, Fox AM, Mueller NR, Merritt EA, Fan E, Parsons M, Van Voorhis WC, Maly DJ. Development of Toxoplasma gondii calcium-dependent protein kinase 1 (TgCDPK1) inhibitors with potent anti-toxoplasma activity. Journal of Medicinal Chemistry. 2012;55(5):2416–26. doi: 10.1021/jm201713h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Castellanos-Gonzalez A, White AC, Jr, Ojo KK, Vidadala RS, Zhang Z, Reid MC, Fox AM, Keyloun KR, Rivas K, Irani A, Dann SM, Fan E, Maly DJ, Van Voorhis WC. A novel calcium-dependent protein kinase inhibitor as a lead compound for treating cryptosporidiosis. Journal of Infectious Diseases. 2013;208(8):1342–8. doi: 10.1093/infdis/jit327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ojo KK, Eastman RT, Vidadala R, Zhang Z, Rivas KL, Choi R, Lutz JD, Reid MC, Fox AM, Hulverson MA, Kennedy M, Isoherranen N, Kim LM, Comess KM, Kempf DJ, Verlinde CL, Su XZ, Kappe SH, Maly DJ, Fan E, Van Voorhis WC. A specific inhibitor of PfCDPK4 blocks malaria transmission: chemical-genetic validation. Journal of Infectious Diseases. 2014;209(2):275–84. doi: 10.1093/infdis/jit522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vidadala RS, Ojo KK, Johnson SM, Zhang Z, Leonard SE, Mitra A, Choi R, Reid MC, Keyloun KR, Fox AM, Kennedy M, Silver-Brace T, Hume JC, Kappe S, Verlinde CL, Fan E, Merritt EA, Van Voorhis WC, Maly DJ. Development of potent and selective Plasmodium falciparum calcium-dependent protein kinase 4 (PfCDPK4) inhibitors that block the transmission of malaria to mosquitoes. Eur J Med Chem. 2014;74:562–73. doi: 10.1016/j.ejmech.2013.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ojo KK, Reid MC, Kallur Siddaramaiah L, Muller J, Winzer P, Zhang Z, Keyloun KR, Vidadala RS, Merritt EA, Hol WG, Maly DJ, Fan E, Van Voorhis WC, Hemphill A. Neospora caninum calcium-dependent protein kinase 1 is an effective drug target for neosporosis therapy. PLoS One. 2014;9(3):e92929. doi: 10.1371/journal.pone.0092929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vidadala RS, Rivas KL, Ojo KK, Hulverson MA, Zambriski JA, Bruzual I, Schultz TL, Huang W, Zhang Z, Scheele S, DeRocher AE, Choi R, Barrett LK, Siddaramaiah LK, Hol WG, Fan E, Merritt EA, Parsons M, Freiberg G, Marsh K, Kempf DJ, Carruthers VB, Isoherranen N, Doggett JS, Van Voorhis WC, Maly DJ. Development of an Orally Available and Central Nervous System (CNS) Penetrant Toxoplasma gondii Calcium-Dependent Protein Kinase 1 (TgCDPK1) Inhibitor with Minimal Human Ether-a-go-go-Related Gene (hERG) Activity for the Treatment of Toxoplasmosis. Journal of Medicinal Chemistry. 2016;59(13):6531–46. doi: 10.1021/acs.jmedchem.6b00760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Van Voorhis WC, Fan E, Maly DJ, Ojo KK, Plymate SR. Bumped Kinase Inhibitor Compositions and Methods for Treating Cancer. WO/2016/123151. WIPO Patent Application. 2016 Aug 04;
- 55.Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochemical Journal. 2007;408(3):297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tandon M, Johnson J, Li Z, Xu S, Wipf P, Wang QJ. New pyrazolopyrimidine inhibitors of protein kinase d as potent anticancer agents for prostate cancer cells. PLoS One. 2013;8(9):e75601. doi: 10.1371/journal.pone.0075601. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.