

Abstract
The MYO1C gene produces three alternatively spliced isoforms, differing only in their N-terminal regions (NTRs). These isoforms, which exhibit both specific and overlapping nuclear and cytoplasmic functions, have different expression levels and nuclear–cytoplasmic partitioning. To investigate the effect of NTR extensions on the enzymatic behavior of individual isoforms, we overexpressed and purified the three full-length human isoforms from suspension-adapted HEK cells. MYO1CC favored the actomyosin closed state (AMC), MYO1C16 populated the actomyosin open state (AMO) and AMC equally, and MYO1C35 favored the AMO state. Moreover, the full-length constructs isomerized before ADP release, which has not been observed previously in truncated MYO1CC constructs. Furthermore, global numerical simulation analysis predicted that MYO1C35 populated the actomyosin·ADP closed state (AMDC) 5-fold more than the actomyosin·ADP open state (AMDO) and to a greater degree than MYO1CC and MYO1C16 (4- and 2-fold, respectively). On the basis of a homology model of the 35-amino acid NTR of MYO1C35 (NTR35) docked to the X-ray structure of MYO1CC, we predicted that MYO1C35 NTR residue Arg-21 would engage in a specific interaction with post-relay helix residue Glu-469, which affects the mechanics of the myosin power stroke. In addition, we found that adding the NTR35 peptide to MYO1CC yielded a protein that transiently mimics MYO1C35 kinetic behavior. By contrast, NTR35, which harbors the R21G mutation, was unable to confer MYO1C35-like kinetic behavior. Thus, the NTRs affect the specific nucleotide-binding properties of MYO1C isoforms, adding to their kinetic diversity. We propose that this level of fine-tuning within MYO1C broadens its adaptability within cells.
Keywords: enzyme mechanism, molecular modeling, molecular motor, myosin, pre-steady-state kinetics, MYO1C, NMI
Introduction
Myosins are molecular motors that utilize ATP binding, hydrolysis, and product release to perform mechanical work along actin filaments. All myosins share a highly conserved motor domain, a lever arm, and a tail domain that exhibits substantial diversity. The mechanochemical transduction pathway of the ATPase cycle and the major structural biochemical intermediates are conserved across the myosin family (1). Importantly, to perform their myriad biological functions, myosins have evolved unique kinetic adaptations by modulating the rate and equilibrium constants of the ATPase cycle (2–4).
Myosin IC (MYO1C),2 a class I myosin, is produced as three splice isoforms that differ only at the N-terminal region (NTR) (Fig. 1) (5). Although they share identical motor domains, three calmodulin-binding IQ motifs containing a nuclear localization signal (6) and a membrane-binding tail domain (Fig. 1A), each isoform has acquired differences in function and nuclear–cytoplasmic partitioning. MYO1CC, the first isoform to be identified, localizes mostly to the cytoplasm and interacts with plasma membrane phosphoinositides via its PH domain; functionally, it participates in the generation of membrane tension, cell migration, vesicle trafficking, signal transduction, and hearing (7–14). MYO1CC nuclear import is regulated by calcium (15), but its nuclear functions remain unknown. MYO1C16 has a 16-amino acid (aa) extension at the NTR, which includes a nucleolar localization signal (16). MYO1C16, previously referred to as nuclear myosin I (or NMI), localizes mostly to the nucleus (17), and its nuclear localization is moderately affected by calcium (15). In the nucleus, MYO1C16 is involved in transcription by all three RNA polymerases, mRNA maturation, chromatin remodeling, and chromosome movement (18–23). MYO1C16 also localizes to the cytoplasm, where, like MYO1CC, it contributes to the regulation of cell membrane tension (24). MYO1C16 knock-out mice do not exhibit any obvious phenotype (25). MYO1C35 has an additional 35-aa extension at the NTR (partially overlapping with MYO1C16 (Fig. 1B)) and also localizes mostly to the nucleus, where it interacts with RNA polymerase II but not RNA polymerase I (5). In mouse, MYO1C35 is expressed at low levels in a tissue-specific pattern, whereas MYO1C16 and MYO1CC are ubiquitously expressed at comparable levels in most tissues (26).
Figure 1.

Alignment of isoforms and domain structure of MYO1C. A, schematic representation of MYO1C domain structures. B, splicing pattern and alignment of the initial sequences of MYO1C isoforms. NCBI RefSeq: NP_001074248.1, NP_001074419.1, NP_203693.3 for MYO1C35, MYO1C16, and MYO1CC, respectively. C, Coomassie Blue-stained SDS-polyacrylamide gel of the human MYO1C35 (lane 1), MYO1C16 (lane 2), and MYO1CC (lane 3). Molecular masses are 121.7, 119.6, and 117.9 kDa for MYO1C35, MYO1C16, and MYO1CC, respectively. The associated calmodulin light chain can be seen in all lanes (∼17 kDa).
Although the biological functions and localization of all three MYO1C isoforms have been thoroughly investigated, only MYO1CC has been characterized enzymatically. MYO1CC is a low-duty-ratio myosin (i.e. it spends most of the ATPase cycle in the weak actin-binding states), despite the fact that its actin attachment lifetime is relatively long (27, 28). It exhibits a weak coupling between ADP and actin binding, and ADP release is coupled to an additional lever arm movement, which facilitates additional work subsequent to the power stroke (29, 30). The force-sensitive transition in the ATPase cycle is the isomerization that follows ATP binding, and the rate-limiting step has been proposed to be a transition that precedes entry to the strong-binding state (28). Because of these properties, MYO1CC may be viewed as a tension sensor or slow transporter (3, 28, 29). The kinetics of the additional two isoforms have not been investigated previously, and the impact of alternative splicing on the enzymology of MYO1C isoforms is not fully understood.
A recently solved crystal structure of the closely related protein Myo1b shows that its NTR interacts with calmodulin bound to the first IQ motif (31). Deletion of nine aa of the NTR of MYO1CC, or replacement of this sequence with residues from the N terminus of Myo1b, dramatically changes the kinetics and tension-sensing properties of MYO1CC (32). This region is shared by all three isoforms of MYO1C, but the functions of the NTR extensions remain to be elucidated.
We investigated how the kinetic properties of the MYO1C isoforms are affected by alternative splicing of the NTR. To this end, we expressed and purified the three full-length human MYO1C isoforms in a human cell line and characterized their steady-state ATPase and nucleotide-binding activities by both kinetic and equilibrium measurements. Our detailed kinetic analysis revealed that the diverse NTRs affect isomerization of the nucleotide pocket. Specifically, the NTR stabilizes both the AMO (open state) and the AMDC (ADP closed state preceding isomerization). The open–closed transitions are dependent on the length of the NTR. Next, we computationally analyzed the interaction between the 35-aa NTR with the myosin heavy chain by molecular dynamic modeling and docking to the recently solved crystal structure of MYO1CC-1IQ (33). Finally, we found that the addition of the NTR35 peptide in trans to the MYO1CC isoform mimicked the ATP-induced acto·MYO1C·ADP dissociation kinetics of the MYO1C35 isoform and stabilized the AMDC state.
Results
N-terminal sequence alignments and purification of full-length human MYO1C isoforms
MYO1C35 and MYO1C16 share 10 aa from exon 1 and an additional 6 or 25 aa from exons −1 or −2, respectively (Fig. 1, A and B). The remaining identical 1028 aa constitute the motor domain, which binds nucleotides and actin; the lever arm, which consists of three IQ motifs for calmodulin light-chain binding; and the PH domain in the tail, which binds phosphoinositides. To better match the cellular environment in which MYO1C functions, we established an expression and purification system for human myosins using a suspension adapted human HEK293SF-3F6 cell line. This system may be of use in future studies aimed at expanding the options for expression of other myosins with more complex architectures. For our experiments, we purified the three human MYO1C isoforms as full-length constructs with calmodulin as their light chain (Fig. 1C) and determined whether they display full calmodulin light-chain binding motif occupancy by preforming actin co-sedimentation followed by SDS-PAGE and densitometry (supplemental Fig. S1) with calmodulin standards to determine light-chain concentration. We found that all MYO1C isoforms bound three molecules of calmodulin (2.9 ± 0.10, 2.75 ± 0.13, and 3.1 ± 0.07 for MYO1C35, MYO1C16, and MYO1CC, respectively).
Actin-activated steady-state ATPase activity of full-length human MYO1C isoforms
Under our reaction conditions, all three isoforms exhibited actin-activated steady-state ATPase activity with hyperbolic dependence on actin concentration, which allowed us to determine the steady-state parameters according to the Michaelis-Menten model (supplemental Fig. S2 and Table S2). Our results revealed that the steady-state kinetic parameters did not indicate whether the different NTRs of these isoforms impact their enzymology. Hence, we reasoned that a detailed kinetic dissection of the ATPase cycle of each isoform might reveal modulation of the rate constants of their ATPase cycle (see Scheme 1).
Scheme 1.

ATP induced an acto·MYO1C population of weakly bound states
The increase in pyrene-labeled actin fluorescence and the decrease in light scattering upon ATP binding to acto·MYO1C isoforms were monitored as a function of [ATP]. The signal arises from the induced weak-binding states (pyrene-actin, Fig. 2A) or dissociation from actomyosin (light scattering, Fig. 2A). For all three isoforms, both signals were best fitted to double-exponential equations, suggesting that the isoforms have similar ATP-binding mechanisms (Fig. 2A). This is explained by actomyosin existing in two states: the fast phase reflects ATP binding to the AMO state, and the slow phase represents the conversion from AMC to AMO (29, 30). The equilibrium for this transition is termed K′α (where K′α = k′−α/k′+α) as shown in Scheme 2. The observed rate constants of the fast phase (kobs,fast) were hyperbolically dependent on [ATP] (Fig. 3B), yielding K′1T and k′+2T (Scheme 2 and Equation 1).
| (Eq. 1) |
Figure 2.

ATP-induced population of weakly bound acto·MYO1C states. A, representative time courses of pyrene fluorescence enhancement and normalized light scattering reduction after rapid mixing of 25 nm acto·MYO1C16 isoform with 0, 0.03125, 0.0625, 0.125, 0.25, 0.5, 1, 2, 4, or 8 mm ATP. Data are averaged transients (n = 3). The data were best fitted to a double-exponential equation. MYO1CC (black), MYO1C16 (red), and MYO1C35 (blue) are shown as solid circles for pyrene fluorescence data and as open circles for light-scattering data. The solid lines for pyrene and dashed lines for light scattering represent the best fits to the data. B, the fast-phase kobs was best fitted to a rectangular hyperbola (Scheme 2 and Equation 1), giving the equilibrium constant for MgATP binding to actomyosin, K′1T, and the kinetic constant for isomerization after MgATP binding, k′+2T. C, slow-phase kobs,slow values plotted as a function of [MgATP]. The slow kobs was best fitted to a rectangular hyperbola (Scheme 2 and Equation 2), yielding the kinetic constant for the closed-to-open isomerization of the ATP-binding pocket, k′+α. D, a rectangular hyperbola was fitted to the data of the ratio between the fast (Afast) and slow (Aslow) time-course amplitudes, giving the closed-to-open nucleotide pocket equilibrium constant, K′α. E, amplitude of ATP-induced dissociation. The error bars of the fitting are within data points.
Scheme 2.

Figure 3.

Global numerical simulation of time-dependent distribution of the reaction intermediates of Scheme 2. A–C, simulated data sets of the ATP-induced population of weakly bound acto·MYO1C states fitted to raw data from MYO1C35, MYO1C16, and MYO1CC. Each graph shows in solid lines the time courses of data collected at 0.03125 (blue), 0.0625 (brown), 0.125 (yellow), 0.25 (purple), 0.5 (green), 1 (light blue), 2 (dark red), 4 (blue), and 8 (orange) mm ATP as presented in Fig. 2A for pyrene-labeled acto·MYO1C ATP-induced dissociation. The solid lines through the data sets are the fitted curves resulting from performing global numerical analysis on the entire set of data for each isoform. The fitting was to the sum of the AM·ATP and A states according to Scheme 2. D–F, time-dependent distribution of biochemical intermediates of the reaction according to the simulation mechanism shown in Scheme 2 for MYO1C35, MYO1C16, and MYO1CC, respectively. Blue, AMC; red, AMO; yellow, AM(ATP); purple, AM·ATP; green, A state. The light blue line represents the sum of the AM·ATP and A states, which reflect the pyrene signal of weakly bound or dissociated states.
The kobs,fast of MYO1C16 and MYO1CC continued to increase above 8 mm MgATP, but the total amplitudes of the fluorescence increase arising from the fast phase reached saturation above 4 mm MgATP (Fig. 2E). This observation suggests the existence of a small population exhibiting faster kinetics. The apparent equilibrium constants for ATP binding, K′1T, as determined by pyrene-actin fluorescence, were similar among isoforms (K′1T,p = 11.5 ± 1.94, 10.3 ± 2.37, and 12.9 ± 3.59 mm−1 for MYO1C35, MYO1C16, and MYO1CC, respectively (Table 1)). However, the apparent equilibrium constants measured by light scattering, K′1,LS, was ∼2-fold smaller for MYO1C16 (K′1,LS = 13.9 ± 2.49, 6.9 ± 1.51, and 14.01 ± 5.3 mm−1 for MYO1C35, MYO1C16, and MYO1CC, respectively (Table 1)). Furthermore, for all isoforms, k′+2T values measured by light scattering were ∼2-fold smaller than those obtained by the pyrene-actin fluorescence assay (k′+2T,LS = 13.5 ± 0.61, 14.4 ± 0.84, and 12.1 ± 1.07 s−1 for MYO1C35, MYO1C16, and MYO1CC, respectively (Table 1)).
Table 1.
ATP-induced acto·MYO1C dissociation measured by pyrene-actin and light scattering
Conditions used were: 20 mm MOPS, pH 7.0, 25 mm KAc, 2 mm MgCl2, 0.2 mm EGTA, and 1 mm DTT at 20 ± 0.1 °C.
| Constant | MYO1C35 |
MYO1C16 |
MYO1CC |
|||
|---|---|---|---|---|---|---|
| Pyrene-actin | Light scattering | Pyrene-actin | Light scattering | Pyrene-actin | Light scattering | |
| k′+α (s−1) | 0.9 ± 0.03 | 1.2 ± 0.11 | 1.0 ± 0.03 | 1.4 ± 0.05 | 1.2 ± 0.05 | 1.2 ± 0.06 |
| k′−α (s−1)a | 0.6 ± 0.03 | 0.9 ± 0.15 | 0.9 ± 0.05 | 1.6 ± 0.14 | 1.8 ± 0.14 | 1.8 ± 0.26 |
| K′α | 1.6 ± 0.04 | 1.3 ± 0.09 | 1.1 ± 0.02 | 0.9 ± 0.04 | 0.7 ± 0.02 | 0.7 ± 0.06 |
| K′1T·k′+2T (μm−1 s−1) | 0.33 ± 0.07 | 0.19 ± 0.04 | 0.29 ± 0.09 | 0.10 ± 0.03 | 0.25 ± 0.09 | 0.17 ± 0.08 |
| K′1T (mm−1) | 11.5 ± 1.94 | 13.9 ± 2.49 | 10.3 ± 2.37 | 6.9 ± 1.51 | 12.9 ± 3.59 | 14.0 ± 5.28 |
| k′+2T (s−1) | 28.3 ± 1.32 | 13.5 ± 0.61 | 27.9 ± 2.06 | 14.4 ± 0.84 | 19.6 ± 1.51 | 12.1 ± 1.07 |
a Calculated.
The observed slow phase in ATP-induced dissociation from actomyosin mostly represents the decrease in the population of the AMC state over time. The [ATP]-dependence of the observed slow-phase rates were fitted to a rectangular hyperbola, yielding k'+α (Fig. 2B) according to Scheme 2 and Equation 2.
| (Eq. 2) |
The rate constant for the nucleotide pocket transition from closed to open, k′+α, did not differ significantly among the MYO1C isoforms in either assay (k′+α,p = 0.9 ± 0.03, 1.0 ± 0.03, and 1.2 ± 0.05 s−1 and k′+α,LS = 1.2 ± 0.11, 1.4 ± 0.05, and 1.2 ± 0.06 s−1 for MYO1C35, MYO1C16, and MYO1CC, respectively (Table 1)). The ratio between the amplitudes of the fast and slow phases (Fig. 2D) represents the equilibrium constant, reflecting the distribution of the closed and open states, K′α (34).
Interestingly, unlike MYO1CC, which favors the closed state, MYO1C35 favors the open state, and MYO1C16 populates both states equally (K′α,p = 1.6 ± 0.04, 1.1 ± 0.02, and 0.7 ± 0.02 and K′α,LS = 1.3 ± 0.09, 0.9 ± 0.04 and 0.7 ± 0.06 for MYO1C35, MYO1C16, and MYO1CC, respectively (Table 1)). The observed changes in K′α in both assays were consistent with an overall shift toward population of the AMO state as the NTR grew longer.
Global numerical curve fitting of ATP-induced population of weakly bound acto·MYO1C states
The explicit solutions for the mechanism shown in Scheme 2 cannot extract the four parameters k′+1T, k′−1T, k′−2T, and k′diss. Therefore, we performed numerical integration (simulation) using KinTek Explorer (35, 36) by globally fitting our time-dependent reaction curves. The kinetic parameters extracted by performing simulation on the complete data sets for each isoform are presented in Fig. 3 and Table 2. The signal represents the sum of the AM·ATP and A states (weak-binding states). As shown in Table 4, the results of the simulation were in good agreement with the experimentally determined parameters K′1T and k′+2T, and the data provided numeric solutions for k′+1T, k′−1T, k′−2T, and K′2T. k′+1T decreased, whereas k′−1T did not differ between the isoforms (k′+1T = 6.6, 5.1, and 4.3 s−1 and k′−1T = 548, 568, and 531 s−1 for MYO1C35, MYO1C16, and MYO1CC, respectively). k′−2T of MYO1C35 was 2- and 8-fold lower than the values for MYO1C16 and MYO1CC, respectively (Table 2). K′2T, the equilibrium constant for isomerization after ATP binding, was 2- and 6-fold higher in MYO1C35 than in MYO1C16 and MYO1CC, respectively (Table 2). Both the forward and reverse rate constants for the nucleotide-binding isomerization from the closed-to-open state, k′+α and k′-α, were smaller than the experimentally determined constants (Table 2). The equilibrium rate constant for the closed-to-open isomerization, K′α, was similar between the experiment and simulation for MYO1CC. However, for MYO1C16 and MYO1C35, the predicted values were 1.5- and 2-fold larger, respectively, than the experimentally measured values (K′α = 3.0, 1.5, and 0.8 for MYO1C35, MYO1C16, and MYO1CC, respectively). Overall, the results were statistically significant in terms of the goodness of the fits per set of each isoform (Fig. 3, A–C). The confidence of the fitting results is expressed as the ratio of χ2/minimum χ2 for the forward versus reverse constants, which assures that the fits reached a global minimum. The signal arising from the summation of the weak-binding states in the simulation allowed determination of the rate of actomyosin dissociation, k′diss. Interestingly, we found that the value for MYO1CC (k′dissC = 1.0 s−1) was 5-fold smaller than the values for MYO1C35 and MYO1C16 (k′diss = 5.1 and 5.7 s−1, respectively). This analysis yielded additional rate constants that describe ATP binding to acto·MYO1C in greater detail and, more importantly, identified specific steps that differed among MYO1C isoforms.
Table 2.
ATP-induced pyrene(acto)·MYO1C dissociation by global fit simulation
Conditions used were: 20 mm MOPS, pH 7.0, 25 mm KAc, 2 mm MgCl2, 0.2 mm EGTA, and 1 mm DTT at 20 ± 0.1 °C.
| Constant | MYO1C35 | MYO1C16 | MYO1CC |
|---|---|---|---|
| k′+α (s−1) | 0.50 | 0.86 | 1.20 |
| k′−α (s−1) | 0.17 | 0.58 | 1.60 |
| K′α | 2.99 | 1.48 | 0.75 |
| K′1T k′+2T (μm−1 s−1)a | 0.33 | 0.24 | 0.19 |
| k′+1T (μm−1s−1) | 6.25 | 5.13 | 4.34 |
| k′−1T (s−1) | 548.00 | 568.00 | 531.00 |
| K′1T (mm−1) | 11.41 | 9.03 | 8.17 |
| k′+2T (s−1) | 29.00 | 26.80 | 23.00 |
| k′−2T (s−1) | 7.79 | 3.97 | 0.97 |
| K′2T | 3.72 | 6.75 | 23.66 |
| k′Diss (s−1) | 5.08 | 5.71 | 1.02 |
a Calculated.
Table 4.
ATP-induced prebound pyrene·acto·MYO1C·ADP dissociation by global fit simulation
| Constant | MYO1C35 | MYO1C16 | MYO1CC |
|---|---|---|---|
| k′+1D (s−1) | 4.5 ± 0.83 | 3.3 ± 0.17 | 3.6 ± 0.13 |
| k′−1D (μm−1·s−1) | 1.7 ± 0.14 | 6.4 ± 0.35 | 4.6 ± 0.17 |
| K′1D (μm) | 2.6 | 0.5 | 0.8 |
| k′+2D (s−1) | 0.9 ± 0.10 | 1.0 ± 0.03 | 1.2 ± 0.01 |
| k′−2D (s−1) | 5.2 ± 0.37 | 2.1 ± 0.12 | 1.3 ± 0.05 |
| K′2D | 0.2 | 0.4 | 0.9 |
| K′AD (K′1D·K2D) (μm) | 0.5 | 0.2 | 0.7 |
| kobs,AMDC (s−1)a | 0.4 ± 0.00 | 0.4 ± 0.00 | 0.5 ± 0.04 |
| AAMDCa | 15.6 ± 0.00 | 12.8 ± 0.01 | 16.9 ± 0.42 |
| kobs,fast,AMDO (s−1)a | 10.9 ± 0.09 | 6.6 ± 0.37 | 5.05 ± 0.25 |
| Afast,AMDOa | 1.02 ± 0.00 | 1.9 ± 0.05 | 7.5 ± 0.24 |
a The parameters obtained by fitting the exponential function to the simulated AMDO and AMDC states shown in Fig. 5.
ADP-binding kinetics to acto·MYO1C isoforms
The rate of ADP release can be determined by measuring the kinetics of ATP-induced dissociation of actomyosin·ADP as a function of [ADP] (37, 38). Fig. 4A shows representative time courses of acto·MYO1C·ADP premixed with 3.75 μm ADP (final concentration) upon rapid mixing with 1 mm ATP. The transient time courses were best fitted to a sum of two exponentials for all three isoforms (supplemental Fig. S3). The fast phase is thought to reflect ATP binding to free actomyosin, whereas the slow phase reflects the fraction of ADP dissociation from the acto·MYO1C·ADP complex (37, 39). Although acto·MYO1CC and acto·MYO1C16 sustained the fast and the slow phases throughout the entire range of [ADP], acto·MYO1C35·ADP prebound to ADP at a concentration of 7.5 μm or higher lost the fast-phase component due to loss of the fast amplitude (Fig. 4, B and D). Similar behavior has been reported for MYO1CC-1IQ and MYO1CC-3IQ (28, 30).
Figure 4.

ADP dissociation determined by ATP-induced dissociation of acto·MYO1C·ADP. A, representative time courses of pyrene-actin fluorescence enhancement after rapid mixing of 2 mm MgATP with an equilibrated mixture of 25 nm pyrene-labeled acto·MYO1C isoforms with 7.5 μm ADP. MYO1CC, MYO1C16, and MYO1C35 are shown in black, red, and blue, respectively. The data were best fitted to a double-exponential equation. Inset, the same data plotted on a logarithmic time scale. Data are averaged transients (n = 3). B and C, fast-phase kobs (B) and slow-phase kobs (C) plotted as a function of [MgADP]. The fast and the slow kobs were best fitted to a rectangular hyperbola (Scheme 3 and Equation 3) yielding Ifast, K′0.5,fast and k′min,fast, and Islow, K′0.5,slow and k′min,slow, respectively (Scheme 3 and Equation 3). D, total amplitudes of the fast and slow kobs are represented by the solid and open circles, respectively. The fast amplitude showed hyperbolic dependence on [MgADP] (solid line through the data). The MYO1C35 fast phase was not observed above 7.5 μm ADP due to the loss of the fast amplitude E, plot of the fraction of amplitudeslow fitted to a rectangular hyperbola (Equation 4). The error bars of the fitting are within the data points.
For all three isoforms, the dependence of both fast and slow kobs on [ADP] exhibited hyperbolic behavior, suggesting that ADP dissociation occurs via at least two transitions preceding the complete dissociation from actomyosin (Fig. 4, B and C). This is described as actomyosin·ADP isomerization from AMDC (ADP closed binding state) to AMDO (ADP open binding state), which has been observed in several other myosins (34, 40). However, for MYO1CC, AMD isomerization has not been kinetically identified in previous studies. A reaction mechanism that accounts for these events is presented in Scheme 3.
Scheme 3.

According to Scheme 3, in the absence of ADP, the signal arises from ATP-induced dissociation of the AMC and AMO states. Preincubation with higher [ADP] increases population of the AMDO and AMDC states, and at saturating ADP, AMDO and AMDC are the predominant states. Thus, the fast and slow kobs values measured upon rapid mixing with 1 mm ATP reflect the summation of all of these states. kobs,fast and kobs,slow are described by Equation 3 (Fig. 4, B and C).
| (Eq. 3) |
The y intercept of kobs,fast (Ifast) at [ADP] = 0 represents the rate constant for nucleotide binding by the AMO state. Indeed, Ifast values (29.4 ± 1.68, 30.6 ± 2.48, and 24.6 ± 1.86 s−1 for MYO1C35, MYO1C16, and MYO1CC, respectively (Table 3)) are very similar to the rate of isomerization after ATP binding, k′+2T (Table 2 and Scheme 3 for 1 mm ATP). At saturating ADP, AMDC and AMDO are the predominant states, and kobs,fast reports the decay of the AMDO state. ADP release was ∼4-fold faster for MYO1C16 than for MYO1CC (k′min,fast = 8.4 ± 3.90 and 2.2 ± 2.69 s−1 for MYO1C16 and MYO1CC, respectively (Table 3)). The fast phase of MYO1C35 was not observed above 7.5 μm ADP due to a loss of the fast amplitude (Fig. 4D). This suggests that, for MYO1C35, AMDC is the predominant state and ADP release occurs sequentially. For MYO1C16 and MYO1CC, the two ADP states co-existed over the entire range of the measured [ADP]. K0.5,fast was 2-fold lower for MYO1C16 than for MYO1CC (K0.5,fast = 0.4 ± 0.16 and 0.8 ± 0.26 μm for MYO1C16 and MYO1CC, respectively). The y intercept of kobs,slow (Islow) represents the rate of AMC-to-AMO isomerization (Fig. 5C). Indeed, Islow values (I slow = 0.9 ± 0.02, 1.5 ± 0.02, and 1.9 ± 0.07 s−1 for MYO1C35, MYO1C16, and MYO1CC, respectively (Table 3)) were very similar to k′+α (Table 3). At saturating ADP, the slow kobs represents the rate of AMDC decay. k′min,slow differed among the isoforms (k′min,slow = 0.4 ± 0.03, 0.5 ± 0.02, and 0.7 ± 0.10 s−1 for MYO1C35, MYO1C16, and MYO1CC, respectively (Table 3)). For MYO1C35 and MYO1C16, K′0.5,slow was similar and 1.4-fold lower than for MYO1CC (K′0.5,slow = 0.15 ± 0.07, 0.16 ± 0.01, and 0.21 ± 0.04 μm for MYO1C35, MYO1C16, and MYO1CC, respectively).
Table 3.
ADP binding to acto·MYO1C measured by kinetic competition with ATP
Conditions used were: 20 mm MOPS, pH 7.0, 25 mm KAc, 2 mm MgCl2, 0.2 mm EGTA, and 1 mm DTT at 20 ± 0.1 °C.
| Constant | MYO1C35 | MYO1C16 | MYO1CC |
|---|---|---|---|
| k′max,fast (s−1) | ≈0 | 8.4 ± 3.90 | 2.2 ± 2.69 |
| K′0.5,fast (μm) | 1.3 ± 0.51 | 0.4 ± 0.16 | 0.8 ± 0.26 |
| Ifast (s−1) | 29.4 ± 1.68 | 30.6 ± 2.48 | 24.6 ± 1.86 |
| k′max,slow (s−1) | 0.4 ± 0.03 | 0.5 ± 0.02 | 0.7 ± 0.10 |
| K′0.5,slow (μm) | 0.15 ± 0.07 | 0.16 ± 0.01 | 0.21 ± 0.04 |
| Islow (s−1) | 0.9 ± 0.02 | 1.5 ± 0.02 | 1.9 ± 0.07 |
| K′AD (μm) | 0.68 ± 0.23 | 0.19 ± 0.03 | 0.12 ± 0.05 |
Figure 5.

Global numerical simulation of time-dependent distribution of reaction intermediates of Scheme 3. A–C, simulated data sets of the ATP-induced population of weakly bound acto·MYO1C·ADP states fitted to raw data from MYO1C35, MYO1C16, and MYO1CC. Each graph shows the time course of data collected after preincubation with 1.25 (blue), 2.5 (brown), 5 (yellow), 7.5 (purple), 10 (green), 20 (light blue), or 30 (dark red) μm prebound ADP mixed rapidly with 1 mm ATP, as presented in A for pyrene-labeled acto·MYO1C35·ADP ATP-induced dissociation. The solid lines through the data sets are fitted curves resulting from performing global numerical analysis on the entire set of data for each isoform. D–F, time-dependent distribution of biochemical intermediates according to the simulation mechanism shown in Scheme 3 for MYO1C35, MYO1C16, and MYO1CC. Blue, AMC; red, AMO; yellow, AM(ATP); purple, AM·ATP; green, A; light blue, AMDC; dark red, AMDO. The light blue line represents the sum of the AM·ATP and A states, which represent the pyrene signal of weakly bound or dissociation states. AMDC and AMDO traces were fitted to single and double-exponential equations, respectively. The fitting parameters, kobs,AMDC, AAMDC, kobs,fast,AMO, and Afast,AMDO, are shown in Table 4.
For all three isoforms, the slow-phase amplitudes remained constant as a function of [ADP] (Fig. 4D) in contrast to kobs,slow, which exhibited hyperbolic dependence on [ADP]. At saturating ADP, both AMDO and AMDC should be fully occupied. Thus, the K′0.5 value for the fraction of the slow amplitude as a function of [ADP] indicates the overall affinity for ADP. The fraction of the slow phase (Fig. 3E) was fitted according to Equation 4.
| (Eq. 4) |
The overall affinity for ADP (reflected by K′AD) was ∼3-fold stronger for MYO1C16 and MYO1CC than for MYO1C35 (0.68 ± 0.23, 0.19 ± 0.03, and 0.12 ± 0.05 μm for MYO1C35, MYO1C16, and MYO1CC, respectively).
Global numerical curve fitting of ATP-induced acto·MYO1C·ADP dissociation
The minimum mechanism for dissociation of prebound ADP from actomyosin·ADP upon ATP binding involves at least five biochemical transitions (Scheme 3). Previously, we analyzed our data only in the defined AM or AMD states in the absence of ADP or under saturating ADP conditions. However, to analyze our data throughout the entire range of [ADP], considering all intermediates, we globally fitted the entire data set according to Scheme 3 (Fig. 5). To constrain the simulation, we used the parameters determined in the ATP-induced dissociation experiments (Table 4). Only four rate constants describing ADP binding and dissociation were determined by the model [k′+1D (s−1), k′−1D (μm−1·s−1), k′+2D (s−1), k′-2D (s−1)]. The fitting iterations to the data sets were allowed to run until they converged to the best possible fitting parameters to reach a global minimum (χ2/degree of freedom < 1.1, S.D. (σ) < 0.85). The good of the fitting results is expressed as the ratio of χ2/minimum χ2 for the forward versus reverse constants, which assures that the fits reached a global minimum (supplemental Fig. S5). The rate constant for ADP release from the AMDO state, k′+1D, was 1.4-fold larger for MYO1C35 than MYO1C16/C (Table 4). The rate for ADP binding for MYO1C35, however, was 3.7- and 2.7-fold smaller than those of MYO1C16 and MYO1CC (Table 4). As a result, the ADP-binding affinities of MYO1C16 and MYO1CC were 5.2- and 3.2-fold stronger than that of MYO1C35 (Table 4). The rate constant for isomerization of AMDC to AMDO, k′+2D, increased gradually as the NTR became shorter (Table 4). The rates for the closed-to-open nucleotide pocket isomerization were very similar to those for the isomerization of AMC to AMO (Table 3 and Scheme 2). By contrast, the reverse rate for the open-to-closed isomerization decreased as the NTR became shorter (Table 4). Consequently, two of the three isoforms populate the AMDC state to a greater extent than the AMDO state but with different ratios. MYO1C35 populates the closed state 5-fold more than the open state; MYO1C16 favors the closed state by 2.5-fold; and MYO1CC populates both states with almost equal probability (Table 4). The overall ADP affinity (K′AD = 0.5, 0.2, and 0.7 μm for MYO1C35, MYO1C16, and MYO1CC, respectively) agreed with the measured K′AD for MYO1C35/16, but was 5-fold smaller than the measured K′AD for the measured MYO1CC.
The simulated progression curves allowed us to follow the decay upon addition of ATP of AMDC and AMDO equilibrated with 30 μm ADP. AMDC decayed exponentially, with kobs and amplitude similar to the measured kobs,slow for all three isoforms (Figs. 4 and 5 and Tables 3 and 4). However, AMDO exhibited double-exponential decay with a fast phase that was not completely consistent with the measured fast phase, even though the fitted curve yielded a fast phase identical to the measured data (Figs. 4 and 5 and Tables 3 and 4). This suggests that the measured slow phase reflects AMDC decay, whereas the measured fast phase reflects a mixture of several states in the system.
Structural homology modeling and molecular dynamic docking of the NTR35 domain with MYO1CC
To gain further insights into how the NTRs impact the structural properties of MYO1C isoforms, we applied structure-based molecular dynamics and docking routines. First, we used PSIPRED version 3.3 to perform a secondary structure prediction of the NTR of MYO1C35, which indicated that this region consists of four β-strands interrupted by three coils (Fig. 6A). A FASTA search against structured proteins revealed that the NTR of MYO1C35 shares 18.4% sequence identity (44.7% sequence similarity) with desulforedoxin in (PDB ID: 1DHG) (Fig. 6B). Remarkably, desulforedoxin adopts a compact β-barrel fold comprising four β-strands, which is similar to the PSIPRED prediction. Therefore, the homology model of MYO1C35-NTR was based on desulforedoxin (PDB ID: 1DHG). In that model, a HPH motif (similar to the WPH motif of Myo1B (31)) is exposed at the tip of the β-barrel. Next, we performed docking experiments of the predicted folded motif of MYO1C35-NTR. For this procedure, we used Maestro in the Schrödinger Software Suite to generate different states of the predicted homology model by molecular dynamics simulations (41, 42). These different conformations of the NTR model were used as ligands for protein-protein docking studies using the crystal structure of MYO1CC (0, 6.4, 8.6, 15.5, and 25 ns after energy minimization) (PDB ID: 4BYF). For this purpose, we used the ClusPro online docking tool for protein-protein docking studies (43). Different constraints were added for the docking procedures. For example, the HPH motif (H18-P19-H20) of the NTR model was involved in the interaction, and the C-terminal Phe-57 of the NTR model exhibited a repulsive interaction. Most of the resulting docking models were out of range of the experimentally solved N terminus. The best hit within a reachable range is shown in Fig. 6D. In this model, the MYO1C35 NTR interacts with the experimentally solved crystal structure. Interestingly, according to this model, the NTR of the MYO1C35 interacts with amino acid region 619–636, which was identified by Schwab et al. (16) as corresponding to one of the two nucleolar localization signals of MYO1C16. The second nucleolar localization signal of MYO1C16 is located on the NTR itself, suggesting that the two are connected. Besides numerous electrostatic and hydrophobic contacts between the N terminus of the crystal structure and the nucleolar localization signal, one specific interaction stands out: residue Arg-21 engages in polar contact with Glu-469 of the loop directly after the relay helix, which could have a mechanical impact on the myosin power stroke. Consistent with this model, our kinetic studies revealed that MYO1C35 has a >2-fold faster AMDO-to-AMDC isomerization and a 3-fold slower ADP-binding rate constant than MYO1C16 and MYO1CC.
Figure 6.
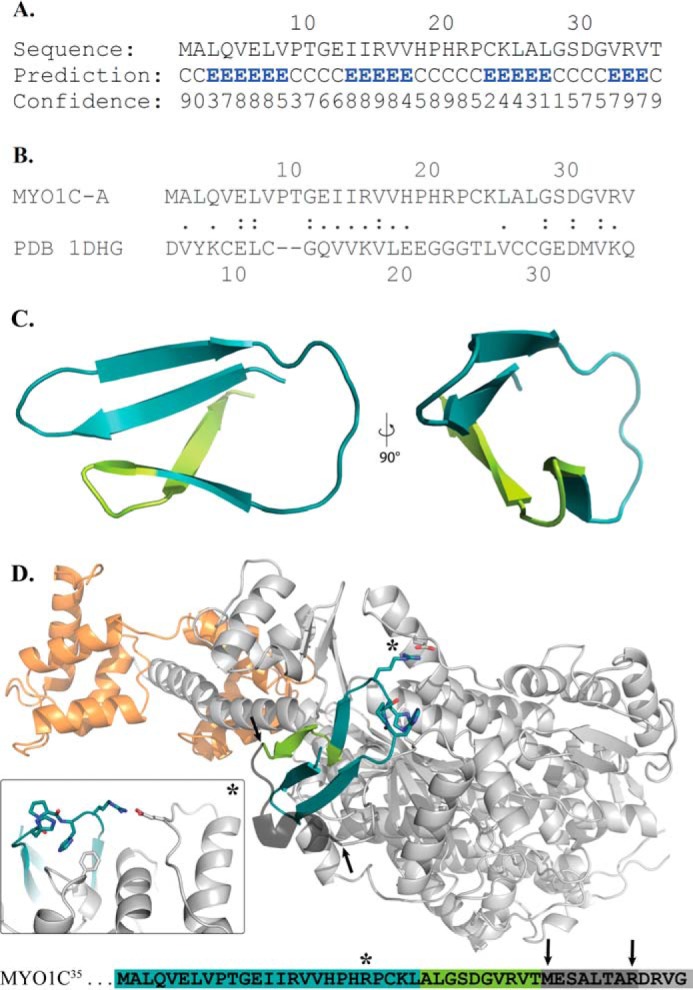
Structural homology modeling and molecular dynamics docking of the NTR extension. A, secondary structure prediction of NTR of MYO1C35. B, FASTA search against structured proteins revealed that the NTR of MYO1C35 shares sequence homology with desulforedoxin. C, homology model of the MYO1C35 NTR from two angles, showing the unclosed β-barrel-like structure. The model was based on desulforedoxin (PDB ID: 1DHG). D, overview of experimentally determined structure of MYO1C (PDB ID: 4BYF) shown in gray. The bound calmodulin is shown in orange and the NTR model in cyan/light green after the best protein-protein docking hit. The black arrows indicate the termini of the model and the solved myosin structure. The connector part of both is built after sequence similarity of nonredundant PDB sequences (52) with YASARA. It shows a qualitative possibility of this connectional part. Inset, a close-up of the contact between the NTR model and the post-relay helix region is marked with a star. The detailed view illustrates the possible salt bridge between Arg-21 and Glu-469 and the interaction of His-20 with Phe-57. At the bottom, an overview of the sequence shows the color code and position markers.
Determination of NTR35, NTR35-R21G, and NTR16 secondary structure by circular dichroism
To test our structural homology modeling, we synthesized three peptides corresponding to the 35-aa NTR (NTR35), NTR35-R21G (a mutant peptide based on the results of molecular dynamic modeling), and 16-aa NTR (NTR16). We then determined their secondary structures by performing circular dichroism (CD) measurements and deconvolution of their spectra (Fig. 7 and supplemental Tables S4 and S5). NTR35 exhibits a strong negative peak at 208 nm. Deconvolution of its spectrum yielded a prediction of 38% anti-parallel β-sheet, 47% unstructured, and 15% turns at 20 °C (Fig. 7 and supplemental Table S5). This is highly similar to the predicted model based on the structural homology presented in Fig. 6C. Point mutation of R21G within this peptide to generate NTR35-R21G shifted the negative peak to 209 nm (Fig. 7). We also determined the secondary structure of the shorter NTR16, which shares 10 aa with NTR35 (Fig. 7). According to the predicted secondary model, ∼43% of the identical amino acids fall within the predicted anti-parallel β-sheet (Fig. 6C).
Figure 7.
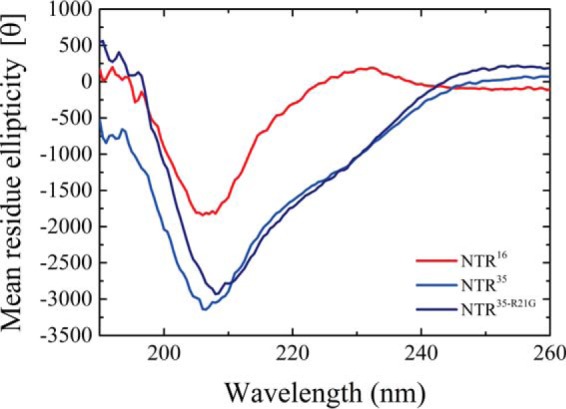
CD spectrum of the NTR peptides. Shown are the CD spectra of NTR16 peptide (red), NTR35 (blue), and NTR35-R21G (dark blue) at 20 °C. The data presented are an average of three measurements.
NTR35 peptide added in trans to MYO1CC induces MYO1C35-like kinetic behavior
The CD measurements of the various NTRs showed that these peptides form independently folded domains. Hence, they may invoke similar effects whether they are presentin trans or covalently attached to the polypeptide chain. We studied the effect of the NTR35 and NTR35-R21G peptides on ATP-induced dissociation of acto·MYO1CC·ADP and compared this effect among the three isoforms (Fig. 8). These experiments were performed in the presence of 20 μm folded peptide, high enough to saturate binding to MYO1CC (both 50 and 100 μm folded peptides yielded similar results). Interestingly, we observed the same kinetic behavior in kobs,fast and kobs,slow of ATP-induced dissociation of acto·MYO1C35·ADP as with acto·MYO1CC·ADP preincubated with NTR35. This remarkable finding demonstrates that NTR35 impacts the nucleotide-dependent transition in trans in the same way as when it is present on a continuous polypeptide chain. NTR35-R21G did not affect MYO1CC kinetic behavior to the same extent, confirming the predicted interaction of Arg-21 with the rest of the myosin heavy chain. Finally, the addition of the NTR peptides in trans influenced the fraction of the slow amplitude (Fig. 8B). This shifted the population of AMDC and AMDO toward the closed states, as predicted by our model.
Figure 8.
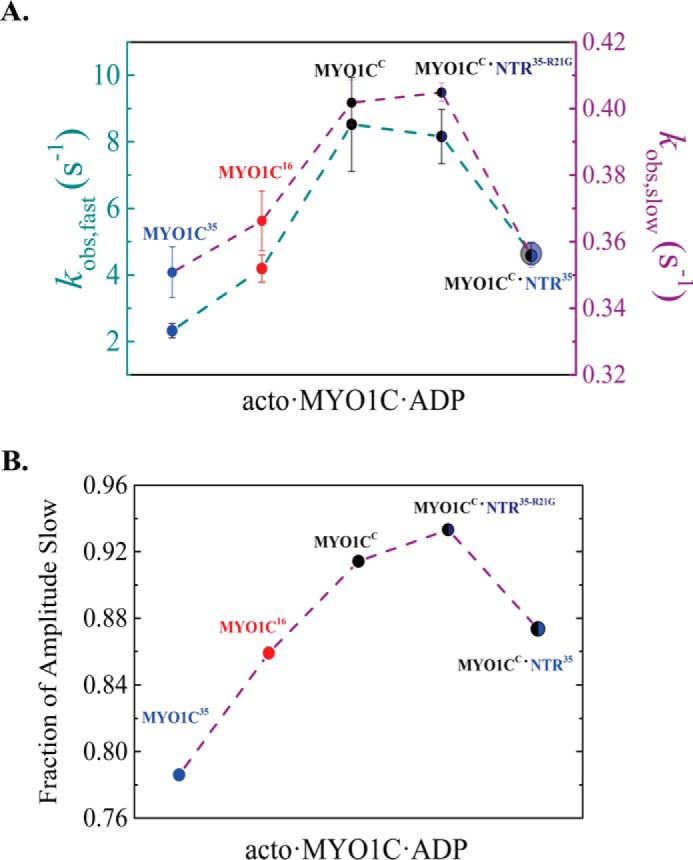
NTR35 peptide added in trans to MYO1CC induces MYO1C35-like kinetic behavior. A, fast-phase (dark cyan) and slow-phase (purple) kobs of the double-exponential fit to transients obtained after rapid mixing of 1 mm MgATP with an equilibrated mixture of 50 nm pyrene-labeled acto·MYO1C isoforms with 2.5 μm ADP. Shown are MYO1CC (black), MYO1C16 (red), MYO1C35 (blue), and MYO1CC (black) in the presence of 20 μm NTR35-R21G (dark blue) or NTR35 (blue). B, fraction of the slow amplitude of the same measurements. Data are from averaged transients (n = 6). The error bars of fitting are within the data points.
Discussion
We performed comparative studies of MYO1C splice isoforms in the context of their full-length proteins with the goal of achieving accurate allosteric awareness as proposed by Preller and Manstein (44). The steady-state parameters did not show dramatic changes in overall ATPase behavior among the isoforms. However, a detailed kinetic analysis revealed intrinsic divergence among the isoforms that either balanced out or had a low impact on kcat and KATPase.
We compared our results with those obtained to date with truncated MYO1CC constructs. We determined the equilibrium constant of MYO1C nucleotide-binding pocket isomerization using both pyrene-actin and light-scattering approaches. Previous studies on the MYO1CC-1IQ/3IQ motor domain with variable lever arm constructs showed that the MYO1CC isoform populates mostly the AMC state (27, 28, 29, 30). Our results supported these findings but also demonstrated that the equilibrium of full-length MYO1CC is shifted from AMC toward AMO by 7-fold relative to the 1IQ construct and 2-fold relative to the 3IQ construct.
ADP-binding kinetics to acto·MYO1C isoforms reveal conservation of ADP isomerization states
ADP-binding kinetics contribute to the dwelling time of myosin in the strong-binding state and hence its effect on the duty ratio (40, 45). Both EM and mechanical measurements show that the ADP release mechanism of MYO1CC is biphasic and depends upon additional movement of the lever arm and load (29, 30). However, kinetic measurements of ATP binding to prebound actomyosin·ADP using the 1IQ and 3IQ MYO1CC constructs could not detect intrinsic isomerization prior to ADP release due to the loss of the fast-phase kobs (27–30). Here, we show for the first time that full-length MYO1C undergo closed-to-open isomerization before ADP is released. This isomerization was also detected using truncated constructs of the closely related Myo1b (32). Therefore, we suggest that the addition of the tail domain shifts the equilibrium toward the AMDO state.
NTR effect on actomyosin nucleotide pocket isomerization
The NTR extensions altered the closed-to-open isomerization of the nucleotide pocket by stabilizing the AMO and AMDC states. As a result, the differences in behavior imply that each isoform has different kinetics depending on the ATP/ADP ratio. Related to this finding, the simulated models (Figs. 3 and 5) demonstrated that the larger the population of the AMO and AMDC states, the larger the population of the AM(ATP) state and the faster it forms, at a given [ATP]. The simulation also revealed differences in k′−2T and k′diss. MYO1CC populated the AM·ATP (weak-binding state) longer than MYO1C35, suggesting that the NTR extensions destabilize the weak-binding state and could be responsible for differences in the tension-sensing features between the two isoforms (32).
Communication between nucleotide- and actin-binding sites
The communication between the nucleotide- and actin-binding sites can be described by a closed thermodynamic square, in which ADP (D) and actin (A) binding to myosin (M) are linked by four equilibrium constants as shown in Scheme 4.
Scheme 4.
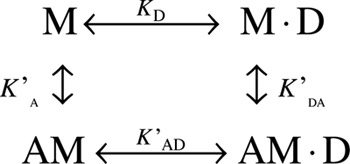
Myosins that generate rapid sliding velocities (e.g. muscle myosins) have large thermodynamic coupling constants (>10) and hence strong coupling between actin and ADP binding. On the other hand, myosins that function as gated/processive or tension sensors (e.g. myosins V, VI, and VII) (3, 40, 45, 46) have small thermodynamic coupling constants (<5). Table 3 and supplemental Fig. S4 and Table S3 show K′A, K′DA, and K′AD for each of the isoforms (see supplemental information text, Fig. S4, Scheme S1, and Equation S1 for a description of the equilibrium-binding experiments). The KD values of each isoform according to Scheme 4 are K′DA/K′A = 1.40, 0.99, and 1.39 for MYO1C35, MYO1C16, and MYO1CC, respectively, and the affinities for ADP in the absence of actin are KD = 0.3, 0.18, and 0.28 μm, respectively. The results support weak thermodynamic coupling between ADP and actin binding, consistent with a role for MYO1C isoforms as tension sensors or slow transporters in ensembles.
Consideration of the structural impact of the NTR in light of current models
Greenberg et al. (32) studied how MYO1CC-3IQ NTR impacts load dependence and kinetics, either by deleting the first nine residues of MYO1CC or replacing them with the first 13 residues of Myo1b. This region is shared by all three MYO1C spliced isoforms. Greenberg et al. (32) performed comprehensive biochemical and mechanical (under load) studies to investigate how such structural changes affect the motor properties of MYO1CC; specifically, the results of the unloaded kinetics revealed that isomerization after nucleotide binding and AMC-to-AMO isomerization are strongly affected by the identity of the NTR, which can alter k′+2, k′+α, and k′−α quite significantly. They found that the addition or deletion of these structural elements affects the active site isomerization by increasing its flexibility. Finally they proposed that the NTR plays an important role in stabilizing the post-power-stroke conformation (32). Our results show that the extended NTRs affect nucleotide pocket isomerization by decreasing both k′+α and k′−α without affecting k′+2. Moreover, unlike MYO1CC, which tended to populate the AMC state in the ATP-induced dissociation experiment, MYO1C35 populated mostly the AMO state, whereas MYO1C16 populated both states equally (Fig. 2). Together, our results indicate that the lengthening of the NTR increases the rigidity of the nucleotide pocket and stabilizes the AMO state. In the prebound ADP measurements, all three isoforms tended to populate the AMDC state but to varying extents (MYO1C35 > MYO1C16 > MYO1CC) (Fig. 4). This suggests that the NTR extensions stabilize the post-power-stroke state, similar to what was observed previously in NTR mutants as well as Myo1b (32). Our results indicate some degree of correlation between ATP- and ADP-binding kinetics in all three isoforms. It may be that these transitions are linked in terms of structural reorganization, i.e. the open-to-closed isomerization of the nucleotide-binding pocket. In addition, consistent with the findings of Greenberg et al. (32), our results support the idea that the nucleotide-binding pocket is affected by the NTR region. We propose that the extended NTRs of the isoforms form a structural domain (Fig. 7) that affects pocket rigidity and stabilizes the AMO and AMDC states.
Higher level of fine regulation by MYO1C NTRs
Our results show changes in the kinetic parameters that may yield additional specific kinetic adaptations for each of the three isoforms. Several studies have suggested that in addition to their distinct functions, some overlap could occur in the event that one isoform is lost. Knock-out mice lacking the NM1 (MYO1C16) start codon (without affecting MYO1CC or MYO1C35) exhibit interchangeability and redundancy of myosin isoforms in the cell nucleus, suggesting that both isoforms can substitute for each other in nuclear processes (25). Partial rescue and functional overlap between closely related MYO1C isoforms are likely to minimize the observed cellular and whole-animal knockdown phenotypes (47). MYO1C16, although displaying specific nuclear functions, localizes to the plasma membrane. Furthermore, knock-out of MYO1C16 has strong effects on the elasticity of the plasma membrane around the actin cytoskeleton, as determined by atomic force microscopy (24). Overall, MYO1C isoforms possess overall nearly identical structural domains and most likely are subject to similar post-translational modifications and binding to similar partners. Thus, different mixtures or ensembles of MYO1C isoforms could serve to fine-tune a specific biological function. Finally, to distinguish between the ensemble effects and redundancy of isoforms, all three knockouts should be compared individually.
Experimental procedures
Reagents
All chemicals and reagents were of the highest purity commercially available. ATP was purchased from Roche Applied Science, and ADP was purchased from Bio Basic (Markham, Ontario, Canada). Nucleotide concentrations were determined by measuring absorbance at 259 nm using ϵ259 = 15,400 m−1 cm−1. In all experiments, 1 molar equivalent of MgCl2 was added to nucleotide solutions immediately before use. N-(1-Pyrene)iodoacetamide (Molecular Probes, Eugene, OR), MOPS, EGTA, apyrase (potato grade VII), and phalloidin were purchased from Sigma-Aldrich. MgCl2·6H2O came from Bio Basic and KCl from Merck (Darmstadt, Germany).
Cell culture
All media reagents were purchased from Sigma. Fetal calf serum, l-glutamine, HEPES-KOH, pH 7.4, penicillin, streptomycin, and amphotericin B were purchased from Biological Industries (Beit Haemek, Israel).
Cloning of full-length human MYO1C isoforms
Full-length human MYO1C isoforms (residues 1–1063, 1044m, and 1028 for MYO1C35, MYO1C16, and MYO1CC, respectively) were cloned into the HaloTag-pFC14K Flexi vector (Promega). Human cDNA (HsCD00365758 clone ID), purchased from the ORFeome Collaboration, was used as the template for cloning human isoforms by primer extension PCR using the primers listed in supplemental Table S1. All constructs were fully sequenced and compared with the published sequences of human MYO1C isoform (NCBI RefSeq NM_001080779.1, NM_001080950.1, and NM_033375.4 for MYO1C35, MYO1C16, and MYO1CC, respectively). Human calmodulin was cloned into pF4A.
Expression and purification of full-length human MYO1C isoforms
Human isoform constructs with co-expressed calmodulin were purified from suspension-adapted HEK293SF-3F6 cells using the Promega HaloTag mammalian expression system. Briefly, 1 liter of suspension HEK293SF-3F6 cells (106 cells/ml) was grown in serum-free EX-CELL medium (Sigma-Aldrich) as reported previously (48) or in proprietary cell culture medium made in-house, co-transfected with pFC14K-MYO1C and pF4A-calmodulin, and harvested after 48 h. The cells were lysed (20 mm MOPS, pH 7, 5 mm MgAc, 2 mm K-EGTA, 150 mm KAc, 0.025% Nonidet P-40, 0.025% Triton X-100, 0.5 mm DTT, Promega protease inhibitor mixture, 1 μm E-64, and 2 μm calmodulin) using a Dounce homogenizer followed by centrifugation at 100,000 × g for 1 h in the presence of 2 mm MgATP (4 °C). The supernatant was incubated with HaloTag beads overnight, washed (20 mm MOPS, pH 7, 5 mm MgAc, 2 mm K-EGTA, 150 mm KAc, 0.0025% Nonidet P-40, and 0.5 mm DTT) with or without 2 mm ATP, and then cleaved from the beads using Promega HaloTEV protease. The eluted protein was dialyzed into storage buffer (50% glycerol, 20 mm MOPS, pH 7, 5 mm MgAc, 2 mm K-EGTA, 75 mm KAc, and 0.5 mm DTT) and stored at −20 °C. Purity was ≥95% for all constructs (Fig. 1C). All purifications and experiments were performed in the presence of 5 μm calmodulin. MYO1C isoform concentrations were determined using the predicted extinction coefficient at 280 nm (ExPASy ProtParam) in 6 m guanidine hydrochloride. Absorption spectra were acquired on a T90+ spectrometer (PG Instruments, Leicestershire, UK) controlled by UWin software.
Expression and purification of other proteins
Actin was purified from rabbit or chicken skeletal muscle (labeled with pyrene if needed) and gel-filtered over Sephacryl S-300 HR (49). Ca2+-actin monomers were converted to Mg2+-actin monomers by the addition of 0.2 mm EGTA and 40 μm MgCl2 (excess over [actin]) immediately prior to polymerization by dialysis against KMg50 buffer (20 mm MOPS, 50 mm KAc, 2 mm MgCl2, 0.2 mm EGTA, and 1 mm DTT, pH 7, at 25 °C). The final dialysis was performed against KMg25 buffer (20 mm MOPS, 25 mm KAc, 2 mm MgCl2, 0.2 mm EGTA, and 1 mm DTT, pH 7 at 25 °C). Phalloidin (1:1 molar ratio) was used to stabilize actin filaments. Actin was purified from rabbit skeletal muscle, labeled with pyrene, and gel-filtered over Sephacryl S-300 HR 28 (49) Ca2+-actin monomers were converted to Mg2+-actin monomers with 0.2 mm EGTA and 50 μm MgCl2 (excess over [actin]) immediately prior to polymerization by dialysis against KMg50 and followed by KMg25 buffer. Phalloidin (1.1 molar equivalent) was used to stabilize the actin filaments. Calmodulin was expressed in bacteria and purified as described (50).
Determination of light-chain calmodulin occupancy to the myosin isoforms
Light-chain calmodulin occupancy of myosin isoforms was performed by actin co-sedimentation of the MYO1C constructs as described elsewhere (51) with minor changes. Briefly, 60 nm MYO1C isoforms was incubated with 1 μm actin in KMg25 buffer for 30 min followed by ultracentrifugation in a TLA55 rotor (Beckman) at 186,000 × g for 25 min at 4 °C. The pellets were then washed gently with the KMg25 buffer and resuspended in protein sample buffer in the presence of 3 mm EGTA, heated for 5 min at 95 °C, resolved by gradient SDS-PAGE (10–20%), and visualized by staining with InstantBlueTM (Expedeon, San Diego, CA). Calmodulin band intensities were quantified with ImageLab software using known quantities of calmodulin resolved on the same gel as standards.
Steady-state ATPase activity
The actin-activated steady-state ATPase activity of MYO1C was measured at 20 ± 0.1 °C in KMg25 buffer supplemented with 2 mm MgATP by monitoring changes in absorption at 340 nm for 10 min at 1-s intervals on a UV-spectrometer (37). The concentration of all myosins was 100 nm. Myosin concentrations were determined as described above and verified by gel densitometry.
Equilibrium fluorescence binding of pyrene-actin to myosin and actomyosin
Fluorescence measurements were performed on a PC1 spectrofluorimeter (ISS Inc., Urbana-Champaign, IL) set up in an L-format configuration using an emission channel monochromator. Samples were equilibrated (60 min at room temperature), measured with λex = 365 nm, and monitored with emission monochromators scanning from 390 to 430 nm with a peak at λem = 409 nm.
Stopped-flow measurements
All experiments were performed in KMg25 buffer on a Hi-Tech Scientific SF-61DX2 stopped-flow apparatus (TgK Scientific Ltd., Bradford-on-Avon, UK) with temperature regulated by a thermostat at 20 ± 0.1 °C. For ATP-induced dissociation experiments, due to high [MgATP], buffer ionic strength (I) was held constant throughout the measurements at the ionic strength of KMg25. Unless noted otherwise, the concentrations stated throughout the text are final concentrations after mixing (i.e. in the observation cell). Light scattering was measured with excitation at 313 nm. Pyrene fluorescence was measured (λex = 365) through a long-pass 400-nm filter. Most time courses shown are those of individual, 2000-point transients collected with the instrument in oversampling mode, where the intrinsic time constant for data acquisition is ≈64 μs. Typically, three time courses were averaged before analysis. Time courses exhibiting fast and slow phases were collected on a logarithmic time scale. Data analysis was performed using the Kinetic Studio software provided with the instrument or with Origin (OriginLab Corp., Northampton, MA). Time courses of changes in signal (fluorescence and light scattering) were fitted to a sum of exponentials according to Equation 5,
| (Eq. 5) |
where F(t) is the signal at time t, F∞ is the final signal value, Ai is the amplitude, ki is the observed rate constant characterizing the i-th relaxation process, and n is the total number of observed relaxations. The value of n was either 1 (single exponential) or 2 (double exponential). Fitting was limited to data beyond 1 ms to account for the instrument dead time and to exclude data acquired during the continuous-flow phase of mixing, as recommended by the manufacturer.
Uncertainties are reported as standard errors in the fits, unless stated otherwise, and were propagated using the general formula shown in Equation 6,
| (Eq. 6) |
where the experimental measurements x1, x2…xn have uncertainties dx1, dx2…dxn and a is a function of x1, x2…xn. The Levenberg-Marquardt algorithm was used to solve the minimization of nonlinear least squares curve fitting.
Nucleotide-binding kinetics
The time courses of nucleotide binding were acquired under pseudo first-order conditions with [nucleotide] ≫ [myosin or actomyosin]. Actomyosin samples were prepared by mixing equimolar amounts of MYO1C and actin filaments or, where specified, with [actin] ≫ [myosin]. Actomyosin samples were treated with apyrase (0.01 unit/ml), used to deplete ATP and ADP from actomyosin when relevant, and equilibrated on ice for 10 min before measurements were made. After mixing, the final concentration of apyrase was 0.005 unit/ml (37).
Circular dichroism measurements
Far-UV CD was observed with an Applied Photophysics PiStar CD spectrometer (Surrey, UK) equilibrated with nitrogen gas, with the temperature regulated by a thermostat at 20 ± 0.1 °C. Changes in ellipticity were followed from 280 to 190 nm in 20 mm MOPS, 25 mm KCl in a 1-mm path-length fused quartz cell with a step size of 0.5 nm and bandwidth of 10 nm. Typically, three scans were averaged prior to analysis. Peptide concentrations were 0.2 mg/ml, corresponding to 54, 56.1, and 115 μm NTR35, NTR35-R21G, and NTR16, respectively (supplemental Table S4). The mean residue ellipticity ([θ], in mdeg·cm2·dmol−1·residue−1) was derived from the raw data (θ, in millidegrees (mdeg)) using the following formula: [θ] = θ × 100/(l × n × c), where l is the path length of the cuvette, c is the molar concentration of the peptides, and n is the number of residues in the peptides (i.e. 35 or 16 residues). The CD spectrum was deconvoluted using the BestSel server (http://bestsel.elte.hu/)3 (53). The normalized root-mean-square deviation represents the goodness of the deconvolution ( NRMSD, supplemental Table S5).
Kinetic simulations and modeling
We used KinTek Explorer (35, 36) to simulate the complex reaction shown in Schemes 3 and 4, utilizing numerical integration and global fitting of a family of data sets to a single model to extract mechanistic information directly from kinetic data. First, the transients were fitted to an analytic function to derive their standard deviation (σ), which was then incorporated into the statistical analysis of the goodness of the fits. The fitting parameters were then loaded into the Ode45 function in Matlab to produce the model and fitting.
Molecular dynamics simulation
For molecular dynamics simulation and energy minimization processes, we used Desmond in the Maestro 11 work suite (41, 42). The OPLS_2005 force field was used in a minimized 10 Å orthorhombic water box with 0.05 m sodium chloride ions. The simulation was performed with the TIP3P water model, including a 0.03-ns quick relaxation step. The simulation time was 25 ns at 300 K and 1 atmosphere.
Protein docking
Protein-protein docking was performed using the ClusPro online docking tool (43). The MYO1CC crystal structure (PDB ID: 4BYF) was used as the receptor molecule. The ligand molecule was a homology model of an NTR in minimized conformation with four additional different plateau states of the molecular dynamics simulation (6.4, 8.6, 15.5, and 25 ns). Different parameters of attraction and repulsion were used, including determined attraction for His-18–Pro-19–His-20 of the NTR and repulsion for Val-31.
Author contributions
A. H. and L. Z. designed and performed the research, analyzed the data, and wrote the article. R. R. performed research, and M. U. developed the methodology to express constructs in suspension-adapted HEK293SF-3F6 cell line. P. Y. A. R. and A. O. S. performed the structural homology modeling, P. Y. A. R. preformed molecular dynamic modeling and docking, and P. Y. A. R., S. G., M. H. T., and D. J. M. analyzed data.
Supplementary Material
Acknowledgments
We thank Maria Poley, Hadar Levi, and Rinat Cohen for technical assistance (A. H. group). We also thank the A. H. group members for their helpful comments on the manuscript.
This work was supported by Marie Curie Career Integration Grant 1403705/11 and Grant 296/13 from the Israeli Scientific Foundation (to A. H.). This work also was supported by the Volkswagen Stiftung, Niedersächsisches Vorab, Joint Lower Saxony-Israeli Research Projects (Grant VWZN3012) (to A. H., M. H. T., and D. J. M.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Figs. S1–S5, Tables S1–S5, Equation S1, Scheme S1, accompanying information, and Ref. 1.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- MYO1C
- myosin IC
- NTR
- N-terminal region
- aa
- amino acid
- AM
- actomyosin
- AMD
- actomyosin·ADP
- PDB
- Protein Data Bank.
References
- 1. Houdusse A., and Sweeney H. L. (2016) How myosin generates force on actin filaments. Trends Biochem. Sci. 41, 989–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. De La Cruz E. M., and Ostap E. M. (2004) Relating biochemistry and function in the myosin superfamily. Curr. Opin. Cell Biol. 16, 61–67 [DOI] [PubMed] [Google Scholar]
- 3. Bloemink M. J., and Geeves M. A. (2011) Shaking the myosin family tree: Biochemical kinetics defines four types of myosin motor. Semin. Cell Dev. Biol. 22, 961–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Heissler S. M., and Sellers J. R. (2016) Kinetic adaptations of myosins for their diverse cellular functions. Traffic 17, 839–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ihnatovych I., Migocka-Patrzalek M., Dukh M., and Hofmann W. A. (2012) Identification and characterization of a novel myosin Ic isoform that localizes to the nucleus. Cytoskeleton (Hoboken) 69, 555–565 [DOI] [PubMed] [Google Scholar]
- 6. Dzijak R., Yildirim S., Kahle M., Novák P., Hnilicová J., Venit T., and Hozák P. (2012) Specific nuclear localizing sequence directs two myosin isoforms to the cell nucleus in calmodulin-sensitive manner. PLoS ONE 7, e30529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Adams R. J., and Pollard T. D. (1989) Binding of myosin I to membrane lipids. Nature 340, 565–568 [DOI] [PubMed] [Google Scholar]
- 8. Tang N., Lin T., and Ostap E. M. (2002) Dynamics of myo1c (myosin-iβ) lipid binding and dissociation. J. Biol. Chem. 277, 42763–42768 [DOI] [PubMed] [Google Scholar]
- 9. Hokanson D. E., Laakso J. M., Lin T., Sept D., and Ostap E. M. (2006) Myo1c binds phosphoinositides through a putative pleckstrin homology domain. Mol. Biol. Cell 17, 4856–4865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nambiar R., McConnell R. E., and Tyska M. J. (2009) Control of cell membrane tension by myosin-I. Proc. Natl. Acad. Sci. U.S.A. 106, 11972–11977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brandstaetter H., Kendrick-Jones J., and Buss F. (2012) Myo1c regulates lipid raft recycling to control cell spreading, migration and Salmonella invasion. J. Cell Sci. 125, 1991–2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bose A., Guilherme A., Robida S. I., Nicoloro S. M., Zhou Q. L., Jiang Z. Y., Pomerleau D. P., and Czech M. P. (2002) Glucose transporter recycling in response to insulin is facilitated by myosin Myo1c. Nature 420, 821–824 [DOI] [PubMed] [Google Scholar]
- 13. Zuo J. (2002) Transgenic and gene targeting studies of hair cell function in mouse inner ear. J. Neurobiol. 53, 286–305 [DOI] [PubMed] [Google Scholar]
- 14. Dumont R. A., Zhao Y. D., Holt J. R., Bähler M., and Gillespie P. G. (2002) Myosin-I isozymes in neonatal rodent auditory and vestibular epithelia. J. Assoc. Res. Otolaryngol. 3, 375–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Maly I. V., and Hofmann W. A. (2016) Calcium-regulated import of myosin IC into the nucleus. Cytoskeleton (Hoboken) 73, 341–350 [DOI] [PubMed] [Google Scholar]
- 16. Schwab R. S., Ihnatovych I., Yunus S. Z., Domaradzki T., and Hofmann W. A. (2013) Identification of signals that facilitate isoform-specific nucleolar localization of myosin IC. Exp. Cell Res. 319, 1111–1123 [DOI] [PubMed] [Google Scholar]
- 17. Nowak G., Pestic-Dragovich L., Hozák P., Philimonenko A., Simerly C., Schatten G., and de Lanerolle P. (1997) Evidence for the presence of myosin I in the nucleus. J. Biol. Chem. 272, 17176–17181 [DOI] [PubMed] [Google Scholar]
- 18. Pestic-Dragovich L., Stojiljkovic L., Philimonenko A. A., Nowak G., Ke Y., Settlage R. E., Shabanowitz J., Hunt D. F., Hozak P., and de Lanerolle P. (2000) A myosin I isoform in the nucleus. Science 290, 337–341 [DOI] [PubMed] [Google Scholar]
- 19. Philimonenko V. V., Zhao J., Iben S., Dingová H., Kyselá K., Kahle M., Zentgraf H., Hofmann W. A., de Lanerolle P., Hozák P., and Grummt I. (2004) Nuclear actin and myosin I are required for RNA polymerase I transcription. Nat. Cell Biol. 6, 1165–1172 [DOI] [PubMed] [Google Scholar]
- 20. Cavellán E., Asp P., Percipalle P., and Farrants A. K. (2006) The WSTF-SNF2h chromatin remodeling complex interacts with several nuclear proteins in transcription. J. Biol. Chem. 281, 16264–16271 [DOI] [PubMed] [Google Scholar]
- 21. Obrdlik A., Louvet E., Kukalev A., Naschekin D., Kiseleva E., Fahrenkrog B., and Percipalle P. (2010) Nuclear myosin 1 is in complex with mature rRNA transcripts and associates with the nuclear pore basket. FASEB J. 24, 146–157 [DOI] [PubMed] [Google Scholar]
- 22. Percipalle P., Fomproix N., Cavellán E., Voit R., Reimer G., Krüger T., Thyberg J., Scheer U., Grummt I., and Farrants A. K. (2006) The chromatin remodelling complex WSTF-SNF2h interacts with nuclear myosin 1 and has a role in RNA polymerase I transcription. EMBO Rep. 7, 525–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chuang C. H., Carpenter A. E., Fuchsova B., Johnson T., de Lanerolle P., and Belmont A. S. (2006) Long-range directional movement of an interphase chromosome site. Curr. Biol. 16, 825–831 [DOI] [PubMed] [Google Scholar]
- 24. Venit T., Kalendová A., Petr M., Dzijak R., Pastorek L., Rohožková J., Malohlava J., and Hozák P. (2016) Nuclear myosin I regulates cell membrane tension. Sci. Rep. 6, 30864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Venit T., Dzijak R., Kalendová A., Kahle M., Rohožková J., Schmidt V., Rülicke T., Rathkolb B., Hans W., Bohla A., Eickelberg O., Stoeger T., Wolf E., Yildirim A. Ö., Gailus-Durner V., et al. (2013) Mouse nuclear myosin I knock-out shows interchangeability and redundancy of myosin isoforms in the cell nucleus. PLoS ONE 8, e61406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sielski N. L., Ihnatovych I., Hagen J. J., and Hofmann W. A. (2014) Tissue specific expression of myosin IC isoforms. BMC Cell Biol. 15, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lin T., Greenberg M. J., Moore J. R., and Ostap E. M. (2011) A hearing loss-associated myo1c mutation (R156W) decreases the myosin duty ratio and force sensitivity. Biochemistry 50, 1831–1838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Greenberg M. J., Lin T., Goldman Y. E., Shuman H., and Ostap E. M. (2012) Myosin IC generates power over a range of loads via a new tension-sensing mechanism. Proc. Natl. Acad. Sci. U.S.A. 109, E2433–E2440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Batters C., Arthur C. P., Lin A., Porter J., Geeves M. A., Milligan R. A., Molloy J. E., and Coluccio L. M. (2004) Myo1c is designed for the adaptation response in the inner ear. EMBO J. 23, 1433–1440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Adamek N., Coluccio L. M., and Geeves M. A. (2008) Calcium sensitivity of the cross-bridge cycle of Myo1c, the adaptation motor in the inner ear. Proc. Natl. Acad. Sci. U.S.A. 105, 5710–5715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shuman H., Greenberg M. J., Zwolak A., Lin T., Sindelar C. V., Dominguez R., and Ostap E. M. (2014) A vertebrate myosin-I structure reveals unique insights into myosin mechanochemical tuning. Proc. Natl. Acad. Sci. U.S.A. 111, 2116–2121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Greenberg M. J., Lin T., Shuman H., and Ostap E. M. (2015) Mechanochemical tuning of myosin-I by the N-terminal region. Proc. Natl. Acad. Sci. U.S.A. 112, E3337–E3344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Münnich S., Taft M. H., and Manstein D. J. (2014) Crystal structure of human myosin 1c, the motor in GLUT4 exocytosis: Implications for Ca2+ regulation and 14–3-3 binding. J. Mol. Biol. 426, 2070–2081 [DOI] [PubMed] [Google Scholar]
- 34. Geeves M. A., Perreault-Micale C., and Coluccio L. M. (2000) Kinetic analyses of a truncated mammalian myosin I suggest a novel isomerization event preceding nucleotide binding. J. Biol. Chem. 275, 21624–21630 [DOI] [PubMed] [Google Scholar]
- 35. Johnson K. A., Simpson Z. B., and Blom T. (2009) FitSpace Explorer: An algorithm to evaluate multidimensional parameter space in fitting kinetic data. Anal. Biochem. 387, 30–41 [DOI] [PubMed] [Google Scholar]
- 36. Johnson K. A., Simpson Z. B., and Blom T. (2009) Global Kinetic Explorer: A new computer program for dynamic simulation and fitting of kinetic data. Anal. Biochem. 387, 20–29 [DOI] [PubMed] [Google Scholar]
- 37. De La Cruz E. M., and Ostap E. M. (2009) Kinetic and equilibrium analysis of the myosin ATPase. Methods Enzymol. 455, 157–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Adamek N., and Geeves M. A. (2014) Use of pyrene-labelled actin to probe actin-myosin interactions: Kinetic and equilibrium studies. EXS 105, 87–104 [DOI] [PubMed] [Google Scholar]
- 39. Lewis J. H., Lin T., Hokanson D. E., and Ostap E. M. (2006) Temperature dependence of nucleotide association and kinetic characterization of myo1b. Biochemistry 45, 11589–11597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nyitrai M., and Geeves M. A. (2004) Adenosine diphosphate and strain sensitivity in myosin motors. Philos. Trans. R Soc. Lond. B Biol. Sci. 359, 1867–1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schrödinger (2017) Maestro, in Release 2017–1, Schrödinger, LLC, New York, NY [Google Scholar]
- 42. Desmond Molecular Dynamics System (2017) Release 2017–1, D. E. Shaw Research, Maestro-Desmond Interoperability Tools, Schrödinger, LLC, New York, NY [Google Scholar]
- 43. Kozakov D., Hall D. R., Xia B., Porter K. A., Padhorny D., Yueh C., Beglov D., and Vajda S. (2017) The ClusPro Web server for protein-protein docking. Nat. Protoc. 12, 255–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Preller M., and Manstein D. J. (2013) Myosin structure, allostery, and mechano-chemistry. Structure 21, 1911–1922 [DOI] [PubMed] [Google Scholar]
- 45. Henn A., and De La Cruz E. M. (2005) Vertebrate myosin VIIb is a high duty ratio motor adapted for generating and maintaining tension. J. Biol. Chem. 280, 39665–39676 [DOI] [PubMed] [Google Scholar]
- 46. Taft M. H., Hartmann F. K., Rump A., Keller H., Chizhov I., Manstein D. J., and Tsiavaliaris G. (2008) Dictyostelium myosin-5b is a conditional processive motor. J. Biol. Chem. 283, 26902–26910 [DOI] [PubMed] [Google Scholar]
- 47. Tyska M. J., Mackey A. T., Huang J. D., Copeland N. G., Jenkins N. A., and Mooseker M. S. (2005) Myosin-1a is critical for normal brush border structure and composition. Mol. Biol. Cell 16, 2443–2457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shneyer B. I., Ušaj M., and Henn A. (2016) Myo19 is an outer mitochondrial membrane motor and effector of starvation-induced filopodia. J. Cell Sci. 129, 543–556 [DOI] [PubMed] [Google Scholar]
- 49. Pardee J. D., and Spudich J. A. (1982) Purification of muscle actin. Methods Enzymol. 85, 164–181 [DOI] [PubMed] [Google Scholar]
- 50. Putkey J. A., Donnelly P. V., and Means A. R. (1987) Bacterial expression vectors for calmodulin. Methods Enzymol. 139, 303–317 [DOI] [PubMed] [Google Scholar]
- 51. Lewis J. H., Greenberg M. J., Laakso J. M., Shuman H., and Ostap E. M. (2012) Calcium regulation of myosin-I tension sensing. Biophys. J. 102, 2799–2807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Michalsky E., Goede A., and Preissner R. (2003) Loops in Proteins (LIP): A comprehensive loop database for homology modelling. Protein Eng. 16, 979–985 [DOI] [PubMed] [Google Scholar]
- 53. Micsonai A., Wien F., Kernya L., Lee Y. H., Goto Y., Réfrégiers M., and Kardos J. (2015) Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy. Proc. Natl. Acad. Sci. USA 112, E3095–E3103 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.