

Abstract
Background
Everolimus significantly improves progression-free survival (PFS) and has been approved to use in aromatase inhibitor pretreated patients with hormone receptor positive advanced breast cancer. Metformin has been shown to inhibit mTOR pathway, with more favorable safety profile, leading to this hypothesis-generating trial to assess whether metformin enhances the efficacy of aromatase inhibitors.
Methods
60 postmenopausal women with hormone receptor positive locally advanced or metastatic breast cancer were randomly assigned 1:1 to aromatase inhibitor (exemestane 25mg/d or letrozole 2.5mg/d depending on the most recent treatment) plus metformin (0.5g bid, orally) or placebo. The primary endpoint was PFS, and secondary endpoints were objective response rate, clinical benefit rate, overall survival and safety.
Results
Median PFS was 4.7 months in the combination group and 6.0 months in the control group (hazard ratio, 1.2; 95% confidence interval [CI], 0.7 to 2.1; P =0.48). ORR was 6.7% in the combination group and 0% in the control group (odds ratio for ORR not available; P =0.99), and CBR was 33.3% and 50.0%, respectively (OR for CBR 0.5; 95% CI, 0.2 to 1.4; P=0.15). No significant difference in overall survival was observed between the combination and control groups (median OS, 30.9 vs. 32.4 months; P = 0.81). Subgroup analyses didn't find any specific population favoring the combination treatment. No substantial difference in incidence or severity of adverse events was seen between the two treatment groups.
Conclusion
This randomized phase II clinical trial failed to show an improved efficacy with the addition of metformin to endocrine therapy, although with excellent tolerability.
Keywords: metastatic breast cancer, hormone receptor positive, endocrine therapy, metformin, pre-treated
INTRODUCTION
The biguanide metformin is one of the backbone drugs in the treatment of hyperglycemia and type 2 diabetes [1]. It exerts its hypoglycemic action by sensitizing peripheral tissues to insulin, increasing insulin-dependent glucose uptake of cells, inhibiting gluconeogenesis, and decreasing glucose absorption in the small intestine [2].
Recently, metformin has being assessed as an anti-cancer agent [3, 4]. Retrospective epidemiological studies based on public databases, comprehensive literature search and meta-analysis compared tumor incidence and cancer-related mortality of diabetic patients taking metformin with those taking other hypoglycemic agents as well as non-diabetic patients. It was concluded that tumor incidence and cancer-related mortality decreased with metformin treatment [5–9]. Possible molecular mechanisms include direct (insulin-independent) and indirect (insulin-dependent) actions of the drug [10]. The direct effects of metformin mainly occur through the induction of AMP-activated protein kinase (AMPK), consequently reducing mammalian target of rapamycin (mTOR) signaling and protein synthesis in cancer cells [11]. The indirect effects are the downstream response to AMPK activation. Metformin inhibits the transcription of key gluconeogenesis genes in the liver and reduces blood glucose and insulin [2]. Thus, it inhibits insulin-induced proliferation of tumor cells [12, 13].
Several clinical studies have confirmed the antitumor effects of metformin in breast cancer patients, especially those with diabetes or other metabolism disorders. A retrospective clinical study showed that patients treated with metformin during neoadjuvant chemotherapy have a higher pathologic complete response (pCR) rate than diabetic and non-diabetes patients not administered metformin [14]. Furthermore, a Chinese phase II randomized clinical trial demonstrated an increased pCR rate in early breast cancer patients after addition of metformin (0.25g tid) to routine chemotherapy regimens (cyclophosphamide + epirubicin + 5-fluorouracil) [15]. In addition to efficacy outcomes, several surrogate markers of relevant signaling pathways have been described in breast cancer. These include Ki67, S6K, 4E-BP-1, AMPK and other key molecules of the AMPK/mTOR pathway [16]. Overall, most studies indicated reduced ki67 and increased TUNEL levels after metformin administration [16, 17].
Emerging evidence demonstrates that PI3K/Akt/mTOR activation is an important mechanism of acquired endocrine resistance [18]. Everolimus, an inhibitor of mTOR, reverses resistance to endocrine therapy, and is effective in patients who experience progression after prior endocrine treatments [19, 20]. BOLERO-2 demonstrated that everolimus combined with exemestane (steroidal AI) improves progression-free survival (PFS) in advanced breast cancer patients previously treated with endocrine therapies, compared with the exemestane monotherapy group [19].
Metformin inhibits the activation of mTOR and relevant proliferation signaling pathways. Since everolimus was not approved by CFDA and unavailable in China at the time of study design, we assessed whether metformin could constitute a substitute for everolimus in the treatment of patients with hormone receptor (HR) positive breast cancer. Thus, we hypothesized that metformin could further enhance the efficacy of aromatase inhibitors, in patients with HR-positive advanced breast cancer.
RESULTS
Patients
In total, 60 patients were randomly assigned, 30 each to the AI plus metformin and AI groups (Figure 1). The eligible patients were enrolled between Jun 17, 2012 and Oct 14, 2014. Baseline characteristics, including treatment history, were well balanced across the treatment groups (Table 1). Median age was 57 years (range, 33-73). A total of 13 (43.3%) patients had visceral metastases in the combination group, and 18 (60.0%) in the control group. 14 (46.7%) and 8 (26.7%) patients had first-line endocrine therapy in the combination and control groups, respectively. The patients were divided by sensitivity to previous endocrine therapy (Table 1). Most patients in both groups showed secondary resistance. In addition, the most recent endocrine therapy included antiestrogen agents (16.7% and 33.3% in the combination and control groups, respectively) and AI (83.3% and 66.7% in the combination and control groups, respectively).
Figure 1. Consort graph.

Table 1. Patient characteristics.
| Characteristic | AI +Met (n = 30)No.(%) | AI (n = 30)No.(%) |
|---|---|---|
| Median age, years (range) | 57.5(33-72) | 56.5(33-73) |
| De novo stage IV | 1(3.3) | 2(6.7) |
| Disease free interval* | ||
| Median(year) | 5.4 | 5.7 |
| Range(year) ≤24 mo-No.(%) >24 mo-No.(%) | 1.8-14.92(6.7)27(90.0) | 0.5-14.05(16.7)23(76.7) |
| Measurable lesion-No.(%) | 19(63.3) | 16(53.3) |
| No. of metastatic sites-No.(%) | ||
| 1 | 12(40.0) | 11(36.7) |
| 2 | 9(30.0) | 12(40.0) |
| ≥3 | 9(30.0) | 7(23.3) |
| Metastatic sites-No.(%) | ||
| Visceral | 13(43.3) | 18(60.0) |
| Lung | 10(33.3) | 14(46.7) |
| Liver | 5(16.7) | 3(10.0) |
| Non-visceral | 17(56.7) | 12(40.0) |
| Bone alone | 5(16.7) | 6(20.0) |
| Most recent endocrine therapy-No.(%) | ||
| Adjuvant endocrine therapy | 14(46.7) | 8(26.7) |
| Endocrine therapy for MBC | 16(53.3) | 22(73.3) |
| Lines of endocrine therapy for MBC-No.(%) | ||
| First line | 14(46.7) | 8(26.7) |
| Second line | 15(50.0) | 20(66.7) |
| Third or more line | 1(3.3) | 2(6.7) |
| Sensitivity to previous endocrine therapy#-No.(%) | ||
| Primary resistance | 3(10.0) | 5(16.7) |
| Secondary resistance | 24(80.0) | 21(70.0) |
| Naïve | 2(6.7) | 2(6.7) |
| Not evaluable | 1(3.3) | 2(6.7) |
| Most recent treatment-No.(%) | ||
| Antiestrogen | 5(16.7) | 10(33.3) |
| AI | 25(83.3) | 20(66.7) |
| Previous lines of chemotherapy for MBC-No.(%) | ||
| 0 | 20(66.7) | 22(73.3) |
| 1 ≥2 | 4(13.3)6(20.0) | 7(23.3)1(3.3) |
*Disease-free interval is defined as the time from diagnosis of breast cancer to first relapse (29 patients in the combination group and 28 patients in single-agent group).
# Primary endocrine resistance is defined as: a relapse while on the first 2 years of adjuvant endocrine therapy, or PD within first 6 months of first-line endocrine therapy for MBC, while on endocrine therapy. Secondary (acquired) endocrine resistance is defined as: a relapse while on adjuvant endocrine therapy but after the first 2 years, or a relapse within 12 months of completing adjuvant endocrine therapy, or PD ≥6 months after initiating endocrine therapy for MBC, while on endocrine therapy.
Efficacy
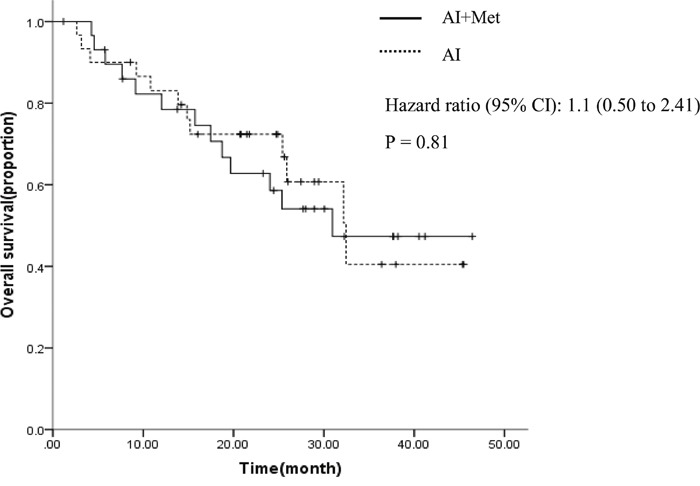
The cutoff date for the study was Jul 10, 2016. All randomized patients were included in this analysis (Figure 1). Efficacy evaluations were listed in Table 2. Treatment with AI plus metformin, compared with placebo plus AI, did not significantly improve PFS. Treatment in the combination group resulted in a median PFS of 4.7 months, and 6.0 months in the control group (hazard ratio [HR], 1.2; 95% CI, 0.7 to 2.1; P =0.48; Figure 2), which is based on intention-to-treat analysis; 49 progression events were obtained, with 23 (76.7%) in the combination group and 26 (86.7%) in the control group.
Table 2. Evaluation of efficacy.
| Variable | AI +Met (n = 30)No.(%) | AI (n = 30)No.(%) |
|---|---|---|
| Progression-free survival | ||
| Events — No. (%) | 23(76.7) | 26(86.7) |
| Duration — mo | ||
| Median | 4.7 | 6.0 |
| 95%CI | 0.3-9.0 | 4.0-7.9 |
| Overall survival | ||
| Events — No. (%) | 13(25.0) | 12(39.3) |
| Duration — mo | ||
| Median | 30.9 | 32.4 |
| 95%CI | NR | 23.8-41.0 |
| Best overall response-No.% | ||
| Complete response | 0(0.0) | 0(0.0) |
| Partial response | 2(6.7) | 0(0.0) |
| Stable disease | 14(46.7) | 22(73.3) |
| Duration of SD ≥24 weeks | 8(26.7) | 15(50.0) |
| Progression disease | 12(40.0) | 7(23.3) |
| NE | 2(6.7) | 1(3.3) |
| ORR | 2(6.7) | 0 |
| CBR | 10(33.3) | 15(50.0) |
ORR was 6.7% (95%CI 0.3-16) in the combination group, and 0% (95%CI not available) in the control group. CBR was 33.3% (95%CI 15-51) in the combination group, and 50.0% (95%CI 31-69) in the control group. AI and metformin combination was not associated with increased ORR (OR not available P=0.99) and CBR (OR=0.50, 95% CI 0.2 to 1.4, P=0.15). Median follow-up was 22.3 months. A total of 13 (43.3%) patients died in the combination group, and 12 (40.0%) in the control group. Median OS times were 30.9 and 32.4 months for the combination and control groups, respectively (HR= 1.1, 95%CI 0.5 to 2.4, P = 0.81, Figure 3).
Figure 2. Progression-free survival curves by treatment arm.
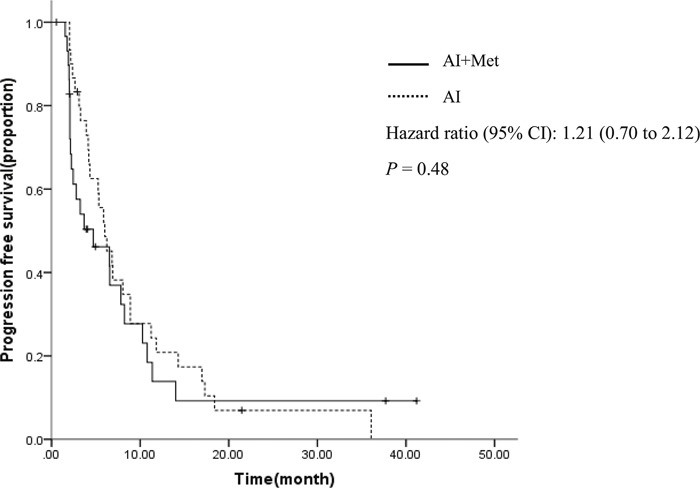
Figure 3. Overall survival curves by treatment arm.
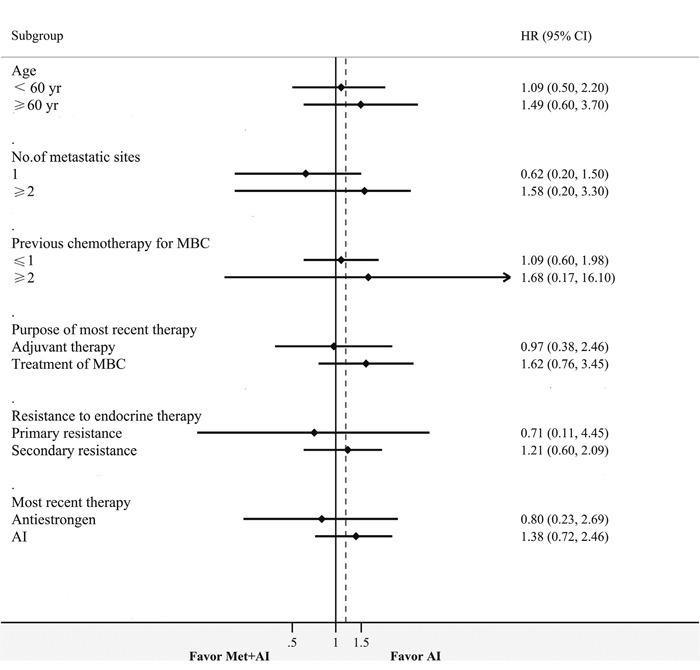
In subgroup analysis, treatment effects were consistent across all predefined subgroups. A numerical PFS increase was obtained in the combination group in patients with primary resistance compared to those with secondary resistance (10.8 vs. 5.8 months, p=0.71). Patients had slightly but non-statistically signifcant PFS increase after last antiestrogen therapy in the combination group compared with the control group (10.8 vs. 4.3 months, p=0.72) (Table 3, Figure 4). Patients with primary resistance benefited from metformin, with risk of progression numerically reduced by 29% (HR=0.7, 95% CI 0.1 to 4.4; Table 3 and Figure 4). A numerical reduction in risk of progression was 20% in patients after last antiestrogen therapy when combined with metformin (HR=0.8, 95% CI 0.2 to 2.7; Table 3, Figure 4). No significant difference was observed, with a limited number of patients in each subgroup.
Table 3. Subgroup analysis.
| N | PFS | HR | 95%CI | P | ||
|---|---|---|---|---|---|---|
| Age | ||||||
| <60 year | AI+Met | 16 | 6.5 | 1.09 | 0.5-2.2 | 0.810 |
| AI | 21 | 6.9 | ||||
| ≥60 year | AI+Met | 14 | 3.2 | 1.49 | 0.6-3.7 | 0.391 |
| AI | 9 | 5.2 | ||||
| No. of metastatic sites | ||||||
| =1 | AI+Met | 12 | 6.5 | 0.62 | 0.2-1.5 | 0.327 |
| AI | 11 | 5.8 | ||||
| ≥2 | AI+Met | 18 | 2.8 | 1.58 | 0.7-3.3 | 0.218 |
| AI | 19 | 6.9 | ||||
| Previous chemotherapy for MBC | ||||||
| ≤1 | AI+Met | 24 | 6.5 | 0.95 | 0.6-2.5 | 0.758 |
| AI | 29 | 6.2 | ||||
| ≥2 | AI+Met | 6 | 2.0 | 1.68 | 0.2-16.1 | 0.649 |
| AI | 1 | 2.3 | ||||
| Purpose of most recent therapy | ||||||
| Adjuvant therapy | AI+Met | 14 | 7.8 | 0.97 | 0.4-2.5 | 0.067 |
| AI | 8 | 5.2 | ||||
| Treatment of MBC | AI+Met | 16 | 2.4 | 1.62 | 0.8-3.4 | 0.211 |
| AI | 22 | 6.2 | ||||
| Resistance to endocrine therapy** | ||||||
| Primary resistance | AI+Met | 3 | 10.8 | 0.71 | 0.1-4.4 | 0.714 |
| AI | 5 | 5.8 | ||||
| Secondary resistance | AI+Met | 24 | 3.2 | 1.21 | 0.6-2.1 | 0.719 |
| AI | 21 | 5.3 | ||||
| Most recent therapy | ||||||
| Antiestrongen | AI+Met | 5 | 10.8 | 0.80 | 0.2-2.7 | 0.722 |
| AI | 10 | 4.3 | ||||
| AI | AI+Met | 25 | 2.8 | 1.38 | 0.7-2.6 | 0.320 |
| AI | 20 | 6.2 | ||||
| Overall | AI+Met | 30 | 4.7 | 1.21 | 0.7-2.1 | 0.487 |
| AI | 30 | 6.0 | ||||
** Primary endocrine resistance is defined as: a relapse while on the first 2 years of adjuvant endocrine therapy, or PD within first 6 months of first-line endocrine therapy for MBC, while on endocrine therapy. Secondary (acquired) endocrine resistance is defined as: a relapse while on adjuvant endocrine therapy but after the first 2 years, or a relapse within 12 months of completing adjuvant endocrine therapy, or PD ≥6 months after initiating endocrine therapy for MBC, while on endocrine therapy.
Figure 4. Forest plot of PFS.
Safety
Median drug exposure times were 3.8 months (range, 1.5 to 32.0 months) in the combination group and 6.0 months (range, 2 to 30.7 months) in the control group. Both combination and monotherapy were well tolerated. No substantial differences in the incidence and severity of adverse events (AEs) were obtained between the two treatment groups (Table 4). The most common AEs were arthralgia (n=5; 16.7%) and headache (n=3; 10.0%) in the combination group, and arthralgia (n=3; 10.0%) in the control group. Grade 3/4 AEs were anemia (n=1; 3.3%) and thrombocytopenia (n=1; 3.3%) in the combination group. No treatment related serious AEs were observed in this study. One patient in the combination group discontinued the treatment for grade 3 anemia and grade 4 thrombocytopenia, which was considered to be related to disease progression. This patient received symptomatic treatment, including blood transfusion, but died of myelosuppression.
Table 4. Adverse event.
| AE | AI+Met | AI | ||||||
|---|---|---|---|---|---|---|---|---|
| Grade 1 | Grade 2 | Grade 3 | Grade 4 | Grade 1 | Grade 2 | Grade 3 | Grade 4 | |
| Anemia | 0 | 0 | 1(3.33%) | 0 | 0 | 0 | 0 | 0 |
| Neutropenia | 0 | 0 | 0 | 0 | 1(3.33%) | 0 | 0 | 0 |
| Thrombocytopenia | 0 | 0 | 0 | 1(3.33%) | 0 | 0 | 0 | 0 |
| Fever | 1(3.33%) | 0 | 0 | 0 | 1(3.33%) | 0 | 0 | 0 |
| Diarrhea | 1(3.33%) | 1(3.33%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Anorexia | 1(3.33%) | 0 | 0 | 0 | 1(3.33%) | 0 | 0 | 0 |
| Fatigue | 1(3.33%) | 0 | 0 | 0 | 2(6.67%) | 0 | 0 | 0 |
| Nausea and vomiting | 2(6.67%) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Abdominal distension | 1(3.33%) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Constipation | 1(3.33%) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Arthralgia | 5(16.7%) | 0 | 0 | 0 | 3(10.0%) | 0 | 0 | 0 |
| Alopecia | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Rash | 0 | 0 | 0 | 0 | 1(3.33%) | 0 | 0 | 0 |
| Edema | 0 | 0 | 0 | 0 | 1(3.33%) | 0 | 0 | 0 |
| Liver enzyme escalation | 2(6.67%) | 0 | 0 | 0 | 2(6.67%) | 0 | 0 | 0 |
| Myalgia | 0 | 0 | 0 | 0 | 1(3.33%) | 0 | 0 | 0 |
| Headache | 3(10.0%) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Skin Pruritus | 0 | 0 | 0 | 0 | 1(3.33%) | 0 | 0 | 0 |
| Cough | 0 | 1(3.33%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Insomnia | 1(3.33%) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Hand-foot syndrome | 0 | 1(3.33%) | 0 | 0 | 0 | 0 | 0 | 0 |
DISCUSSION
This phase II randomized clinical trial failed to show an improved efficacy after metformin addition. PFS was 4.7 months in the AI and metformin group, and 6.0 months in the control group, with no significant difference. In subgroup analysis, no favorable PFS for the combination group was observed. Similarly, no significant increase was found in ORR, CBR and OS. As expected, the incidence rates of AEs were identical in two groups.
Few studies have confirmed efficacy outcome data for metformin in breast cancer patients. Only two studies indicated that metformin improves pCR rate when combined with chemotherapy in the neoadjuvant setting. A retrospective study showed that diabetic patients with concurrent metformin in neoadjuvant chemotherapy have a higher pCR rate than diabetic and non-diabetic patients not administered metformin (24% vs. 8% and 16%, respectively, p = 0.02) [14]. Another Chinese phase II randomized clinical trial showed increased pCR after metformin addition (20% vs 6.7%, p=0.0119) [15].
However, most previous studies focused on surrogate biomarkers of cell proliferation, cell apoptosis, or AMPK/mTOR signaling affected by metformin as neoadjuvant therapy. The majority of these studies showed reduced Ki67 levels, increased TUNEL staining [17], increased mean AMPK scores, and decreased pAKT scored [16]. Nevertheless, other trials reported conflicting outcomes. A phase II RCT of 200 patients found no significant difference in Ki67 comparing metformin treatment with controls. Interestingly, metformin significantly decreases ki-67 in women with HOMA (homeostatic model assessment, to evaluate status of insulin resistance) > 2.8 or high body mass index (BMI), indicating that metformin's effects may differ according to insulin resistance and the metabolic characteristics of patients [21]. The above translational studies mainly indicated that metformin exerts antitumor activity by inhibiting cell proliferation and promoting apoptosis, although further studies are required for confirmation. To this end, numerous clinical trials are ongoing, involving metformin as monotherapy (NCT01101438, NCT01905046), or combined with anti-tumor agents such as docetaxel, epirubicin and cyclophosphamide (NCT01929811), letrozole (NCT01589367), ganitumab (NCT01042379), and sirolimus (NCT02145559).
Though all pre-specified subgroups showed no significant differences, a numerical PFS increase in the combination group was observed in patients with primary resistance and those progressing on last antiestrogen therapy. Patients with primary resistance seemed to be more responsive to metformin, although there was a limited number of patients in this subgroup. Previous studies indicate that metformin inhibits the proliferation of endocrine-resistant breast cancer cell lines, and these effects may be more pronounced in patients with primary resistance. First, metformin could induce apoptosis and inhibit proliferation mediated by AMPK signaling in endocrine therapy resistant HR-positive breast cancer cell lines [22]. Secondly, the mechanisms of endocrine resistance differ between primary and secondary resistance. Primary resistance mainly includes lack of ERα expression, while secondary resistance is associated with a plethora of mechanisms, and increased crosstalk receptor tyrosine kinase signaling (EGFR, ERBB2, IGF1R and PI3K pathways) may play an important role [23, 24]. Most importantly, with more crosstalk proliferation pathways involved in secondary resistance, growth inhibition and apoptosis induction by metformin may be weakened, or even eliminated. Breast cancer with secondary resistance often shows induced IGF1R signaling [25], which may also explain metformin resistance [26]. Activation of the IGFR-1/IRS-1 axis results in elevated cell survival signals, thus counteracting the antitumor activity of metformin [26].
Patients progressing on last antiestrogen therapy had somewhat prolonged PFS in the combination group. This was consistent with subgroup analysis data in BOLERO-2. Previous studies suggested that mTOR inhibitors may have favorable effects in antiestrogen resistant breast cancer compared with the AI resistant counterpart. A pre-clinical study compared the growth inhibition effects of everolimus in TAM-R (tamoxifen resistance model) and MCF7-X (estrogen deprivation resistance model), and found higher inhibition rates and lower half maximal inhibitory concentrations in the TAM-R group compared with the MCF7-X model. Besides its inhibition of mTOR, metformin decreases basal and insulin-stimulated aromatase expression, which is an important mechanism of its usefulness in polycystic ovary syndrome [27]. Metformin may synergistically act with aromatase inhibitors by further inhibiting aromatase. Changing another mechanism of endocrine therapy, from antiestrogen to aromatase inhibition, had higher antitumor effects in patients who had prior antiestrogen therapy compared with those who had prior steroidal AI. These effects were enhanced by metformin. However, the above interpretations are mostly based on preliminary studies, and the number of patients in every subgroup is too small to detect differences. Therefore, additional clinical studies are required to confirm these findings.
The current study showed a negative outcome, and was incapable to confirm an enhanced anti-tumor activity after metformin addition. This could be attributed to the following reasons. On the one hand, we administered metformin at the conventional dose as used in type 2 diabetes, since 0.5g bid PO proves to be a safe dose after decades of clinical use among type 2 diabetes. Previous studies showed that much higher concentrations of metformin are needed in order to exert its direct effects on AMPK–mTOR [28–31]. Results from two xenograft models reported that the human equivalent of 1500–2250 mg/day is needed to inhibit tumorigenesis [32–34]. The above studies administered metformin in the neoadjuvant setting at doses between 1500 mg/d-2000 mg/d as a monotherapy or in combination [14, 16, 21, 35, 36]. But pitifully, there were not any clinical study we could refer to when we designed this study in 2011. Therefore, we chose a safe dose of metformin in this exploratory study. The dose in this study seems insufficient to yield full anti-tumor activity and reverse resistance to endocrine therapy. Actually, in our Center, further dose-escalation study to confirm optimal dose of metformin combined with aromatase inhibitors among ER+HER2- advanced breast cancer patients is ongoing.
One the other hand, indirect, insulin-dependent effects may play a more important role in anticancer effects of metformin compared with direct, insulin-independent effects. In addition, the majority of studies indicated that metformin is most effective in patients with high BMI and insulin resistance [21]. In the current study, the BMIs of patients were relatively normal (median 22.1, range 18.7-26.5) and few had metabolic disorders like diabetes. Therefore, the benefits from insulin-dependent effects may be limited in the current patient population. The main limitation of this study is that the HOMA index and other metabolic markers were not assessed, making it difficult to accurately identify which subtype of patients can benefit from such treatment. Moreover, overactivation of the IGF1R pathway is one of the key mechanisms of acquired resistance among HR-positive patients [25]. Enhanced IGF1R activity promotes cell survival and weakens the antitumor activity of metformin [26].
This study was a pioneer work, providing preliminary data of the effects of metformin plus AI in postmenopausal patients with hormone receptor positive metastatic breast cancer. Overall, the results suggested that metformin does not enhance efficacy in terms of PFS, ORR, CBR and OS, and does not increase the incidence rates of adverse events, when combined with AI. Patients with primary resistance and progressing on last antiestrogen therapy showed a trend of prolonged PFS. However, these data should be interpreted in the context of limited statistical power provided by a relatively small sample size. Further studies with sufficient metformin amounts, measurements of metabolic markers, appropriate populations, and larger sample sizes are warranted in the future.
MATERIALS AND METHODS
Study design and treatments
This was a prospective, open-label, randomized, single-center, phase II trial comparing aromatase inhibitors plus metformin and aromatase inhibitors plus placebo in HR-positive postmenopausal patients with metastatic breast cancer pretreated with endocrine therapy. Eligible patients were randomly assigned 1:1 to one of the following 2 treatment arms: aromatase inhibitor (letrozole 2.5 mg/d orally or exemestane 25 mg/d orally) plus metformin (0.5g bid, orally) or aromatase inhibitor plus placebo. The dose of metformin administered reflected the conventional dosage used in type 2 diabetes. Letrozole was administered to patients experiencing progression on/after tamoxifen or steroid aromatase inhibitor (SAI) treatment. Exemestane was administered to patients with previous non-steroid aromatase inhibitor (NSAI) treatment. Four weeks were defined as a treatment cycle. The patients received treatment until disease progression, intolerable toxicity, or voluntary refusal. The current study was performed in accordance with the Declaration of Helsinki and consistent with the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) Good Clinical Practice. The study protocol, patient consent forms, and information sheets were approved by the relevant independent ethics committees and institutional review boards. All patients provided written informed consent before enrolment (ClinicalTrials.gov identifier: NCT01654185).
Patients
Eligible patients were postmenopausal women with locally advanced or metastatic, pathologically confirmed ER and/or PgR positive breast cancer. Patients who experienced progression after prior endocrine therapy in adjuvant or metastatic settings were allowed to be enrolled. Measurable lesions were not necessary in eligible patients. Patients treated with chemotherapy for advanced disease were also allowed. In addition, all eligible patients were required to have an ECOG score of 0 to 1, evidence of adequate organ function, and life expectancy of at least 3 months.
Exclusion criteria were: HER2 overexpression (3+ status by immunohistochemistry or amplification ratio ≥2.0 by fluorescence in situ hybridization); disease progression on chemotherapy for advanced breast cancer, or less than 2 week washout period for the last endocrine therapy after chemotherapy; life-threatening visceral metastases or central nervous system metastases; radiotherapy within 4 weeks of randomization (palliative radiotherapy for bone metastasis within 2 weeks was permitted); current or prior malignancy (except breast cancer, adequately treated skin cancer, or in situ carcinoma of the cervix); treatment with other experimental drugs before randomization; long-term systemic steroid therapy; prior or present metformin use for blood glucose control; age ≥70 with renal hypofunction or any severe concomitant conditions.
Efficacy and tolerability
The primary endpoint was progression free survival (PFS), defined as the time from randomization to objective disease progression or death for any cause before documented disease progression. Secondary end points were objective response rate (ORR, proportion of patients with complete or partial response), clinical benefit rate (CBR, proportion of all patients with complete response, partial response, or stable disease for at least 24 weeks), overall survival (OS, time interval from random assignment to death in follow-up), and tolerability.
Tumor assessment was performed by computed tomography, spiral computed tomography, or magnetic resonance imaging, at baseline and every two cycles until disease progression or death occurred, according to RECIST 1.1. Safety and tolerability were assessed at each cycle; incidence and frequency of adverse events (AEs) were recorded throughout the study. AEs were graded using the National Cancer Institute Common Terminology Criteria for Adverse Events (version 4.0).
Statistical analysis
Sample size calculation was based on the primary endpoint of PFS, assuming exponential progression times. To detect a prolongation of 1.38 months in median PFS for AI plus metformin over AI, at a two-sided significance level of 5% with 80% power, at least 30 patients in each arm were required within 12 months, with 10% dropout rate.
For the primary endpoint of PFS, Kaplan-Meier plots revealed median PFS estimates for each treatment group. The primary analysis was an unadjusted log-rank test. The treatment effect was estimated using COX proportional hazards model and expressed as hazard ratio of AI plus metformin versus AI plus placebo. Subgroup analysis used the Cox proportional hazards model, stratified by the following pre-defined covariates: age (<60y vs. ≥60y), number of metastatic sites (1 vs. ≥2), lines of previous chemotherapy (≤1 vs. ≥2), purpose of the most recent therapy (adjuvant therapy vs. treatment of MBC), resistance to endocrine therapy (primary vs. secondary resistance), and last endocrine therapy before the study treatment (antiestrogen vs. aromatase inhibitor). According to the ESO-ESMO 3rd international consensus guidelines for advanced breast cancer (ABC3), primary endocrine resistance is defined as a relapse in the first 2 years of adjuvant endocrine therapy, or PD within the initial 6 months of first-line endocrine therapy for MBC, while on endocrine therapy. Secondary (acquired) endocrine resistance is defined as a relapse while on adjuvant endocrine therapy but after the first 2 years or within 12 months of completing adjuvant endocrine therapy, or PD ≥6 months after initiating endocrine therapy for MBC, while on endocrine therapy. Efficacy analyses were performed according to the intention-to-treat principle, with all randomized patients included for the primary endpoint.
ORR and CBR were assessed by the logistic regression model. Results were expressed as odds ratio of AI plus metformin versus AI plus placebo, with the corresponding 95% CI and P value. OS was evaluated by unadjusted log-rank test as described for PFS analysis. The log-rank test was performed when approximately 50% of the patients had died, which occurred at the time of the present PFS analysis. The two study arms were compared in incidence rates of certain pre-specified categories of adverse events by two-sided Fisher's exact test at nominal significance of P =0.05.
Acknowledgments
We are grateful to the patients who participated in this trial. We acknowledge Xiaoyu Chen, Enying Cao and Yunhua Lu for their great work of patients’ follow-up visit.
Abbreviations
- HR
hazard ratio
- CI
confidence interval
- CR
complete response
- PR
partial response
- SD
stable disease
- PD
progression disease
- PFS
progression-free survival
- CBR
clinical benefit rate
- OR
odds ratio
- ORR
objective response rate
- OS
overall survival
- mTOR
mammalian target of rapamycin
- AMPK
AMP-activated protein kinase
- pCR
pathologic complete response
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
- PI3K
phosphatidylinositol 3-kinase
- CFDA
China Food and Drug Administration
- HR-positive
hormone receptor (HR) positive
- AI
aromatase inhibitor
- AE
adverse event
- HOMA
homeostatic model assessment
- BMI
body mass index
Author contributions
Biyun Wang and Xichun Hu conceived, designed, performed the study and revised the manuscript. Yannan Zhao and Chengcheng Gong performed statistical analysis, wrote the manuscript and checked the English grammars in manuscript. Zhonghua Wang, Jian Zhang, Leiping Wang, Sheng Zhang, Jun Cao, Zhonghua Tao and Ting Li enrolled the patients and performed the study.
CONFLICTS OF INTEREST
The authors report no conflicts of interest in this work.
FUNDING
National Natural Science Foundation of China (Grant No. 81302300).
Shanghai Natural Science Foundation of China (Grant No. 12ZR1406300).
REFERENCES
- 1.Bailey CJ, Turner RC. Metformin. N Engl J Med. 1996;334:574–579. doi: 10.1056/NEJM199602293340906. [DOI] [PubMed] [Google Scholar]
- 2.Grzybowska M, Bober J, Olszewska M. [Metformin - mechanisms of action and use for the treatment of type 2 diabetes mellitus] Postepy Hig Med Dosw (Online) 2011;65:277–285. doi: 10.5604/17322693.941655. [Article in Polish] [DOI] [PubMed] [Google Scholar]
- 3.Ben SI, Le Marchand-Brustel Y, Tanti JF, Bost F. Metformin in cancer therapy: a new perspective for an old antidiabetic drug? Mol Cancer Ther. 2010;9:1092–1099. doi: 10.1158/1535-7163.MCT-09-1186. [DOI] [PubMed] [Google Scholar]
- 4.Rotella CM, Monami M, Mannucci E. Metformin beyond diabetes: new life for an old drug. Curr Diabetes Rev. 2006;2:307–315. doi: 10.2174/157339906777950651. [DOI] [PubMed] [Google Scholar]
- 5.Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. BMJ. 2005;330:1304–1305. doi: 10.1136/bmj.38415.708634.F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Libby G, Donnelly LA, Donnan PT, Alessi DR, Morris AD, Evans JM. New users of metformin are at low risk of incident cancer: a cohort study among people with type 2 diabetes. Diabetes Care. 2009;32:1620–1625. doi: 10.2337/dc08-2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Decensi A, Puntoni M, Goodwin P, Cazzaniga M, Gennari A, Bonanni B, Gandini S. Metformin and cancer risk in diabetic patients: a systematic review and meta-analysis. Cancer Prev Res (Phila) 2010;3:1451–1461. doi: 10.1158/1940-6207.CAPR-10-0157. [DOI] [PubMed] [Google Scholar]
- 8.Landman GW, Kleefstra N, van Hateren KJ, Groenier KH, Gans RO, Bilo HJ. Metformin associated with lower cancer mortality in type 2 diabetes: ZODIAC-16. Diabetes Care. 2010;33:322–326. doi: 10.2337/dc09-1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bowker SL, Majumdar SR, Veugelers P, Johnson JA. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin: response to Farooki and Schneider. Diabetes Care. 2006;29:1990–1991. doi: 10.2337/dc06-0997. [DOI] [PubMed] [Google Scholar]
- 10.Dowling RJ, Goodwin PJ, Stambolic V. Understanding the benefit of metformin use in cancer treatment. BMC Med. 2011;9:33. doi: 10.1186/1741-7015-9-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dowling RJ, Zakikhani M, Fantus IG, Pollak M, Sonenberg N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 2007;67:10804–10812. doi: 10.1158/0008-5472.CAN-07-2310. [DOI] [PubMed] [Google Scholar]
- 12.Algire C, Zakikhani M, Blouin MJ, Shuai JH, Pollak M. Metformin attenuates the stimulatory effect of a high-energy diet on in vivo LLC1 carcinoma growth. Endocr Relat Cancer. 2008;15:833–839. doi: 10.1677/ERC-08-0038. [DOI] [PubMed] [Google Scholar]
- 13.Goodwin PJ, Pritchard KI, Ennis M, Clemons M, Graham M, Fantus IG. Insulin-lowering effects of metformin in women with early breast cancer. Clin Breast Cancer. 2008;8:501–505. doi: 10.3816/CBC.2008.n.060. [DOI] [PubMed] [Google Scholar]
- 14.Jiralerspong S, Palla SL, Giordano SH, Meric-Bernstam F, Liedtke C, Barnett CM, Hsu L, Hung MC, Hortobagyi GN, Gonzalez-Angulo AM. Metformin and pathologic complete responses to neoadjuvant chemotherapy in diabetic patients with breast cancer. J Clin Oncol. 2009;27:3297–3302. doi: 10.1200/JCO.2009.19.6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jinhua X, Difa L. Comparison of FEC plus metformin and FEC alone in neoadjuvant chemotherapy among early breast cancer patients. Pract J Cancer. 2011;1:92–93. [Google Scholar]
- 16.Hadad SM, Coates P, Jordan LB, Dowling RJ, Chang MC, Done SJ, Purdie CA, Goodwin PJ, Stambolic V, Moulder-Thompson S, Thompson AM. Evidence for biological effects of metformin in operable breast cancer: biomarker analysis in a pre-operative window of opportunity randomized trial. Breast Cancer Res Treat. 2015;150:149–155. doi: 10.1007/s10549-015-3307-5. [DOI] [PubMed] [Google Scholar]
- 17.Niraula S, Dowling RJ, Ennis M, Chang MC, Done SJ, Hood N, Escallon J, Leong WL, McCready DR, Reedijk M, Stambolic V, Goodwin PJ. Metformin in early breast cancer: a prospective window of opportunity neoadjuvant study. Breast Cancer Res Treat. 2012;135:821–830. doi: 10.1007/s10549-012-2223-1. [DOI] [PubMed] [Google Scholar]
- 18.Johnston SR. Clinical efforts to combine endocrine agents with targeted therapies against epidermal growth factor receptor/human epidermal growth factor receptor 2 and mammalian target of rapamycin in breast cancer. Clin Cancer Res. 2006;12:1061s–1068s. doi: 10.1158/1078-0432.CCR-05-2125. [DOI] [PubMed] [Google Scholar]
- 19.Baselga J, Campone M, Piccart M, Burris HR, Rugo HS, Sahmoud T, Noguchi S, Gnant M, Pritchard KI, Lebrun F, Beck JT, Ito Y, Yardley D, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med. 2012;366:520–529. doi: 10.1056/NEJMoa1109653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bachelot T, Bourgier C, Cropet C, Ray-Coquard I, Ferrero JM, Freyer G, Abadie-Lacourtoisie S, Eymard JC, Debled M, Spaeth D, Legouffe E, Allouache D, El Kouri C, Pujade-Lauraine E. Randomized phase II trial of everolimus in combination with tamoxifen in patients with hormone receptor-positive, human epidermal growth factor receptor 2-negative metastatic breast cancer with prior exposure to aromatase inhibitors: a GINECO study. J Clin Oncol. 2012;30:2718–2724. doi: 10.1200/JCO.2011.39.0708. [DOI] [PubMed] [Google Scholar]
- 21.Bonanni B, Puntoni M, Cazzaniga M, Pruneri G, Serrano D, Guerrieri-Gonzaga A, Gennari A, Trabacca MS, Galimberti V, Veronesi P, Johansson H, Aristarco V, Bassi F, et al. Dual effect of metformin on breast cancer proliferation in a randomized presurgical trial. J Clin Oncol. 2012;30:2593–2600. doi: 10.1200/JCO.2011.39.3769. [DOI] [PubMed] [Google Scholar]
- 22.Berstein LM, Yue W, Wang JP, Santen RJ. Isolated and combined action of tamoxifen and metformin in wild-type, tamoxifen-resistant, and estrogen-deprived MCF-7 cells. Breast Cancer Res Treat. 2011;128:109–117. doi: 10.1007/s10549-010-1072-z. [DOI] [PubMed] [Google Scholar]
- 23.Musgrove EA, Sutherland RL. Biological determinants of endocrine resistance in breast cancer. Nat Rev Cancer. 2009;9:631–643. doi: 10.1038/nrc2713. [DOI] [PubMed] [Google Scholar]
- 24.Turner NC, Neven P, Loibl S, Andre F. Advances in the treatment of advanced oestrogen-receptor-positive breast cancer. Lancet. 2017;389:2403–2414. doi: 10.1016/S0140-6736(16)32419-9. [DOI] [PubMed] [Google Scholar]
- 25.Massarweh S, Osborne CK, Creighton CJ, Qin L, Tsimelzon A, Huang S, Weiss H, Rimawi M, Schiff R. Tamoxifen resistance in breast tumors is driven by growth factor receptor signaling with repression of classic estrogen receptor genomic function. Cancer Res. 2008;68:826–833. doi: 10.1158/0008-5472.CAN-07-2707. [DOI] [PubMed] [Google Scholar]
- 26.Vazquez-Martin A, Oliveras-Ferraros C, Del BS, Martin-Castillo B, Menendez JA. If mammalian target of metformin indirectly is mammalian target of rapamycin, then the insulin-like growth factor-1 receptor axis will audit the efficacy of metformin in cancer clinical trials. J Clin Oncol. 2009;27:e207–e209. e210. doi: 10.1200/JCO.2009.24.5456. [DOI] [PubMed] [Google Scholar]
- 27.Rice S, Pellatt L, Ramanathan K, Whitehead SA, Mason HD. Metformin inhibits aromatase via an extracellular signal-regulated kinase-mediated pathway. Endocrinology. 2009;150:4794–4801. doi: 10.1210/en.2009-0540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zakikhani M, Dowling R, Fantus IG, Sonenberg N, Pollak M. Metformin is an AMP kinase-dependent growth inhibitor for breast cancer cells. Cancer Res. 2006;66:10269–10273. doi: 10.1158/0008-5472.CAN-06-1500. [DOI] [PubMed] [Google Scholar]
- 29.Algire C, Amrein L, Bazile M, David S, Zakikhani M, Pollak M. Diet and tumor LKB1 expression interact to determine sensitivity to anti-neoplastic effects of metformin in vivo. Oncogene. 2011;30:1174–1182. doi: 10.1038/onc.2010.483. [DOI] [PubMed] [Google Scholar]
- 30.Liu B, Fan Z, Edgerton SM, Deng XS, Alimova IN, Lind SE, Thor AD. Metformin induces unique biological and molecular responses in triple negative breast cancer cells. Cell Cycle. 2009;8:2031–2040. doi: 10.4161/cc.8.13.8814. [DOI] [PubMed] [Google Scholar]
- 31.Memmott RM, Mercado JR, Maier CR, Kawabata S, Fox SD, Dennis PA. Metformin prevents tobacco carcinogen--induced lung tumorigenesis. Cancer Prev Res (Phila) 2010;3:1066–1076. doi: 10.1158/1940-6207.CAPR-10-0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang X, Wullschleger S, Shpiro N, McGuire VA, Sakamoto K, Woods YL, McBurnie W, Fleming S, Alessi DR. Important role of the LKB1-AMPK pathway in suppressing tumorigenesis in PTEN-deficient mice. Biochem J. 2008;412:211–221. doi: 10.1042/BJ20080557. [DOI] [PubMed] [Google Scholar]
- 33.Anisimov VN, Berstein LM, Egormin PA, Piskunova TS, Popovich IG, Zabezhinski MA, Kovalenko IG, Poroshina TE, Semenchenko AV, Provinciali M, Re F, Franceschi C. Effect of metformin on life span and on the development of spontaneous mammary tumors in HER-2/neu transgenic mice. Exp Gerontol. 2005;40:685–693. doi: 10.1016/j.exger.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 34.Anisimov VN, Egormin PA, Bershtein LM, Zabezhinskii MA, Piskunova TS, Popovich IG, Semenchenko AV. Metformin decelerates aging and development of mammary tumors in HER-2/neu transgenic mice. Bull Exp Biol Med. 2005;139:721–723. doi: 10.1007/s10517-005-0389-9. [DOI] [PubMed] [Google Scholar]
- 35.Kalinsky K, Crew KD, Refice S, Xiao T, Wang A, Feldman SM, Taback B, Ahmad A, Cremers S, Hibshoosh H, Maurer M, Hershman DL. Presurgical trial of metformin in overweight and obese patients with newly diagnosed breast cancer. Cancer Invest. 2014;32:150–157. doi: 10.3109/07357907.2014.889706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cazzaniga M, DeCensi A, Pruneri G, Puntoni M, Bottiglieri L, Varricchio C, Guerrieri-Gonzaga A, Gentilini OD, Pagani G, Dell'Orto P, Lazzeroni M, Serrano D, Viale G, Bonanni B. The effect of metformin on apoptosis in a breast cancer presurgical trial. Br J Cancer. 2013;109:2792–2797. doi: 10.1038/bjc.2013.657. [DOI] [PMC free article] [PubMed] [Google Scholar]