

Abstract
The development of multidrug resistance (MDR) is one of the major challenges to the success of traditional chemotherapy treatment in cancer patients. Most studies to date have focused on strategies to reverse MDR following its development. However, agents utilizing this approach have proven to be of limited clinical use, failing to demonstrate an improvement in therapeutic efficacy with almost no significant survival benefits observed in cancer clinical trials. An alternative approach that has been applied is to prevent or delay MDR prior or early in its development. Recent investigations have shown that preventing the emergence of MDR at the onset of chemotherapy treatment, rather than reversing MDR once it has developed, may assist in overcoming drug resistance. In this review, we focus on a number of novel strategies used by small-molecule inhibitors to prevent the development of MDR. These agents hold great promise for prolonging the efficacy of chemotherapy treatment and improving the clinical outcomes of patients with cancers that are susceptible to MDR development.
Keywords: drug resistance, MDR, prevention, Pgp, Pgp inhibitor
INTRODUCTION
Multidrug resistance (MDR) is a phenomenon by which, after exposure to a chemotherapeutic agent, cancer cells develop resistance, and simultaneous cross-resistance, to a wide range of functionally and structurally unrelated chemotherapeutic drugs [1, 2]. Intrinsic or acquired MDR is one of the main reasons for chemotherapy failure, leading to the recurrence of malignant tumors and ultimately, patient relapse or death [3]. Various mechanisms have been attributed to MDR, such as enhanced drug efflux, increased DNA damage repair, reduced apoptosis, elevated autophagy, and/or altered drug metabolism [2, 4–6]. In order to improve the efficacy of chemotherapy, strategies to reverse MDR have been studied extensively over the past few decades. Three generations of MDR reversing agents, categorized according to their characteristics and chronology, have been developed, including verapamil, valspodar (PSC833), biricodar (VX710), tariquidar (XR9576) and laniquidar (R101933) [7]. A number of clinical trials of the above agents have been conducted in various different cancers types [8–11]. Unfortunately, almost no substantial survival benefits have been established, which has largely limited their widespread clinical application [12]. Recently, several studies have demonstrated that drug combinations can selectively kill resistant cells, while protecting normal cells [13–15]. For example, chemotherapeutic drugs that induce apoptosis in both normal and cancer cells, by the activation of caspases, can be used in combination with caspase inhibitors, which abrogate the chemotherapy-induced apoptosis. As a consequence of this, sensitive cells can be protected, while drug resistant cells undergo apoptosis [16]. This may be a result of enhanced drug pump expression in drug resistant cells, which export caspase inhibitors out of the MDR cells. However, these protective and selective effects may not be achieved if the normal cells express drug pumps, or if the drug resistant cells are deficient of these drug efflux proteins, thereby limiting the clinical application. The majority of previous research has solely focused on the reversal of MDR rather than prevention measures. Understanding the underlying molecular mechanisms in the evolution of MDR during the course of chemotherapy may aid in the design of novel strategies for overcoming MDR. In recent years, studies have shifted focus onto new strategies for the prevention of MDR emergence in cancer [17–20]. Several novel small-molecule inhibitors, including PSC833, VX710, XR9576, and NSC23925 have recently been confirmed as prophylactics that can prevent the induction of MDR [19, 21, 22] (Figure 1). Some of these have been applied in various clinic trails for the treatment of multiple cancer types, such as ovarian cancer, lung cancer, and multiple myeloma [9, 10, 23–25]. In this review, we summarize different model systems that prevent the development of MDR in cancer, and describe several potential mechanisms of specific agents to block drug resistance in cancers.
Figure 1. Schematic of the mechanisms involved in the prevention of drug resistance.

(1) Drug efflux is increased by the overexpression of ABC transporters during chemotherapy. Curcuminoids and NSC77037 can decrease NF-κB transcription [53, 97], while PSC833 and BVDU can suppress the ABCB1 gene amplification [29, 48]. VX710, XR9576, PSC833, NSC23925, Bexarotene, NS-398, Dexrazoxane, Curcuminoid, and P85 directly inhibit the protein expression of P-glycoprotein (Pgp) [19, 27, 33, 34]. (2) Anticancer drugs can become conjugated to glutathione (GSH), leading to their inactivation. This is catalyzed by Glutathione S-transferase (GST), through utilization of the energy derived from ATP hydrolysis. Ethacrynic acid (EA), P85 and selenium compounds can inhibit GST activity [28, 30, 42], and selenium compounds even can decrease the level of GSH [43]. (3) Apoptosis can be initiated through two signaling pathways, the intrinsic pathway (mitochondrial-mediated) and the extrinsic pathway (death receptor-mediated). NSC23925 can inhibit the expression of Bcl-xL, an anti-apoptotic protein, thereby promoting apoptosis [21].
Strategies to establish model systems for preventing the development of MDR in vitro and in vivo
In order to investigate strategies for the prevention of MDR in human cancer, it is necessary to establish a model of the process in which resistance develops after exposure of sensitive tumor cells to chemotherapeutic drugs. In recent years, a variety of experimental approaches have been applied to set up drug-resistant models and to evaluate the efficacy of small-molecule inhibitors on preventing the development of MDR in different cancers.
Establishment of drug-resistant cell lines in vitro
There are three main treatment schemes used to generate MDR cell line model systems. The first scheme requires progressive dose-dependent drug treatment, whereas the second and third schemes are time-dependent only. In the first scheme, sensitive parental cancer cells are exposed to a stepwise increase in the concentration of chemotherapeutic drug to develop MDR cell lines [19, 21, 22, 26–31]. Using this approach, cell lines with several-hundred-fold greater resistance to the chemotherapeutic drug, relative to the sensitive cell lines, can be established [21]. In the second scheme, sensitive parental cancer cells are exposed to a fixed concentration chemotherapeutic drug for a continuous and lengthy period of time [28, 32, 33]. Eventually these cancer cells will present with stable growth in the concentration of anticancer drug that would result in death of 80–90% of parental cells [33]. The third scheme requires intermittent drug treatment, i.e., sensitive parental cancer cells are exposed to repeated treatment cycles [34–37]. For example, in one study of the human breast cancer cell line, MDA-MB-231, resistance in cells was established by a 10-day treatment cycle, which included 3-days of bexarotene treatment followed by 7-days with control medium. At the end of each treatment cycle, cancer cells are harvested and re-planted into a new flask, and then re-exposed to further treatment cycles [34] (Figure 2A). In addition to studying the mechanisms of drug resistance through the comparison of sensitive and resistant cancer cells, the potential mechanisms of novel therapeutics can also be evaluated through similar experimental models. For example, the early and late stage effects of drug treatment in the development of drug resistance can be examined using chemotherapeutic drug treatment alone or in combination with small-molecule inhibitors in sensitive parental cancer cell lines (Figure 2B).
Figure 2. Schematic summarizing how small-molecule inhibitors may prevent the development of drug resistance in vitro.

(A) There are three main treatment schemes used to generate drug-resistant cell lines. In the first scheme, drug treatment is dose-dependent, whereas in the second and third the drug treatments are time-dependent. The first scheme is a progressive-concentration drug treatment, where sensitive parental cancer cells are exposed to gradually increasing concentrations of anticancer drugs [21]. The second scheme is continuous drug treatment, whereby sensitive parental cancer cells are exposed to a fixed concentration of drug for a continuous period of time [33]. In the third scheme, drug treatment is intermittent, with sensitive parental cancer cells exposed to repeated treatment cycles [34]. (B) In contrast to the anti-cancer agents alone causing chemotherapy resistance in these three schemes, in the presence of small-molecule inhibitors, resistance cannot develop during the drug exposure.
Establishment of drug-resistant xenograft tumor models in vivo
Cancer cell lines in vitro lose many of their in vivo features, because they fail to address the impact of environmental signals that are present in tumors [38]. Therefore, xenograft tumor models grown in physiologically-relevant tumor microenvironments, are able to mimic the oxygen, nutrient, and hormone levels of the patient's primary tumor site, as well as maintain the genetic and epigenetic abnormalities, thereby more closely representing patient tumor growth patterns [39, 40]. As a consequence of this, responses to anticancer agents will more closely represent those seen in the patient [41].
In this approach, sensitive parental cancer cells in the log-phase of growth are harvested and implanted subcutaneously into right and left axial regions of 3-to-4-week-old nude mice [21]. When tumor volumes are measurable, chemotherapeutic drug treatment is commenced. There are two well established time-dependent treatment schemes. The first involves intermittent drug treatment [21, 22, 26], e.g., mice treated intraperitoneally with paclitaxel 25 mg/kg twice a week for 3 weeks followed by a treatment-free interval of 2 weeks [21]. This 5 week cycle is then continued for the duration of the experiment (Figure 3A). In contrast to this, the second scheme involves continuous drug treatment [31, 34–36, 42–45], e.g., mice can be treated intraperitoneally with paclitaxel 20 mg/kg once a week for 6 weeks [35]. The selected concentration of the cytotoxic agent is fixed at the maximum tolerated dose, which is known to cause < 10% weight loss during the experiments (Figure 3A). Moreover, the efficacy and prevention of resistance development by co-treatment of chemotherapeutic drugs with small-molecule inhibitors, both of which are initiated at the onset of first treatment, can be assessed by the measured tumor volumes (Figure 3B).
Figure 3. Small-molecule inhibitors prevent the development of drug resistance in vivo.
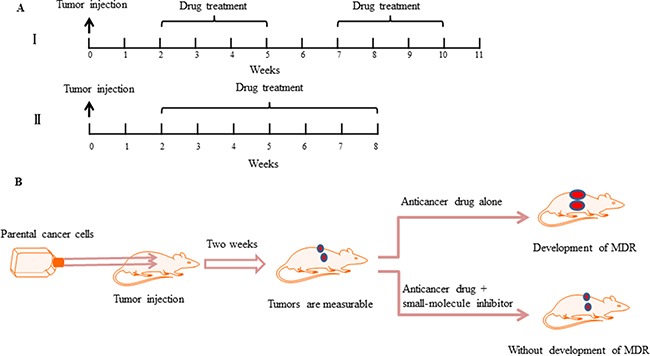
(A) Chemotherapy drug treatment schedule in a drug-resistant xenograft tumor model. Tumor cells are implanted subcutaneously into the left and right axial regions of 3-to-4-week-old nude mice. When the tumor volumes can be measured, two different administration methods of anticancer drugs are applied. I. The mice are exposed to a single drug concentration for 3 weeks followed by a treatment-free interval of 2 weeks. A second round of the same treatment is then continued [21]. II. The mice are exposed to a single drug concentration for six weeks, without a treatment-free interval [35]. (B) Schematic demonstrating that compared with anticancer drug treatments alone, combining a small-molecule inhibitor can prevent the development of drug resistance and prevent tumor growth.
Prevention of MDR emergence in cancer cells both in vitro and in vivo
Pgp inhibitors
Previous studies have confirmed that MDR both in cancer cell lines and human tumor tissues is most often associated with the overexpression of the ATP-binding cassette transporter, P-glycoprotein (Pgp), also known as multidrug resistance protein 1 (MRP1), which is encoded by the ABCB1 gene [46]. Pgp is an ATP-dependent drug-efflux pump, which exports a structurally and functionally diverse set of chemotherapy drugs from the inside of cancer cells to the outside, resulting in decreased intracellular drug accumulation [47]. While Pgp inhibitors have been shown to re-sensitize MDR cells to chemotherapeutic drugs in vitro, almost no survival benefits were found in clinical trials [10, 12]. This reversal strategy has thus far proven inadequate in dealing with the major clinical problem of drug resistance, and as such a new approach is needed. Perhaps, instead of reversing resistance after it has already occurred, it may be more effective to prevent or delay the occurrence of MDR with the initial chemotherapeutic drug treatment [18, 20, 48].
Several experiments have indicated that there exist a number of Pgp inhibitors that may prevent or delay the development of MDR. In parental pediatric rhabdomyosarcoma cells, which were exposed to a stepwise increase in vincristine concentration, the addition of the Pgp inhibitors, PSC833, VX710 and XR9576, prevented the development of resistance in a period of over 40 weeks [19]. In the absence of the inhibitors, vincristine resistance developed rapidly. Furthermore, all resistant cell lines expressed strikingly elevated ABCB1 mRNA and Pgp protein levels compared with the parental cell lines that were treated in combination with the Pgp inhibitors [19] (Figure 1). In line with this, another study reported that doxorubicin (Dox) co-treatment with PSC 833 decreased cellular resistance to Dox by suppressing activation of the ABCB1 gene and the emergence of the MDR phenotype in human uterine sarcoma cells [48]. However, these inhibitors are substrates of the Pgp transporter, acting mainly through competitive inhibition of Pgp-mediated drug efflux, and as such they lack specific target sites [7]. Consequently, clinical toxicities associated with their use at the required concentrations to inhibit Pgp function have prohibited their widespread use. For example, PSC833 and VX710 have shown unexpected pharmacokinetic interactions with paclitaxel, the major adverse effects including myelosuppression and non-hematologic toxicities [1, 49]. Therefore, the development of more potent and specific drugs that act to prevent the development of MDR is required for cancer patient treatment.
NSC23925
NSC23925 (2-(4-methoxyphenyl)-4-quinolinyl) (2-piper-idinyl) methanol) is a long-acting methoxyphenyl piperidinyl compound, which has been identified as a noncompetitive, selective and effective inhibitor of Pgp [50]. When the effect of NSC23925 at reverse drug resistance in MDR ovarian cancer cell lines was examined, the potency of NSC23925 was been shown to be 20 and 50 fold greater than that of verapamil and Cyclosporin A (CsA), respectively [50]. Our previous studies have demonstrated that NSC23925 could prevent the introduction of paclitaxel resistance in ovarian cancer and osteosarcoma both in vitro and in vivo [21, 22, 26]. Ovarian cancer cells were cultured with stepwise increased concentration of paclitaxel in combination with or without NSC23925 [22]. After over half a year of drug treatment, cells that were exposed to paclitaxel alone grew stably in culture medium in the presence of 0.3 μM paclitaxel, without showing any signs toxicity [22]. However, cells selected with paclitaxel in combination with NSC23925 could not survive in the medium with more than 0.001 μM paclitaxel. Pgp was markedly overexpressed in all resistant cell lines, but was not expressed at detectible levels in combination treated cells. These observations indicated that NSC23925 prevented the emergence of paclitaxel resistance through specifically inhibiting the expression of Pgp, which was further confirmed in xenograft mouse models. The combination treatment remarkably prevented tumor growth compared with control saline treatment and paclitaxel treatment alone [22]. Besides the change in expression of Pgp, several apoptotic-related proteins were also detected to have changed expression after treatment in mouse tumor tissues. Anti-apoptotic proteins, such as survivin, Bcl-xL and MCL-1, showed significantly lower expression levels in the presence of NSC23925. CD44 and integrin β3, which are cancer stem cell markers, also displayed significantly decreased expression in combination-treatment tumor samples [21]. These findings suggest that co-treatment of paclitaxel with NSC23925 enhances apoptosis, which may be contributed to the prevention of paclitaxel resistance (Figure 1). The toxicity of NSC23925, determined by evaluating body weight, blood cell count and histology of organs in mouse models, showed no remarkable toxicities when exposed to NSC23925 alone or in combination with paclitaxel over the full duration of the experiment [21]. In all treatment conditions, the mice appeared to tolerate the treatments well.
NSC77037
NSC77037, also known as Tetrandrine or CBT-1, is a bisbenzylisoquinoline alkaloid compound, which was isolated from the tuberous root of Stephania tetrandra [51]. The mechanism of NSC77037 is similar to that of NSC23925, which is to directly stimulate Pgp ATPase activity and inhibit the function of Pgp [52]. In combination with Dox, NSC77037 has been shown to prevent Dox induced MDR in human leukemia K562 cells by markedly inhibiting the ABCB1 gene transcription [53]. K562 cells were exposed to Dox alone or in combination with three different concentrations of NSC77037. The results showed that NSC77037 could prevent Dox induced ABCB1 expression and Pgp function in a dose-dependent manner. Several studies have shown that the inducible transcription factor, NF-κB, can upregulate ABCB1 gene expression [54, 55]. Interestingly, while exposure of K562 cells to Dox alone elevated the expression of NF-κB in the nucleus and nuclear localization of the active protein, the Dox-NSC77037 combination decreased total and nuclear NF-κB expression as well as attenuated its DNA-binding activity, through which it inhibited the overexpression of the downstream gene ABCB1 [53] (Figure 1).
Recently, NSC77037 was examined in a phase I clinical trial in combination with Dox to treat patients with advanced cancer [56]. Compared to other Pgp inhibitors, NSC77037 did not significantly alter the pharmacokinetics of Dox and the side effects including moderate nausea and occasional vomiting were mild. The antitumor efficacy was encouraging in the study, 25 of 34 patients were evaluable for response and 5 patients demonstrated tumor shrinkage [56]. It is currently being assessed in Phase II clinical trials of the NSC77037 in combination with paclitaxel for the treatment of solid tumors [57]. NSC77037, with lack of toxicity, could inhibit Pgp-mediated efflux of rhodamine 123 from peripheral blood mononuclear cells and normal liver to a degree, which was achieved by using other Pgp inhibitors [57].
Dexrazoxane
Dexrazoxane, is a bis-dioxopiperazine compound, which is hydrolyzed to form a chelating agent analogous to ethylenediaminetetraacetic acid (EDTA) [58]. Many preclinical studies have shown that dexrazoxane significantly protects against Dox-induced cardiotoxicity, a major drawback of Dox [59, 60]. Moreover, clinical observation of advanced breast cancer patients treated with the FAC scheme (5-fluorouracil, Dox and cyclophosphamide) plus dexrazoxane, has shown increased overall median survival rates [61]. In contrast to Pgp inhibitors, dexrazoxane has not been described as a modulator of MDR, because it could not reverse MDR once the resistance has been formed [27]. But dexrazoxane has been shown to significantly delay the emergence of drug resistance in the human leukemia cell line K562 [27]. K562 cells that were exposed to gradually increasing Dox concentrations for 7 months in the presence of dexrazoxane, showed a marked reduction in the development of Dox resistance, compared with cells treated with Dox alone. This was reported to be a result of dexrazoxane-mediated suppression of the Dox induced upregulation of the ABCB1 gene and Pgp protein [27] (Figure 1).
Bexarotene
Bexarotene, also known as LGD1069 or Targretin, is a selective retinoid X receptor ligand, which has been used as an efficacious chemopreventive and chemotherapeutic agent in preclinical mice breast cancer models [62, 63]. The primary side effects of bexarotene include hypertriglyceridemia and hypothyroidism, which can be reversed or managed with corresponding drugs [64]. Bexarotene has shown great potential for chemotherapy-based combination because of its non-overlapping side effect profile with the majority of cytotoxic agents and lack of toxicity. Combination with bexarotene can prevent and reverse acquired drug resistance in several cancer cell lines both in vitro and in vivo, including advanced prostate cancer, breast cancer, and non-small cell lung cancer (NSCLC) [34–36]. For example, when the breast cancer cells, MDA-MB-231, were exposed to paclitaxel alone they demonstrated a sigmoidal concentration-dependent growth inhibition, whereas the combination of paclitaxel and bexarotene showed limited growth inhibition during the experiment [34]. Repeated exposure of cells to paclitaxel alone resulted in the development of acquired MDR. In parallel, co-treatement with paclitaxel/bexarotene delayed the growth of cells for an additional 2 months and the cells remained chemosensitive towards all the tested anticancer drugs. Similar to paclitaxel, the combination of bexarotene with other chemotherapeutic drugs, such as Dox and cisplatin, also has been shown to prevent and delay the development of MDR [34, 36]. There are several mechanisms of bexarotene that have been attributed to its preventive MDR effect [34–36]. Firstly, while resistant cells express increased levels of ABCB1 and Pgp, they are almost undetectable in the parental cancer cells and in the co-treated cells (Figure 1). Secondly, compared with anticancer drug treatment alone, bexarotene in combination markedly suppressed the invasiveness and angiogenic potential of cancer cells. Thirdly, bexarotene maintained the genomic integrity of tumor cells by interfering with the acquisition of spontaneous mutations. These in vitro findings were further confirmed in mice xenograft tumor models [34–36]. In combination with a cytotoxic agent, bexarotene has been shown to dramatically decrease tumor growth compared with the cytotoxic agent alone. More recently, a study demonstrated that bexarotene could prevent and reverse gemcitabine resistance in NSCLC cells by modulating ribonucleotide reductase M1 (RRM1) gene amplification both in vitro and in vivo [37]. The RRM1 gene is located in the chr11p15.5 locus, which highly expressed in gemcitabine resisitant cells, as determined by microarray gene analysis. Therefore, RRM1 might be supressed by cell exposure to bexarotene. A phase I/II clinical trial has shown that bexarotene in combination with cisplatin or vinorelbine can prolong the survival of advanced NSCLC patients [65].
Ethacrynic acid
Ethacrynic acid (EA) is a FDA-approved specific inhibitor of glutathione S-transferase (GST), which has been shown to enhance the cytotoxicity of anticancer drugs and reverse drug resistance [66–68]. Overexpression of GST is often regarded as a contributor to the development of cytotoxic agent resistance [67]. In a study of ovarian cancer cells that developed resistance following either exposure to low dose concentration of melphalan for 7 days, or incrementally increased dosing of melphalan for over a year, these resistant cell lines showed elevated levels of GST activity and mRNA compared with their parental cells [28] (Figure 1). However, the 7-day melphalan-resistant cells quickly reverted back to a drug sensitive phenotype in the absence of treatment within 2 weeks, indicating that the resistance was not a stable characteristic. In contrast to this, the one-year treated cell line maintained a permanent resistant phenotype, even when melphalan treatment was withdrawn. These results provide a possible explanation for enhanced efficacy of intermittent chemotherapy treatment in contrast to continuous drug delivery in clinic [69]. In addition, the development of melphalan resistance could be prevented by co-incubation with EA during the 7-day melphalan treatment in ovarian cancer cells. Moreover, this suppression of melphalan-induced resistance by EA developed in a dose-dependent manner through a reduction in GST gene expression [28].
Selenium compounds
Selenium is an essential dietary trace element which has been extensively studied for its anticarcinogenic activity [70]. Selenium compounds, including selenite and selenomethionine, are capable of chemopreventive effectiveness through enhancing the efficacy of standard cytotoxic agents while reducing the side-effects of chemotherapy [71, 72]. Selenium compounds have been shown to prevent the emergence of drug resistance to melphalan, cisplatin, and carboplatin in ovarian cancer, both in vitro and in vivo [32, 42–45]. Interestingly, studies have shown that, selenium compounds can suppress the amplification of the GST gene, which is induced during the development of melphalan resistance [32, 42]. Selenium compounds are also able to inhibit levels of the antioxidant, glutathione (GSH), which is known to be induced during cisplatin treatment [43]. Multiple chemotherapeutic agents, such as the platinum class of drugs, are conjugated to GSH, leading to inactivation of these drugs [73] (Figure 1). Previous studies have demonstrated that elevated GSH level is correlated with platinum drug-induced resistance [74, 75]. Furthermore, inclusion of selenite compounds with cisplatin treatment prolonged and enhanced the efficacy of chemotherapy treatment, resulting in reduced growth of ovarian tumor xenografts [44]. Notably, when cells from platinum resistant or sensitive tumors were transplanted into new animals, the derivative tumors retained the drug resistant or sensitive phenotype of the tumors from which they originated [43–45]. These results support the hypothesis that prevention of resistance may result from a genetic or epigenetic effect of selenium compounds.
BVDU
(E)-5-(2-Bromovinyl)-2-deoxyuridine (BVDU, RP101) is well established as a highly potent and selective anti-viral agent, inhibiting both herpes simplex virus type 1 (HSV-1) and varicella-zoster virus (VZV) [76]. BVDU has also been shown to suppress 2-amino-6-mercaptopurine-induced SV40 viral amplification in Chinese hamster cells and abolished triethylene melamine (TEM)-induced mitotic recombination in yeast [77]. Importantly, gene amplification and recombination may be regarded as contributors to acquired chemoresistance. Gene recombination is functionally related to amplification, indeed the first step of both processes are the same [78, 79]. In a study by Fahrig et al., mouse leukemia cell lines were exposed to 4 weeks of increasing concentrations of Dox in the presence or absence of BVDU [29]. In the cells treated with BVDU, Dox resistance did not develop, which was likely due to the suppression of ABCB1 gene amplification and expression [29] (Figure 1). Similarly, BVDU has also been reported to prevent drug resistance to methotrexate (MTX), by suppressing the chemotherapy-induced amplification of the dihydrofolate reductase (Dhfr) gene in 3T6 cells [80]. Further studies also revealed that in combination, BVDU strengthened cytotoxicity of all tested chemotherapy agents, including DOX, mitomycin C, mitoxantrone, glufosfamide, and cisplatin, by inducing apoptosis in vitro and dramatically enhanced tumor regression in vivo [80]. While the anticancer drugs alone resulted in significant body weight loss in rats, this was partly inhibited with BVDU co-treatment. This may indicate BVDU co-treatment can reduce nonspecific toxicity and optimized antitumor efficiency. There are several possible mechanisms explaining the modulation by BVDU. Firstly, BVDU enhances the activity of NAD(P)H: quinone oxidoreductase 1 (NQO1), a FAD containing quinone reductase, which is frequently decreased in MDR cancer cells [81]. Secondly, BVDU inhibits the expression of the oncogene DDX1 and the DNA repair associated enzymes, UBE2N and APEX. Thirdly, BVDU inhibits cell survival pathways, involving STAT3 and JUN-D, leading to promotion of apoptotic pathways [82, 83]. Lastly, during recovery, BVDU promotes upregulation of microfilamental proteins and suppression of ATP generating proteins [80].
NS-398
N-[2-(cyclohexyloxy)4-nitrophenyl]-methanesulfonamide (NS-398), is a specific inhibitor of Cyclooxygenase-2 (COX-2), an enzyme required for the formation of prostanoids [84]. Reports have shown that COX-2 is upregulated in a wide range of solid tumors, promoting angiogenesis, invasiveness and anti-apoptotic activity [85, 86]. Selected COX-2 inhibitors, such as meloxicam and rofecoxib, have been reported to reverse MDR by reducing the expression of ABC-transporter proteins [87, 88]. Similar to this, treatment with NS-398 alone has been shown to not affect cell proliferation in parental breast cancer cells, but is able to prevent or reduce the development of the chemoresistance phenotype in breast cancer cells [33]. In a study of breast cancer cells exposed to low dose Dox for 10 days or 2 months in order to develop drug resistance, demonstrated both cell lines developed enhanced levels of Pgp and COX-2 [33]. The resistance formed in the 10-day Dox treated cells was shown to be reversible following either drug withdrawal or with NS-398 co-treatment. Whereas, in the 2 month-resistance model, the “permanent” resistance that was established with Dox treatment, did not occur with NS-398 treatment, and consequently, the cytotoxic effects of Dox remained potent. The application of NS-398 in combination is believed to have prevented the development of Dox resistance, through suppression of Pgp and COX-2 activity (Figure 1). Therefore, by decreasing Pgp, significantly more intracellular drug accumulation and retention was observed in vitro.
Curcuminoids
Curcuminoids, are a class of natural phenolic coloring compounds, which are isolated from the rhizomes of Curcuma longa [89]. Curcuminoids show a broad range of biological activities, including potent anti-inflammatory, anti-oxidant and anti-tumor properties [90, 91]. Increasing evidence indicates that curcuminoids act to suppress proliferation, metastasis, invasion and angiogenesis of human cancers, through interactions with several intracellular signal transduction pathways [92, 93]. Experimental results and clinical trials have demonstrated that curcumin (Cur) is non-toxic even at high concentrations [94]. Furthermore, curcuminoids have the potential to block acquired resistance induced by adriamycin (ADM), through the downregulation of ABCB1 mRNA and Pgp protein [95]. Curcuminoids contain at least three active forms: curcumin (Cur), demethoxycurcumin (D-Cur) and bisdemethoxycurcumin (BD-Cur) [96]. The anti-chemoresistance activity of these three forms of curcumin has been compared in a study by Xu et al. and the possible mechanisms of activity explored [97]. In this study, human leukemia cells were pretreated with or without Cur, D-Cur and BD-Cur for 24 h, followed by addition of ADM, and then co-incubated for another 72 h. The results revealed that curcuminoids significantly inhibited the expression of ABCB1 and Pgp. D-Cur was the most active drug, followed by BD-Cur, with Cur showing the least activity. Western blot analysis was also applied to explore the effect of curcuminoids on NF-κB transcriptional activity, as ABCB1 gene expression is known to be regulated by NF-κB [55] (Figure 1). BD-Cur also proved to have the greatest activity in suppressing the nuclear translocation of NF-κB, followed by Cur and then D-Cur, which interestingly did not correlate with the trend observed for the preventive activity of the three curcuminoids [97].
Pluronic P85
Pluronic compounds are a remarkable example of polymers, which hold tremendous potential for the advancement of drug delivery applications, due to a number of preferable characteristics, such as biocompatibility, tunable chain length and a number of other properties [98–100]. Pluronic compounds are an A-B-A amphiphilic block copolymers, containing hydrophilic poly (ethylene oxide) (PEO) blocks and hydrophobic poly (propylene oxide) (PPO) blocks [101]. In MDR cancer cells exposed to Pluronics, drug sensitivity to anticancer drugs was reinstated [102, 103]. Because of the lipid-like amphiphilic character, Pluronics can effectively move into cellular membranes and significantly inhibit drug-efflux transporter proteins, such as Pgp, leading to increased intracellular drug accumulation [104]. Pluronic compounds have been shown to induce intracellular ATP depletion, which prevents the activity of Pgp [103]. Pluronic P85 has also been demonstrated to prevent the development of MDR in the human breast carcinoma cell line, MCF7 [30]. MCF7 cells were exposed to increasing concentrations of Dox with or without P85 for 305 days in vitro. In the cells treated with 1000 ng/ml of Dox alone, stable growth in culture medium was eventually obtained, whereas Dox-P85 co-treated cells were unable to survive in the culture medium supplemented with more than 10 ng/ml of Dox [30]. P85 was shown to prevent Dox resistance through inhibition of ABCB1 and Pgp expression, and it also simultaneously suppressed expression of the GST pi gene, which is also a relevant indicator of chemotherapy resistance (Figure 1). Furthermore, DNA microarray analysis revealed that co-treatment with P85 abolished alterations of genes that are involved with drug metabolism, apoptosis, stress response, transcriptional factors and tumorigenesis [30]. Therefore, application of Pluronic P85 in the clinic might potentially enhance therapeutic outcomes in patients with breast cancer. Similar effects of P85 have also been reported in leukemia cells both in vitro and in vivo [31]. The mechanisms of P85 on MDR prevention in leukemia were reportedly similar to that in breast cancer.
Conclusion and future perspective
MDR is a major obstacle that severely limits the efficacy of clinical chemotherapy in the treatment of cancer. Experimental drug resistance cancer models have contributed to the identification of many of the underlying mechanisms involved in the development of MDR. Many attempts have been conducted in clinic trials to overcome drug resistance, but have been met with limited success. Preventing the development of drug resistance holds great value as a novel strategy for anticancer treatment [17, 19, 22, 35]. Recent studies have demonstrated that several small-molecule inhibitors, including Pgp inhibitors, are capable at preventing the development of MDR when co-treated with cytotoxic drugs in different in vitro and in vivo model systems (Table 1). Preventing or delaying the emergence of drug resistance is likely to enhance the effectiveness of chemotherapy and improve clinic outcomes for patients with cancer. This review highlights that a pilot study is necessary to optimize the concentrations required for long-term exposure to chemotherapeutic agents and small-molecule inhibitors to prevent MDR development and consequently poor patient treatment outcomes. Further studies on the mechanisms of prevention of MDR and clinical testing of these agents are needed to determine their efficacy in the treatment of human cancers.
Table 1. Small-molecule inhibitors prevent the development of drug resistance in human cancer.
| Compound | Functional mechanism | Cytotoxic agent used in combination | Cancer type | Reference |
|---|---|---|---|---|
| VX710 | inhibits the expression of ABCB1 mRNA and Pgp | vincristine | rhabdomyosarcoma | [19] |
| XR9576 | inhibits the expression of ABCB1 mRNA and Pgp | vincristine | rhabdomyosarcoma | [19] |
| PSC833 | inhibits the expression of ABCB1 mRNA and Pgp | vincristine | rhabdomyosarcoma | [19] |
| suppresses the activation of ABCB1 gene; inhibits the appearance of MDR phenotype |
doxorubicin | uterine sarcoma | [48] | |
| NSC23925 | suppresses the expression of Pgp and enhances apoptosis | palitaxel | ovarian cancer | [21, 22] |
| inhibits the expression of Pgp | palitaxel | osteosarcoma | [26] | |
| NSC77037 | inhibits the expression of ABCB1 gene and Pgp; decreases NF-kB transcription and protein activity |
doxorubicin | leukemia | [53] |
| Dexrazoxane | inhibits the expression of ABCB1 mRNA and Pgp | doxorubicin | leukemia | [27] |
| Bexarotene | inhibits the expression of ABCB1 mRNA and Pgp; suppresses angiogenic and invasiveness potential; |
paclitaxel; doxorubicin; cisplatin | advanced prostate cancer; breast cancer; non-small cell lung cancer |
[34–36] |
| decreases spontaneous mutation rate suppresses the amplification of RRM1 gene |
gemcitabine | non-small cell lung cancer | [37] | |
| Ethacrynic acid | inhibits the levels of GST activity and mRNA | melphalan | ovarian cancer | [28] |
| Selenium compounds | inhibit the expression of GST mRNA and enzyme activity | melphalan | ovarian cancer | [32, 42] |
| inhibit the level of GSH | cisplatin | ovarian cancer | [43] | |
| maintain the sensitivity following transplantation | cisplatin; carboplatin | ovarian cancer | [43–45] | |
| prolong and enhance the efficacy of chemotherapy treatment | cisplatin | ovarian cancer | [44] | |
| BVDU | inhibits the amplification of ABCB1 gene | doxorubicin | leukemia; fibrosarcoma; mammary adenocarcinomas | [29, 80] |
| suppresses the amplification of Dhfr gene enhances the activity of NQO1; |
methotrexate | fibrosarcoma | [80] | |
| inhibits the expression of oncogenes and DNA repair enzymes; suppresses the proteins of survival pathways or ATP generation; |
doxorubicin; mitomycin C; mitoxantrone | hepatosarcoma | [80] | |
| up-regulates microfilamental proteins | ||||
| NS-398 | inhibits the expression of Pgp and COX-2 | doxorubicin | breast cancer | [33] |
| Curcuminoids | inhibits the expression of ABCB1 mRNA and Pgp; suppress the nuclear translocation of NF-kB |
adriamycin | leukemia | [95, 97] |
| Pluronic P85 | inhibits the expression of ABCB1 mRNA and Pgp; abolishes the alterations of genes implicated in drug resistance |
doxorubicin | breast cancer; leukemia | [30, 31] |
| suppresses the level of GST pi mRNA | doxorubicin | breast cancer | [30] |
ACKNOWLEDGMENTS AND FUNDING
This work was supported by a Joint Research Fund devoted to clinical pharmacy and precision medicine (Z.D and Q.K). Support has also been provided by the National Natural Science Foundation of China (No.: 81402266 and 81372875), the Gattegno and Wechsler funds. Dr.Wang is supported by a scholarship from the China Scholarship Council.
Footnotes
CONFLICTS OF INTEREST
The authors declare that they have no competing interests.
REFERENCES
- 1.Yusuf RZ, Duan Z, Lamendola DE, Penson RT, Seiden MV. Paclitaxel resistance: molecular mechanisms and pharmacologic manipulation. Curr Cancer Drug Targets. 2003;3:1–19. doi: 10.2174/1568009033333754. [DOI] [PubMed] [Google Scholar]
- 2.Szakacs G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5:219–234. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- 3.Liang XJ, Chen C, Zhao Y, Wang PC. Circumventing tumor resistance to chemotherapy by nanotechnology. Methods Mol Biol. 2010;596:467–488. doi: 10.1007/978-1-60761-416-6_21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li Y, Jiang W, Hu Y, Da Z, Zeng C, Tu M, Deng Z, Xiao W. MicroRNA-199a-5p inhibits cisplatin-induced drug resistance via inhibition of autophagy in osteosarcoma cells. Oncol Lett. 2016;12:4203–4208. doi: 10.3892/ol.2016.5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kachalaki S, Ebrahimi M, Mohamed Khosroshahi L, Mohammadinejad S, Baradaran B. Cancer chemoresistance; biochemical and molecular aspects: a brief overview. Eur J Pharm Sci. 2016;89:20–30. doi: 10.1016/j.ejps.2016.03.025. [DOI] [PubMed] [Google Scholar]
- 6.Salehan MR, Morse HR. DNA damage repair and tolerance: a role in chemotherapeutic drug resistance. Br J Biomed Sci. 2013;70:31–40. doi: 10.1080/09674845.2013.11669927. [DOI] [PubMed] [Google Scholar]
- 7.Palmeira A, Sousa E, Vasconcelos MH, Pinto MM. Three decades of P-gp inhibitors: skimming through several generations and scaffolds. Curr Med Chem. 2012;19:1946–2025. doi: 10.2174/092986712800167392. [DOI] [PubMed] [Google Scholar]
- 8.Baekelandt M, Lehne G, Trope CG, Szanto I, Pfeiffer P, Gustavssson B, Kristensen GB. Phase I/II trial of the multidrug-resistance modulator valspodar combined with cisplatin and doxorubicin in refractory ovarian cancer. J Clin Oncol. 2001;19:2983–2993. doi: 10.1200/JCO.2001.19.12.2983. [DOI] [PubMed] [Google Scholar]
- 9.Fracasso PM, Brady MF, Moore DH, Walker JL, Rose PG, Letvak L, Grogan TM, McGuire WP. Phase II study of paclitaxel and valspodar (PSC 833) in refractory ovarian carcinoma: a gynecologic oncology group study. J Clin Oncol. 2001;19:2975–2982. doi: 10.1200/JCO.2001.19.12.2975. [DOI] [PubMed] [Google Scholar]
- 10.Seiden MV, Swenerton KD, Matulonis U, Campos S, Rose P, Batist G, Ette E, Garg V, Fuller A, Harding MW, Charpentier D. A phase II study of the MDR inhibitor biricodar (INCEL, VX-710) and paclitaxel in women with advanced ovarian cancer refractory to paclitaxel therapy. Gynecol Oncol. 2002;86:302–310. doi: 10.1006/gyno.2002.6762. [DOI] [PubMed] [Google Scholar]
- 11.Lhomme C, Joly F, Walker JL, Lissoni AA, Nicoletto MO, Manikhas GM, Baekelandt MM, Gordon AN, Fracasso PM, Mietlowski WL, Jones GJ, Dugan MH. Phase III study of valspodar (PSC 833) combined with paclitaxel and carboplatin compared with paclitaxel and carboplatin alone in patients with stage IV or suboptimally debulked stage III epithelial ovarian cancer or primary peritoneal cancer. J Clin Oncol. 2008;26:2674–2682. doi: 10.1200/JCO.2007.14.9807. [DOI] [PubMed] [Google Scholar]
- 12.Kaye SB. Reversal of drug resistance in ovarian cancer: where do we go from here? J Clin Oncol. 2008;26:2616–2618. doi: 10.1200/JCO.2008.16.2123. [DOI] [PubMed] [Google Scholar]
- 13.Blagosklonny MV. Targeting cancer cells by exploiting their resistance. Trends Mol Med. 2003;9:307–312. doi: 10.1016/s1471-4914(03)00111-4. [DOI] [PubMed] [Google Scholar]
- 14.Blagosklonny MV. Antagonistic drug combinations that select against drug resistance: from bacteria to cancer. Cancer Biol Ther. 2007;6:1013–1014. doi: 10.4161/cbt.6.7.4340. [DOI] [PubMed] [Google Scholar]
- 15.Seebacher NA, Richardson DR, Jansson PJ. A mechanism for overcoming P-glycoprotein-mediated drug resistance: novel combination therapy that releases stored doxorubicin from lysosomes via lysosomal permeabilization using Dp44mT or DpC. Cell Death Dis. 2016;7:e2510. doi: 10.1038/cddis.2016.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blagosklonny MV. Treatment with inhibitors of caspases, that are substrates of drug transporters, selectively permits chemotherapy-induced apoptosis in multidrug-resistant cells but protects normal cells. Leukemia. 2001;15:936–941. doi: 10.1038/sj.leu.2402127. [DOI] [PubMed] [Google Scholar]
- 17.Frenkel GD, Caffrey PB. A prevention strategy for circumventing drug resistance in cancer chemotherapy. Curr Pharm Des. 2001;7:1595–1614. doi: 10.2174/1381612013397186. [DOI] [PubMed] [Google Scholar]
- 18.Sikic BI, Fisher GA, Lum BL, Halsey J, Beketic-Oreskovic L, Chen G. Modulation and prevention of multidrug resistance by inhibitors of P-glycoprotein. Cancer Chemother Pharmacol. 1997;40:S13–19. doi: 10.1007/s002800051055. [DOI] [PubMed] [Google Scholar]
- 19.Cocker HA, Tiffin N, Pritchard-Jones K, Pinkerton CR, Kelland LR. In vitro prevention of the emergence of multidrug resistance in a pediatric rhabdomyosarcoma cell line. Clin Cancer Res. 2001;7:3193–3198. [PubMed] [Google Scholar]
- 20.Germann UA, Harding MW. Chemosensitizers to overcome and prevent multidrug resistance? J Natl Cancer Inst. 1995;87:1573–1575. doi: 10.1093/jnci/87.21.1573. [DOI] [PubMed] [Google Scholar]
- 21.Yang X, Shen J, Gao Y, Feng Y, Guan Y, Zhang Z, Mankin H, Hornicek FJ, Duan Z. Nsc23925 prevents the development of paclitaxel resistance by inhibiting the introduction of P-glycoprotein and enhancing apoptosis. Int J Cancer. 2015;137:2029–2039. doi: 10.1002/ijc.29574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang X, Feng Y, Gao Y, Shen J, Choy E, Cote G, Harmon D, Zhang Z, Mankin H, Hornicek FJ, Duan Z. NSC23925 prevents the emergence of multidrug resistance in ovarian cancer in vitro and in vivo. Gynecol Oncol. 2015;137:134–142. doi: 10.1016/j.ygyno.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 23.Baer MR, George SL, Dodge RK, O’Loughlin KL, Minderman H, Caligiuri MA, Anastasi J, Powell BL, Kolitz JE, Schiffer CA, Bloomfield CD, Larson RA. Phase 3 study of the multidrug resistance modulator PSC-833 in previously untreated patients 60 years of age and older with acute myeloid leukemia: Cancer and Leukemia Group B Study 9720. Blood. 2002;100:1224–1232. [PubMed] [Google Scholar]
- 24.Kelly RJ, Draper D, Chen CC, Robey RW, Figg WD, Piekarz RL, Chen X, Gardner ER, Balis FM, Venkatesan AM, Steinberg SM, Fojo T, Bates SE. A pharmacodynamic study of docetaxel in combination with the P-glycoprotein antagonist tariquidar (XR9576) in patients with lung, ovarian, and cervical cancer. Clin Cancer Res. 2011;17:569–580. doi: 10.1158/1078-0432.CCR-10-1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gandhi L, Harding MW, Neubauer M, Langer CJ, Moore M, Ross HJ, Johnson BE, Lynch TJ. A phase II study of the safety and efficacy of the multidrug resistance inhibitor VX-710 combined with doxorubicin and vincristine in patients with recurrent small cell lung cancer. Cancer. 2007;109:924–932. doi: 10.1002/cncr.22492. [DOI] [PubMed] [Google Scholar]
- 26.Yang X, Yang P, Shen J, Osaka E, Choy E, Cote G, Harmon D, Zhang Z, Mankin H, Hornicek FJ, Duan Z. Prevention of multidrug resistance (MDR) in osteosarcoma by NSC23925. Br J Cancer. 2014;110:2896–2904. doi: 10.1038/bjc.2014.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sargent JM, Williamson CJ, Yardley C, Taylor CG, Hellmann K. Dexrazoxane significantly impairs the induction of doxorubicin resistance in the human leukaemia line, K562. Br J Cancer. 2001;84:959–964. doi: 10.1054/bjoc.2001.1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caffrey PB, Zhu M, Zhang Y, Chinen N, Frenkel GD. Rapid development of glutathione-S-transferase-dependent drug resistance in vitro and its prevention by ethacrynic acid. Cancer Lett. 1999;136:47–52. doi: 10.1016/s0304-3835(98)00307-3. [DOI] [PubMed] [Google Scholar]
- 29.Fahrig R, Steinkamp-Zucht A, Schaefer A. Prevention of adriamycin-induced mdr1 gene amplification and expression in mouse leukemia cells by simultaneous treatment with the anti-recombinogen bromovinyldeoxyuridine. Anticancer Drug Des. 2000;15:307–312. [PubMed] [Google Scholar]
- 30.Batrakova EV, Kelly DL, Li S, Li Y, Yang Z, Xiao L, Alakhova DY, Sherman S, Alakhov VY, Kabanov AV. Alteration of genomic responses to doxorubicin and prevention of MDR in breast cancer cells by a polymer excipient: pluronic P85. Mol Pharm. 2006;3:113–123. doi: 10.1021/mp050050g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sharma AK, Zhang L, Li S, Kelly DL, Alakhov VY, Batrakova EV, Kabanov AV. Prevention of MDR development in leukemia cells by micelle-forming polymeric surfactant. J Control Release. 2008;131:220–227. doi: 10.1016/j.jconrel.2008.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caffrey PB, Zhu M, Frenkel GD. Prevention of the development of melphalan resistance in vitro by selenite. Biol Trace Elem Res. 1998;65:187–195. doi: 10.1007/BF02789095. [DOI] [PubMed] [Google Scholar]
- 33.Zatelli MC, Luchin A, Tagliati F, Leoni S, Piccin D, Bondanelli M, Rossi R, degli Uberti EC. Cyclooxygenase-2 inhibitors prevent the development of chemoresistance phenotype in a breast cancer cell line by inhibiting glycoprotein p-170 expression. Endocr Relat Cancer. 2007;14:1029–1038. doi: 10.1677/ERC-07-0114. [DOI] [PubMed] [Google Scholar]
- 34.Yen WC, Lamph WW. The selective retinoid X receptor agonist bexarotene (LGD1069, Targretin) prevents and overcomes multidrug resistance in advanced breast carcinoma. Mol Cancer Ther. 2005;4:824–834. doi: 10.1158/1535-7163.MCT-05-0018. [DOI] [PubMed] [Google Scholar]
- 35.Yen WC, Corpuz MR, Prudente RY, Cooke TA, Bissonnette RP, Negro-Vilar A, Lamph WW. A selective retinoid X receptor agonist bexarotene (Targretin) prevents and overcomes acquired paclitaxel (Taxol) resistance in human non-small cell lung cancer. Clin Cancer Res. 2004;10:8656–8664. doi: 10.1158/1078-0432.CCR-04-0979. [DOI] [PubMed] [Google Scholar]
- 36.Yen WC, Lamph WW. A selective retinoid X receptor agonist bexarotene (LGD1069, Targretin) prevents and overcomes multidrug resistance in advanced prostate cancer. Prostate. 2006;66:305–316. doi: 10.1002/pros.20347. [DOI] [PubMed] [Google Scholar]
- 37.Tooker P, Yen WC, Ng SC, Negro-Vilar A, Hermann TW. Bexarotene (LGD1069, Targretin), a selective retinoid X receptor agonist, prevents and reverses gemcitabine resistance in NSCLC cells by modulating gene amplification. Cancer Res. 2007;67:4425–4433. doi: 10.1158/0008-5472.CAN-06-4495. [DOI] [PubMed] [Google Scholar]
- 38.Yamada KM, Cukierman E. Modeling tissue morphogenesis and cancer in 3D. Cell. 2007;130:601–610. doi: 10.1016/j.cell.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 39.Hamilton TC, Young RC, Louie KG, Behrens BC, McKoy WM, Grotzinger KR, Ozols RF. Characterization of a xenograft model of human ovarian carcinoma which produces ascites and intraabdominal carcinomatosis in mice. Cancer Res. 1984;44:5286–5290. [PubMed] [Google Scholar]
- 40.Tentler JJ, Tan AC, Weekes CD, Jimeno A, Leong S, Pitts TM, Arcaroli JJ, Messersmith WA, Eckhardt SG. Patient-derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol. 2012;9:338–350. doi: 10.1038/nrclinonc.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kerbel RS. Human tumor xenografts as predictive preclinical models for anticancer drug activity in humans: better than commonly perceived-but they can be improved. Cancer Biol Ther. 2003;2:S134–139. [PubMed] [Google Scholar]
- 42.Caffrey PB, Frenkel GD. Treatment of human ovarian tumor xenografts with selenite prevents the melphalan-induced development of drug resistance. Anticancer Res. 1998;18:3017–3020. [PubMed] [Google Scholar]
- 43.Caffrey PB, Frenkel GD. Selenium compounds prevent the induction of drug resistance by cisplatin in human ovarian tumor xenografts in vivo. Cancer Chemother Pharmacol. 2000;46:74–78. doi: 10.1007/s002800000127. [DOI] [PubMed] [Google Scholar]
- 44.Caffrey PB, Frenkel GD. Selenite enhances and prolongs the efficacy of cisplatin treatment of human ovarian tumor xenografts. In Vivo. 2012;26:549–552. [PubMed] [Google Scholar]
- 45.Caffrey PB, Frenkel GD. Prevention of carboplatin-induced resistance in human ovarian tumor xenografts by selenite. Anticancer Res. 2013;33:4249–4254. [PubMed] [Google Scholar]
- 46.Glavinas H, Krajcsi P, Cserepes J, Sarkadi B. The role of ABC transporters in drug resistance, metabolism and toxicity. Curr Drug Deliv. 2004;1:27–42. doi: 10.2174/1567201043480036. [DOI] [PubMed] [Google Scholar]
- 47.Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- 48.Beketic-Oreskovic L, Duran GE, Chen G, Dumontet C, Sikic BI. Decreased mutation rate for cellular resistance to doxorubicin and suppression of mdr1 gene activation by the cyclosporin PSC 833. J Natl Cancer Inst. 1995;87:1593–1602. doi: 10.1093/jnci/87.21.1593. [DOI] [PubMed] [Google Scholar]
- 49.Tamaki A, Ierano C, Szakacs G, Robey RW, Bates SE. The controversial role of ABC transporters in clinical oncology. Essays Biochem. 2011;50:209–232. doi: 10.1042/bse0500209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Duan Z, Choy E, Hornicek FJ. NSC23925, identified in a high-throughput cell-based screen, reverses multidrug resistance. PLoS One. 2009;4:e7415. doi: 10.1371/journal.pone.0007415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dai CL, Xiong HY, Tang LF, Zhang X, Liang YJ, Zeng MS, Chen LM, Wang XH, Fu LW. Tetrandrine achieved plasma concentrations capable of reversing MDR in vitro and had no apparent effect on doxorubicin pharmacokinetics in mice. Cancer Chemother Pharmacol. 2007;60:741–750. doi: 10.1007/s00280-007-0420-0. [DOI] [PubMed] [Google Scholar]
- 52.Susa M, Choy E, Yang C, Schwab J, Mankin H, Hornicek F, Duan Z. Multidrug resistance reversal agent, NSC77037, identified with a cell-based screening assay. J Biomol Screen. 2010;15:287–296. doi: 10.1177/1087057109359422. [DOI] [PubMed] [Google Scholar]
- 53.Shen H, Xu W, Chen Q, Wu Z, Tang H, Wang F. Tetrandrine prevents acquired drug resistance of K562 cells through inhibition of mdr1 gene transcription. J Cancer Res Clin Oncol. 2010;136:659–665. doi: 10.1007/s00432-009-0704-3. [DOI] [PubMed] [Google Scholar]
- 54.Amiri KI, Richmond A. Role of nuclear factor-kappa B in melanoma. Cancer Metastasis Rev. 2005;24:301–313. doi: 10.1007/s10555-005-1579-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Seebacher NA, Richardson DR, Jansson PJ. Glucose modulation induces reactive oxygen species and increases P-glycoprotein-mediated multidrug resistance to chemotherapeutics. Br J Pharmacol. 2015;172:2557–2572. doi: 10.1111/bph.13079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oldham RK, Reid WK, Preisler HD, Barnett D. A phase I and pharmacokinetic study of CBT-1 as a multidrug resistance modulator in the treatment of patients with advanced cancer. Cancer Biother Radiopharm. 1998;13:71–80. doi: 10.1089/cbr.1998.13.71. [DOI] [PubMed] [Google Scholar]
- 57.Kelly RJ, Robey RW, Chen CC, Draper D, Luchenko V, Barnett D, Oldham RK, Caluag Z, Frye AR, Steinberg SM, Fojo T, Bates SE. A pharmacodynamic study of the P-glycoprotein antagonist CBT-1(R) in combination with paclitaxel in solid tumors. Oncologist. 2012;17:512. doi: 10.1634/theoncologist.2012-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Creighton AM, Hellmann K, Whitecross S. Antitumour activity in a series of bisdiketopiperazines. Nature. 1969;222:384–385. doi: 10.1038/222384a0. [DOI] [PubMed] [Google Scholar]
- 59.Lipshultz SE, Rifai N, Dalton VM, Levy DE, Silverman LB, Lipsitz SR, Colan SD, Asselin BL, Barr RD, Clavell LA, Hurwitz CA, Moghrabi A, Samson Y, et al. The effect of dexrazoxane on myocardial injury in doxorubicin-treated children with acute lymphoblastic leukemia. N Engl J Med. 2004;351:145–153. doi: 10.1056/NEJMoa035153. [DOI] [PubMed] [Google Scholar]
- 60.Bjelogrlic SK, Radic J, Radulovic S, Jokanovic M, Jovic V. Effects of dexrazoxane and amifostine on evolution of Doxorubicin cardiomyopathy in vivo. Exp Biol Med (Maywood) 2007;232:1414–1424. doi: 10.3181/0705-RM-138. [DOI] [PubMed] [Google Scholar]
- 61.Swain SM, Whaley FS, Gerber MC, Ewer MS, Bianchine JR, Gams RA. Delayed administration of dexrazoxane provides cardioprotection for patients with advanced breast cancer treated with doxorubicin-containing therapy. J Clin Oncol. 1997;15:1333–1340. doi: 10.1200/JCO.1997.15.4.1333. [DOI] [PubMed] [Google Scholar]
- 62.Gottardis MM, Bischoff ED, Shirley MA, Wagoner MA, Lamph WW, Heyman RA. Chemoprevention of mammary carcinoma by LGD1069 (Targretin): an RXR-selective ligand. Cancer Res. 1996;56:5566–5570. [PubMed] [Google Scholar]
- 63.Wu K, Zhang Y, Xu XC, Hill J, Celestino J, Kim HT, Mohsin SK, Hilsenbeck SG, Lamph WW, Bissonette R, Brown PH. The retinoid X receptor-selective retinoid, LGD1069, prevents the development of estrogen receptor-negative mammary tumors in transgenic mice. Cancer Res. 2002;62:6376–6380. [PubMed] [Google Scholar]
- 64.Duvic M, Hymes K, Heald P, Breneman D, Martin AG, Myskowski P, Crowley C, Yocum RC, Bexarotene Worldwide Study G. Bexarotene is effective and safe for treatment of refractory advanced-stage cutaneous T-cell lymphoma: multinational phase II–III trial results. J Clin Oncol. 2001;19:2456–2471. doi: 10.1200/JCO.2001.19.9.2456. [DOI] [PubMed] [Google Scholar]
- 65.Khuri FR, Rigas JR, Figlin RA, Gralla RJ, Shin DM, Munden R, Fox N, Huyghe MR, Kean Y, Reich SD, Hong WK. Multi-institutional phase I/II trial of oral bexarotene in combination with cisplatin and vinorelbine in previously untreated patients with advanced non-small-cell lung cancer. J Clin Oncol. 2001;19:2626–2637. doi: 10.1200/JCO.2001.19.10.2626. [DOI] [PubMed] [Google Scholar]
- 66.Musdal Y, Hegazy UM, Aksoy Y, Mannervik B. FDA-approved drugs and other compounds tested as inhibitors of human glutathione transferase P1–1. Chem Biol Interact. 2013;205:53–62. doi: 10.1016/j.cbi.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 67.Bernig T, Ritz S, Brodt G, Volkmer I, Staege MS. Glutathione-S-transferases and Chemotherapy Resistance of Hodgkin’s Lymphoma Cell Lines. Anticancer Res. 2016;36:3905–3915. [PubMed] [Google Scholar]
- 68.Hansson J, Berhane K, Castro VM, Jungnelius U, Mannervik B, Ringborg U. Sensitization of human melanoma cells to the cytotoxic effect of melphalan by the glutathione transferase inhibitor ethacrynic acid. Cancer Res. 1991;51:94–98. [PubMed] [Google Scholar]
- 69.Saltz L. Planned treatment interruptions and chemotherapy-free intervals in the treatment of metastatic colorectal cancer: time to start stopping? Semin Oncol. 2006;33:S28–32. doi: 10.1053/j.seminoncol.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 70.Jung HJ, Seo YR. Current issues of selenium in cancer chemoprevention. BioFactors. 2010;36:153–158. doi: 10.1002/biof.81. [DOI] [PubMed] [Google Scholar]
- 71.Qi Y, Fu X, Xiong Z, Zhang H, Hill SM, Rowan BG, Dong Y. Methylseleninic acid enhances paclitaxel efficacy for the treatment of triple-negative breast cancer. PloS one. 2012;7:e31539. doi: 10.1371/journal.pone.0031539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hu YJ, Chen Y, Zhang YQ, Zhou MZ, Song XM, Zhang BZ, Luo L, Xu PM, Zhao YN, Zhao YB, Cheng G. The protective role of selenium on the toxicity of cisplatin-contained chemotherapy regimen in cancer patients. Biol Trace Elem Res. 1997;56:331–341. doi: 10.1007/BF02785304. [DOI] [PubMed] [Google Scholar]
- 73.Meijer C, Mulder NH, Timmer-Bosscha H, Sluiter WJ, Meersma GJ, de Vries EG. Relationship of cellular glutathione to the cytotoxicity and resistance of seven platinum compounds. Cancer Res. 1992;52:6885–6889. [PubMed] [Google Scholar]
- 74.Johnson SW, Laub PB, Beesley JS, Ozols RF, Hamilton TC. Increased platinum-DNA damage tolerance is associated with cisplatin resistance and cross-resistance to various chemotherapeutic agents in unrelated human ovarian cancer cell lines. Cancer Res. 1997;57:850–856. [PubMed] [Google Scholar]
- 75.Meijer C, Mulder NH, Hospers GA, Uges DR, de Vries EG. The role of glutathione in resistance to cisplatin in a human small cell lung cancer cell line. Br J Cancer. 1990;62:72–77. doi: 10.1038/bjc.1990.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.De Clercq E. (E)-5-2-bromovinyl-2′-deoxyuridine BVDU. Med Res Rev. 2005;25:1–20. doi: 10.1002/med.20011. [DOI] [PubMed] [Google Scholar]
- 77.Fahrig R, Steinkamp-Zucht A. Induction or suppression of SV40 amplification by genotoxic carcinogens, non-genotoxic carcinogens or tumor promoters. Mutat Res. 1996;356:217–224. doi: 10.1016/0027-5107(96)00061-9. [DOI] [PubMed] [Google Scholar]
- 78.Wahl GM. The importance of circular DNA in mammalian gene amplification. Cancer Res. 1989;49:1333–1340. [PubMed] [Google Scholar]
- 79.Schimke RT, Sherwood SW, Hill AB, Johnston RN. Overreplication and recombination of DNA in higher eukaryotes: potential consequences and biological implications. Proc Natl Acad Sci USA. 1986;83:2157–2161. doi: 10.1073/pnas.83.7.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fahrig R, Heinrich JC, Nickel B, Wilfert F, Leisser C, Krupitza G, Praha C, Sonntag D, Fiedler B, Scherthan H, Ernst H. Inhibition of induced chemoresistance by cotreatment with (E)-5-(2-bromovinyl)-2′-deoxyuridine (RP101) Cancer Res. 2003;63:5745–5753. [PubMed] [Google Scholar]
- 81.Oh ET, Park HJ. Implications of NQO1 in cancer therapy. BMB Rep. 2015;48:609–617. doi: 10.5483/BMBRep.2015.48.11.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JE., Jr Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 83.Weitzman JB, Fiette L, Matsuo K, Yaniv M. JunD protects cells from p53-dependent senescence and apoptosis. Mol Cell. 2000;6:1109–1119. doi: 10.1016/s1097-2765(00)00109-x. [DOI] [PubMed] [Google Scholar]
- 84.Masferrer JL, Zweifel BS, Manning PT, Hauser SD, Leahy KM, Smith WG, Isakson PC, Seibert K. Selective inhibition of inducible cyclooxygenase 2 in vivo is antiinflammatory and nonulcerogenic. Proc Natl Acad Sci USA. 1994;91:3228–3232. doi: 10.1073/pnas.91.8.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Patel VA, Dunn MJ, Sorokin A. Regulation of MDR-1 (P-glycoprotein) by cyclooxygenase-2. J Biol Chem. 2002;277:38915–38920. doi: 10.1074/jbc.M206855200. [DOI] [PubMed] [Google Scholar]
- 86.Gasparini G, Longo R, Sarmiento R, Morabito A. Inhibitors of cyclo-oxygenase 2: a new class of anticancer agents? Lancet Oncol. 2003;4:605–615. doi: 10.1016/s1470-2045(03)01220-8. [DOI] [PubMed] [Google Scholar]
- 87.Chen SF, Zhang ZY, Zhang JL. Meloxicam increases intracellular accumulation of doxorubicin via downregulation of multidrug resistance-associated protein 1 (MRP1) in A549 cells. Genet Mol Res. 2015;14:14548–14560. doi: 10.4238/2015.November.18.18. [DOI] [PubMed] [Google Scholar]
- 88.Zatelli MC, Luchin A, Piccin D, Tagliati F, Bottoni A, Vignali C, Bondanelli M, degli Uberti EC. Cyclooxygenase-2 inhibitors reverse chemoresistance phenotype in medullary thyroid carcinoma by a permeability glycoprotein-mediated mechanism. J Clin Endocrinol Metab. 2005;90:5754–5760. doi: 10.1210/jc.2005-1362. [DOI] [PubMed] [Google Scholar]
- 89.Jeenger MK, Shrivastava S, Yerra VG, Naidu VG, Ramakrishna S, Kumar A. Curcumin: a pleiotropic phytonutrient in diabetic complications. Nutrition. 2015;31:276–282. doi: 10.1016/j.nut.2014.06.015. [DOI] [PubMed] [Google Scholar]
- 90.Morzycki JW, Rarova L, Gruz J, Sawczuk T, Kielczewska U, Siergiejczyk L, Wojtkielewicz A. Synthesis of Aromatic Retinoids and Curcuminoids and Evaluation of their Antiproliferative, Antiradical, and Anti-inflammatory Activities. Chemistry Open. 2016;5:339–350. doi: 10.1002/open.201600027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Aggarwal BB, Sung B. Pharmacological basis for the role of curcumin in chronic diseases: an age-old spice with modern targets. Trends Pharmacol Sci. 2009;30:85–94. doi: 10.1016/j.tips.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 92.Shi M, Cai Q, Yao L, Mao Y, Ming Y, Ouyang G. Antiproliferation and apoptosis induced by curcumin in human ovarian cancer cells. Cell Biol Int. 2006;30:221–226. doi: 10.1016/j.cellbi.2005.10.024. [DOI] [PubMed] [Google Scholar]
- 93.Kunnumakkara AB, Anand P, Aggarwal BB. Curcumin inhibits proliferation, invasion, angiogenesis and metastasis of different cancers through interaction with multiple cell signaling proteins. Cancer Lett. 2008;269:199–225. doi: 10.1016/j.canlet.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 94.Epstein J, Sanderson IR, Macdonald TT. Curcumin as a therapeutic agent: the evidence from in vitro, animal and human studies. Br J Nutr. 2010;103:1545–1557. doi: 10.1017/S0007114509993667. [DOI] [PubMed] [Google Scholar]
- 95.Xu D, Tian W, Shen H. Curcumin prevents induced drug resistance: a novel function? Chin J Cancer Res. 2011;23:218–223. doi: 10.1007/s11670-011-0218-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chen J, Li L, Su J, Chen T. Natural borneol enhances bisdemethoxycurcumin-induced cell cycle arrest in the G2/M phase through up-regulation of intracellular ROS in HepG2 cells. Food Funct. 2015;6:740–748. doi: 10.1039/c4fo00807c. [DOI] [PubMed] [Google Scholar]
- 97.Xu D, Tian W, Shen H. P-gp upregulation may be blocked by natural curcuminoids, a novel class of chemoresistance-preventing agent. Mol Med Rep. 2013;7:115–121. doi: 10.3892/mmr.2012.1106. [DOI] [PubMed] [Google Scholar]
- 98.Petersen LK, Narasimhan B. Combinatorial design of biomaterials for drug delivery: opportunities and challenges. Expert Opin Drug Deliv. 2008;5:837–846. doi: 10.1517/17425247.5.8.837. [DOI] [PubMed] [Google Scholar]
- 99.Iyer AK, Singh A, Ganta S, Amiji MM. Role of integrated cancer nanomedicine in overcoming drug resistance. Adv Drug Deliv Rev. 2013;65:1784–1802. doi: 10.1016/j.addr.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 100.Duncan R. The dawning era of polymer therapeutics. Nat Rev Drug Discov. 2003;2:347–360. doi: 10.1038/nrd1088. [DOI] [PubMed] [Google Scholar]
- 101.Adams ML, Lavasanifar A, Kwon GS. Amphiphilic block copolymers for drug delivery. J Pharm Sci. 2003;92:1343–1355. doi: 10.1002/jps.10397. [DOI] [PubMed] [Google Scholar]
- 102.Kabanov AV, Alakhov VY. Pluronic block copolymers in drug delivery: from micellar nanocontainers to biological response modifiers. Crit Rev Ther Drug Carrier Syst. 2002;19:1–72. doi: 10.1615/critrevtherdrugcarriersyst.v19.i1.10. [DOI] [PubMed] [Google Scholar]
- 103.Batrakova EV, Li S, Elmquist WF, Miller DW, Alakhov VY, Kabanov AV. Mechanism of sensitization of MDR cancer cells by Pluronic block copolymers: Selective energy depletion. Br J Cancer. 2001;85:1987–1997. doi: 10.1054/bjoc.2001.2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kabanov AV, Batrakova EV, Alakhov VY. Pluronic block copolymers for overcoming drug resistance in cancer. Adv Drug Deliv Rev. 2002;54:759–779. doi: 10.1016/s0169-409x(02)00047-9. [DOI] [PubMed] [Google Scholar]