

Summary
STIM1 in the endoplasmic reticulum and CRACM1 in the plasma membrane are essential molecular components for controlling the store-operated CRAC current [1–4]. CRACM1 proteins multimerize and bind STIM1 [5, 6], and the combined overexpression of STIM1 and CRACM1 reconstitutes amplified CRAC currents [7–10]. Mutations in CRACM1 determine the selectivity of CRAC currents, demonstrating that CRACM1 forms the CRAC channel’s ion-selective pore [11, 5, 6], but the CRACM1 homologs CRACM2 and CRACM3 are less well characterized [7, 12]. Here, we show that both CRACM2 and CRACM3, when overexpressed in HEK293 cells stably expressing STIM1, potentiate ICRAC to current amplitudes 15–20 times larger than native ICRAC. A nonconducting mutation of CRACM1 (E106Q) acts as a dominant negative for all three CRACM homologs, suggesting that they can form heteromultimeric channel complexes. All three CRACM homologs exhibit distinct properties in terms of selectivity for Ca2+ and Na+, differential pharmacological effects in response to 2-APB, and strikingly different feedback regulation by intracellular Ca2+. Each of the CRAC channel proteins’ specific functional features and the potential heteromerization provide for flexibility in shaping Ca2+ signals, and their characteristic biophysical and pharmacological properties will aid in identifying CRAC-channel species in native cells that express them.
Results and Discussion
In many cell types, store-operated Ca2+ entry represents the primary mechanism underlying long-lasting elevations in intracellular Ca2+, elevations that follow IP3-mediated release of Ca2+ from intracellular stores [13–16]. Previous investigations have identified CRACM1 (or Orai1) as the calcium-release-activated calcium (CRAC) channels in the plasma membrane [3, 4]. There are three mammalian homologous CRAC channel proteins, CRACM1, CRACM2, and CRACM3, and all homologs are widely expressed at the mRNA level [12].
CRACM Homologs Represent Store-Operated Channels
To assess the functional properties of CRACM proteins, we overexpressed all three CRACM species in HEK293 cells that stably overexpress STIM1 and measured CRAC currents in response to store depletion. Upon store depletion with IP3, all CRACM homologs produced large membrane currents (Figure 1A) with inwardly rectifying current-voltage (I/V) relationships characteristic of ICRAC (Figure 1B). Although our data substantiate that CRACM2 represents a store-operated channel, they are at variance with the reported inability of CRACM3 to increase store-operated Ca2+ entry or CRAC currents [7]. We should note, however, that our own attempts with the commercial CRACM3 vector used by Mercer et al. also failed to produce enhanced CRAC currents when expressed in STIM1-expressing cells (data not shown). However, after subcloning the CRACM3 sequence into another vector (see the Experimental Procedures in the Supplemental Data online), we consistently observed significant CRACM3 currents.
Figure 1.
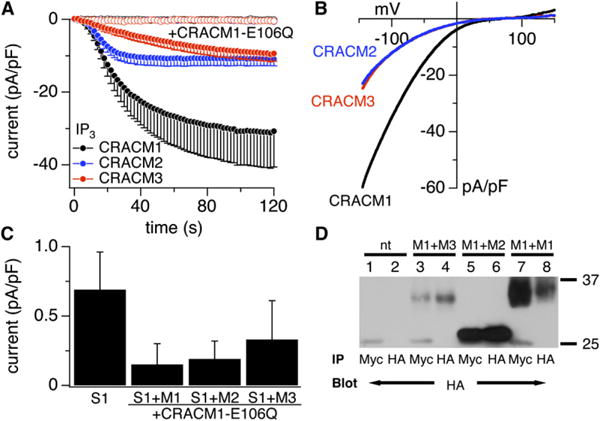
All CRACM Homologs Produce Store-Operated Currents
(A) Average CRAC current densities at −80 mV induced by IP3 (20 μM) in stable STIM1-expressing HEK293 cells transiently overexpressing CRACM1 (black, n = 12), CRACM2 (blue, n = 7), and CRACM3 (red, n = 9). Open symbols represent cells that were cotransfected with the WT constructs of the three homologs plus the dominant negative E106Q mutant of CRACM1 (CRACM1-E106Q + CRACM1, n = 6; + CRACM2, n = 6; + CRACM3, n = 7). [Ca2+]i was clamped to near zero with 20 mM BAPTA. Error bars indicate SEM.
(B) Average current-voltage (I/V) relationships of CRAC currents extracted from representative HEK293 cells shown in (A) obtained at 120 s. Data represent leak-subtracted current densities (pA/pF) evoked by 50 ms voltage ramps from −150 to +150 mV corresponding to CRACM1 (black, n = 11), CRACM2 (blue, n = 6), and CRACM3 (red, n = 9).
(C) Average CRAC current densities at −80 mV in cells expressing STIM1 alone (n = 13) or additionally with CRACM1-E106Q + CRACM1/CRACM2/CRACM 3; data points correspond to currents analyzed from (A) at 120 s. Error bars indicate SEM.
(D) Coimmunoprecipitation of CRACM1 with CRACM2 and CRACM3. Wild-type HEK293 cells were cotransfected with CRACM1-Myc in combination with HA-CRACM1, HA-CRACM2, or HA-CRACM3. Lanes 1 and 2 show nontransfected HEK293 cells. Lanes 3 and 4 show that CRACM1-Myc can co-IP HA-CRACM3, HA-CRACM2 (lanes 5 and 6), and HA-CRACM1 (lanes 7 and 8). The resulting immune complexes were immunoblotted with HA antibody, thus revealing bands with molecular weights of ~33, ~28, and ~31 kDa for CRACM1, CRACM2, and CRACM3, respectively.
The average current amplitudes of CRACM2 and CRACM3 at −80 mV were approximately 3-fold smaller than the corresponding amplitude of CRACM1, but they are still 15- to 20-fold larger than native CRAC currents in wild-type or STIM1-expressing HEK293 cells. The differences in amplitudes may be due to different expression levels but could also reflect differences in single-channel conductance or open probability. The activation kinetics of the CRACM homologs were distinctly different, with half-maximal activation times (±SEM) of CRACM1 at 35 ± 7 s (n = 12), CRACM2 at 21 ± 3 s (n = 8), and CRACM3 at 63 ± 7 s (n = 9). These were unrelated to current magnitude because they were preserved when analyzing currents with matched amplitudes (Figure 2) or at various [Ca2+]i (see Figure 3). We also examined whether CRACM currents were activated when preventing store refilling with 20 mM BAPTA in the pipette. Indeed, all three CRACM species produced CRAC-like currents with a characteristic delay that presumably reflects the time needed to deplete stores through leak pathways (Figure S1A). Under these conditions, CRAC currents developed with a similar time course, thus indicating that store depletion is likely to be the rate-limiting step for CRAC activation. The I/V relationships (Figure S1B) confirm that these currents also have the typical shape of CRAC currents. These results demonstrate that all three CRACM homologs can generate amplified store-operated CRAC currents and that they possess characteristic kinetics of activation, thus possibly indicating differences in the binding or interaction with STIM1.
Figure 2.
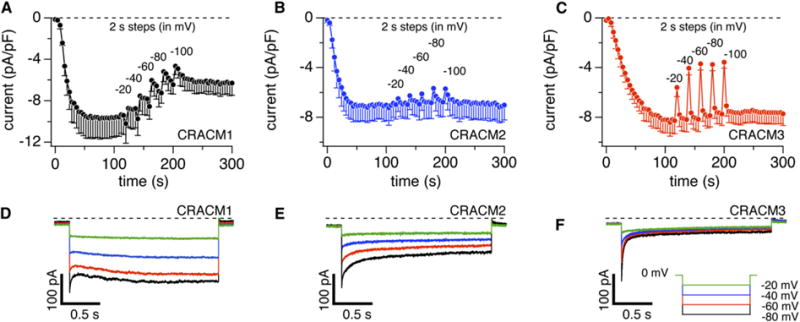
CRACM Homologs Have Distinct Fast and Slow Ca2+-Dependent-Inactivation Properties
(A) Average CRAC current densities at −80 mV induced by IP3 (20 μM) with 10 mM EGTA in stable STIM1-expressing HEK293 cells transiently overexpressing CRACM1 (n = 3; total n = 8 and three cells with the smallest current densities were averaged to approximate the lower current densities of CRACM2 and CRACM3). CRAC currents were monitored continuously by voltage ramps spanning −100 mV to +100 mV over 50 ms delivered at a rate of 0.5 Hz. After CRAC currents were fully activated (120 s), rectangular voltage pulses of 2 s duration were delivered to various negative voltages (see [D]–[F]) interspaced by 10 ramps. Error bars indicate SEM.
(B) Experimental protocol as described in (A), but for CRACM2-expressing cells. Note the minor fast inactivation and virtual absence of slow inactivation. Error bars indicate SEM.
(C) Experimental protocol as described in (A), but for CRACM3-expressing cells. Note the significant fast inactivation and virtual absence of slow inactivation. Error bars indicate SEM.
(D) Average CRAC currents evoked by step pulses (2 s duration) to −20 mV (green), −40 mV (blue), −60 mV (red), and −80 mV (black) in cells expressing CRACM1 (n = 3, same cells as in [A]). At the beginning of each pulse, 2.5 ms were blanked out so that residual capacitative artifacts could be eliminated.
(E) Average CRAC currents evoked by step pulses from −20 mV to −80 mV in cells expressing CRACM2 (n = 4, same cells as in [B]).
(F) Average CRAC currents evoked by step pulses from −20 mV to −80 mV in cells expressing CRACM3 (n = 5, same cells as in [C]).
Figure 3.
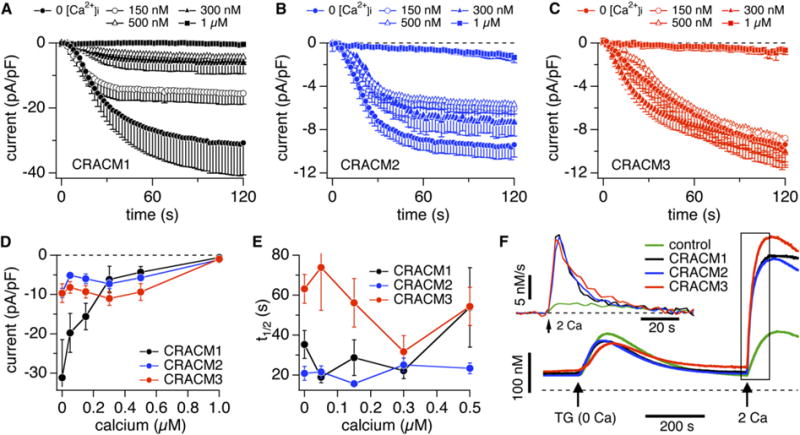
CRACM1, but Not CRACM2 or CRACM3, Is Inhibited by Increased [Ca2+]i
(A) Average CRAC-current densities at −80 mV induced by IP3 (20 μM) in stable STIM1-expressing HEK293 cells transiently overexpressing CRACM1 and perfused with increasing [Ca2+]i (n = 5–12). Error bars indicate SEM.
(B) Experimental protocol as described in (A), but for CRACM2-expressing cells (n = 5–8). Error bars indicate SEM.
(C) Experimental protocol as described in (A), but for CRACM3-expressing cells (n = 9–15). Error bars indicate SEM.
(D) Average current densities of CRACM1 (black), CRACM2 (blue), and CRACM3 (red) at −80 mV extracted at 120 s (150 s for CRACM3) from the cells shown in (A)–(C) and plotted versus [Ca2+]i. Error bars indicate SEM.
(E) Half-maximal activation time of CRACM1 (black, n = 5–12), CRACM2 (blue, n = 5–8), and CRACM3 (red, n = 9–15) plotted versus [Ca2+]i. Data were derived from the cells shown in (A)–(C). All cells had similar series resistances in the range of 4–6 MU. Error bars indicate SEM.
(F) Average changes in [Ca2+]i induced by store depletion in stable STIM1-expressing HEK293 cells transfected with empty vector (green, n = 14), or transiently overexpressing CRACM1 (black, n = 23), CRACM2 (blue, n = 39), or CRACM3 (red, n = 27). The arrows indicate application of thapsigargin (2 μM) in Ca2+-free solution to induce store depletion and readmission of 2 mM Ca2+ to probe Ca2+ entry. The inset represents rates of [Ca2+]i obtained by differentiating the trace segment enclosed by the rectangle.
CRACM Homologs Form Heteromeric Channels
Given that all three homologs produced store-operated channels and CRACM1 has been shown to form multimeric channel complexes, we used a nonconducting CRACM1 (E106Q) pore mutation that confers a dominant-negative phenotype on native CRAC channels [5] to assess whether CRACM1 can assemble into heteromeric channel complexes with CRACM2 and CRACM3. Figures 1A and 1C illustrate that the co-overexpression of CRACM1-E106Q in equal amounts with the three wild-type homologs essentially abolished CRAC currents, suggesting that the CRACM1 pore mutant indeed confers a dominant-negative effect. Coimmunoprecipitation experiments confirmed that CRACM1 can form stable heteromeric complexes with both of its homologs (Figure 1D).
CRACM Homologs Differ in Ca2+-Dependent Inactivation
Native CRAC currents are regulated by [Ca2+]i and subject to both fast and slow Ca2+-dependent inactivation [14, 17–20]. Fast inactivation, occurring in the millisecond range, is believed to result from Ca2+ binding to the channel itself [17, 18, 20], whereas slow inactivation over tens of seconds may result from store refilling or regulatory mechanisms through cellular-feedback mechanisms on the channel [21, 22, 19]. Figure 2 illustrates IP3-induced CRAC currents with intracellular solutions that contained 10 mM EGTA, which is slower in chelating Ca2+ than BAPTA and therefore less efficient in suppressing fast Ca2+-dependent inactivation [17, 20]. If sufficient Ca2+ accumulates intracellularly, it may overpower the buffering capacity and then reveal slow Ca2+-dependent processes as well. CRAC currents were monitored continuously by voltage ramps spanning −100 mV to +100 mV over 50 ms delivered at a rate of 0.5 Hz. After CRAC currents were fully activated, we delivered rectangular voltage pulses of 2 s duration and increasing hyperpolarizations so as to increase Ca2+ entry. Figures 2A–2C illustrate that each hyperpolarizing pulse caused a fast drop in CRACM1-current amplitude that slowly, but not completely, recovered before the next pulse was delivered. The fast drop in current is due to fast inactivation, and the recovery is likely to be the net result of two opposing effects, recovery of channels from fast inactivation and slow inactivation proceeding over tens of seconds (see also Figure 3). In the case of CRACM1, the slow inactivation resulting from the five hyperpolarizing pulses resulted in ~50% reduction in CRAC current over a period of ~100 s. The same experimental protocol performed in cells expressing CRACM2 or CRACM3 revealed only fast inactivation of currents with no significant slow inactivation (Figures 2B and 2C). CRACM2 appeared fairly resistant to Ca2+-induced inactivation in general, with only a small component of fast inactivation, whereas CRACM3 displayed a much greater degree of fast inactivation. In both cases, recovery from fast inactivation was essentially complete within 20 s.
Figures 2D–2F illustrate averages of high-resolution CRAC currents produced by the hyperpolarizing pulses in (A)–(C), revealing the degree of fast Ca2+-dependent inactivation of the three homologs. CRACM3 currents exhibit a striking Ca2+-dependent inactivation that at −80 mV is characterized by a predominant exponential decay by ~80% with a time constant of τ = 17 ms and a very small slow component of τ2 = 130 ms. We confirmed that this dramatic inactivation of CRACM3 is in fact entirely due to Ca2+ in experiments in which we delivered a hyperpolarizing voltage pulse to −80 mV in the presence of 10 mM Ca2+ and after switching to DVF solution. This revealed a rapidly inactivating current while Ca2+ was present, and a sustained, noninactivating current when divalent cations were absent (Figure S2). CRACM2 exhibits moderately quick Ca2+-dependent inactivation, decaying with two time constants of τ1 = 80 ms and τ2 = 900 ms that both contribute in roughly equal amounts to total fast inactivation of ~50%. CRACM1 exhibits complex behavior that may reflect three Ca2+-dependent feedback effects and therefore cannot be readily assessed quantitatively in terms of time constants. Presumably, this channel quickly inactivates in a similar manner as CRACM2 with two fast inactivation time courses [17, 18, 20]; however, it appears that the second phase of fast inactivation is partially masked by a slower wave of reactivation. This reactivation is most pronounced at the more negative voltage pulses and appears to be both Ca2+ and voltage dependent, as it was significantly attenuated, but not abolished, when exposing cells to DVF solutions (Figure S2B). In the absence of Ca2+, both CRACM1 and CRACM3 currents still increase slightly, probably because of voltage-dependent facilitation. The slow inactivation of CRACM1 currents is not obvious in the recordings shown in Figure 2D because it occurs over tens of seconds (see Figure 2A). However, slow inactivation is reflected by the lower initial current amplitudes induced by the most negative pulses.
To assess the slow Ca2+-dependent inactivation of CRAC currents quantitatively, we perfused cells with 20 mM BAPTA and appropriate amounts of CaCl2 so that free [Ca2+]i was clamped to defined levels between 0 and 1 μM. Figure 3A shows that [Ca2+]i dose-dependently inhibited CRACM1 currents but had little or no significant effect on CRACM2 or CRACM3 (Figures 3B and 3C). The absence of significant slow inactivation seen with CRACM2 or CRACM3 is likely to be of some importance in the physiological context because intermediate [Ca2+]i levels occurring physiologically (300– 500 nM) would tend to maintain activity of CRACM2 and CRACM3 channels, whereas CRACM1 currents would be significantly reduced. Only at 1 μM [Ca2+]i were the CRACM2 and CRACM3 currents suppressed almost as strongly as those carried by CRACM1. It remains to be determined whether the inhibitory effect seen at this high concentration reflects direct channel inhibition, is due to decreased coupling of STIM1 and CRACM proteins, or is caused by decreased IP3 efficacy and refilling of stores.
We also examined the effect of [Ca2+]i on the kinetics of CRAC-current activation by determining the time to half-maximal activation (t1/2). We found this parameter to be predominantly independent of [Ca2+]i for CRACM2 and CRACM1, which both had similarly fast activation kinetics (Figure 3E). At low [Ca2+]i levels, CRACM3 currents activated significantly slower than those of the other homologs, but they accelerated at intermediate [Ca2+]i of 150–300 nM (Figure 3E).
Slow Ca2+-dependent inactivation would be expected to at least partially affect the amount of Ca2+ entry observed in intact cells, where [Ca2+]i increases because of CRAC-channel activity. We assessed and compared this by monitoring fura-2 signals in cells overexpressing the various CRACM proteins, and we subjected them to a standard protocol where store-depletion was induced by thapsigargin in the absence of extracellular Ca2+; this was followed by readmission of 2 mM Ca2+ for probing store-operated Ca2+ entry (Figure 3F). In empty-vector-transfected cells, Ca2+ readmission caused a moderate increase in [Ca2+]i by store-operated entry through endogenous CRAC channels. Cells overexpressing CRACM homologs produced significantly greater [Ca2+]i changes that are even more impressive when analyzing the rate of Ca2+ entry by differentiation of the fura-2 signals (see inset in Figure 3F). Although CRACM1 is capable of generating 3-fold larger currents compared to CRACM2 or CRACM3 when [Ca2+]i is buffered to near zero (see Figure 1A), all three homologs achieve similar absolute levels in [Ca2+]i and initial rates of Ca2+ entry when assessed by fura-2 in intact cells. Although [Ca2+]i signals in intact cells are complex and subject to numerous feedback mechanisms, slow Ca2+-dependent inactivation may account at least partially for the relatively lesser increase in [Ca2+]i observed with CRACM1. Thus, the [Ca2+]i signals obtained in intact cells, where global [Ca2+]i increases into the range of 300–500 nM, are comparable to the amplitudes of CRAC currents observed when clamping global [Ca2+]i to defined levels of that range (see Figure 3D).
CRACM Homologs Differ in Selectivity
Previous work on CRACM1 has identified critical residues in three regions that affect selectivity of the channel. Glutamate residue 106 in transmembrane (TM) segment 1 [11, 5, 6] and glutamate residue 190 in TM 3 [11, 5] are thought to form a ring of negatively charged amino acids lining the pore of the channel. Both of these residues are conserved identically in all three CRACM homologs and are therefore unlikely to account for differential selectivity. However, we have previously identified a third region, located in the loop between TM 1 and TM 2, that affects selectivity of CRACM1 [5]. This region has three key aspartate residues (D110/D112/D114) that we have proposed to form a second ring of negative charges that coordinate a second Ca2+ ion to the CRACM1 pore, and those residues differ in the three homologs (CRACM2: E110/Q112/Q114; CRACM3: E110/D112/E114). We therefore analyzed and compared the selectivity profiles of all three proteins with respect to Ca2+, Ba2+, and Na+ permeation (Figure 4). In the presence of 10 mM extracellular Ca2+, all three homologs generated large inward currents at −80 mV (Figure 4A) and exhibited similar inwardly rectifying I/V relationships (Figures 4B and 4C). When removing extracellular Ca2+, inward currents were suppressed to the same degree in the three channel species (Figures 4A–4C), demonstrating that they share similarly high Ca2+ selectivity and discriminate against Na+ ions as long as Mg2+ ions (2 mM) are present.
Figure 4.
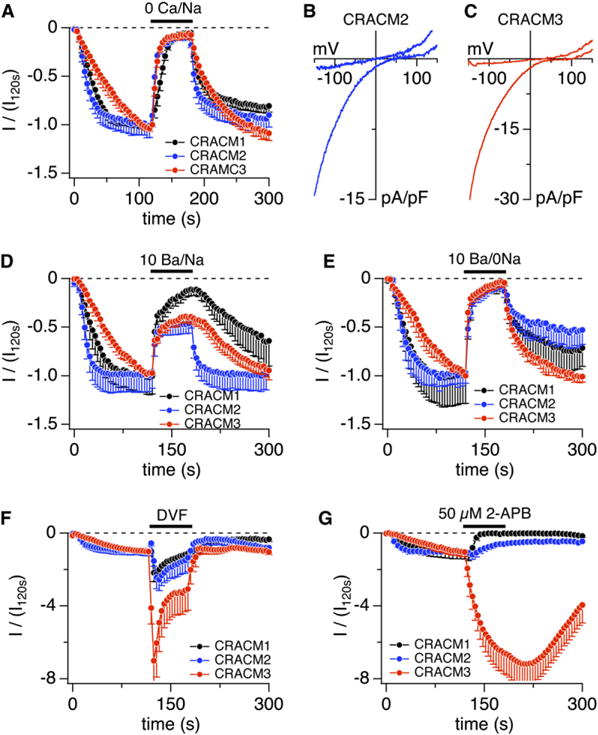
CRACM Homologs Have Distinct Ion Selectivity and Pharmacology
(A) Average normalized CRAC currents at −80 mV induced by IP3 (20 μM) in stable STIM1-expressing HEK293 cells transiently overexpressing CRACM1 (black, n = 12, data taken from [5]) CRACM2 (blue, n = 8), or CRACM3 (red, n = 10). Currents of individual cells were normalized to the current before solution change at 120 s (I/I120s). [Ca2+]i was clamped to near zero with 20 mM BAPTA. The bar indicates application of nominally Ca2+-free external solution. Error bars indicate SEM.
(B) Average I/V relationships of CRACM2 currents extracted from representative cells shown in (A) obtained at 120 s and 180 s (n = 7). Data represent leak-subtracted current densities (pA/pF) evoked by 50 ms voltage ramps from −150 to +150 mV.
(C) Average I/V relationships of CRACM3 currents extracted from representative cells shown in (A) at 120 s and 180 s into the experiment (n = 9).
(D) Average normalized CRAC currents (I/I120s) at −80 mV induced by IP3 (20 μM) in stable STIM1-expressing HEK293 cells transiently overexpressing CRACM1 (black, n = 5), CRACM2 (blue, n = 7), or CRACM3 (red, n = 10). The bar indicates application of an external solution containing 10 mM Ba2+ in the presence of Na+. Error bars indicate SEM.
(E) Average normalized CRAC currents (I/I120s) at −80 mV induced by IP3 (20 μM) in stable STIM1-expressing HEK293 cells transiently overexpressing CRACM1 (black, n = 9; data taken from [5]), CRACM2 (blue, n = 6), or CRACM3 (red, n = 6). The bar indicates application of an external solution containing 10 mM Ba2+ with external Na+ being replaced by TEA+. Error bars indicate SEM.
(F) Average normalized CRAC currents (I/I120s) at −80 mV induced by IP3 (20 μM) in stable STIM1-expressing HEK293 cells transiently overexpressing CRACM1 (black, n = 3, data taken from [5]), CRACM2 (blue, n = 5), or CRACM3 (red, n = 10). The bar indicates application of divalent-free external solution. Error bars indicate SEM.
(G) Average normalized CRAC currents (I/I120s) at −80 mV induced by IP3 (20 μM) in stable STIM1-expressing HEK293 cells transiently over-expressing CRACM1 (black, n = 8), CRACM2 (blue, n = 4), or CRACM3 (n = 9). The bar indicates application of external solution containing 50 μM 2-APB. Error bars indicate SEM.
We next tested whether the CRACM homologs might exhibit different selectivities for Ba2+ ions. Figure 4D illustrates that equimolar substitution greatly reduces inward currents in CRACM1, suggesting that this protein can discriminate Ca2+ ions against Ba2+. Remarkably, in cells overexpressing CRACM2 or CRACM3, there remains significantly more inward current when Ba2+ is used as charge carrier, and this finding at first glance would indicate higher Ba2+ permeation. However, because Na+ ions remain present in the extracellular solution, there is also the possibility that Na+ might contribute to inward current when Ba2+ is present. Indeed, when performing the same experiments as in Figure 4D, but additionally replacing Na+ with TEA, the inward currents through all three homologs were essentially abolished (Figure 4E); this indicated that Na+ ions or a mixture of Na+ and Ba2+ may be carrying the current seen in Figure 4D. Native CRAC currents in Jurkat T cells and RBL cells have been considered to carry Ba2+ ions [23, 18]; however, this was determined in solutions in which both Na+ and Ba2+ were present. We re-examined Ba2+ permeation in Jurkat T cells by replacing 10 mM Ca2+ equimolarly with Ba2+ in the presence and absence of Na+ and find that significant inward currents through native CRAC channels are only recorded when both ions are present and are absent when Ba2+ is used as the sole charge carrier (see Figure S3).
To further assess the selectivity of CRACM channels, we tested for possible differences in Na+ permeation in divalent-free solutions and 10 mM EDTA. Under these conditions, CRAC channels become permeable to Na+ [17], thus typically generating a 2-fold increase in inward current in HEK293 cells overexpressing CRACM1 (Figure 4F). The fact that the same experimental protocol produces slightly larger CRACM2 currents, whereas CRACM3 generates a significantly larger monovalent current again suggests that CRACM homologs exhibit slightly different selectivities for Na+ ions. Although mutational analysis is required to identify the contributions of the amino acid residues responsible for these differences, it seems likely that the 110/112/114 residues may be involved because those have been determined to contribute to monovalent permeation [5] and they are different in the three homologs.
CRACM Homologs Differ in Pharmacology
Finally, we tested for pharmacological differences between the CRACM homologs. 2-APB has been found to potentiate CRAC currents at low concentrations (%5 μM) and inhibit them at high concentrations (R10 μM) [24–26]. We previously demonstrated that CRACM1 is indeed completely inhibited by 50 μM 2-APB [8] (see Figure 4G). However, CRACM2 appears to be significantly less sensitive because the same concentration reduced the current only by approximately 50%. The most striking effect, however, was observed with CRACM3, which was not inhibited at all and instead greatly potentiated by 50 μM 2-APB. Although the mechanism of action of 2-APB remains unknown and it cannot be considered a specific pharmacological tool for CRAC channels, the compound clearly has differential effects on the three homologs. If these effects also apply to native CRACM homomeric channels, it may currently represent the best pharmacological tool to identify endogenous CRAC-channel species expressed in various cell types.
In summary, our data present a comprehensive characterization of the three CRACM channels and reveal distinct biophysical properties such as activation kinetics, selectivity, Ca2+-dependent inactivation, and pharmacology (Table 1). Finally, we demonstrate that the three homologs can form heteromeric channel complexes that may endow cells to express tailor-made CRAC channels for specific Ca2+ signaling needs. The specific properties of CRACM channels described here may serve as a reference for future studies aimed at classifying the CRAC-channel composition of native cell types as well as guidance for site-directed-mutagenesis studies designed to localize the sites responsible for the differences in functional and pharmacological properties of the CRACM channels.
Table 1.
Properties of the Mammalian CRACM Proteins
| CRACM1 | CRACM2 | CRACM3 | |
|---|---|---|---|
| Store-operated | Yes | Yes | Yes |
| Activation time (t1/2) | 35 ± 7 s | 21 ± 3 s | 63 ± 7 s |
| Ca2+-dependent inactivation (fast) | Moderate | Moderate | Strong |
| Ca2+-dependent inactivation (slow) | Strong | None | None |
| Ca2+-dependent reactivation | Yes | No | No |
| Selectivity | Ca2+ >> Na+, Ba2+ | Ca2+ >> Na+, Ba2+ | Ca2+ >> Na+, Ba2+ |
| Monovalent permeation in DVF solutions | Moderate | Moderate | Strong |
| 2-APB effect at 50 μM | Block | Reduction | Potentiation |
Supplementary Material
Acknowledgments
We thank M. Bellinger for help with cell culture. This work was supported in part by National Institutes of Health grants R01-AI050200 (R.P.). C.P. was supported by a fellowship from the Deutsche Forschungsgemeinschaft (PE-1478/1-1).
Footnotes
Supplemental Data
Experimental Procedures and three figures are available at http://www.current-biology.com/cgi/content/full/17/9/794/DC1/.
References
- 1.Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169:435–445. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, Stauderman KA, Cahalan MD. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437:902–905. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, Rao A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–185. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- 4.Vig M, Peinelt C, Beck A, Koomoa DL, Rabah D, Koblan-Huberson M, Kraft S, Turner H, Fleig A, Penner R, et al. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science. 2006;312:1220–1223. doi: 10.1126/science.1127883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vig M, Beck A, Billingsley JM, Lis A, Parvez S, Peinelt C, Koomoa DL, Soboloff J, Gill DL, Fleig A, et al. CRACM1 multimers form the ion-selective pore of the CRAC channel. Curr Biol. 2006;16:2073–2079. doi: 10.1016/j.cub.2006.08.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yeromin AV, Zhang SL, Jiang W, Yu Y, Safrina O, Cahalan MD. Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature. 2006;443:226–229. doi: 10.1038/nature05108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mercer JC, Dehaven WI, Smyth JT, Wedel B, Boyles RR, Bird GS, Putney JW., Jr Large store-operated calcium selective currents due to co-expression of Orai1 or Orai2 with the intracellular calcium sensor, Stim1. J Biol Chem. 2006;281:24979–24990. doi: 10.1074/jbc.M604589200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peinelt C, Vig M, Koomoa DL, Beck A, Nadler MJ, Koblan-Huberson M, Lis A, Fleig A, Penner R, Kinet JP. Amplification of CRAC current by STIM1 and CRACM1 (Orai1) Nat Cell Biol. 2006;8:771–773. doi: 10.1038/ncb1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soboloff J, Spassova MA, Tang XD, Hewavitharana T, Xu W, Gill DL. Orai1 and STIM reconstitute store-operated calcium channel function. J Biol Chem. 2006;281:20661–20665. doi: 10.1074/jbc.C600126200. [DOI] [PubMed] [Google Scholar]
- 10.Zhang SL, Yeromin AV, Zhang XH, Yu Y, Safrina O, Penna A, Roos J, Stauderman KA, Cahalan MD. Genome-wide RNAi screen of Ca2+ influx identifies genes that regulate Ca2+ release-activated Ca2+ channel activity. Proc Natl Acad Sci USA. 2006;103:9357–9362. doi: 10.1073/pnas.0603161103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A, Hogan PG. Orai1 is an essential pore subunit of the CRAC channel. Nature. 2006;443:230–233. doi: 10.1038/nature05122. [DOI] [PubMed] [Google Scholar]
- 12.Gwack Y, Srikanth S, Feske S, Cruz-Guilloty F, Oh-Hora M, Neems DS, Hogan PG, Rao A. Biochemical and functional characterization of Orai family proteins. J Biol Chem. 2007 doi: 10.1074/jbc.M609630200. in press. Published online February 9, 2007. [DOI] [PubMed] [Google Scholar]
- 13.Penner R, Matthews G, Neher E. Regulation of calcium influx by second messengers in rat mast cells. Nature. 1988;334:499–504. doi: 10.1038/334499a0. [DOI] [PubMed] [Google Scholar]
- 14.Hoth M, Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 1992;355:353–356. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- 15.Parekh AB, Penner R. Store depletion and calcium influx. Physiol Rev. 1997;77:901–930. doi: 10.1152/physrev.1997.77.4.901. [DOI] [PubMed] [Google Scholar]
- 16.Parekh AB, Putney JW., Jr Store-operated calcium channels. Physiol Rev. 2005;85:757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- 17.Hoth M, Penner R. Calcium release-activated calcium current in rat mast cells. J Physiol. 1993;465:359–386. doi: 10.1113/jphysiol.1993.sp019681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zweifach A, Lewis RS. Rapid inactivation of depletion-activated calcium current (ICRAC) due to local calcium feedback. J Gen Physiol. 1995;105:209–226. doi: 10.1085/jgp.105.2.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parekh AB. Slow feedback inhibition of calcium release-activated calcium current by calcium entry. J Biol Chem. 1998;273:14925–14932. doi: 10.1074/jbc.273.24.14925. [DOI] [PubMed] [Google Scholar]
- 20.Fierro L, Parekh AB. Fast calcium-dependent inactivation of calcium release-activated calcium current (CRAC) in RBL-1 cells. J Membr Biol. 1999;168:9–17. doi: 10.1007/s002329900493. [DOI] [PubMed] [Google Scholar]
- 21.Parekh AB, Penner R. Depletion-activated calcium current is inhibited by protein kinase in RBL-2H3 cells. Proc Natl Acad Sci USA. 1995;92:7907–7911. doi: 10.1073/pnas.92.17.7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zweifach A, Lewis RS. Slow calcium-dependent inactivation of depletion-activated calcium current. Store-dependent and -independent mechanisms. J Biol Chem. 1995;270:14445–14451. doi: 10.1074/jbc.270.24.14445. [DOI] [PubMed] [Google Scholar]
- 23.Hoth M. Calcium and barium permeation through calcium release-activated calcium (CRAC) channels. Pflugers Arch. 1995;430:315–322. doi: 10.1007/BF00373905. [DOI] [PubMed] [Google Scholar]
- 24.Braun FJ, Broad LM, Armstrong DL, Putney JW., Jr Stable activation of single Ca2+ release-activated Ca2+ channels in divalent cation-free solutions. J Biol Chem. 2001;276:1063–1070. doi: 10.1074/jbc.M008348200. [DOI] [PubMed] [Google Scholar]
- 25.Prakriya M, Lewis RS. Potentiation and inhibition of Ca2+ release-activated Ca2+ channels by 2-aminoethyldi-phenyl borate (2-APB) occurs independently of IP3 receptors. J Physiol. 2001;536:3–19. doi: 10.1111/j.1469-7793.2001.t01-1-00003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hermosura MC, Monteilh-Zoller MK, Scharenberg AM, Penner R, Fleig A. Dissociation of the store-operated calcium current ICRAC and the Mg-nucleotide-regulated metal ion current MagNuM. J Physiol. 2002;539:445–458. doi: 10.1113/jphysiol.2001.013361. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.