

Abstract
In many Gram-negative bacteria, the peptidoglycan synthase PBP1A requires the outer membrane lipoprotein LpoA for constructing a functional peptidoglycan required for bacterial viability. Previously, we have shown that the C-terminal domain of Haemophilus influenzae LpoA (HiLpoA) has a highly conserved, putative substrate-binding cleft between two α/β lobes. Here, we report a 2.0 Å resolution crystal structure of the HiLpoA N-terminal domain. Two subdomains contain tetratricopeptide-like motifs that form a concave groove, but their relative orientation differs by ∼45° from that observed in an NMR structure of the Escherichia coli LpoA N domain. We also determined three 2.0–2.8 Å resolution crystal structures containing four independent full-length HiLpoA molecules. In contrast to an elongated model previously suggested for E. coli LpoA, each HiLpoA formed a U-shaped structure with a different C-domain orientation. This resulted from both N-domain twisting and rotation of the C domain (up to 30°) at the end of the relatively immobile interdomain linker. Moreover, a previously predicted hinge between the lobes of the LpoA C domain exhibited variations of up to 12°. Small-angle X-ray scattering data revealed excellent agreement with a model calculated by normal mode analysis from one of the full-length HiLpoA molecules but even better agreement with an ensemble of this molecule and two of the partially extended normal mode analysis-predicted models. The different LpoA structures helped explain how an outer membrane-anchored LpoA can either withdraw from or extend toward the inner membrane-bound PBP1A through peptidoglycan gaps and hence regulate the synthesis of peptidoglycan necessary for bacterial viability.
Keywords: crystal structure, gram-negative bacteria, peptidoglycan, protein domain, small-angle X-ray scattering (SAXS), conformational flexibility, normal mode analysis, peptidoglycan synthase PBP1A, transpeptidase
Introduction
There is an urgent need to define new drug targets to cope with the growing number of antibiotic-resistant bacteria (1). Since the discovery of penicillin, the proteins involved in the maintenance of the bacterial cell wall have remained important targets for antibiotic development. The cell wall, located between the inner and outer membranes of Gram-negative bacteria, is composed of peptidoglycan (PG),4 a glycan made up of strands with repeating oligopeptide–disaccharide units. The pentapeptide groups from newly synthesized strands form peptide cross-links with acceptor peptides from existing strands, resulting in a netlike macromolecule (sacculus). This sacculus, by surrounding the entire cell, provides resistance to turgor (osmotic pressure), maintains cell shape, and is essential for cell viability (2–4).
Peptidoglycan in most Gram-negative bacteria is polymerized in the periplasm from peptide-substituted disaccharides (muropeptide) by RodA, a SEDS family inner membrane transglycosylase (5, 6), and the transglycosylase (TG) domains of inner membrane-anchored, class A penicillin-binding proteins, PBP1A and PBP1B (7). Transpeptidases (TPs) form peptide cross-links between nascent PG strands and existing strands of the sacculus. These include class B PBP TPs (5) associated with the SEDS enzyme and the TP domains of PBP1A and PBP1B. With the discovery that RodA synthesizes a significant portion of PG, the exact role of PBP1A and PBP1B is not clear, but they may be important for PG repair or forming the PG at the septum that forms during cell division (8). In Escherichia coli, either PBP1A or PBP1B is essential for growth (9).
LpoA is a two-domain outer membrane lipoprotein and is encoded in the genomes of many proteobacteria (8, 10). In E. coli, EcLpoA was shown to tightly bind to EcPBP1A and activate its transpeptidase and to be essential in a mutant that lacked PBP1B (8, 10, 11). Moreover, the presence of the LpoA C domain was required for these activities. In Vibrio cholera, deletion of LpoA caused growth defects in minimal media. Such mutants also showed less virulence in vivo (12). In the Haemophilus influenzae bacterium, the orthologous gene lpoA (previously yraM or HI1655) was shown to be essential for cell growth in vitro (13). The likely reason is that the ponA-encoded PBP1A in H. influenzae is also essential for cellular growth, as indicated by high-density transposon mutagenesis experiments (14, 15).
Experimentally determined structures have previously been reported for the individual N- and C-terminal domains of LpoA. We described the 1.35 Å resolution crystal structure of the C-terminal domain of H. influenzae LpoA (LpoA-C; residues 257–575) (16). This domain shares a homologous fold, but low sequence identity, with periplasmic substrate-binding proteins involved in the high-affinity import of small molecules by ABC-like transporters (17). The bilobate structure of LpoA-C adopts an “open” conformation, with a large cleft between the two lobes, typical of unliganded substrate-binding proteins (16). In periplasmic substrate-binding proteins, ligand molecules tightly bind in the cleft and stabilize a closed-cleft conformation (18). Because the residues exposed in the LpoA-C cleft are highly conserved in all LpoA sequences and form nonpolar as well as polar surfaces, this cleft probably binds another molecule, possibly PBP1A or peptidoglycan. Truncation experiments confirmed that the EcLpoA C domain was sufficient for the activation of the EcPBP1A transpeptidase in vitro (8).
The N-terminal domain of H. influenzae LpoA (LpoA-N), which includes residues 26–256, was predicted, based on significant sequence similarity to other periplasmic proteins of known structure, to contain repeats of the tetratricopeptide (TPR)-like motif, a helix-turn-helix motif commonly involved in protein–protein interactions (16). This prediction was more or less borne out by the recent NMR structure of E. coli LpoA-N, which indeed showed adjacent helix-turn-helix motifs, but without the usual curvature associated with TPR-containing domains (19).
Knowledge of these structures by themselves appears to be insufficient for fully understanding how LpoA promotes PG synthesis. As the cell grows, PG hydrolases and amidases form holes in the sacculus to expose acceptor peptides for cross-linking to new strands (reviewed in Refs. 7 and 21), and turgor may cause these gaps to widen (22). Whereas movement of PBP1A is known to be controlled by the cytoskeletal actin-like protein MreB and associated inner membrane proteins MreC, MreD, RodA, and RodZ, what directs PBP1A specifically to the regions requiring new PG strands is unknown. As an outer membrane lipoprotein, LpoA would be in a position to sense the integrity of PG (2, 7). Only at gaps or holes could LpoA extend its C domain through the PG and activate PBP1A to insert and cross-link new PG strands. Consistent with this proposal, recent studies of E. coli LpoB (an activator of PBP1B but structurally unrelated to LpoA) predicted that its N-terminal ∼50 residues are unstructured and probably long enough to position the LpoB globular domain so that it can interact with PBP1B (23, 24). Moreover, from small-angle X-ray scattering and NMR data, Jean et al. (19) proposed that full-length E. coli LpoA is a relatively rigid rod that is narrow enough to fit through the gaps and pores in the PG and that it is about 150 Å long, a sufficient length to interact with PBP1A. But it remains unknown how such an elongated and rigid protein would be accommodated in the absence of a suitable PG hole and how this LpoA structure could adapt to different distances between the inner and outer membrane in different species.
To clarify what structural elements of LpoA will allow it to pass through gaps in the PG and interact with PBP1A, we set out to provide experimentally determined structural data for an intact full-length LpoA. Here we present structures from three crystal forms of full-length H. influenzae LpoA, determined to resolutions of 2.0, 2.8, and 2.6 Å, as well as the crystal structure of its N domain (LpoA-N) to a resolution of 2.0 Å. Unlike the E. coli model (19), the full-length LpoA was not elongated in the crystals but forms a variable jaw-like structure. Also, the N domain assumed the canonical superhelical twist associated with other TPR-containing proteins. These structures provide insight into the possible role of the N domain and how conformational flexibility allows LpoA to adapt to different PG architectures.
Results
The N-terminal domain of LpoA
The crystal structure of the H. influenzae LpoA N-terminal domain (LpoA-N), containing LpoA residues 33–253 and selenomethionine in place of Met, was refined to an Rwork/Rfree of 0.18/0.22 for data extending to 1.95 Å resolution (see Table 1 and “Experimental procedures”). The crystal structure includes two independent LpoA-N molecules with residues 33–248 and 33–249, respectively. The two molecules superpose with an RMSD of 0.49 Å for 202 of 215 residues. The molecules are arranged N-to-C along a pseudo-43-screw axis (supplemental Fig. S1). The two molecules make intermolecular polar interactions between non-conserved residues on helices H2–H4 and H11-H12.
Table 1.
Crystallographic data and refinement
| LpoA-N (SeMet, PDB: 4P29) | LpoAOrt (PDB: 5KCN) | LpoAMon (SeMet, PDB: 5VBG) | LpoAMon2 (SeMet, PDB: 5VAT) | |
|---|---|---|---|---|
| Beamline | APS DND-CAT 5ID | APS LS-CAT 21ID-G | APS LS-CAT 21ID-D | APS LS-CAT 21ID-D |
| Wavelength (Å) | 0.9793 | 0.9786 | 0.9793 | 1.0781 |
| Resolution range (Å) | 40.49–1.95 (2.02–1.95)a | 29.67–1.97 (2.04–1.97) | 35.98–2.8 (2.9–2.8) | 34.37–2.6 (2.69–2.6) |
| Space group | P212121 | P212121 | P21 | P21 |
| Unit cell | ||||
| a, b, c (Å) | 48.03, 51.18, 198.61 | 65.87, 68.43, 128.40 | 67.46, 69.35, 73.58 | 84.94, 72.7, 120.57 |
| α, β, γ (degrees) | 90, 90, 90 | 90, 90, 90 | 90, 110.18, 90 | 90, 94.42, 90 |
| Molecules/asymmetric unit | 2 | 1 | 1 | 2 |
| Total reflections observed | 369,694 | 272,803 (23,046) | 44,230 (1,319) | 107,514 (10,036) |
| Unique reflections | 36,225 (3,587)b | 41,937 (4,049) | 26,165 (1,065)b | 72,310 (7,177)b |
| Multiplicity | 5.4 (4.7) | 6.5 (5.7) | 1.7 (1.2) | 1.5 (1.4) |
| Data completeness (%) | 98.7 (99.9) | 99 (97) | 84.8 (34.8) | 81.8 (81.8) |
| Mean I/σ(I) | 10.4 (3.1) | 23.70 (10.37) | 6.08 (1.81) | 6.22 (2.15) |
| Wilson B-factor | 39.50 | 31.09 | 50.55 | 47.95 |
| Rmerge | 0.07 (0.39) | 0.057 (0.16) | 0.0811 (0.254) | 0.0772 (0.351) |
| Rmeas | NAc | 0.063 (0.17) | 0.113 (0.354) | 0.109 (0.495) |
| CC½ | NA | 0.995 (0.987) | 0.988 (0.927) | 0.982 (0.667) |
| CC* | NA | 0.999 (0.997) | 0.997 (0.981) | 0.995 (0.894) |
| Reflections used in refinement | 36,224 (3,585) | 41,902 (4,048) | 26,126 (1,064) | 72,308 (7,177)b |
| Reflections used for R-free | 1,808 (178) | 2,021 (196) | 1,371 (58) | 3,153 (299)b |
| Rwork | 0.184 (0.290) | 0.177 (0.207) | 0.205 (0.311) | 0.224 (0.330) |
| Rfree | 0.219 (0.336) | 0.214 (0.241) | 0.246 (0.364) | 0.256 (0.373) |
| CCwork | NA | 0.957 (0.937) | 0.944 (0.715) | 0.940 (0.659) |
| CCfree | NA | 0.931 (0.922) | 0.938 (0.561) | 0.951 (0.578) |
| Non-hydrogen atoms | 3,720 | 4,605 | 4,277 | 8,591 |
| Protein | 3,449 | 4,209 | 4,251 | 8,454 |
| Ligands | 28 | 1 | 2 | 0 |
| Solvent | 243 | 395 | 24 | 137 |
| Protein residues | 431 | 538 | 543 | 1,082 |
| RMSD bonds (Å) | 0.005 | 0.004 | 0.002 | 0.002 |
| RMSD angles (degrees) | 0.74 | 0.63 | 0.45 | 0.54 |
| Ramachandran favored (%) | 98 | 98 | 98 | 98.23 |
| Ramachandran allowed (%) | 1.9 | 1.7 | 2.6 | 1.77 |
| Ramachandran outliers (%) | 0 | 0 | 0 | 0 |
| Rotamer outliers (%) | 1.9 | 0.22 | 0 | 0.22 |
| Clash score | 1.43 | 1.79 | 1.06 | 2.85 |
| Average B-factor (Å2) | 56.68 | 48.90 | 51.41 | 66.69 |
| Protein | 56.76 | 49.36 | 51.46 | 67.08 |
| Ligands | 70.64 | 29.80 | 41.87 | |
| Solvent | 53.84 | 44.03 | 42.83 | 42.56 |
| TLS groups | 8 | 5 | 3 | 5 |
a Statistics for the highest-resolution shell are shown in parentheses.
b Bijovet-related reflections are counted independently.
c NA, not available.
The structure is predominantly helical, consisting of 14 α-helices (Fig. 1). Near the N terminus of the structure, there are two approximately antiparallel α-helices, H1 and H2, connected by a 5-residue β-like strand. These helices do not contact each other, but both straddle helix H3 lying beneath them. The H2-H3 loop (positions 53–58) in each molecule has poor electron density, but the path of the main chain could be discerned in molecule A. Helical pairs H3 and H4, H5 and H6, and H7 and H8 as well as H9 form 3½ tetratricopeptide repeat-like (TPR-like) helix-turn-helix motifs. They pack against each other in a canonical superhelical fashion to provide the protein with concave and convex surfaces (Fig. 1B). The 6-residue helix H10 is approximately perpendicular to H9 and is not part of a TPR-like motif. H11 and H12 form a TPR-like motif but do not participate in superhelical packing, as do the other helix-turn-helix motifs. The short C-terminal helices H13 and H14 precede what in the full-length LpoA molecule is the linker strand (residues 251–257) between the N and C domains (see below).
Figure 1.

Solvent-accessible residues of LpoA-N are mostly nonconserved. A and B, schematic representation of the HiLpoA-N structure (chain A only). Helices of each TPR-like motif have a unique color, except for gray (H1-H2, H13-H14) and green (H9-H10), which are not TPR-like motifs. Views are perpendicular into the groove (A) and a side view (B). C, contact surface of the LpoA-N structure with coloring calculated by Consurf (62) from a sequence alignment of 74 putative LpoA amino acid sequences taken from the RP75 (March 2014) set of representative genomes (25) in Pfam PF04348 and aligned with Muscle in Jalview (61). Alignments are available in the supplemental materials. Sequences >96% identical or <200 residues in length were excluded. Front and back views of the domain are shown. Cyan surface, residues that are most variable; magenta, residues that are the most conserved. The black arrow points to a small highly conserved pocket (see “Results”). Note that surface accessibilities of certain residues may differ in LpoA-N structures from other species. D, same as C, but the input to Consurf was an alignment of only the seven sequences from the family Pasteurellaceae. E, same as C, but the input to Consurf was an alignment of 51 sequences from the family Enterobacteriaceae. Yellow surfaces, residues that the Consurf algorithm was not able to assign a statistically significant conservation score.
The structure of HiLpoA-N can be considered as composed of two subdomains. Subdomain 1, composed of residues 33–148 (H1–H7), contains two three-helix bundles, H3–H5 and H5–H7, each having nonpolar residues at its core. In subdomain 2, residues 149–249, nonpolar side chains from H11 interact with those from H8–H10, and these helices interact with residues downstream to form a second nonpolar core that includes three Trp residues and other aromatic residues. Other than contacts between H7 and H8, there are few interactions between subdomain 1 and subdomain 2, which allows for flexibility within the N domain, as discussed below.
This division of LpoA-N into two distinct subdomains is also suggested by a comparison of the HiLpoA-N crystal structure with the NMR structure of the E. coli LpoA N domain (19). HiLpoA-N and EcLpoA-N have sequences that are 30% identical, and both form similar TPR-like motifs. However, whereas the structures of residues 33–149 and of 150–248 of HiLpoA-N are similar, respectively, to the equivalent regions in EcLpoA-N (Fig. 2, A and B), the orientation of these two regions relative to one another in the HiLpoA-N structure differs markedly from that in the EcLpoA-N structure, by a ∼45° rotation (Fig. 2C). This difference is consistent with the EcLpoA-N structure not showing the typical superhelical conformation of TPR-containing domains (19).
Figure 2.

The LpoA-N TPR-like domain has a typical superhelical twist in contrast to the EcLpoA-N NMR structure. The α-carbon traces of EcLpoA-N (PDB entry 2MYH) (19) are shown in cyan and HiLpoA-N in magenta. A, subdomain 1 of EcLpoA-N (residues 31–146) was superposed onto the corresponding region of HiLpoA-N. The RMSD of the Cα positions was calculated to be 1.6 Å for 94 of the 116 superposed residues selected by “super” in PyMOL. B, the latter portion of EcLpoA-N (residues 151–248) was superposed onto the corresponding region of the HiLpoA-N structure, with an RMSD = 1.7 Å for 98 of the 114 residues superposed. C, overlay of the HiLpoA-N (magenta) and the EcLpoA-N structures (cyan) after fitting their subdomains 1 as in A. The view is along the rotation axis (black circle) that relates the C-terminal portions (subdomains 2) of the two N-domain structures and passes through the center of helix H7 at residue 139 of HiLpoA-N.
Most solvent-exposed residues of LpoA-N are not conserved
To identify the surfaces of LpoA-N that are conserved, we aligned the RP75 subset of LpoA sequences from the Pfam family PF04348 (accessed March 2014) (25, 26) that were >200 residues long. This alignment resulted in 74 sequences from 40 genera of γ-proteobacteria. This analysis, when including all of the sequences, revealed the LpoA N domain to have few conserved solvent-exposed residues (Fig. 1C). A small pocket on the convex side of LpoA-N surrounded by the highly conserved residues His-235, Ala-236, and Trp-204 was also found (arrow in Fig. 1C). The lack of conservation of the surface of the N-terminal domain stands in marked contrast to the C domain (see below). In the context of the full-length HiLpoA structure, the C domain has 4 times the percentage of solvent-exposed area considered highly conserved (magenta in Fig. 1C) compared with the N domain. The C domain has been shown in E. coli to be functionally indispensable for cell growth in an LpoA-dependent cell growth assay and for binding to EcPBP1A (8, 10). Also, this lack of conservation anywhere on the LpoA N-domain surface contrasts with an assessment that only included sequences from the Pasteurellaceae or Enterobacteriaceae families (Fig. 1, D and E). This latter assessment did reveal highly conserved regions, on the concave side of the N domain, including the loop between H3 and H4 (residues 76–78 and 81), the surface of H2 and H3, and the H9–H14 interface, including residues Gln-172, Asp-176, and Lys-243.
LpoA-N forms a groove like TPR-containing peptide-binding proteins
The packing of TPR-like motifs in LpoA-N creates a groove on the concave surface directly over H7 with side chains from H3 and H5 and H8 and H9 forming the walls of the groove (Figs. 1B and 3 (A and B)). This groove is closed at one end by residues Asp-41, Arg-136, and Arg-170, which are located on the H1-H2 connector, H7, and H8, respectively (Fig. 3A). Arg-136 and Arg-71 are conserved in several bacterial families, and these two residues form a pocket directly under the H1-H2 connector at the closed end of the groove (Fig. 3C). A sulfate ion is bound in the pocket of the LpoA-N chain A molecule. Electron density at the same position in chain B could not be identified and was modeled as glycerol. Electrostatic calculations showed the LpoA-N cleft to have an overall positive charge (Fig. 3B), with the basic residues Arg-158, Arg-170, Arg-75, Arg-106, and Lys-177 facing this groove (Fig. 3C). Glu-146, Asn-147, and Arg-158, also point into the groove and are mostly conserved in Pasteurellaceae and Enterobacteriaceae.
Figure 3.

Groove on the concave surface of LpoA-N. A, enlarged view of polar side chains that are exposed in the concave groove of the N domain. B, solvent contact surface of the N-domain groove, viewed as in A, and colored by electrostatic potential as calculated by APBS (20). The range of the potentials shown (red to blue) is −4 to 4 e−/Å3. C, sulfate (orange/red) bound in the groove of the LpoA-N structure and ligated directly by three Arg and through water to another Arg. Side-chain sticks are colored according to their degree of conservation calculated with sequences only selected from the Pasteurellaceae and Enterobacteriaceae families. D, the TPR-containing domain of NtrR (top, PDB entry 4GPK, residues 68–301) bound to its cognate peptide NtrX (magenta, residues 25–32) and LpoA-N (bottom). The first helix-turn-helix (red) motif of LpoA-N was oriented with the second helix-turn-helix motif (blue) of NtrR. For reference, the location of the NtrX peptide in the context of LpoA-N (in the groove) is shown in gray sticks.
A comparison of the crystal structure of LpoA-N with other proteins suggests that the groove in LpoA-N may be suitable for binding peptides or proteins. To find structural homologs of the LpoA-N structure, we compared it with other protein structures with DALI (27). All of the homologs superposed onto the TPR-like motifs (H3–H9) of HiLpoA-N but often had more repeats than did LpoA-N. No significant structural homologs were found for the non-TPR region (helices H10–H14) of LpoA-N. The highest scoring homologs included members of a family of cytoplasmic quorum-sensing receptors from Gram-positive bacteria (28) that bind small imported peptides. The RapI receptor (PDB entry 4ILA) without bound peptide displayed the highest sequence identity (21%) with LpoA-N (139 residues superposed with an RMSD of 3.1 Å). NprR (PDB entry 4GPK) from Bacillus cerius is another member of this transcription regulator family and had the highest Z-score (13.0; a measure of structural similarity) in the DALI search. This structure contains the signaling peptide from NprX bound in the concave cleft of its TPR domain (29). When LpoA-N was superposed onto NprR by aligning the first TPR motif (H3 and H4) of LpoA-N to the second TPR motif of NprR, the peptide-binding cleft of NprX coincided with the groove of LpoA-N (Fig. 3D). Similar to LpoA-N, the NprR-binding groove also contains polar side chains, which in NprR interact with the main chain of the bound NprX peptide.
Full-length LpoA is not extended, and the linker mostly interacts with the C domain
Structures of the full-length HiLpoA were determined from three different crystal forms. The orthorhombic crystal structure (LpoAOrt), with one molecule in the asymmetric unit, included residues 34–573 and was refined to 2.0 Å resolution with an Rwork/Rfree = 0.17/0.22 (Fig. 4A and Table 1). We also refined structures with data from two different monoclinic crystal forms of LpoA: LpoAMon to 2.8 Å with Rwork/Rfree = 0.21/0.25, and LpoAMon2 with two molecules in the asymmetric unit to 2.6 Å and Rwork/Rfree = 0.22/0.26. Whereas detailed differences between these HiLpoA structures relevant to the flexibility of the molecule were observed and are discussed later, here we first describe the structure of LpoA in the orthorhombic form (LpoAOrt), being of highest resolution.
Figure 4.

Full-length structure of LpoAOrt. A, schematic representation of LpoAOrt structure with the N domain colored yellow, C domain in blue, and linker (residues 251–256) in green. The black arrow indicates the view in B and C. B, schematic representation of LpoAOrt highlighting the C domain and its N lobe (blue) and C lobe (orange). The view is looking directly into the C-domain cleft (see arrow in A). C, solvent contact surface representation of LpoAOrt in the same view as B and colored according to sequence conservation (same color scheme as Fig. 1C). Highly conserved residues, suggesting a functional binding site, were observed in the cleft between the two lobes of the C domain. D, electronegative region on “top” of the C domain facing the N domain. The view is in the direction of the red arrow in C. E, residues forming the electronegative surface in D. Residues 345–350 are shown in green and are discussed under “Results.” F, cross-section of LpoA showing how the electronegative region at the top of the C domain is opposite the N-domain electropositive groove. The protein surface is colored by electrostatics.
The N domain and C-terminal domain (C domain, residues 257–575) are arranged to form a U-shaped (jaw-like) structure, with the longest dimension being 91 Å between residues 91 and 505 in LpoAOrt, and a width ranging from 35 to 75 Å (Fig. 4A; see also “LpoA flexibility suggested by structure comparisons, normal mode analysis, and small-angle X-ray scattering”). We estimated the maximum length of HiLpoAOrt to be ∼105 Å, on the assumption that residues 26–32 (not present in the crystallized protein) are unstructured as they are in the EcLpoA-N NMR structure (19).
The N domain is joined to the first β-strand of the C domain through a 7-residue linker polypeptide, residues 251–257 (Fig. 5B). The linker is in an extended conformation with residues 252–257 primarily in a crevice on the side of the C domain and Phe-251 making van der Waals contacts with both N and C domains (Fig. 5B). About 50% of the surface area of the linker is exposed to bulk solvent. There are only a few direct hydrogen bonds between the linker residues and the rest of the protein (see the legend to Fig. 5), with other polar interactions mediated by water molecules. The C domain also interacts directly with the N domain through van der Waals interactions (buried surface area = 270 Å2) and at least six water-mediated polar interactions (Fig. 5C).
Figure 5.
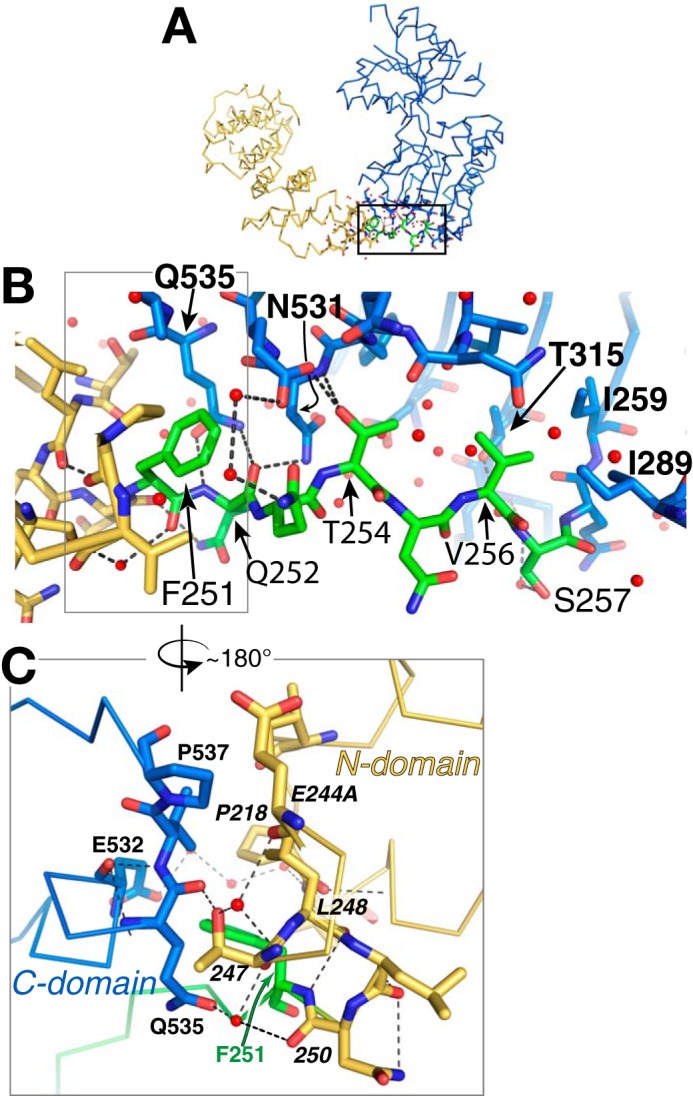
LpoAOrt linker interacts primarily with the C domain. A, full-length LpoAOrt showing a rectangle corresponding to the enlarged view of B. B, this view of LpoAOrt shows linker residues 251–257 colored green, the N domain in gold, and the C domain in blue. Red spheres, ordered water molecules. Black dashes, hydrogen bonds between the linker and the rest of the protein. Observed linker–protein interactions include hydrogen bonds from the Gln-252 carbonyl oxygen to the Asn-531 and Gln-535 (C domain) side chains and from the Phe-251 amide to the Gln-535 side chain. Also, the Thr-254 hydroxyl contributes hydrogen bonds to both the Glu-532 side chain and the main chain of residue 531. All other polar interactions with the linker were observed to be mediated by water molecules. Val-256 was found to make nonpolar interactions with the side chains of residues 259, 289, and 315 from the C domain. C, the boxed region in B is enlarged and rotated ∼180° to show residue Phe-251 centered at the small nonpolar interface of the N and C domains and making nonpolar contacts with Phe-218, Val-219, and Leu-248 from the N domain and residues 535–536 from the C domain.
The C-domain structures in LpoAOrt and LpoA-C are very similar
The C-terminal domain of the LpoAOrt structure is very similar to the LpoA-C (PDB entry 3CKM, C-terminal domain only) structure determined previously (supplemental Fig. S2) (16). The domain consists of two lobes (N lobe and C lobe) connected by three polypeptide strands and separated by a large cleft (Fig. 4B). Each lobe superposes on the corresponding lobe of the 3CKM structure with an RMSD of 0.3 Å. Based on the concentration of highly conserved residues between the lobes (Fig. 4C), we previously proposed it to be a ligand-binding cleft (16). Two regions of LpoAOrt differed significantly from the 3CKM structure of LpoA-C. Residues 425–434 have a different conformation, which is probably due to different crystal packing. One loop near the cleft (residues 345–350) was reported to have ambiguous density in the previous 3CKM structure but is well-resolved in LpoAOrt and adopts a conformation different from that previously modeled in 3CKM (green in Fig. 4E and supplemental Fig. S3). Consequently, this opened a path from the top of the binding cleft into a partially conserved and electronegative region at the “top” of the C domain, which faces the N-domain groove described above (Fig. 4, D–F). Differences between the C domain in the four full-length structures are discussed below.
LpoA flexibility suggested by structure comparisons, normal mode analysis, and small-angle X-ray scattering
By comparing the four full-length crystal structures of LpoA, we observed at least three modes of flexibility: twisting of the TPR-like motifs in the N domain, hinge bending between the two lobes of the C domain, and C-domain rotation at the end of the interdomain linker. Surprisingly, most of the linker region as defined above assumes a very similar conformation in all LpoA crystal structures (supplemental Fig. S4).
To assess the conformational variability of the LpoA N domain, we superposed subdomain 1 (i.e. residues 33–148 (H1–H7)) of each of the two N domains in the LpoA-N crystal and the N domains from LpoAOrt and LpoAMon onto each other and calculated the rotations necessary to superpose each of the subdomain 2 regions (residues 149–246) (Fig. 6A). They all showed 4–5° rotations, but along different axes, implying a general flexibility between these two subdomains. Such rotations within the N domain, although relatively small in magnitude, can lead to considerable effects on the position of the C domain. For example, the twisting of the N domain in the LpoAMon structure is responsible for rotating the C domain (N lobe) by 4.4°, effectively shifting it an average of 5 Å in the direction of the linker in comparison with the LpoAOrt structure (supplemental Fig. S5). No change in the linker conformation was observed.
Figure 6.

Comparison of LpoA domain structures from different crystal forms. A, N domain twists and flexes. Overlay of the N domains of chain B of LpoA-N (cyan), LpoAOrt (magenta), and LpoAMon (orange) on that of chain A of LpoA-N (gray), based on least-squares fittings of their subdomain 1 (residues 33–149, left half of the figure). Rotations of the first three N-domain structures by about 4.7, 4.0, and 4.3°, respectively, around the correspondingly colored rotation axes yielded the best superpositions of their subdomains 2 (residues 150–248, right half of the figure) onto that of chain A of LpoA-N. This rotation axis in LpoAOrt (magenta) passes through residue 148 and is approximately parallel to H5, whereas that in LpoAMon (orange) is almost parallel to H8. The rotation axis for chain B of LpoA-N (cyan) runs through the center of subdomain 2 and is almost perpendicular to the LpoAMon axis. B, hinge bending in C domain. Shown is an overlay of the C domain of LpoAMon2 chain B (green) on that of LpoAMon (orange), after fitting their N lobes (residues 360–488 and 560–573) to each other. The C lobe of the LpoAMon2 chain B C domain was observed to be rotated, here by 12.1° around the green axis, with respect to the LpoAMon C lobe. Supplemental Fig. S2 shows two other C-domain comparisons. C, the consequence of the two lobes of the C domain spreading apart in B is that the LpoAMon2 chain B (bottom; green solvent contact surface) has a more direct pathway (black arrow) to the electronegative region on top of the C domain (red arrow; also see Fig. 4E). In contrast, in the LpoAMon (top, orange) C-domain structure, access to the electronegative region is blocked. This may affect the site accessibility to a bound polypeptide or PG strand.
Previously, we reported that normal mode calculations of LpoA-C (PDB entry 3CKM) indicated that a hinge region between the two lobes of the C-terminal domain would allow the cleft between the two lobes to open and close, similar to differences actually observed in multiple crystal forms of the Leu/Ile/Val-binding protein and other periplasmic binding proteins (16, 18). Comparing the two lobes of the C domain in all full-length crystal structures shows that the maximum difference is between the C domains of LpoAMon and LpoAMon2 chain B, where the cleft opening increases by 12.1° around an axis passing near the hinge region between the two lobes (Fig. 6B; also see supplemental Fig. S2B). Notably, residues Arg-393 and Lys-322, which block access to the electronegative region on the top side of the C domain in the LpoAOrt and LpoAMon structures (Fig. 4E), are farther apart in LpoAMon2 chain B, extending the putative binding cleft region (Fig. 6C).
Results of normal mode analyses (NMA) often reflect actual protein dynamics observed in solution (30–33). To examine the potential for flexibility in full-length LpoAOrt, we calculated the normal mode-derived motions of LpoAOrt with an arbitrary amplitude of 500 (34) and compared the starting structure with the final structure. In the final NMA structure (NMA(LpoAOrt); Fig. 7A, left), the N-domain superhelix was slightly unwound, and the C domain was rotated ∼25° with respect to the starting conformation.
Figure 7.

LpoA crystal structures progressively extend until they reach the NMA prediction. A, overlay of LpoAOrt (gray surface representation) on the extended model (magenta) predicted by normal mode calculations by ElNèmo (34) for mode 7 and amplitude = 500. The overlay was based on fitting residues 33–148 (subdomain 1) of the N domain of LpoAOrt to that of the normal mode model and revealed the largest intramolecular distance change (23 Å) between LpoAOrt and the predicted structure (right; helices (cylinders) and β-strands (arrows)). B, comparison of the four crystal structures and two models calculated by NMA (mode 7, amplitude = 500). Subdomains 1 of the N domain of each structure are identically oriented. The RMSDs of the Cα coordinates for each crystal structure compared with the NMA(LpoAOrt) model were 6.3 Å for LpoAMon, 5.5 Å for LpoAOrt, 3.6 Å for LpoAMon2 molecule A, and 2.5 Å for LpoAMon2 molecule B, showing that the latter structure is most similar to the NMA results. The NMA(LpoAOrt) had a calculated radius of gyration (32.4 Å) very close to that determined directly from the SAXS data (32.6 Å). A similar figure with the molecules rotated ∼90° is shown in supplemental Fig. S6A. The image at the far right is one of several hypothetical EcLpoA models, constructed by putting the two domains end-to-end, that was consistent with the Rg from SAXS data (19). The image is from a supplementary figure published in Ref. 19 and reproduced with permission. The multiple ensemble search of the HiLpoA crystal structures and NMA models also suggests that HiLpoA samples can sample extended structures, such as the NMA(LpoMon2B) model. C, the linker regions (green) of LpoAOrt (black) on LpoAMon2B (red) were superposed to reveal that the C domain of LpoAMon2B rotates around an axis (red solid arrow) approximately parallel to the linker but with the pivot point at the end of the linker. The view is approximately perpendicular to the rotation axis. D, superposing the N domain of LpoAOrt (gray) to that of LpoAMon2B (red) shows a relative displacement between their C domains as large as 33 Å at residue 453. E, N domains of LpoAMon2B (red) and NMA(LpoAMon2B) (purple) superposed to show how NMA(LpoAMon2B) is more extended and rotates an additional 25° from LpoAMon2B. The orientation is slightly different from that in D, and domains are not shown to scale.
To assess whether the conformation predicted by the normal mode analysis actually occurs experimentally, we compared the NMA(LpoAOrt) prediction with the two molecules (chains A and B) in the asymmetric unit of the LpoAMon2 crystal form. Each of these two molecules (green and red in Fig. 7B and supplemental Fig. S6A) was observed to adopt a conformation on the path to a more extended structure similar to that predicted by the above normal mode calculation. After superposing subdomain 1 (residues 33–148) of the N domains, there were two components of the conformational change between LpoAOrt and the LpoAMon2 structures. In the N-terminal domain, subdomain 2 (residues 149–250) of both molecules from LpoAMon2 was observed to be rotated 5° with respect to subdomain 1, resulting in a 2–3 Å movement of the C domain (not shown); second, the C-terminal domains of the two molecules (A and B) of the LpoAMon2 asymmetric unit were rotated by 20 and 30°, respectively, relative to the LpoAOrt structure (Fig. 7B). In the latter case, the net rotation is around an axis almost parallel to the linker strand (Fig. 7C). As in NMA(LpoAOrt), the conformational change was observed to be mediated primarily by slight changes in the main-chain dihedral angles of residues 256–259 at the end of the linker (Fig. 7C). The resulting largest difference between LpoAOrt and LpoAMon2 molecule B was at residue 453 of the C domain, which moves 33 Å (Fig. 7D). The largest change in an intramolecular distance was 25 Å between residues 54 and 454 of the two structures (supplemental Fig. S6B). The longest dimension in the LpoAMon2 molecule B structure, at 99 Å, is 8 Å greater than that measured in the LpoAOrt structure (supplemental Fig. S6C). The consequence of this conformational change is that the center of the putative binding groove (around residue 360) in the C domain tilts and moves 13 Å away from the N terminus, which in the periplasm would be toward the inner membrane. To further quantify the similarity, we calculated the RMSDs of all of the full-length structures superposed onto the extended structure predicted by normal mode analysis (Fig. 7B). Molecule B of the LpoAMon2 structure was most similar to the extended structure predicted by the NMA of LpoAOrt with an RMSD of 2.5 Å. Repeating the NMA calculation (with amplitude of 500) starting from molecule B of the LpoAMon2 structure resulted in NMA(LpoAMon2) with another ∼25° rotation of the C-domain movement relative to LpoAMon2 and the longest dimension of the protein now extending 116 Å (at maximum length excluding residues 26–32) and a predicted radius of gyration (Rg) of 34.9 Å (Fig. 7, B and E).
To further validate the LpoA structures, we measured small-angle scattering curves at four concentrations of LpoA(29–575) (supplemental Fig. S7A). The small-angle X-ray scattering (SAXS) data show that LpoA is monomeric in solution. We estimated the molecular weight from SAXS data in relative scale using two methods. The molecular weights are 57,489 and 63,859 from methods of correlation volume (35) and apparent volume (36), respectively. The estimations were made with scattering data extending to q = 0.25 Å−1 (q = 4πsinθ/λ). The theoretical Mr was 60,518. The Guinier plots (Fig. 8A) show that the intensity at zero angle is proportional to the protein concentration, and the experimental Rg decreases with increasing concentration. Such an effect can be eliminated by extrapolation to infinite dilution (zero concentration). The Rg at infinite dilution is 32.6 ± 0.6 Å in reciprocal space from the Guinier plot (Fig. 8B) and 33.1 ± 0.3 Å in real space calculated by the GNOM software (37).
Figure 8.

Small-angle X-ray scattering data. A, Guinier plots of X-ray small-angle scattering intensities measured at four concentrations of LpoA. Shown are the Rg values derived from the linear portion of the Guinier plot. The reciprocal lattice vector length q is 4πsinθ/λ (wave number). The plot was in the q range, where Rg × qmax is ≤1.3. B, this Guinier plot obtained after data extrapolation to zero protein concentration gives a radius of gyration of 32.65 Å. C, the Porod-Debye plot does not have the typical Porod plateau, indicating a flexible protein structure (40). D, SAXS data are plotted as a dimensionless Kratky plot based on Rg (41). See “Results” for significance. E, predicted scattering from the NMA(LpoAOrt) structure (red) fit best to the experimental scattering curve (black) with χ = 1.25. The lower section of the graph shows the residual, calculated as the ratio between the two curves. F, same as E, but the scattering curve (red) for a 3-model ensemble (LpoAOrt, NMA(LpoAOrt), NMA(LpoAMon2)) is fit to the observed data (black) with χ = 0.80. G, the three structures constituting the ensemble. The most extended structure in the ensemble, NMA(LpoAMon2B), has a 45% volume fraction, suggesting that it is a significant conformer in solution. H, pairwise distance–distribution plot, P(r). The maximal distance (Dmax) of 118 Å derived from this is consistent with the most extended structure considered here, NMA(LpoAMon2B), with maximum interatomic distance of 116.7 Å.
Predicted Rg values for each of the four LpoA models in the crystal structures and for the models predicted by normal mode analysis of LpoAOrt and LpoAMon2B were calculated with Crysol (38) (Fig. 7B). The Rg(calc) for the NMA of LpoAOrt (Rg(calc) = 32.4 Å) was closest to the Rg from the SAXS experimental data. Predicted scattering profiles for the models were fit to the observed data with FoXS (39) that included parameters for a hydration layer, excluded volume adjustment, and implicit hydrogens and are shown in Table 2 (left). Again, NMA (LpoAOrt) fit best with χ = 1.25 (Fig. 8E).
Table 2.
Results of fitting each LpoA structure to the observed SAXS data with FoXS (64) (left); an ensemble of three models chosen by multiple ensemble search in FoXS gave the best fit (right)
Also see Fig. 8, E and F.

The HiLpoA SAXS results are in contrast to the SAXS analysis of EcLpoA (19). After comparing the distance distribution function P(r) derived from EcLpoA SAXS data with those calculated from various models, Jean et al. (19) showed that, in comparison with other models, a fully extended EcLpoA was consistent with an observed Rg = 42.2 Å (Fig. 7B, far right). Although NMA(LpoAOrt) best fit the HiLpoA SAXS data, two observations suggested that the molecule samples other conformations, perhaps more elongated than that observed in the crystal structures. The Porod-Debye plot (Fig. 8C) calculated from the SAXS data did not show the Porod-Plateau, which suggested that the LpoA protein in solution is more flexible (40). Second, an Rg-based, dimensionless Kratky plot (Fig. 8D) (41) shows an asymmetric bell shape and the peak position (1.97, 1.21) shifted to higher values with respect to a globular protein (1.73, 1.11). This indicated that the protein has structured domains linked by a flexible segment and can adopt a more elongated shape than a globular protein.
Consistent with the above, we obtained a better fit (χ = 0.80) to the HiLpoA SAXS experimental data when an ensemble of three models (42) were considered together, each with a specific volume fraction: LpoAOrt 0.27, NMA(LpoAOrt) 0.28, and NMA(LpoAMon2) 0.45 (Table 2 (right) and Fig. 8 (F and G)). Note that the structure with the highest volume fraction, NMA(LpoAMon2), is the most elongated, with a C-domain rotation of about 40–50° compared with the LpoAOrt conformation, yet this structure by itself does not fit the experimental data as well as NMA(LpoAOrt) (χ = 2.18 versus 1.25; Table 2, left). This means that the ensemble may be a better interpretation of the SAXS data with the closed and more extended structures existing simultaneously in solution. The presence of an extended structure like NMA(LpoAMon2), with a maximum interatomic distance of 116.7 Å, is also consistent with the Dmax value of 118 Å determined from the pairwise distance–distribution plot, P(r) (Fig. 8H). This also would support the ability to adopt an extended conformation in vivo for interaction with PBP1A.
Discussion
This paper reports crystal structures for the N domain and full-length HiLpoA as well as alternative conformations suggested by normal mode calculations and consistent with the SAXS analysis. The U-shaped (or L-shaped) arrangement of the two domains in the full-length HiLpoA structures presented here is very different from the elongated model proposed for EcLpoA in solution (see below; Fig. 7B) (19). In HiLpoAOrt, the nonspecific interactions between the linker and C domain may restrain the position of the linker (along the side of the C domain) and result in the observed orientation of the two domains in the HiLpoA full-length structures. Despite the differences in domain orientation observed in the different full-length crystal structures, the linkers have very similar structures. Whereas the linker is the same length in all LpoA homologs, its sequence is not highly conserved. Phe-251, which makes nonpolar contacts with both domains of HiLpoA, is only present in Pasteurellaceae and some Enterobacteriaceae sequences. Several sequence families (including the Enterobacteriaceae and Shewanellaceae) have proline at position 253 (HiLpoA numbering), which, when modeled in HiLpoA, does not affect the linker conformation but might affect its flexibility. Similarly, species from Shewanella, Pseudomonas, and Vibrio have a Pro at 256 in place of Val. In summary, the linker sequence may regulate the conformational flexibility of LpoA, perhaps reflecting the peptidoglycan architecture of a particular species.
Need for LpoA flexibility
During PBP1A-mediated PG synthesis (at least in E. coli), LpoA must extend its C domain a sufficient distance toward the inner membrane to interact with the ODD domain (also called the OB domain due to its oligosaccharide-binding-like domain fold (43)) and TP domains of PBP1A (8). The LpoA architecture must provide conformational flexibility for at least two reasons. First, the width of the periplasm, the thickness of the peptidoglycan, and the distance of PG from the outer membrane probably differ between families of bacteria (e.g. see Ref. 44). Noting that the width of the periplasm in E. coli was shown to be 200 Å (44), Jean et al. (19) proposed, based on small-angle X-ray scattering data, that the EcLpoA full-length structure is extended with an overall length of 145 Å. They reasoned that this length would allow the C domain to bind to the PBP1A ODD domain. However, this model may not hold for other species because periplasm width varies among species. Analyses by atomic force and electron microscopy of the envelope of Aggregatibacter actinomycetem-comitans, a species in the same Pasteurellaceae family as H. influenzae, revealed that the outer membrane is highly rugose (ruffled) (45). The distance between the relatively smooth inner membrane and the outer membrane fluctuates by as much as 150 Å. According to the authors, this feature is also present in H. influenzae (45).
The second reason for flexibility is to differentiate between intact PG and the presence of gaps, as suggested previously (22). Intact sacculus has holes/pores of about 40 Å in diameter. Due to the elasticity of PG, these holes would enlarge during cell growth, resulting in gaps requiring repair by a PG synthase (22, 46). It was proposed that the larger gaps would allow LpoA to extend through the sacculus and bind to PBP1A, resulting in a localized activation of PBP1A and insertion of new PG strands to repair the hole (7, 22) (Fig. 9). Because LpoA appears randomly distributed throughout the outer membrane (OM) (8), it is conceivable that in regions of intact PG, LpoA would need to “curl up” or assume a more compact conformation to fit in the space between the OM and PG (22) (Fig. 9). This would argue for a non-rigid LpoA molecule (i.e. one with conformational flexibility).
Figure 9.
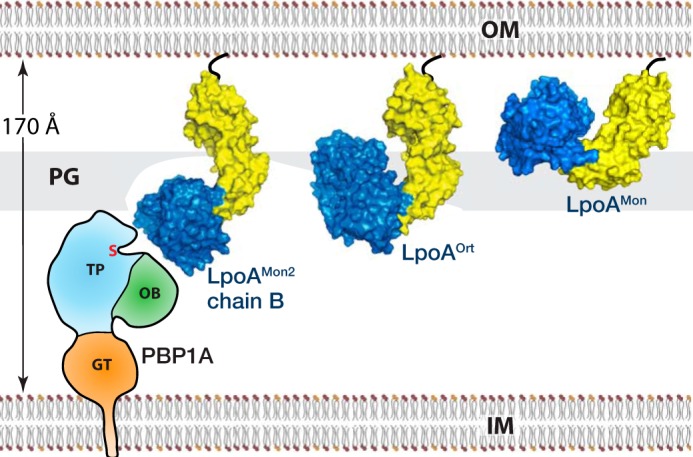
Model of how LpoA might interact with PBP1A. The three different LpoA crystal structures suggest how the conformational dynamics of LpoA could allow it to fit through holes in the peptidoglycan and interact with the peptidoglycan synthase PBP1A (schematic representation of A. baumannii PBP1A (PDB entry 3UDF)). Shown from right to left, LpoAMon or a more compact structure would rotate to fit above the PG, here assumed to be at 60 Å below the OM. A structure similar to LpoAOrt could detect the presence of a hole in the PG, and a more extended conformation like that observed for LpoAMon2 chain B or NMA(LpoAMon2B) would be long enough to interact with PBP1A. IM, inner membrane; OB, oligosaccharide binding–like domain (also referred to as ODD); GT, glycosyltransferase domain; S, catalytic serine.
N-domain flexibility
From comparing the crystal forms of HiLpoA, we observed three examples of flexibility: twisting and flexing of the N domain, rotation of the entire C domain with respect to the N domain, and hinge motion between the lobes of the C domain (Figs. 6 and 7). All of the structures showed small but significant flexing of the N domain (Fig. 6A). In the normal mode analysis of LpoAOrt, relative twisting of the TPR-like motifs changes the length of the N domain, causing changes in the position of the C domain with only minor changes in the main-chain torsion angles of the interdomain linker. The low sequence conservation of the N domain may reflect its ability to adopt a conformation or length specific to the cell wall structure of a particular species. The HiLpoA-N structure suggests that the N domain may have the important role in the positioning of the C domain at the appropriate location to interact with PBP1A. This role in C-domain positioning is also supported by the observation that the N-domain sequences of LpoA from several genera are shorter (e.g. Proteus, Providencia, Morganella, and Acinetobacter), which may correlate with different cell wall morphologies.
The LpoA N-domain structure, which adopts a crescent-shaped form typical of other TPR-containing domains, is strikingly different from the N-domain structure of EcLpoA determined by NMR (Fig. 2C) (19). This may be due to differences in sequence between HiLpoA-N and EcLpoA-N or reflect a difference in the crystal versus solution environment. Perhaps the conformational difference in EcLpoA-N reflects particular aspects of the periplasm (width?) or PG structure that are different from other Gram-negative bacteria, as mentioned above. For example, in E. coli, the Lpp outer membrane lipoprotein interacts both covalently and noncovalently with the PG, effectively fixing the PG at a relatively fixed distance below the outer membrane. However, H. influenzae lacks this protein, so the distance from the outer membrane to PG may vary.
The N-domain groove on the concave face of the domain resembles peptide- or protein-binding sites found on other TPR-like proteins. Recent reports suggest that LpoA may bind other polypeptides. CpoB is a periplasmic protein that localizes to the septum, where it interacts with E. coli PBP1B, LpoB, and the Tol proteins, and is necessary for PBP1B function. Gray et al. (47) showed that the N domain of LpoA could rescue the function of CpoB when PBP1B was the only functioning synthase. Because both CpoB and LpoA-N contain TPR-like motifs, the authors suggested that LpoA-N may interact with other PG-associated proteins or perhaps link to the Tol protein (47). Additionally, Dörr et al. (48) discovered in V. cholera that the periplasmic protein CsiV bound to both LpoA (specific domain not known) and PG and regulated the ability of LpoA to activate the PBP1A transpeptidase. CsiV is encoded in a subset of species that encode LpoA, but not in H. influenzae or E. coli. Perhaps there is another protein in E. coli or H. influenzae that serves this role.
Both molecules in the LpoAMon2 structure when compared with LpoAOrt show a 5° rotation within the N domain, which has the effect of translating the C domain (similar to movement observed in LpoAMon; supplemental Fig. S5B). In addition, the C domain rotates 20–30° around an axis parallel to the linker region, which in the cell positions the C domain farther away from the outer membrane and potentially closer to PBP1A. Interestingly, in the LpoAMon2 chain B structure, the observed C-domain rotation does not affect linker conformation significantly (supplemental Fig. S4) but is a result of main-chain torsion angle differences at residues 257 and 259 following the linker (Fig. 7C). This is consistent with the NMR measurements of full-length EcLpoA, which suggested a relatively rigid linker (19). The presence of four conformations in three crystal forms suggests that LpoA is sampling many different conformations in solution.
The SAXS results confirm that an ensemble of structures varying from the LpoAOrt to the NMA(LpoAMon2B) conformation may occur in solution, although in vivo, we would expect the distance between the outer membrane and peptidoglycan layer, as well as the thickness of the peptidoglycan, to limit the number of conformations actually sampled. The protein dynamics may also be limited by molecular crowding due to the high concentration of proteins in the periplasm. The movements are probably necessary for LpoA binding to PBP1A. In the absence of LpoA, the movement of PBP1A in the periplasm is fast and diffusive (49). When LpoA activates PBP1A catalytic activity or binds to PBP1A, the net mobility of PBP1A decreases (49). This is probably necessary for productive catalytic function and may be a major regulatory function of LpoA (49).
At least in E. coli, the LpoA C domain is crucial for the binding and activating of PBP1A (8, 10, 16). Superpositions of C-domain pairs LpoAMon on LpoAOrt and LpoAMon on LpoAMon2 showed a movement of the two lobes of the C domain and a narrowing or opening of the cleft between them (Fig. 6B and supplemental Fig. S2B). This observation suggests that LpoA-C (as well as the full-length structure) can sample multiple conformations in solution similar to that observed for LivJ, a structural homolog of LpoA-C (18). This may reflect an induced fit model for PBP1A (or possibly PG) binding. Although ABC transporter substrate-binding proteins often completely close the cleft on substrate binding, we suspect that the C-domain cleft of LpoA may not completely close because there is a highly conserved salt bridge between Arg-515 in the N lobe and Asp-488 (Glu in some species) in the C lobe that may restrain the open position of the two lobes (supplemental Fig. S8).
We estimate that the groove on the concave side of the N domain would lie ∼50 Å below the outer membrane (Fig. 9). Here it might make interactions with peptidoglycan. Consistent with this proposal is the finding that EcLpoA bound to peptidoglycan sacculi purified from E. coli (8). For the domain configuration observed in the LpoA full-length crystal structures, we expect LpoA (in the LpoAMon2 chain B or NMA(LpoAMon2)) to extend about 115 Å below the outer membrane, positioning the center of the C domain just below the presumed location of the peptidoglycan layer. The model depicted in Fig. 9 assumes a periplasm width of 170 Å and a HiPBP1A model with dimensions similar to the known structure of Acinetobacter baumannii PBP1A (43). Future structural studies of a PBP1A-LpoA complex will help clarify how the two proteins interact.
Experimental procedures
Plasmid construction
Oligonucleotides were purchased from Sigma Genosys Ltd. and Integrated DNA Technologies. Plasmids and PCR products were purified with kits from Qiagen or Promega. Restriction enzymes and polymerases were used as recommended by the manufacturers (Invitrogen, Promega, and New England Biolabs). Plasmid constructs were sequenced at the University of Michigan DNA Sequencing Core.
PCR primers and details of the plasmid constructs are shown in Tables 3 and 4. Genomic DNA isolated from the H. influenzae Rd strain (ATCC number 9008) (50) served as the PCR template. For plasmid pLpoAN-His6, the gene fragment corresponding to the N-terminal domain (residues 33–253) of H. influenzae LpoA was amplified by PCR with primers LpoA(33)_F and LpoA(253)_R, digested with NcoI and SalI, and ligated into pETBlueTM-2 (Novagen) cut with NcoI and XhoI. Similarly, pLpoA-His6, encoding LpoA residues 33–573, was constructed with primers LpoA(33)_F and LpoA(573)_R. Mutagenesis of pLpoA-His6 with QuikChange (Stratagene) and DNA oligonucleotides LpoA_delH6_F and LpoA_delH6_R resulted in pLpoA encoding the full-length molecule without the His6 tag: Met-Ala-LpoA(33–575). For the SAXS experiments, the pMCSG7-LpoA(29–575) expression plasmid encoding a cleavable N-terminal His6 tag was prepared by amplifying H. influenzae lpoA with PCR primers F_LIC_yraM(29) and R_LIC_STP_yraM(575) and cloning the product into pMCSG7 by ligation-independent cloning (51).
Table 3.
Oligonucleotide primer sequences
| Name | Sequence | Use |
|---|---|---|
| LpoA(33)_F | CATGCCATGGCGAATTTCACGCAAACCTTACAA | PCR |
| LpoA(253)_R | GCCGACGTCGACTTGTTGGAAATTAAGCAATGTAAG | PCR |
| LpoA(573)_R | GCCGACGTCGACAACTGGTACAATTGCACCATC | PCR |
| F_LIC_yraM(29) | TACTTCCAATCCAATGCACTACTTGGTAGCAATTTCACG | PCR |
| R_LIC_STP_yraM(575) | TTATCCACTTCCAATGTTAGTTTGCAACTGGTACAATTG | PCR |
| LpoA_delH6_F | GATGGTGCAATTGTACCAGTTGCCAACTAACACCACCAC | QuikChange |
| LpoA_delH6_R | GTGGTGGTGTTAGTTGGCAACTGGTACAATTGCACCATC | QuikChange |
| pETBlueUp | TCACGACGTTGTAAAACGAC | Sequencing |
| pETBlueDOWN | GTTAAATTGCTAACGCAGTCA | Sequencing |
| nt746 | CAGCGAATACTGGCG | Sequencing |
| nt1139 | GGCCCATTACTAAAACAA | Sequencing |
| nt1520 | GCAGAAATGAAAGGTTAT | Sequencing |
Table 4.
Bacterial strains and plasmids
| Name | Description/Protein expressed | Parent plasmid/PCR primers | Source/Reference |
|---|---|---|---|
| Strains | |||
| H. influenzae Rd | Source of genomic DNA | Ref. 50 | |
| NovaBlue | Novagen | ||
| OrigamiTM(DE3)/pLacI | Expression host strain | Novagen | |
| TunerTM(DE3)/pLacI | Expression host strain | Novagen | |
| Expression plasmids | |||
| pETBlue-2TM | T7 promoter expression | Novagen | |
| pLpoAN-His6 | MA-LpoA(33–253)-VE-His6 | pETBlue-2/LpoA(33)_F, LpoA(253)_R | This work |
| pLpoA-His6 | MA-LpoA(33–573)-VE-His6 | pETBlue-2/LpoA(33)_F, LpoA(573)_R | This work |
| pLpoA | MA-LpoA(33–575) | pLpoA-His6/LpoA_delH6_F, LpoA_delH6_R | This work (primers for QuikChange mutagenesis) |
| pMCSG7 | T7 promoter expression, LIC | pET21a | Ref. 51, kind gift of Dr. Mark Donnelly |
| pMCSG7-LpoA(29–575) | His6-(TeV_site)-NA-LpoA (residues 29–575) | pMCSG7/F_LIC_yraM(29), R_LIC_STP_yraM(575) | This work (expressed protein for SAXS measurements) |
LpoA-N
To express LpoA-N, E. coli TunerTM(DE3)/pLacI cells (Novagen) were transformed with pLpoAN-His6 (encoding MA-LpoA(33–253)-VE-His6). Typically, 1 liter of lysogeny broth (LB) supplemented with ampicillin (100 μg/ml) was inoculated with a 10-ml starter culture and incubated at 37 °C with shaking at 250 rpm until the apparent A600 reached 0.7. Isopropyl β-d-1-thiogalactopyranoside (IPTG) was added to a final concentration of 1 mm, and growth was continued for an additional 3 h at 37 °C. Cells were harvested by centrifugation at 3000 × g for 10 min. Selenomethionine (SeMet)-containing LpoA-N was expressed similarly, except before induction with IPTG, the cells were centrifuged, washed, and resuspended in M9 minimal medium (52) supplemented with amino acids (except Met) and 80 mg/ml SeMet. Both wild-type LpoA-N and LpoA-N(SeMet) were purified in a single step on an Ni2+-nitrilotriacetic acid-agarose column (Qiagen), as described previously (16). The purified protein was dialyzed into 20 mm Tris-HCl, pH 8.0, 0.1% β-mercaptoethanol, 1 mm EDTA, 0.1 mm benzamidine and concentrated to 10 mg/ml before storage at −20 °C.
Orthorhombic crystals of YraM-N and YraM-N(SeMet) were grown in hanging drops consisting of 2 μl of 10 mg/ml protein solution and 2 μl of precipitant (30% polyethylene glycol monomethyl ether 5000, 0.2 m ammonium sulfate, 0.1 m MES, pH 6.5) equilibrated against 1 ml of the precipitant by vapor diffusion. Crystals grew to a maximum size of 0.6 × 0.4 × 0.1 mm within a week. Crystals were harvested into precipitant solution containing 10% glycerol, mounted on loops, and immediately frozen in liquid nitrogen.
Native and heavy atom derivative intensity data from LpoA-N crystals were collected on a Rigaku R-Axis II detector and phased by multiple isomorphous replacement with SOLVE and RESOLVE (53). Electron density maps revealed two molecules per asymmetric unit, and an initial model was built with O (54). Diffraction images from LpoA-N(SeMet) crystals were collected at the absorption peak wavelength of 0.979 Å on a MAR-CCD detector at station 5ID, DND-CAT, Advanced Photon Source, Argonne National Laboratory. Reflection intensities to 1.95 Å resolution were integrated and scaled with d*TREK (55). Unit cell dimensions and statistics for the SeMet crystals are shown in Table 1. Structure refinement in PHENIX (56) with TLS parameters produced a model with Rwork/Rfree = 0.19/0.22 and excellent geometry. One sulfate was modeled in the electropositive pocket of molecule A in the LpoA-N structure (Fig. 3C), whereas density tentatively modeled as glycerol was observed in the corresponding pocket of molecule B. A sulfate was also observed to interact with Arg-148 of each molecule.
Full-length LpoA crystal structures
His-tagged full-length LpoA expressed from TunerTM(DE3)/pLacI cells transformed with pLpoA-His6 was susceptible to proteolysis and failed to produce crystals at 4 or 22 °C. To prepare LpoA protein without a His6 tag, 1 liter of Terrific Broth supplemented with 100 μg/ml ampicillin and 33 μg/ml chloramphenicol was inoculated overnight with a 10-ml starter culture of OrigamiTM(DE3)/pLacI previously transformed with pLpoA (Table 4). Cultures were grown at 37 °C with shaking at 250 rpm to an A600 = 0.6–0.8, transferred to room temperature, and induced with 0.2 mm IPTG. Cells were harvested 16 h later and lysed by applying sonication. LpoA was purified by carrying out ammonium sulfate fractionation at 30 and 50% and then dialyzed against 50 mm Tris-HCl, pH 8.0, overnight to remove salt. The protein was loaded successively onto Source Q and then Mono Q anion-exchange columns and eluted with a 0–1 m NaCl gradient in 50 mm Tris-HCl, pH 8. The LpoA peak eluted at ∼150 mm NaCl. Peak fractions were concentrated and loaded on a HiPrepTM Superdex 75 16/60 (GE Biosciences) gel filtration column, where LpoA eluted as a single peak with an apparent molecular mass of 60 kDa. LpoA in 150 mm NaCl, 50 mm Tris-HCl, pH 8.0, was concentrated to 33 mg/ml as determined by absorbance at 280 nm with an extinction coefficient calculated by ProtParam (http://web.expasy.org/protparam) and used immediately for crystallization.
This full-length LpoA formed clusters of branched needles under many conditions using vapor diffusion at both 22 and 4 °C. To minimize the growth of these clusters, we added 0.2 μl of 30% xylitol (Hampton Research) to hanging drops containing 1 μl of concentrated protein and 1 μl of precipitant (25% (w/v) PEG 1500, 0.1 m MMT buffer (20 mm dl-malic acid, 40 mm MES, 40 mm Tris-HCl), pH 4.0). The trays containing these hanging drops were incubated at 4 °C for 2 days and then transferred in an insulated box to 22 °C. Here, the drops produced rod-shaped orthorhombic crystals, which diffracted X-rays to a dmin of 1.97 Å. Intensity data from these crystals, after being frozen, were collected at beamline 21ID-G at LS-CAT (Advanced Photon Source, Argonne, IL) and processed with HKL-2000 (57). These data were phased by the molecular replacement method with Phaser (58) using the previously determined LpoA-C (PDB entry 3CKM) and the LpoA-N structure as search models. Refinement with PHENIX and manual refitting with Coot resulted in final Rwork and Rfree values of 0.17 and 0.21, respectively (56, 59). The final model included residues 34–573, with poor electron density for residues 55–58 and 430–433. Residues 473–478, which were well-ordered in the LpoA-C structure (PDB entry 3CKM), were observed to be disordered in LpoAOrt, probably due to the different arrangements of the molecules in their respective crystals. The two models also differed in the 345–350 loop above the cleft and between the two lobes. In the LpoA-C structure, density was ambiguous, and residues 346–349 were omitted from the refinement calculations (16). In LpoAOrt, this loop was well-resolved but with a different conformation than in the LpoA-C structure (supplemental Fig. S4).
Monoclinic crystals of LpoAMon(SeMet) were grown using similar procedures as above, but with 8% PEG 4000, 0.1 m sodium acetate trihydrate, pH 4.6, as the precipitant with added xylitol. These crystals were transferred to a precipitant solution containing 15% glycerol and then frozen in liquid nitrogen. These frozen crystals did not diffract as well as the orthorhombic crystals but provided a 90% complete data set to 2.8 Å resolution (Table 1). The structure was solved by molecular replacement as above and refined with PHENIX. Restraining the structure to the orthorhombic structure in the final cycles resulted in an Rwork/Rfree = 0.21/0.23 with excellent geometry.
Crystals of another monoclinic form, LpoAMon2, were grown from conditions similar to the above monoclinic crystal, and they showed different unit cell dimensions and two molecules per asymmetric unit. These crystals yielded an incomplete data set to a resolution of 2.6 Å that was phased as above with Phaser in PHENIX. The structure was refined with PHENIX to an Rwork/Rfree = 0.23/0.29 (Table 1).
Software used for analyzing the structures included PyMOL for preparing the figures (60), Jalview for sequence alignments (61), Consurf for the calculation of sequence homology (62), and ElNémo for normal mode calculations (34). Coordinates were deposited in the Protein Data Bank (63).
Small-angle X-ray scattering
For the SAXS experiment, full-length HiLpoA protein with an N-terminal His6 tag was expressed from Origami (DE3) cells transformed with pMCSG7-LpoA(29–575). Protein was purified on a TALON® (Clontech) cobalt affinity column, digested with TeV to remove the His6 tag, rerun on the cobalt column, and, after dialysis and concentration, loaded on a Superdex-75 column. Fractions were evaluated by SDS-PAGE (supplemental Fig. S7A), pooled, and frozen at −80 °C. Protein was buffer-exchanged into 50 mm Tris-HCl, 150 mm NaCl, 10% glycerol, pH 7.3, and dilutions were prepared in the same buffer for SAXS. The final purified protein contained residues S-N-A-HiLpoA(29–575) and migrated at ∼60,000 on an SDS-polyacrylamide gel (supplemental Fig. S7A), consistent with the predicted molecular weight of 60,518.
Preliminary scattering profiles were measured on the in-house SAXS instrument at the SAXS core (NCI, National Institutes of Health). Subsequently, small-angle scattering from buffer (50 mm Tris-HCl, 150 mm NaCl, 10% glycerol, pH 7.3) and four LpoA protein dilutions (8.8, 4.4, 2.2, and 1.1 mg/ml) were measured at the 12-ID-B beamline of the Advanced Photon Source (APS), Argonne National Laboratory, with photon energy of 14 keV and an off-center Pilatus 2 M detector. Simultaneous wide-angle X-ray scattering also was recorded, and the total q range covered was ∼0.005 < q < 2.8 Å−1. A total of 30 sequential data frames were recorded for each buffer and sample solution with an exposure time of 0.75–2 s to minimize radiation damage and obtain a good signal/noise ratio. The 2D images were corrected and reduced to 1D scattering profiles using the Matlab software package at the beamlines. The 1D SAXS profiles were grouped by sample and averaged, and background scattering due to the buffer was subtracted (supplemental Fig. S7B). The scattering intensity profiles were extrapolated to infinite dilution, to remove the scattering contribution due to interparticle interactions, and to low scattering angle following the results of the Guinier plot (Fig. 8, A and B). Both intensity at zero scattering angle (I0) and Rg calculated from each Guinier plot (Fig. 8A) were dependent on protein concentration, as expected. The Rg was calculated for each of the six models with the Crysol software (38). Theoretical scattering profiles were generated from the six models and compared with the experimental SAXS data at q < 0.5 Å−1 using the FoXS software (64). The minimal ensemble search of the six structures to improve the fit to the observed data was calculated by the FoXS server (http://modbase.compbio.ucsf.edu/foxs)5 (64).
Author contributions
Protein expression, crystallization, and structure determination were performed by K. S., J. V., and B. T. Structure refinement was performed by K. S., J. V., and M. A. S. SAXS data collection and analyses were performed by L. F. and M. A. S. K. S, J. V., L. F., and M. A. S. prepared the manuscript. M. A. S. was responsible for project direction. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Dr. Brian Akerley for support from the beginning of this project and for reading the manuscript. We thank Mykola Murskyj for help with cloning and members of the Saper laboratory for advice. For the SAXS experiments, we acknowledge use of the SAXS Core facility of the Center for Cancer Research, NCI/National Institutes of Health, and thank Dr. Xiaobing Zuo for expert support. We thank the outstanding synchrotron support staff at LS-CAT and DND-CAT. We also benefitted from discussions with Tobias Dörr, Matthew Larson, Kathleen Wisser, Erik R. P. Zuiderweg, and Charles Brooks III. We also thank Ilan Rosenshine and Susan Buchanan for hosting M. A. S. in their laboratories. We thank the software authors for writing and sharing the software necessary for this project. Jerry Brown provided expert editing. This research used resources of the Advanced Photon Source (APS), a United States Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory (ANL) under Contract DE-AC02-06CH11357. The SAXS beamline 12-ID-B at APS was allocated under the PUP-24152 agreement between NCI/National Institutes of Health and ANL. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817). The LpoA-N data were measured at the DuPont-Northwestern-Dow Collaborative Access Team (DND-CAT) located at Sector 5 of APS. DND-CAT was supported by E. I. DuPont de Nemours & Co., The Dow Chemical Company, and the State of Illinois.
This work was supported by the Philip Morris External Research Program, National Institutes of Health (NIAID R21 AI099984), and in part by a Lady Davis Fellowship (M. A. S.) from the Hebrew University of Jerusalem. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This paper is dedicated to the memory of our colleague and mentor Professor Martha L. Ludwig.
This article contains supplemental Figs. S1–S8, three sequence alignment files, and PDB files of the two NMA-derived models.
The atomic coordinates and structure factors (codes 4P29, 5KCN, 5VBG, and 5VAT) have been deposited in the Protein Data Bank (http://wwpdb.org/).
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- PG
- peptidoglycan
- LpoA
- generic reference or the full-length protein from H. influenzae
- LpoA-N
- N-terminal domain
- LpoA-C
- C-terminal domain
- LpoAOrt
- LpoAMon, and LpoAMon2, crystal structures of the orthorhombic, monoclinic, and second monoclinic HiLpoA crystal forms, respectively
- LpoAMon2B
- chain B of the LpoAMon2 crystal structure
- EcLpoA
- E. coli LpoA
- PBP1A and PBP1B
- bifunctional PG synthase penicillin-binding protein 1A and 1B, respectively
- SeMet
- selenomethionine
- SAXS
- small-angle X-ray scattering
- TG
- transglycosylase
- TP
- transpeptidase
- TPR
- tetratricopeptide
- PDB
- Protein Data Bank
- RMSD
- root mean square deviation
- NMA
- normal mode analyses
- OM
- outer membrane
- IPTG
- isopropyl β-d-1-thiogalactopyranoside.
References
- 1. World Health Organization (2014) Antimicrobial resistance: global report on surveillance 2014. World Health Organization, Geneva, Switzerland [Google Scholar]
- 2. Turner R. D., Vollmer W., and Foster S. J. (2014) Different walls for rods and balls: the diversity of peptidoglycan. Mol. Microbiol. 91, 862–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Egan A. J., and Vollmer W. (2013) The physiology of bacterial cell division. Ann. N.Y. Acad. Sci. 1277, 8–28 [DOI] [PubMed] [Google Scholar]
- 4. Lovering A. L., Safadi S. S., and Strynadka N. C. (2012) Structural perspective of peptidoglycan biosynthesis and assembly. Annu. Rev. Biochem. 81, 451–478 [DOI] [PubMed] [Google Scholar]
- 5. Cho H., Wivagg C. N., Kapoor M., Barry Z., Rohs P. D., Suh H., Marto J. A., Garner E. C., and Bernhardt T. G. (2016) Bacterial cell wall biogenesis is mediated by SEDS and PBP polymerase families functioning semi-autonomously. Nat. Microbiol. 10.1038/nmicrobiol.2016.172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Meeske A. J., Riley E. P., Robins W. P., Uehara T., Mekalanos J. J., Kahne D., Walker S., Kruse A. C., Bernhardt T. G., and Rudner D. Z. (2016) SEDS proteins are a widespread family of bacterial cell wall polymerases. Nature 537, 634–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Typas A., Banzhaf M., Gross C. A., and Vollmer W. (2012) From the regulation of peptidoglycan synthesis to bacterial growth and morphology. Nat. Rev. Microbiol. 10, 123–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Typas A., Banzhaf M., van den Berg van Saparoea B., Verheul J., Biboy J., Nichols R. J., Zietek M., Beilharz K., Kannenberg K., von Rechenberg M., Breukink E., den Blaauwen T., Gross C. A., and Vollmer W. (2010) Regulation of peptidoglycan synthesis by outer-membrane proteins. Cell 143, 1097–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yousif S. Y., Broome-Smith J. K., and Spratt B. G. (1985) Lysis of Escherichia coli by β-lactam antibiotics: deletion analysis of the role of penicillin-binding proteins 1A and 1B. J. Gen. Microbiol. 131, 2839–2845 [DOI] [PubMed] [Google Scholar]
- 10. Paradis-Bleau C., Markovski M., Uehara T., Lupoli T. J., Walker S., Kahne D. E., and Bernhardt T. G. (2010) Lipoprotein cofactors located in the outer membrane activate bacterial cell wall polymerases. Cell 143, 1110–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lupoli T. J., Lebar M. D., Markovski M., Bernhardt T., Kahne D., and Walker S. (2014) Lipoprotein activators stimulate Escherichia coli penicillin-binding proteins by different mechanisms. J Am. Chem. Soc. 136, 52–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dörr T., Möll A., Chao M. C., Cava F., Lam H., Davis B. M., and Waldor M. K. (2014) Differential requirement for PBP1a and PBP1b in in vivo and in vitro fitness of Vibrio cholerae. Infect. Immun. 82, 2115–2124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wong S. M., and Akerley B. J. (2003) Inducible expression system and marker-linked mutagenesis approach for functional genomics of Haemophilus influenzae. Gene 316, 177–186 [DOI] [PubMed] [Google Scholar]
- 14. Wong S. M., Bernui M., Shen H., and Akerley B. J. (2013) Genome-wide fitness profiling reveals adaptations required by Haemophilus in coinfection with influenza A virus in the murine lung. Proc. Natl. Acad. Sci. U.S.A. 110, 15413–15418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gawronski J. D., Wong S. M., Giannoukos G., Ward D. V., and Akerley B. J. (2009) Tracking insertion mutants within libraries by deep sequencing and a genome-wide screen for Haemophilus genes required in the lung. Proc. Natl. Acad. Sci. U.S.A. 106, 16422–16427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vijayalakshmi J., Akerley B. J., and Saper M. A. (2008) Structure of YraM, a protein essential for growth of Haemophilus influenzae. Proteins 73, 204–217 [DOI] [PubMed] [Google Scholar]
- 17. Berntsson R. P., Smits S. H., Schmitt L., Slotboom D. J., and Poolman B. (2010) A structural classification of substrate-binding proteins. FEBS Lett. 584, 2606–2617 [DOI] [PubMed] [Google Scholar]
- 18. Trakhanov S., Vyas N. K., Luecke H., Kristensen D. M., Ma J., and Quiocho F. A. (2005) Ligand-free and -bound structures of the binding protein (LivJ) of the Escherichia coli ABC leucine/isoleucine/valine transport system: trajectory and dynamics of the interdomain rotation and ligand specificity. Biochemistry 44, 6597–6608 [DOI] [PubMed] [Google Scholar]
- 19. Jean N. L., Bougault C. M., Lodge A., Derouaux A., Callens G., Egan A. J., Ayala I., Lewis R. J., Vollmer W., and Simorre J. P. (2014) Elongated structure of the outer-membrane activator of peptidoglycan synthesis LpoA: implications for PBP1A stimulation. Structure 22, 1047–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Baker N. A., Sept D., Joseph S., Holst M. J., and McCammon J. A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sobhanifar S., King D. T., and Strynadka N. C. (2013) Fortifying the wall: synthesis, regulation and degradation of bacterial peptidoglycan. Curr. Opin. Struct. Biol. 23, 695–703 [DOI] [PubMed] [Google Scholar]
- 22. Turner R. D., Hurd A. F., Cadby A., Hobbs J. K., and Foster S. J. (2013) Cell wall elongation mode in Gram-negative bacteria is determined by peptidoglycan architecture. Nat. Commun. 4, 1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Egan A. J., Jean N. L., Koumoutsi A., Bougault C. M., Biboy J., Sassine J., Solovyova A. S., Breukink E., Typas A., Vollmer W., and Simorre J. P. (2014) Outer-membrane lipoprotein LpoB spans the periplasm to stimulate the peptidoglycan synthase PBP1B. Proc. Natl. Acad. Sci. U.S.A. 111, 8197–8202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. King D. T., Lameignere E., and Strynadka N. C. (2014) Structural insights into the lipoprotein outer-membrane regulator of penicillin-binding protein 1B. J. Biol. Chem. 289, 19245–19253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen C., Natale D. A., Finn R. D., Huang H., Zhang J., Wu C. H., and Mazumder R. (2011) Representative proteomes: a stable, scalable and unbiased proteome set for sequence analysis and functional annotation. PLoS One 6, e18910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Finn R. D., Bateman A., Clements J., Coggill P., Eberhardt R. Y., Eddy S. R., Heger A., Hetherington K., Holm L., Mistry J., Sonnhammer E. L., Tate J., and Punta M. (2014) Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Holm L., and Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Parashar V., Jeffrey P. D., and Neiditch M. B. (2013) Conformational change-induced repeat domain expansion regulates Rap phosphatase quorum-sensing signal receptors. PLoS Biol. 11, e1001512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zouhir S., Perchat S., Nicaise M., Perez J., Guimaraes B., Lereclus D., and Nessler S. (2013) Peptide-binding dependent conformational changes regulate the transcriptional activity of the quorum-sensor NprR. Nucleic Acids Res. 41, 7920–7933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Alexandrov V., Lehnert U., Echols N., Milburn D., Engelman D., and Gerstein M. (2005) Normal modes for predicting protein motions: a comprehensive database assessment and associated Web tool. Protein Sci. 14, 633–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Doruker P., Jernigan R. L., and Bahar I. (2002) Dynamics of large proteins through hierarchical levels of coarse-grained structures. J. Comput. Chem. 23, 119–127 [DOI] [PubMed] [Google Scholar]
- 32. Tama F. (2003) Normal mode analysis with simplified models to investigate the global dynamics of biological systems. Protein Pept. Lett. 10, 119–132 [DOI] [PubMed] [Google Scholar]
- 33. Krebs W. G., Alexandrov V., Wilson C. A., Echols N., Yu H., and Gerstein M. (2002) Normal mode analysis of macromolecular motions in a database framework: developing mode concentration as a useful classifying statistic. Proteins 48, 682–695 [DOI] [PubMed] [Google Scholar]
- 34. Suhre K., and Sanejouand Y. H. (2004) ElNémo: a normal mode web server for protein movement analysis and the generation of templates for molecular replacement. Nucleic Acids Res. 32, W610–W614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rambo R. P., and Tainer J. A. (2013) Accurate assessment of mass, models and resolution by small-angle scattering. Nature 496, 477–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fischer H., de Oliveira Neto M., Napolitano H. B., Polikarpov I., and Craievich A. F. (2010) Determination of the molecular weight of proteins in solution from a single small-angle X-ray scattering measurement on a relative scale. J. Appl. Crystallogr. 43, 101–109 [Google Scholar]
- 37. Svergun D. I. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 [Google Scholar]
- 38. Svergun D., Barberato C., and Koch M. H. J. (1995) CRYSOL - a program to evaluate X-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768–773 [Google Scholar]
- 39. Schneidman-Duhovny D., Hammel M., Tainer J. A., and Sali A. (2013) Accurate SAXS profile computation and its assessment by contrast variation experiments. Biophys. J. 105, 962–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rambo R. P., and Tainer J. A. (2011) Characterizing flexible and intrinsically unstructured biological macromolecules by SAS using the Porod-Debye law. Biopolymers 95, 559–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Durand D., Vivès C., Cannella D., Pérez J., Pebay-Peyroula E., Vachette P., and Fieschi F. (2010) NADPH oxidase activator p67(phox) behaves in solution as a multidomain protein with semi-flexible linkers. J. Struct. Biol. 169, 45–53 [DOI] [PubMed] [Google Scholar]
- 42. Pelikan M., Hura G. L., and Hammel M. (2009) Structure and flexibility within proteins as identified through small angle X-ray scattering. Gen. Physiol. Biophys. 28, 174–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Han S., Caspers N., Zaniewski R. P., Lacey B. M., Tomaras A. P., Feng X., Geoghegan K. F., and Shanmugasundaram V. (2011) Distinctive attributes of β-lactam target proteins in Acinetobacter baumannii relevant to development of new antibiotics. J. Am. Chem. Soc. 133, 20536–20545 [DOI] [PubMed] [Google Scholar]
- 44. Matias V. R., Al-Amoudi A., Dubochet J., and Beveridge T. J. (2003) Cryo-transmission electron microscopy of frozen-hydrated sections of Escherichia coli and Pseudomonas aeruginosa. J. Bacteriol. 185, 6112–6118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Azari F., Nyland L., Yu C., Radermacher M., Mintz K. P., and Ruiz T. (2013) Ultrastructural analysis of the rugose cell envelope of a member of the Pasteurellaceae family. J. Bacteriol. 195, 1680–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Demchick P., and Koch A. L. (1996) The permeability of the wall fabric of Escherichia coli and Bacillus subtilis. J. Bacteriol. 178, 768–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gray A. N., Egan A. J., Van't Veer I. L., Verheul J., Colavin A., Koumoutsi A., Biboy J., Altelaar M. A., Damen M. J., Huang K. C., Simorre J. P., Breukink E., den Blaauwen T., Typas A., Gross C. A., and Vollmer W. (2015) Coordination of peptidoglycan synthesis and outer membrane constriction during Escherichia coli cell division. Elife 10.7554/eLife.07118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dörr T., Lam H., Alvarez L., Cava F., Davis B. M., and Waldor M. K. (2014) A novel peptidoglycan binding protein crucial for PBP1A-mediated cell wall biogenesis in Vibrio cholerae. PLoS Genet. 10, e1004433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lee T. K., Meng K., Shi H., and Huang K. C. (2016) Single-molecule imaging reveals modulation of cell wall synthesis dynamics in live bacterial cells. Nat. Commun. 7, 13170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Akerley B. J., Rubin E. J., Novick V. L., Amaya K., Judson N., and Mekalanos J. J. (2002) A genome-scale analysis for identification of genes required for growth or survival of Haemophilus influenzae. Proc. Natl. Acad. Sci. U.S.A. 99, 966–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Stols L., Gu M., Dieckman L., Raffen R., Collart F. R., and Donnelly M. I. (2002) A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein Expr. Purif. 25, 8–15 [DOI] [PubMed] [Google Scholar]
- 52. Sambrook J., Fritsch E. F., and Maniatis T. (1989) Molecular cloning: a laboratory manual, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 53. Terwilliger T. C. (2002) Automated structure solution, density modification and model building. Acta Crystallogr. D Biol. Crystallogr. 58, 1937–1940 [DOI] [PubMed] [Google Scholar]
- 54. Jones T. A., Zou J. Y., Cowan S. W., and Kjeldgaard M. (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47, 110–119 [DOI] [PubMed] [Google Scholar]
- 55. Pflugrath J. W. (1999) The finer things in X-ray diffraction data collection. Acta Crystallogr. D Biol. Crystallogr. 55, 1718–1725 [DOI] [PubMed] [Google Scholar]
- 56. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 58. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. DeLano W. L. (2015) The PyMOL Molecular Graphics System, version 1.8, Schrödinger, LLC, New York [Google Scholar]
- 61. Troshin P. V., Procter J. B., and Barton G. J. (2011) Java bioinformatics analysis web services for multiple sequence alignment–JABAWS:MSA. Bioinformatics 27, 2001–2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Celniker G., Nimrod G., Ashkenazy H., Glaser F., Martz E., Mayrose I., Pupko T., and Ben-Tal N. (2013) ConSurf: using evolutionary data to raise testable hypotheses about protein function. Isr. J. Chem. 53, 199–206 [Google Scholar]
- 63. Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., and Bourne P. E. (2000) The Protein Data Bank. Nucleic Acids Res. 28, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Schneidman-Duhovny D., Hammel M., Tainer J. A., and Sali A. (2016) FoXS, FoXSDock and MultiFoXS: single-state and multi-state structural modeling of proteins and their complexes based on SAXS profiles. Nucleic Acids Res. 44, W424–W429 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.