

Abstract
Mitochondrially derived peptides represent a new class of circulating signalling molecules. Humanin, the first member of this class, has been shown to have several metabolic effects such as reducing weight gain and visceral fat and increasing glucose‐stimulated insulin release. The discovery of several other new members, such as MOTS‐c and SHLP1–6, has further added to this group. These new peptides have also been found to affect metabolism with MOTS‐c potently decreasing weight gain in mice on a high‐fat diet. This review covers the basic biology of this class of peptides and discusses the relevance to organismal metabolism.
Keywords: insulin, metabolism, mitochondria, neuroendocrine, oxidative metabolism
Abbreviations
- AICAR
5‐aminoimidazole‐4‐carboxamide ribonucleotide
- AMPK
AMP‐activated protein kinase
- CNTFR
ciliary neurotrophic factor receptor
- ERK
extracellular signal‐regulated kinases
- HFD
high‐fat diet
- HNG
S14G humanin
- IGFBP3
insulin‐like growth factor binding protein 3
- JAK
Janus kinase
- MCP‐1
monocyte chemoattractant protein‐1
- MDP
mitochondrially derived peptide
- MOTS‐c
mitochondrial open reading frame of the 12S rRNA‐c
- RANKL
receptor activator of nuclear factor‐κB ligand
- SHLP
small humanin‐like peptide
- STAT
signal transducer and activator of transcription
The discovery of humanin nearly two decades ago has ushered in a new interest in mitochondrial biology that has combined several nascent fields of research (small open reading frame and alternative reading frame biology) with the more established field of mitochondrial biology (Yen et al. 2013; Xiao et al. 2016). More recently we have discovered that humanin is but the first of several mitochondrially derived peptides that are found within small, alternative reading frames of the mitochondrial genome (Fig. 1; Hashimoto et al. 2001b; Guo et al. 2003; Ikonen et al. 2003). Humanin has a number of different cytoprotective and metaboloprotective effects while the recently discovered small humanin‐like peptides (SHLP) 1–6 have similar but distinct properties compared with humanin (Cobb et al. 2016). The discovery of another mitochondrially derived peptide (MDP), namely mitochondrial open reading frame of the 12S rRNA‐c (MOTS‐c), as an exercise mimetic and activator of AMP‐activated protein kinase (AMPK) suggests that these peptides will have an important role in metabolism and could be used as future therapeutics (Lee et al. 2015).
Figure 1. The human mitochondrial genome.
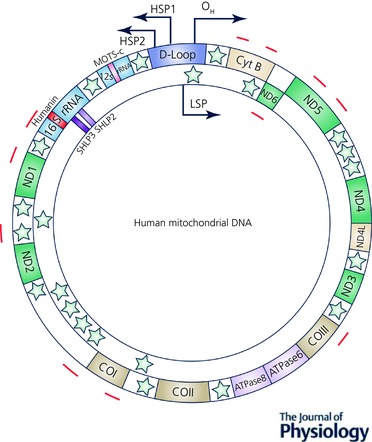
Relative locations of genes are represented as coloured blocks including humanin (red), MOTS‐c (yellow), SHLP2 and SHLP3 (pink and magenta, respectively). Twenty‐two tRNAs encoded from mitochondria are represented as green stars. Potential epigenetic modulation CpG sites in the mtDNA are marked in brown and the coordinates were obtained from Yu et al. 2017.
Humanin
As the first MDP to be discovered, humanin has been found to play a diverse role in a number of different processes. It was first discovered in a screen for proteins that could confer a protective effect from amyloid‐β, a possible cause of Alzheimer's disease. It was later independently found to have both an anti‐apoptotic effect and the ability to bind insulin‐like growth factor binding protein 3 (IGFBP3; Guo et al. 2003; Ikonen et al. 2003). As will be discussed in more detail below, humanin and its analogues have effects on metabolism, increasing glucose‐stimulated insulin release and decreasing body weight gain and visceral fat (Kuliawat et al. 2013; Gong et al. 2015).
In the past year, several new papers have further established the positive effects of humanin in several different domains. Thummasorn et al. have confirmed that humanin treatment can protect against ischaemia–reperfusion injury and shown that this may be due to a decrease in reactive oxygen species generation (Muzumdar et al. 2010; Zhao et al. 2012; Thummasorn et al. 2016). Two other papers have shown the importance of humanin in neurocognition by showing that it can prevent diazepam‐induced memory dysfunction as well as act as an anxiolytic agent (Murakami et al. 2016; Zhao et al. 2016). Gidlund et al. (2016) discovered that muscle humanin levels were increased during resistance training compared to control and aerobic exercise (Nordic walking), but circulating levels were not affected. The authors believed that this change could be due to humanin's role in glucose metabolism. Looking at the cardiovascular aspects of humanin, Nikolakopoulos et al. (2017) examined a population of pre‐eclampsia patients where it was found that humanin levels were elevated; they suggested that this could be in response to cardiovascular stress.
Because of the small size of humanin, thorough investigation into the importance of each amino acid residue has already been conducted (reviewed in Yen et al. 2013). Both phenylalanine‐6 and lysine‐21 are required for humanin binding to IGFBP3 (Ikonen et al. 2003). Changing serine‐14 to a glycine (S14G humanin; HNG) increases the potency of the peptide although the mechanism is still unknown (Hashimoto et al. 2001a). Other residues have been found to be required for secretion of the peptide into the extracellular matrix (Yamagishi et al. 2003). Upon secretion, humanin is believed to activate two different receptors. The first receptor described was the ciliary neurotrophic factor receptor (CNTFR)–gp130 (IL6ST)–the interleukin 27 receptor subunit alpha (WSX1) tripartite receptor that then activates Janus kinase (JAK), signal transducer and activator of transcription (STAT), AKT and extracellular signal‐regulated kinases (ERK) (Hashimoto et al. 2009; Kim et al. 2016; Fig. 2). The formyl peptide receptor like (FPRL) 1 and FPRL2 receptors have also been shown to be activated by humanin and they also signal through ERK (Harada et al. 2004; Ying et al. 2004). As the first MDP discovered, humanin has been the most comprehensively investigated and both structural and functional aspects have been described. Its function as a cytoprotective, anti‐apoptotic peptide has been thoroughly investigated, while its function in cognition is still being examined.
Figure 2. MDP signalling.
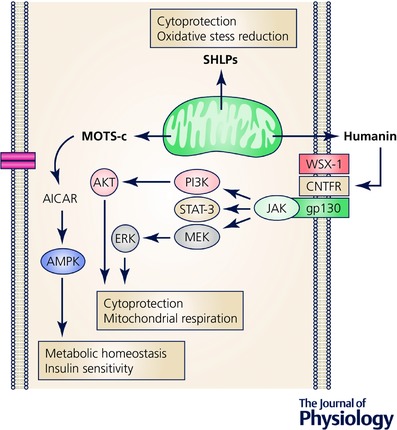
Schematic illustration of humanin‐, MOTS‐c‐ and SHLP‐mediated signalling pathways and biological functions. MEK, mitogen‐activated protein kinase kinase; PI3K, phosphoinositide 3‐kinase.
Newly discovered mitochondrially derived peptides: MOTS‐c and SHLPs
In addition to humanin, an in silico search of the mitochondrial genome revealed several additional potential MDPs. The mitochondrially derived peptide MOTS‐c that was recently discovered by Lee et al. (2015, 2016) is a 16 amino acid peptide located in the 12S rRNA gene. It regulates both insulin sensitivity and metabolic homeostasis via AMPK, increases 5‐aminoimidazole‐4‐carboxamide ribonucleotide (AICAR) levels and activates AMPK in HEK293 cells (Fig. 2; Lee et al. 2015). Reduction of this AMPK activation by chemical compounds or siRNA abolishes the enhanced glucose‐stimulated glycolytic response (Lee et al. 2015). In vivo MOTS‐c infusion significantly increased glucose clearance and insulin‐stimulated glucose disposal rate in glucose‐tolerance test and clamp studies. MOTS‐c further prevents high fat diet (HFD)‐induced obesity and insulin resistance in CD‐1 mice, as well as preventing HFD‐induced obesity independent of caloric intake in C57BL/6J mice (Lee et al. 2015). Supporting the fact that MOTS‐c activates AMPK, Ming et al. (2016) showed that MOTS‐c inhibits receptor activator of nuclear factor‐κB ligand (RANKL)‐induced osteoclast formation via AMPK activation in vitro and suppresses ovariectomy‐induced bone loss in mice (Ming et al. 2016). More recently, the m.1382A>C polymorphism, which is unique to the Northeast Asian population and causes a Lys14Gln replacement, was found to be correlated to longevity in Japanese people through its putative endocrine action, though further studies will be needed to elucidate the actual mechanism (Fuku et al. 2015).
Cobb et al. (2016) recently reported another six small humanin‐like peptides, small humanin‐like peptides (SHLP) 1–6, within the same 16S rRNA gene in which humanin is located (Fig. 1). SHLP2 and SHLP3 demonstrated similar protective effects as humanin and both improved mitochondrial metabolism by increasing oxygen consumption rate and reducing apoptosis and reactive oxygen species (ROS) in NIT‐1 and 22RV1 cells. Just as humanin increases mitochondrial biogenesis, SHLP2 and SHLP3 may also increase both mitochondrial biogenesis and oxygen consumption rate. Alternatively, this increase in oxygen consumption rate could be due to increased uncoupling. Further studies on whether MDPs modulate mitochondrial uncoupling will give us a better understanding of the cause of this increase in oxygen consumption rate. Because mitochondrial oxygen consumption is coupled to ATP production, the increase in energy production and its TCA cycle metabolites may enhance mitochondrial metabolism. Intracerebral infusion of SHLP2 increases glucose uptake and suppresses hepatic glucose production, suggesting that it functions as an insulin sensitizer both peripherally and centrally (Cobb et al. 2016). Further supporting their role as insulin sensitizers, both SHLP2 and SHLP3 also enhance 3T3‐L1 pre‐adipocyte differentiation. Another in vivo effect of SHLP2 injection is to increases leptin levels but with no effect on the inflammatory markers interleukin 6 and monocyte chemoattractant protein‐1 (MCP‐1). On the other hand, SHLP3 elevated both metabolic and inflammatory biomarkers (Cobb et al. 2016). Similar to humanin, the circulating levels of MOTS‐c and SHLP2 decline with age (Lee et al. 2015; Cobb et al. 2016), indicating that they are potential regulators of ageing.
With the addition of MOTS‐c and SHLP1–6, this class of peptides continues to grow exponentially. Unsurprisingly, because these peptides come from the mitochondrial genome, they are involved in common processes such as apoptosis and metabolism, but even though there is some overlapping function, clearly each MDP also has a unique signalling signature leading to an individual response. Future studies have yet to be conducted to determine the signalling pathways involved.
Mitochondria, mitochondrial peptides and metabolism
Cellular bioenergetics
Mitochondria are the primary energy source for all cellular functions. Mitochondria couple the oxidation of nutritional substrates to ATP synthesis. Carbohydrates, fats and proteins are broken down to glucose, free fatty acids and amino acids that can be utilized by mitochondria to produce ATP. Glycolysis generates 2 ATP whereas mitochondrial oxidation of pyruvate derived from glucose and palmitate derived from fatty acids generates 31.5 and 113 ATP, respectively (Mookerjee et al. 2015). These metabolic intermediates are translocated into the mitochondrial matrix and then enter the tricarboxylic acid (TCA) cycle and oxidative phosphorylation. The TCA cycle generates NADH and FADH2 that are fed into the electron transport chain at complex I and II, respectively, to provide electrons. The electron transport chain complexes transfer electrons to oxygen and concomitantly pump protons across the inner mitochondrial membrane to generate an electrochemical proton gradient. This proton‐motive force is then utilized for ATP synthesis and active transport processes in the mitochondria. As mitochondria are the primary source of cellular ATP, mitochondrial quality control mechanisms are required for cellular fitness. For example, mitophagy removes damaged mitochondria that lose their membrane potential (Ashrafi & Schwarz, 2013). In addition to mitochondrial turnover, mitochondrial peptides are produced to preserve essential functions related to energy production. Humanin directly affects mitochondrial bioenergetics by increasing basal oxygen consumption rate, maximum respiration, respiration capacity and ATP production in retinal pigment epithelial (hRPE) cells (Sreekumar et al. 2016). Thus, humanin protected hRPE cells from oxidative damage (tert‐butyl hydroperoxide treatment) perhaps by inhibiting the alteration of mitochondrial bioenergetics by maintaining ATP production and mitochondrial reserve capacity. Increased mitochondrial biogenesis is one possible mechanism for how cells optimize mitochondrial bioenergetics when it is required, and humanin increases the copy number of mtDNA, the number of mitochondria and the expression level of mitochondrial transcription factors, suggesting that humanin increases mitochondrial biogenesis (Sreekumar et al. 2016). Although seemingly contradictory to the previous study, humanin suppresses the increase in mtDNA copy number in serum‐deprived lymphocytes (Kariya et al. 2003). One possible explanation for this opposite result is that humanin treatment improved basal metabolic activity of the mitochondria in serum‐deprived cells. Therefore, the compensatory increase in mtDNA copy number was offset by improved quality of mitochondria. Humanin‐treated cells showed higher metabolic activity and mtDNA copy number could be decreased to maintain cellular homeostasis.
The humanin analogue HNG also increases ATP production and mitochondrial reserve capacity. In addition, HNG increases the mitochondrial membrane potential in H9c2 myoblast cells, and it rescues the loss of membrane potential in response to H2O2 treatment (Klein et al. 2013). Since increased mitochondrial membrane potential is linked to elevated cellular ATP production, in turn, the cellular ATP level is elevated in the presence of HNG in H9c2 cells. Substitution of alanine for Phe6 completely negated the interaction between humanin or HNG and IGFBP3 and generated an enhanced form of humanin (HNF6A or HNGF6A) (Ikonen et al. 2003). HNGF6A regulates glucose metabolism and energy production. HNGF6A promoted the glucose‐induced GLUT2 transporter translocation to the plasma membrane, and increased glucose oxidation and ATP production in βTC3 cells (Kuliawat et al. 2013). HNGF6A also increased the mitochondrial membrane potential in the cells. Similar to humanin, both SHLP2 and SHLP3 increase mitochondrial respiration and ATP production (Cobb et al. 2016).
MOTS‐c, which is encoded in the 12S rRNA region of mitochondria, has also been reported to influence mitochondrial metabolism. MOTS‐c administration increased glucose uptake and glycolysis, whereas it suppressed mitochondrial respiration in cultured cells and skeletal muscle. This resembles a Crabtree effect, namely decreased mitochondrial oxygen consumption rate in response to high glucose uptake (Lee et al. 2015). Furthermore, AMPK and sirtuin 1 (SIRT1) siRNA reduce the glucose‐stimulated glycolytic response, which suggests that AMPK and SIRT1 play roles in MOTS‐c actions on cellular bioenergetics.
Mitochondrially derived peptides are produced in the mitochondria and play an important role in energy production. Exogenously applied humanin localizes to the mitochondria, so it may directly modulate mitochondrial membrane potential to enhance mitochondrial respiration. MOTS‐c may regulate mitochondrial respiration by activation of signalling pathways mediated by AMPK or SIRT1. However, the detailed mechanism concerning the role of MDPs in energy production still needs to be studied.
Amino acid, lipid, and nucleotide metabolism
In addition to producing ATP, mitochondria play important roles in amino acid, lipid and nucleotide metabolism. Thus, the TCA cycle metabolites are utilized for building macromolecules. For example, α‐ketoglutarate and oxaloacetate can be transported into the cytosol and are utilized for de novo protein and nucleotide synthesis (Bohovych & Khalimonchuk, 2016). In addition, citrate can be transported into the cytosol and is utilized for protein acetylation as well as de novo fatty acid synthesis (Wellen et al. 2009; Buchakjian & Kornbluth, 2010). One of the mitochondrial peptides, MOTS‐c, is closely associated with amino acid and lipid metabolism. MOTS‐c modulates the one‐carbon metabolism cycle and purine biosynthesis. It also activates AMPK by blocking de novo purine biosynthesis, resulting in an accumulation of endogenous AICAR. Moreover, MOTS‐c affects fatty acid metabolism via the AICAR–AMPK pathway. As AMPK is the cellular signalling hub for balancing fuel usage and energy demand, MOTS‐c stimulates carnitine shuttles, reduces levels of essential fatty acids and increases the β‐oxidation intermediates (Lee et al. 2015). In addition, MOTS‐c increases metabolite levels of NAD+, glycolysis and the pentose phosphate pathway. Mitochondria modulate amino acid and lipid metabolism in response to cellular homeostatic alteration and metabolic demand. Understanding whether cellular energy demand and metabolic stress can equally regulate MDP expression is an avenue of research that will provide an insight into MDP biology.
Systemic glucose homeostasis and adiposity
Going from the biochemical and cellular level to the physiological level, ATP and metabolites generated from mitochondrial respiration modulate insulin secretion in pancreatic β‐cells. In β‐cells, glucose‐stimulated ATP production increases the ATP/ADP ratio, resulting in closing of ATP‐dependent K+ channels in β‐cells, leading to membrane depolarization and activation of the voltage‐dependent calcium channel (Muoio & Newgard, 2008). The resulting increase in cytoplasmic calcium concentration leads to exocytosis of insulin in pancreatic β‐cells.
Patients with mitochondrial DNA mutations exhibit impaired β‐cell function, and inhibition of mitochondrial metabolism can inhibit glucose‐stimulated insulin secretion (Suzuki et al. 1997; Maechler & Wollheim, 2001). In addition to ATP, metabolites from mitochondrial metabolism including malonyl‐CoA, long‐chain acyl‐CoA and NADPH modulate insulin secretion by inhibiting ATP‐dependent K+ channels (Maechler et al. 1997). Additionally, glutamate is generated in the mitochondria from α‐ketoglutarate and directly stimulates insulin exocytosis (Maechler & Wollheim, 1999).
Mitochondria are closely associated with insulin function as well as insulin secretion. For example, abnormal morphology, decreased mitochondrial number, decreased mitochondrial oxidative enzymes and lower ATP production were commonly found in insulin‐resistant metabolic tissues including skeletal muscle, liver and adipose (Kim et al. 2008; Oropeza et al. 2015). Elevated circulating free fatty acids accumulating in these tissues will also decrease insulin‐stimulated glucose disposal (Boden et al. 1994; Shah et al. 2002; Boden, 2005). This impaired insulin signalling is a major cause of insulin resistance because it not only affects insulin‐stimulated glucose metabolism in skeletal muscle but also impairs other actions of insulin in diverse tissues including liver, adipose tissue and heart. Therefore, glucose and lipid metabolism in the mitochondria plays a critical role in insulin signalling and glucose homeostasis.
Mitochondrial bioenergetics and metabolism are closely associated with insulin signalling and glucose homeostasis (Pagel‐Langenickel et al. 2008; Auger et al. 2015). As MDPs modulate cellular bioenergetics and metabolism in vitro, they also show systemic regulation of metabolism in vivo. Moving from in vitro to in vivo systems, the typical dosage administered to rodents is possibly supraphysiological. It should be noted that the half‐life of humanin and probably most MDPs is in the minutes range and so after an hour, circulating levels of these MDPs would return to baseline (Chin et al. 2013). Multiple animal model studies administering humanin and its analogues support the crucial role of humanin in glucose homeostasis. Intracerebroventricular administration of humanin led to increased insulin sensitivity in the liver and muscle, causing a reduction of hepatic glucose production and increased insulin‐mediated AKT signalling and fatty acid metabolism signalling (Muzumdar et al. 2009). These effects were modulated by humanin‐mediated STAT‐3 activation in the hypothalamus. Peripheral administration of humanin also enhanced peripheral glucose uptake and suppressed hepatic glucose production. HNGF6A, HNG and humanin, but not HNF6A, show insulin‐sensitizing effects. HNGF6A increased glucose‐stimulated insulin secretion in isolated islets from both normal and db/db mice and in mouse pancreatic cells (βTC3) (Kuliawat et al. 2013). Elevated ATP production and the activity of aspartate aminotransferases of the malate–aspartate NADH shuttle are key mechanisms of HNGF6A regulation of insulin secretion. A hyperglycaemic clamp study found that HNGF6A enhanced glucose‐stimulated insulin secretion in young Sprague–Dawley rats (Muzumdar et al. 2009). Additionally, HNGF6A significantly lowers blood glucose in Zucker diabetic fatty rats. The direct effect of HNGF6A on isolated islets and βTC3 cells suggests that HNGF6A mitigates some of the metabolic abnormalities present in islets in type 2 diabetes (Hoang et al. 2010; Kuliawat et al. 2013). Recently, a new role of humanin in lipid metabolism was revealed. Intraperitoneal administration of HNG decreased body weight gain, visceral fat and hepatic triglyceride accumulation in high fat diet‐fed mice (Gong et al. 2015). Increased energy expenditure was examined in HNG‐injected mice, partially explaining the decrease in body weight gain and visceral fat in these mice. The decrease in hepatic triglyceride accumulation is caused by increased activity of hepatic microsomal triglyceride transfer protein and increased hepatic triglyceride secretion. Vagotomy was performed on mice to investigate the role of the hypothalamus in hepatic triglyceride secretion. When these mice were injected with HNG, the surgery blocked humanin's effect of both intravenous and intracerebroventricular infusion on hepatic triglyceride secretion. These results suggest that the effect of humanin is mediated through the hypothalamus as the vagus nerve serves as an efferent connection from the hypothalamus to the liver, but not by a neuroendocrine signal.
SHLP2 and SHLP3 have insulin‐sensitizing effects in vitro and in vivo. Both SHLP2 and SHLP3 accelerated 3T3‐L1 cell (a murine pre‐adipocyte cell line) differentiation in the presence of insulin (Cobb et al. 2016). This suggests that SHLP2 and 3 promote cellular differentiation and enhance insulin sensitivity in adipose tissue. Furthermore, SHLP2, but not SHLP3, enhances the insulin‐sensitizing effect of hepatic glucose production suppression and increased glucose disposal in peripheral tissues. Both ATP and mitochondrial respiration metabolites are equally important for insulin secretion. Although both SHLP2 and SHLP3 enhance ATP production, their modulation of different metabolites could be the mechanism differentiating their distinct effects in vivo. Further investigation to address the mechanism is required.
MOTS‐c enhances whole‐body insulin sensitivity, acting primarily through the muscle. MOTS‐c increases the insulin‐stimulated glucose disposal rate, an indicator of enhanced skeletal muscle insulin sensitivity, but does not alter the rate of hepatic glucose production (Lee et al. 2015). Insulin‐mediated AKT signalling is elevated in the muscle isolated from MOTS‐c‐injected C57BL/6J mice, and differentiated L6 rat myotubes overexpressing MOTS‐c have accelerated glucose uptake and enhanced glucose‐stimulated and maximum glycolytic rate. The role of MOTS‐c in enhancing insulin sensitivity and glucose homeostasis has also been examined in high fat diet‐fed CD‐1 mice. MOTS‐c‐treated HFD‐fed mice showed reduced weight gain but did not show any difference in food intake. This result suggests that MOTS‐c may increase the metabolic rate of these mice and experiments using metabolic cages found that HFD‐fed mice treated with MOTS‐c showed increased respiratory exchange ratio, reflecting increased glucose utilization. This result suggests that MOTS‐c may increase the metabolic rate of these mice and experiments using metabolic cages found that HFD‐fed mice treated with MOTS‐c showed increased energy expenditure and respiratory exchange ratio, reflecting increased glucose utilization. Hepatic lipid accumulation was dramatically reduced in HFD‐fed mice treated with MOTS‐c and MOTS‐c prevented HFD‐induced hyperinsulinaemia, indicating improved glucose homeostasis. Moreover, MOTS‐c promoted AMPK activation and GLUT4 expression in the skeletal muscles of HFD‐fed mice.
Humanin's physiological effects are well established and changes in glucose utilization and insulin sensitization have been found. Hypothalamic signalling is central to these effects as is STAT3 signalling. In contrast, the physiological effects of MOTS‐c and the SHLPs have yet to be thoroughly established. While there are hints of a mechanism, much more research will be required to discover the signalling pathways activated by these MDPs.
Signalling centre
The two functions of mitochondria, to generate ATP and to support biosynthesis, are balanced to meet cellular needs. Clearly, mitochondria receive signals in response to stress and metabolic changes (anterograde signalling) (Quirós et al. 2016), but emerging data suggest that mitochondria are also actively sending signals back to the cytosol and nucleus (retrograde signalling) (Picard et al. 2013). The mitochondrial unfolded protein response is one of the retrograde signalling pathways that increase mitochondrially localized chaperones and proteases to recover mitochondrial protein homeostasis (Nargund et al. 2012; 2015). Interestingly, the mitochondrial unfolded response also modulates cellular metabolism including increased glycolysis and decreased expression of TCA cycle and oxidative phosphorylation genes, potentially to reduce mitochondrial stress and alter cellular metabolism to promote survival (Nargund et al. 2015; Quirós et al. 2016). In addition to the mitochondrial unfolded protein response, mitochondrially derived peptides are emerging as another retrograde signal in response to cellular stress (Lee et al. 2013). Although MDPs play important roles in the regulation of cellular energetics and systemic metabolism, whether MDPs can modulates gene expression and whether there is epigenetic modification in the nucleus remain unexplored. Investigation into the role of MDPs in the nucleus will give us a more comprehensive understanding of MDP signalling.
Conclusions
Mitochondria are metabolic hubs within cells that alter cellular functions in response to cellular stress. Emerging evidence shows that mitochondrially derived peptides are retrograde signalling molecules. These peptides regulate mitochondrial bioenergetics and mitochondrial metabolism; subsequently, they modulate systemic insulin sensitivity and glucose homeostasis. In addition to their intracellular effects, MDPs are found in the circulation and represent a novel method for the mitochondria to signal to the CNS and other peripheral tissues. Although relatively recently compared to humanin, other MDPs are being discovered and their role in metabolism is still emerging. Many more studies will be required to elucidate the mechanism of action of each of these MDPs. Further translational studies will also be required to test how MDPs can be diagnostic markers and potent therapeutics for metabolic diseases including type 2 diabetes.
Additional information
Competing interests
P.C. is a consultant and stockholder of CohBar Inc. K.Y. has consulted for CohBar Inc.
Author contributions
All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This research was supported by a Glenn Foundation Award and National Institute of Health Grants to P.C. (1R01AG 034430, 1R01GM 090311, 1R01ES 020812, and 1R21DK 089447) and a Ellison/AFAR postdoctoral fellowship award to S.J.K.
Biographies
Su‐Jeong Kim received her PhD in biology at Yonsei University. Her PhD thesis was focused on the cytoprotective roles of PARK7 during neuronal cell death in various Parkinson's disease models. She is currently a postdoctoral fellow in the laboratory of Pinchas Cohen at USC Leonard Davis School of Gerontology. She has identified humanin‐mediated signalling pathways that regulate autophagy and cytoprotection. Her research focuses on understanding the roles of mitochondrially derived peptides including humanin in healthspan and lifespan.

Kelvin Yen is a research assistant professor at the Leonard Davis School of Gerontology at the University of Southern California. He received his PhD at the Icahn School of Medicine at Mount Sinai, New York, in the lab of Charles Mobbs. He has spent nearly 20 years researching the biology of ageing, with his most recent research focusing on the physiological role of mitochondrially derived peptides and how they affect ageing and age‐related diseases.
This review was presented at the symposium “Mitochondrial signaling and inter‐organelle crosstalk” which took place at The Integrative Biology of Exercise VII in Phoenix, Arizona, USA, 2–4 November 2016.
References
- Ashrafi G & Schwarz TL (2013). The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ 20, 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auger C, Alhasawi A, Contavadoo M & Appanna VD (2015). Dysfunctional mitochondrial bioenergetics and the pathogenesis of hepatic disorders. Front Cell Dev Biol 3, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boden G (2005). Free fatty acids and insulin secretion in humans. Curr Diab Rep 5, 167–170. [DOI] [PubMed] [Google Scholar]
- Boden G, Chen X, Ruiz J, White JV & Rossetti L (1994). Mechanisms of fatty acid‐induced inhibition of glucose uptake. J Clin Invest 93, 2438–2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohovych I & Khalimonchuk O (2016). Sending out an SOS: mitochondria as a signaling hub. Front Cell Dev Biol 4, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchakjian MR & Kornbluth S (2010). The engine driving the ship: metabolic steering of cell proliferation and death. Nat Rev Mol Cell Biol 11, 715–727. [DOI] [PubMed] [Google Scholar]
- Chin Y‐P, Keni J, Wan J, Mehta H, Anene F, Jia Y, Lue Y‐H, Swerdloff R, Cobb LJ, Wang C & Cohen P (2013). Pharmacokinetics and tissue distribution of humanin and its analogues in male rodents. Endocrinology 154, 3739–3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb LJ, Lee C, Xiao J, Yen K, Wong RG, Nakamura HK, Mehta HH, Gao Q, Ashur C, Huffman DM, Wan J, Muzumdar R, Barzilai N & Cohen P (2016). Naturally occurring mitochondrial‐derived peptides are age‐dependent regulators of apoptosis, insulin sensitivity, and inflammatory markers. Aging 8, 796–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuku N, Pareja‐Galeano H, Zempo H, Alis R, Arai Y, Lucia A & Hirose N (2015). The mitochondrial‐derived peptide MOTS‐c: a player in exceptional longevity? Aging Cell 14, 921–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gidlund EK, Walden F von, Venojärvi M, Risérus U, Heinonen OJ, Norrbom J & Sundberg CJ (2016). Humanin skeletal muscle protein levels increase after resistance training in men with impaired glucose metabolism. Physiol Rep 4, e13063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Z, Su K, Cui L, Tas E, Zhang T, Dong HH, Yakar S & Muzumdar RH (2015). Central effects of humanin on hepatic triglyceride secretion. Am J Physiol Endocrinol Metab 309, E283–E292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo B, Zhai D, Cabezas E, Welsh K, Nouraini S, Satterthwait AC & Reed JC (2003). Humanin peptide suppresses apoptosis by interfering with Bax activation. Nature 423, 456–461. [DOI] [PubMed] [Google Scholar]
- Harada M, Habata Y, Hosoya M, Nishi K, Fujii R, Kobayashi M & Hinuma S (2004). N‐Formylated humanin activates both formyl peptide receptor‐like 1 and 2. Biochem Biophys Res Commun 324, 255–261. [DOI] [PubMed] [Google Scholar]
- Hashimoto Y, Kurita M, Aiso S, Nishimoto I & Matsuoka M (2009). Humanin inhibits neuronal cell death by interacting with a cytokine receptor complex or complexes involving CNTF receptor alpha/WSX‐1/gp130. Mol Biol Cell 20, 2864–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto Y, Niikura T, Ito Y, Sudo H, Hata M, Arakawa E, Abe Y, Kita Y & Nishimoto I (2001a). Detailed characterization of neuroprotection by a rescue factor humanin against various Alzheimer's disease‐relevant insults. J Neurosci 21, 9235–9245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto Y, Niikura T, Tajima H, Yasukawa T, Sudo H, Ito Y, Kita Y, Kawasumi M, Kouyama K, Doyu M, Sobue G, Koide T, Tsuji S, Lang J, Kurokawa K & Nishimoto I (2001b). A rescue factor abolishing neuronal cell death by a wide spectrum of familial Alzheimer's disease genes and Aβ. Proc Natl Acad Sci USA 98, 6336–6341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang PT, Park P, Cobb LJ, Paharkova‐Vatchkova V, Hakimi M, Cohen P & Lee K‐W (2010). The neurosurvival factor Humanin inhibits beta‐cell apoptosis via signal transducer and activator of transcription 3 activation and delays and ameliorates diabetes in nonobese diabetic mice. Metab Clin Exp 59, 343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonen M, Liu B, Hashimoto Y, Ma L, Lee K‐W, Niikura T, Nishimoto I & Cohen P (2003). Interaction between the Alzheimer's survival peptide humanin and insulin‐like growth factor‐binding protein 3 regulates cell survival and apoptosis. Proc Natl Acad Sci USA 100, 13042–13047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kariya S, Takahashi N, Hirano M & Ueno S (2003). Humanin improves impaired metabolic activity and prolongs survival of serum‐deprived human lymphocytes. Mol Cell Biochem 254, 83–89. [DOI] [PubMed] [Google Scholar]
- Kim J‐A, Wei Y & Sowers JR (2008). Role of mitochondrial dysfunction in insulin resistance. Circ Res 102, 401–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Guerrero N, Wassef G, Xiao J, Mehta HH, Cohen P & Yen K (2016). The mitochondrial‐derived peptide humanin activates the ERK1/2, AKT, and STAT3 signaling pathways and has age‐dependent signaling differences in the hippocampus. Oncotarget 7, 46899–46912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein LE, Cui L, Gong Z, Su K & Muzumdar R (2013). A humanin analog decreases oxidative stress and preserves mitochondrial integrity in cardiac myoblasts. Biochem Biophys Res Commun 440, 197–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuliawat R, Klein L, Gong Z, Nicoletta‐Gentile M, Nemkal A, Cui L, Bastie C, Su K, Huffman D, Surana M, Barzilai N, Fleischer N & Muzumdar R (2013). Potent humanin analog increases glucose‐stimulated insulin secretion through enhanced metabolism in the β cell. FASEB J 27, 4890–4898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Yen K & Cohen P (2013). Humanin: a harbinger of mitochondrial‐derived peptides? Trends Endocrinol Metab 24, 222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Kim KH & Cohen P (2016). MOTS‐c: A novel mitochondrial‐derived peptide regulating muscle and fat metabolism. Free Radic Biol Med 100, 182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Zeng J, Drew BG, Sallam T, Martin‐Montalvo A, Wan J, Kim SJ, Mehta H, Hevener AL, de Cabo R & Cohen P (2015). The mitochondrial‐derived peptide MOTS‐c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metab 21, 443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maechler P, Kennedy ED, Pozzan T & Wollheim CB (1997). Mitochondrial activation directly triggers the exocytosis of insulin in permeabilized pancreatic beta‐cells. EMBO J 16, 3833–3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maechler P & Wollheim CB (1999). Mitochondrial glutamate acts as a messenger in glucose‐induced insulin exocytosis. Nature 402, 685–689. [DOI] [PubMed] [Google Scholar]
- Maechler P & Wollheim CB (2001). Mitochondrial function in normal and diabetic β‐cells. Nature 414, 807–812. [DOI] [PubMed] [Google Scholar]
- Ming W, Lu G, Xin S, Huanyu L, Yinghao J, Xiaoying L, Chengming X, Banjun R, Li W & Zifan L (2016). Mitochondria related peptide MOTS‐c suppresses ovariectomy‐induced bone loss via AMPK activation. Biochem Biophys Res Commun 476, 412–419. [DOI] [PubMed] [Google Scholar]
- Mookerjee SA, Goncalves RLS, Gerencser AA, Nicholls DG & Brand MD (2015). The contributions of respiration and glycolysis to extracellular acid production. Biochim Biophys Acta 1847, 171–181. [DOI] [PubMed] [Google Scholar]
- Muoio DM & Newgard CB (2008). Molecular and metabolic mechanisms of insulin resistance and β‐cell failure in type 2 diabetes. Nat Rev Mol Cell Biol 9, 193–205. [DOI] [PubMed] [Google Scholar]
- Murakami M, Nagahama M, Maruyama T & Niikura T (2016). Humanin ameliorates diazepam‐induced memory deficit in mice. Neuropeptides 62, 65–70. [DOI] [PubMed] [Google Scholar]
- Muzumdar RH, Huffman DM, Atzmon G, Buettner C, Cobb LJ, Fishman S, Budagov T, Cui L, Einstein FH, Poduval A, Hwang D, Barzilai N & Cohen P (2009). Humanin: a novel central regulator of peripheral insulin action. PLoS One 4, e6334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzumdar RH, Huffman DM, Calvert JW, Jha S, Weinberg Y, Cui L, Nemkal A, Atzmon G, Klein L, Gundewar S, Ji SY, Lavu M, Predmore BL & Lefer DJ (2010). Acute humanin therapy attenuates myocardial ischemia and reperfusion injury in mice. Arterioscler Thromb Vasc Biol 30, 1940–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nargund AM, Fiorese CJ, Pellegrino MW, Deng P & Haynes CM (2015). Mitochondrial and nuclear accumulation of the transcription factor ATFS‐1 promotes OXPHOS recovery during the UPRmt . Mol Cell 58, 123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM & Haynes CM (2012). Mitochondrial import efficiency of ATFS‐1 regulates mitochondrial UPR activation. Science 337, 587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolakopoulos P, Tzimagiorgis G, Goulis DG, Chatzopoulou F, Zepiridis L & Vavilis D (2017). Serum humanin concentrations in women with pre‐eclampsia compared to women with uncomplicated pregnancies. J Matern Fetal Neonatal Med (in press; https://doi.org/10.1080/14767058.2017.1285885). [DOI] [PubMed] [Google Scholar]
- Oropeza D, Jouvet N, Bouyakdan K, Perron G, Ringuette L‐J, Philipson LH, Kiss RS, Poitout V, Alquier T & Estall JL (2015). PGC‐1 coactivators in β‐cells regulate lipid metabolism and are essential for insulin secretion coupled to fatty acids. Mol Metab 4, 811–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagel‐Langenickel I, Bao J, Joseph JJ, Schwartz DR, Mantell BS, Xu X, Raghavachari N & Sack MN (2008). PGC‐1alpha integrates insulin signaling, mitochondrial regulation, and bioenergetic function in skeletal muscle. J Biol Chem 283, 22464–22472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard M, Shirihai OS, Gentil BJ & Burelle Y (2013). Mitochondrial morphology transitions and functions: implications for retrograde signaling? Am J Physiol Regul Integr Comp Physiol 304, R393–R406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirós PM, Mottis A & Auwerx J (2016). Mitonuclear communication in homeostasis and stress. Nat Rev Mol Cell Biol 17, 213–226. [DOI] [PubMed] [Google Scholar]
- Shah P, Vella A, Basu A, Basu R, Adkins A, Schwenk WF, Johnson CM, Nair KS, Jensen MD & Rizza RA (2002). Effects of free fatty acids and glycerol on splanchnic glucose metabolism and insulin extraction in nondiabetic humans. Diabetes 51, 301–310. [DOI] [PubMed] [Google Scholar]
- Sreekumar PG, Ishikawa K, Spee C, Mehta HH, Wan J, Yen K, Cohen P, Kannan R & Hinton DR (2016). The mitochondrial‐derived peptide humanin protects RPE cells from oxidative stress, senescence, and mitochondrial dysfunction. Invest Ophthalmol Vis Sci 57, 1238–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Iizuka T, Kobayashi T, Nishikawa T, Atsumi Y, Kadowaki T, Oka Y, Kadowaki H, Taniyama M, Hosokawa K, Asahina T & Matsuoka K (1997). Diabetes mellitus associated with the 3243 mitochondrial tRNALeu(UUR) mutation: insulin secretion and sensitivity. Metab Clin Exp 46, 1019–1023. [DOI] [PubMed] [Google Scholar]
- Thummasorn S, Apaijai N, Kerdphoo S, Shinlapawittayatorn K, Chattipakorn SC & Chattipakorn N (2016). Humanin exerts cardioprotection against cardiac ischemia/reperfusion injury through attenuation of mitochondrial dysfunction. Cardiovasc Ther 34, 404–414. [DOI] [PubMed] [Google Scholar]
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR & Thompson CB (2009). ATP‐citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao J, Kim SJ, Cohen P & Yen K (2016). Humanin: Functional interfaces with IGF‐I. Growth Horm IGF Res 29, 21–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagishi Y, Hashimoto Y, Niikura T & Nishimoto I (2003). Identification of essential amino acids in Humanin, a neuroprotective factor against Alzheimer's disease‐relevant insults. Peptides 24, 585–595. [DOI] [PubMed] [Google Scholar]
- Yen K, Lee C, Mehta H & Cohen P (2013). The emerging role of the mitochondrial‐derived peptide humanin in stress resistance. J Mol Endocrinol 50, R11–R19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying G, Iribarren P, Zhou Y, Gong W, Zhang N, Yu Z‐X, Le Y, Cui Y & Wang JM (2004). Humanin, a newly identified neuroprotective factor, uses the G protein‐coupled formylpeptide receptor‐like‐1 as a functional receptor. J Immunol 172, 7078–7085. [DOI] [PubMed] [Google Scholar]
- Yu D, Du Z, Pian L, Li T, Wen X, Li W, Kim S‐J, Xiao J, Cohen P, Cui J, Hoffman AR & Hu J‐F (2017). Mitochondrial DNA hypomethylation is a biomarker associated with induced senescence in human fetal heart mesenchymal stem cells. Stem Cells Int https://doi.org/10.1155/2017/1764549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Sonada S, Yoshikawa A & Ohinata K (2016). Rubimetide, humanin, and MMK1 exert anxiolytic‐like activities via the formyl peptide receptor 2 in mice followed by the successive activation of DP1, A2A, and GABAA receptors. Peptides 83, 16–20. [DOI] [PubMed] [Google Scholar]
- Zhao S‐T, Huang X‐T, Zhang C & Ke Y (2012). Humanin protects cortical neurons from ischemia and reperfusion injury by the increased activity of superoxide dismutase. Neurochem Res 37, 153–160. [DOI] [PubMed] [Google Scholar]