

Abstract
The translational regulator cytosolic polyadenylation element–binding protein 2 (CPEB2) has two isoforms, CPEB2A and CPEB2B, derived by alternative splicing of RNA into a mature form that either includes or excludes exon 4. Previously, we reported that this splicing event is highly dysregulated in aggressive forms of breast cancers, which overexpress CPEB2B. The loss of CPEB2A with a concomitant increase in CPEB2B was also required for breast cancer cells to resist cell death because of detachment (anoikis resistance) and metastasize in vivo. To examine the mechanism by which CPEB2 isoforms mediate opposing effects on cancer-related phenotypes, we used next generation sequencing of triple negative breast cancer cells in which the isoforms were specifically down-regulated. Down-regulation of the CPEB2B isoform inhibited pathways driving the epithelial-to-mesenchymal transition and hypoxic response, whereas down-regulation of the CPEB2A isoform did not have this effect. Examining key nodes of these pathways showed that CPEB2B induced the expression of regulatory DNA trans-factors (e.g. HIF1α and TWIST1). Specifically, CPEB2B functioned as a translational activator of TWIST1 and HIF1α. Functional studies showed that specific down-regulation of either HIF1α or TWIST1 inhibited the ability of CPEB2B to induce the acquisition of anoikis resistance and drive metastasis. Overall, this study demonstrates that CPEB2 alternative splicing is a major regulator of key cellular pathways linked to anoikis resistance and metastasis.
Keywords: alternative splicing, breast cancer, epithelial-mesenchymal transition (EMT), hypoxia-inducible factor (HIF), mRNA, CPEB2, cytoplasmic polyadenylation, epithelial-to-mesenchymal transition, hypoxic response
Introduction
Breast cancer currently has the second highest mortality rate of all types of cancers (1). Although most breast cancers are well-controlled via first-line treatments, patients whose cancers escape these therapies are at high risk for metastasis and associated mortality. Hence, studies which elucidate the mechanisms of tumor formation and metastasis are necessary to combat this deadly disease (2, 3).
Regarding these mechanisms, a multitude of acquired characteristics are necessary for cancer cells to form tumors, invade the surrounding tissues, and, eventually, colonize distant organs (4, 5). One early characteristic is the ability to survive independent of attachment (i.e. anoikis resistance (AnR))3 (6). Our laboratory has recently delineated the RNA alternative splicing (AS) events, which are induced by AnR. One such event, the AS of cytoplasmic polyadenylation element–binding protein 2 (CPEB2) was demonstrated by our laboratory to enhance metastasis of triple negative breast cancer (TNBC) cells. Specifically, the CPEB2B isoform, which is produced by inclusion of exon 4 into the mature CPEB2 mRNA, was observed to be dramatically up-regulated in aggressive forms of human breast cancer and expression of this CPEB2 isoform dramatically enhanced the metastasis of TNBC cells in murine orthotopic models (7). This pro-neoplastic function was in stark contrast to the anti-neoplastic function of the CPEB2A isoform, which is produced by exclusion of exon 4 from the mature CPEB2 mRNA.
CPEB2 is a member of the family of cytosolic polyadenylation element–binding proteins (CPEB), which is comprised of four members (CPEB1–4) that function as regulators of protein translation via modulation of cytoplasmic RNA polyadenylation. In general, CPEB family members are considered to be suppressors of mRNA translation and anti-neoplastic in nature (8, 9). How the inclusion of a small exon (30 amino acids encoded by exon 4) into the mature CPEB2 mRNA to produce the CPEB2B isoform induces an opposite function from other CPEB family members is currently unknown.
In these studies, the cellular mechanisms by which the CPEB2B isoform imparts the pro-neoplastic effects were elucidated. Specifically, these studies demonstrate that CPEB2B drives AnR via induction of epithelial-to-mesenchymal transition (EMT) and hypoxic response pathways. We further show that CPEB2B plays an antagonistic role against CPEB2A by alleviating the translational inhibition of HIF1α and TWIST1 imparted by the CPEB2A splice variant. Thus, these studies show a major regulatory role for the AS of CPEB2 in the EMT and hypoxia pathways, and these studies demonstrate the critical role for AS in regulating specific cellular processes related to stress signaling.
Results
CPEB2A and CPEB2B regulate EMT and hypoxic response pathways in an opposing fashion
Our laboratory previously reported that the splice variants, CPEB2A and B, produced by alternative inclusion/exclusion of exon 4 (Fig. 1A) (7), have contrasting functions on the acquisition of AnR and the metastatic ability of TNBC cells. Specifically, expression of the exon 4–included isoform, CPEB2B, correlated with AnR resistance, high-grade metastatic breast cancer, and a dramatically enhanced metastatic rate in stark contrast to CPEB2A, the exon 4–excluded isoform (7). These published findings supported the hypothesis that CPEB2 isoforms regulated key cell survival pathways in a contrasting manner. To explore this hypothesis, we utilized next generation sequencing (NGS) coupled to specific down-regulation of CPEB2 splice variants in AnR TNBC cells (Fig. 1B). Down-regulation of the CPEB2B isoform in MDA-MB-231 cells, in stark contrast to down-regulation of the CPEB2A isoform, led to suppression of cellular pathways driving both cell growth/proliferation and EMT/hypoxic response. Specifically, CPEB2A was observed as a negative regulator of transcriptomic signatures associated with both the EMT and the hypoxic response pathways as well as tumor suppressor pathways, whereas CPEB2B was observed to act as a major enhancer of these pathways as well as oncogenic signaling (Fig. 1, C and D).
Figure 1.

CPEB2A and CPEB2B regulate EMT/hypoxia pathways in opposing ways. A, exon structure of the CPEB2 gene. B—D, MDA-MB-231 (anoikis resistant) cells were treated with siRNA targeted toward either CPEB2A or CPEB2B. Cells were then subjected to immunoblot analysis (B) with the indicated antibodies as probes or subjected to next generation sequencing. C and D, heat maps based on the differentially regulated overlapping hypoxia/EMT and motility pathways identified by Ingenuity Pathway Analysis.
Ingenuity Pathway Analysis (IPA) demonstrated that cellular signaling nodes linked to TWIST1/SNAI1 and HIF1α-regulated pathways were central to the contrasting functions of CPEB2 splice variants (Fig. 2A). Interestingly, differences in the mRNA levels of these DNA trans-factors were not observed in these NGS data. Because CPEB2 has been implicated as a translational repressor of TWIST1 and HIF1α (10, 11), our findings coupled to these published reports formed the premise that CPEB2A is the suppressing form of CPEB2 on TWIST1 protein expression as reported by others (9, 10), whereas CPEB2B has the previously unexplored function of activating the expression of these DNA trans-factors in response to cellular stress or the acquisition of AnR. In line with this premise, we observed that both HIF1α and TWIST1 were up-regulated in AnR MDA-MB-231 cells, which correlated with a decrease in the CPEB2A/CPEB2B ratio (Fig. 2B). Furthermore, specific modulation of this splice variant ratio using siRNA affected the expression of HIF1α and TWIST1 at the protein level in parental MDA-MB-231 cells. Specifically, down-regulation of CPEB2A using siRNA (Fig. 2C) or ectopic expression of CPEB2B (Fig. 2, D and G) (FLAG-tagged CPEB2) to decrease the CPEB2A/B ratio up-regulated HIF1α and TWIST1 expression at the protein level. In contrast, down-regulation of CPEB2B using siRNA (Fig. 2C) or ectopic expression of CPEB2A (Fig. 2, D and G) (FLAG-tagged CPEB2) to increase the CPEB2A/B ratio dramatically down-regulated expression of these two proteins. These effects also translated to other members of the EMT pathway (N-cadherin, E-cadherin) and other factors, which were identified as part of the next generation sequencing screen (Fig. 2, C—E). Down-regulation of TWIST1 or HIF1α using siRNA blocked the expression of hypoxic response and EMT genes induced by CPEB2B expression in parental MDA-MB-231 cells (Fig. 2F). Taken together, these results confirm that hypoxic and EMT pathways are regulated by CPEB2 AS via modulation of the expression of key DNA trans-factors regulating these pathways. Furthermore, these data show that the CPEB2A isoform is the negative regulator of TWIST1 and HIF1α expression, in complete contrast to the CPEB2B isoform.
Figure 2.

CPEB2A and CPEB2B confer their respective functions via HIF1α and TWIST1 signaling. A, Ingenuity Pathway Analysis (IPA) was used to determine the genes linked to either TWIST1 or HIF1α, which were found to be dysregulated in the NGS experiment described in Fig. 1. B, parental and anoikis-resistant clones of MDA-MB-231 or MDA-MB-468 cells were subjected to immunoblot for the indicated proteins in the hypoxia/EMT pathway. C and D, parental MDA-MB-231 or MDA-MB-468 cells were subjected to either RNAi directed toward either CPEB2A or CPEB2B, or CPEB2A or CPEB2B ectopic expression, then subjected to immunoblot and probed with the indicated antibodies in the hypoxia/EMT pathways. E, confirmation of two factors shown to be regulated by CPEB2A/B knock down via qPCR using commercial primers. F, TWIST1 and HIF1α RNAi was then used to “rescue” the effects of CPEB2B on N-cadherin and E-cadherin. G, expression of CPEB2A and CPEB2B was confirmed via FLAG immunoblot. *, p < 0.05 assessed via ANOVA and Tukey's honest significant difference (HSD) post hoc test. Error bars represent standard deviation.
CPEB2A and CPEB2B regulate TWIST1 and HIF1α at the translational level
A large body of research has demonstrated that HIF1α is regulated via a decrease in ubiquitination and subsequent increase in protein stability after a hypoxic stress (12). Our results suggested an additional component in regard to HIF1α expression, specifically the regulation of HIF1α translation/polyadenylation by CPEB2 isoforms. Therefore, we undertook studies aimed at determining whether CPEB2 AS regulated TWIST1 and HIF1α at either the level of transcription, protein stability, or translation. TWIST1 and HIF1α mRNA levels are not affected by ectopic expression of CPEB2A and B; additionally, the protein stability of these DNA trans-factors were also not affected (data not shown). To determine whether mRNA transcription was affected by CPEB2 AS, nascent RNA transcripts were biotinylated and pulled down with streptavidin. Again, no significant differences because of CPEB2 AS were observed in the rate of nascent mRNA formation (data not shown). To investigate the translation of TWIST1 and HIF1α, l-azidohomoalanine, a methionine mimetic, was used to pulse both CPEB2A and CPEB2B overexpressing cells (Fig. 3A) for nascent protein labeling with biotin. As shown in Fig. 3B, nascent TWIST1 and HIF1α protein levels were up-regulated by CPEB2B, but CPEB2A expression induced the opposite effect. Taken together, these results indicate that CPEB2A/B regulation of the hypoxia/EMT axis occurs at the level translation.
Figure 3.

CPEB2A and CPEB2B act to either enhance or decrease HIF1α and TWIST1 levels via an increase in nascent protein levels. A, shows the input of either CPEB2A or CPEB2B-FLAG after labeling but prior to immunoprecipitation. B, click chemistry was used to label nascent proteins in MDA-MB-231 cells ectopically expressing either CPEB2A or CPEB2B. Biotin-labeled proteins were then precipitated using streptavidin beads and proteins thus isolated were probed using the indicated antibodies.
TWIST1 and HIF1α are required for the acquisition of anoikis resistance and increased metastatic rate induced by CPEB2B
Our data strongly suggest that CPEB2B-mediated increases in AnR occur via dysregulated EMT and hypoxic response signaling. Therefore, we performed experiments to determine whether HIF1α and TWIST1 signaling were important for CPEB2 AS-mediated AnR. Using MDA-MB-231 cells stably overexpressing FLAG-CPEB2B, we found that these cells were protected from cell death induced by detachment when compared with a stable vector control (pcDNA 3.1) (Fig. 4, A and B). Importantly, RNAi directed toward either HIF1α or TWIST1 significantly blocked the acquisition of AnR imparted by CPEB2B expression without effects on basal anoikis sensitivity in parental MDA-MB-231 cells (Fig. 4, A and B).
Figure 4.

CPEB2A and CPEB2B differentially regulate tumorigenesis and tumor size via TWIST1 and HIF1α. A and B, MDA-MB-231 cells were stably transfected with either empty vector or a plasmid construct containing CPEB2B, then subjected to siRNA toward either HIF1α or TWIST1. Cells were then either subjected to immunoblot for the indicated antibodies (lower sections) or exposed to polyHEMA-coated surfaces (or control surfaces) for 24 h, stained for annexin V/PI and analyzed via FACS (upper graphs). All experiments are representative of at least three biological replicates. C and D, MDA-MB-231 (C) and MDA-MB-468 (D) cells were stably transfected with CPEB2B as in (A and B), then subjected to commercially available shRNA directed against HIF1α or shRNA directed against TWIST1 (C and D). E, concomitantly, MDA-MB-231 cells were stably transfected with CPEB2A (CPA), then transduced with adenovirus to ectopically express either TWIST1 or HIF1α. 300,000 cells were implanted into the T1 fat pad of NOD scid gamma mice and after 20–40 days, primary tumor volumes were determined at a single time point. *, p < 0.05 compared to controls and #, p < 0.05 compared to rescue as determined by ANOVA and Tukey's honest significant difference (HSD) post hoc tests. Error bars represent standard deviation.
The requirement of HIF1α and TWIST1 for tumor growth in vivo was then assessed. Indeed, shRNA targeted toward TWIST1 and HIF1α significantly inhibited the enhanced tumor size induced by CPEB2B ectopic expression (Fig. 4, C and D). Inversely, when CPEB2A was ectopically expressed in MDA-MB-231 cells, the volume of primary tumors was significantly reduced in comparison to vector-expressing controls. In addition, rescuing the expression of TWIST1 and HIF1α using adenovirus vectors resulted in a recovery of primary tumor volumes to control levels (Fig. 4E). Finally, when primary tumor sizes were allowed to reach relatively equivalent volumes, enhanced lung metastasis was observed for the CPEB2B overexpressing cells, however shRNA to TWIST1 significantly decreased visible tumor lesions on the lungs (Fig. 5). Interestingly, tumors in which HIF1α was down-regulated were unable to produce primary tumors of significant volume to assess the effects on metastatic capability (data not shown). These results demonstrate that the effects of CPEB2 AS on AnR and subsequent enhancement of metastasis require TWIST1 signaling, whereas AnR and primary tumor growth is strongly associated with the HIF1α DNA trans-factor, but the association of this DNA trans-factor with the metastatic rate is inconclusive at this time.
Figure 5.

CPEB2A and CPEB2B regulate the metastasis of TNBC cells in an opposing fashion via TWIST1. A—D, MDA-MB-231 cells stably expressing CPEB2B were transduced with lentivirus to stably express shRNA directed toward TWIST1 or HIF1α. 250,000 cells were implanted into the T1 fat pad of NOD scid gamma mice, then primary tumors were all allowed to reach roughly 2000 mm3. Tumors were dissected out and measured (D) (black bar = 1 cm), lungs were harvested (B), gross lesions were counted (A, and shown by arrows in B), and lungs were subjected to H&E staining (C). D, lungs were also subjected to sectioning followed by H&E staining. Scale bar indicates the following lengths according to magnification: 4×, 1.0 mm; 20×, 200 μm. * = p < 0.05 compared with controls. # = p < 0.05 compared with rescue.
Discussion
Our laboratory previously identified and characterized the requirement of CPEB2 AS in driving the acquisition of AnR and subsequent enhancement of the metastasis of TNBC (7). In this study, these initial findings were extended to determining the mechanism by which CPEB2B, a novel “pro-metastatic” isoform of CPEB2, induced AnR. Interestingly, CPEB2A and CPEB2B, in an opposing fashion, were found to affect the mRNA levels of various genes central to the motility/hypoxia/EMT axis, but not the mRNA levels of the key nodes in these pathways, HIF1α and TWIST1. In regard to the expression of these key nodes, we found that ectopic expression of CPEB2A decreases the nascent protein synthesis of these DNA trans-factors with no effect on protein stability or transcriptional expression. However, and surprisingly, ectopic expression of CPEB2B enhanced the translation of these proteins.
In regard to the translational regulation of these pathway nodes, CPEB2 has been reported as a translational repressor of target mRNAs such as TWIST1, but our studies demonstrate that specifically the CPEB2A isoform of the CPEB2 gene is a repressor of translation. On the other hand, the CPEB2B isoform is a translational activator of these same target mRNAs demonstrating a major regulatory role for CPEB2 AS in cellular stress responses. How the inclusion of an exon encoding 30 additional amino acids adjacent to the low complexity domain is providing such a drastic functional change is unknown. Based on the repressor complex for the CPEB family member, CPEB1, one can hypothesize that CPEB2B, in contrast to CPEB2A, does not possess the ability to form a stable repressor complex with known interaction partners (e.g. bind PARN, GLD2), and thus, when CPEB2B binds a target CPE RNA cis-element (CPE site) for a specific mRNA, cytoplasmic polyadenylation commences followed by efficient translation of the target mRNA (8, 13, 14). Currently, our preliminary data do not show drastic differences between the isoforms in this regard (data not shown), but the composition of the repressor complex may be different among the CPEB family members as CPEB1 is the most characterized in this regard. Furthermore, the inclusion of exon 4 may simply inactivate the translational repressing function of CPEB2 while retaining the RNA-binding specificity and capability. Thus, CPEB2B, although inactive as a translational repressor, would compete with CPEB2A for binding target RNAs and allow both cytoplasmic polyadenylation and the subsequent translation to proceed.
The above hypothesized mechanisms require the inference that CPEB2A and B bind to the same CPE sites, and thus compete for the binding of target RNAs. In this regard, both our published findings and this study support a straightforward, competition-based system (7). For example, direct modulation of one CPEB2 splice variant induces an enhancement of the opposing function of the other variant (e.g. down-regulation of CPEB2B induces the loss of TWIST1 expression whereas down-regulation of CPEB2A in the same cell lines induces the expression of TWIST1). Because the inclusion of exon 4 is not within the RNA-binding domains and the ratio of CPEB2A/B is associated with function, the premise of a competition-based mechanism is supported as both isoforms likely share the same RNA-binding capacity. Unpublished data from our laboratory also support this mechanism as both CPEB2A and B will bind specifically to the same CPE sites of the TWIST1 3′UTR.4 Additional future studies to determine effects on repressor complex formation and target mRNA polyadenylation are needed to validate this proposed mechanism.
Another interesting finding from this study is that the translation of HIF1α is a key mechanism affecting the expression of this DNA trans-factor. As our data demonstrate, the expression of HIF1α, a major early marker in the hypoxia response pathway, is induced at least partially via translation in these studies, and is responsible for at least some of the tumor formation/tumor growth induced by the “B” isoform of CPEB2. The reigning paradigm is that in response to hypoxia, the proteolytic degradation of HIF1α is inhibited, which is the driving force in increasing HIF1α expression (15, 16). Our study found that increased translation of HIF1α was the major regulating mechanism for the expression of this DNA trans-factor in response to detachment-induced cell stress. It is therefore intriguing to surmise that during other cell stress cascades (e.g. hypoxia), enhanced translation of HIF1α via CPEB2B expression, acts as a cooperative mechanism with the inhibition of proteolytic degradation to drive HIF1α expression. Indeed, if this premise is validated, CPEB2 AS may emerge as a master regulator of a number of cellular stress responses.
Of note, the culmination of the findings in this study coupled to our previous report (7) suggest that targeting the CPEB2B isoform with new therapeutics would have profound effects on suppressing cell survival and stress pathways, and thus dramatically limit metastatic potential. As CPEB2A and B are likely competing for binding to the same mRNAs, specifically targeting CPEB2B may be difficult. On the other hand, therapies which target the CPEB2 splicing mechanism to suppress inclusion of exon 4 would likely inhibit these key survival mechanisms related to cancer metastasis. These types of therapies would be predicted to have limited side effects as normal, nontransformed cells do not normally express CPEB2B.
The present study also demonstrates major roles for both HIF1α and TWIST1 in TNBC tumor maintenance/growth (HIF1α) and metastasis (TWIST1). To date, the literature on the role of TWIST1 and HIF1α in TNBC has been limited, with HIF1α even playing a paradoxical role as a modifier of the tumor suppressing function of PRC2 (17–19). In this study, a major role was identified for TWIST1 in both the AnR and downstream metastasis of TNBC, as down-regulation of this DNA trans-factor led to significant loss of AnR and, subsequently, a reduction in the metastatic rate enhanced by CPEB2B expression. Down-regulation of HIF1α, on the other hand, led to a profound effect on tumor growth, because even after 60 days, tumors did not reach the size of their control shRNA counterparts, and indeed, some tumors regressed (data not shown). Hence, the requirement for HIF1α expression may be limited to tumor growth/maintenance, whereas the requirement for TWIST1 expression leads to TNBC metastasis. The requirement for these two related cellular pathways suggests a link between CPEB2 AS and possibly the inducible nitric oxide synthase (iNOS) system. For example, iNOS inhibitors also significantly reduce the tumor growth and lung metastases of TNBC cells, which correlated with the impairment of HIF1α and TWIST1 expression and signaling (20, 21). Thus, one can speculate that iNOS may regulate CPEB2 AS or vice versa. Indeed, iNOS has been linked to breast cancer progression along with TWIST1. Furthermore, HIF1α has also been linked to iNOS via a normoxic pathway induced by chemotherapy, an efficacy-limiting event (22). Thus, validation of a signaling link between CPEB2 AS and iNOS would logically suggest that the targeting of CPEB2 AS in combination with current first-line therapies may greatly improve initial efficacy and overall durability of these treatments.
In conclusion, our data indicate that CPEB2A and CPEB2B have opposing effects on the hypoxia/EMT/cell motility signaling axis (Fig. 6). Furthermore, the induction of these pathways are critical for the ability of CPEB2B to impart AnR and, thereby, a higher metastatic rate to breast cancer cells. Lastly, our study shows that the ability of CPEB2B to activate these pathways is via enhancement of the translation of TWIST1 and HIF1α mRNAs. Overall, this study when coupled to our previous study showing the high expression of CPEB2B in aggressive breast cancers suggests that this splicing event is a plausible therapeutic target for metastatic breast cancer.
Figure 6.
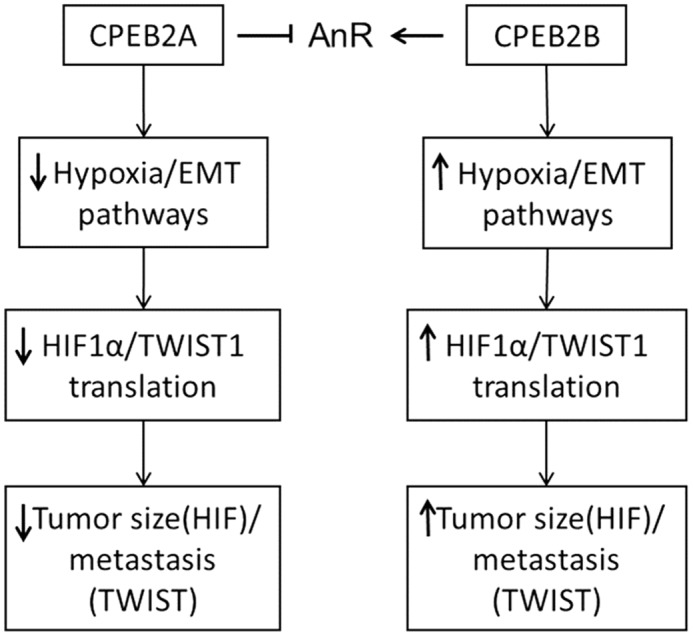
CPEB2A and CPEB2B regulate tumor growth/metastasis of TNBC cells in an opposing fashion via antagonistic effects on the hypoxia and EMT pathway.
Materials and methods
Cell culture and reagents
MDA-MB-231 parental and MDA-MB-468 parental cell lines (purchased from American Type Culture Collection (ATCC)) were maintained in RPMI medium (Invitrogen). All cell lines were supplemented with 10% fetal bovine serum (FBS) (Invitrogen) and 1% penicillin/streptomycin (Invitrogen). All cell lines were maintained in a 95% air/5% CO2 incubator at 37 °C. Cells were passaged once every 3 to 5 days (∼90% confluence), and all experiments were performed during the first 12 passages. Antibodies were purchased from Cell Signaling Technology with the exception of anti-CPEB2 (Santa Cruz Biotechnology) and anti-HIF1α (Abcam) (7, 23–27).
Selection of AnR subpopulation of TNBC cell lines
Poly(2-hydroxyethylmethacrylate) (polyHEMA) solution was prepared to a final concentration of 20 mg/ml in 100% methanol. Tissue culture plates (10 cm) were coated with 4 ml polyHEMA solution which was allowed to evaporate, then a second coating was applied. Plates were rinsed with sterile PBS prior to cell culture to remove any residual methanol. Selection for AnR took place over the course of 4 to 8 weeks. Initially, 1 × 107 MDA-MB-231 or MDA-MB-468 cells were added to the polyHEMA-coated dishes in normal media. Cell debris and dead cells were removed by centrifugation every 3 days until the culture was established, then on a 3-day cycle cellular aggregates were disrupted via pipette to ensure culture homogeneity.
Vectors and RNAi
Custom siRNA toward CPEB2A and CPEB2B utilized in this study was described previously (7). Validated siRNA and shRNA lentiviruses toward HIF1α and TWIST1 were purchased from Sigma-Aldrich. Validated siRNA (used in Fig. 4, A and B) was purchased from Thermo-Fisher (Dharmacon). CPEB2A and CPEB2B shuttle vectors were custom designed and purchased from OriGene Technologies using sequences described previously (7). Shuttle sequences were then cloned into FLAG-tagged vectors (Clontech) and sequenced to confirm.
Next generation sequencing
FASTQ files collected for this experiment were deposited in the Sequence Read Archive (SRA) database (PRJNA278375). Transcript-level expression was calculated using the CLC Genomics Workbench. Mapping was performed using the gene and known transcripts to annotate differentially expressed transcripts. These were reported as reads per kilobase per million (RPKM). Gene-level expression was determined by summing RPKM expression of all the transcripts, then transcript-specific RPKM values were used to calculate the proportion of RPKM transcript/RPKM gene. The final analysis consisted of evaluating the p value for one-sample Student's t tests using a standard α = 0.05 cutoff to determine whether the transcript-specific expression was significantly different from zero. Secondary analysis was performed using CASPER, and the resulting isoform expression differences were read into the R programming environment. Expression comparisons were made using the p values adjusted to false discovery rate, the q-value method. Finally, combining p values across the two independent comparisons was evaluated using Fisher's method. Ingenuity Pathway Analysis was then used to query the most highly up- and down-regulated pathways in the data set.
Transfection (plasmid)
Plasmid transfections were accomplished using the Effectene system (Qiagen) according to manufacturer's instructions as described previously (7, 22–26) or Amaxa Nucleofector (Kit V).
Transfection (RNAi)
Transfections were undertaken using the Amaxa Nucleofector Kit V according to manufacturer's instructions and as described previously (7).
Western blotting
Total protein (10–20 μg) was electrophoretically separated on 7.5–10% polyacrylamide gels. Samples were transferred electrophoretically to PVDF membranes, then probed with the appropriate antibody as described previously (7, 22–26).
Quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR)
Primer/probe sets were described previously (7). PCR was performed as described (22–26). cDNA was synthesized using the Superscript III kit (Life Technologies) and manufacturer's instructions. Samples were then amplified using a Bio-Rad CFX Connect qPCR machine and calculated using the standard curve method.
Competitive quantitative RT-PCR
cDNA was synthesized as described above for qRT-PCR. cDNA samples were subjected to traditional PCR as described (7, 22–26) using primers located on either side of exon 4 of the CPEB2 gene as described previously (7).
Nascent protein labeling
Protocol was adapted from Su Hui Teo et al. (28). Cells were incubated 2 h in methionine-free medium. Then, 50 μm l-azidohomoalanine was added and cells were incubated a further 6 h. Proteins were washed two times in PBS, then lysed in lysis buffer (1% SDS in 50 mm Tris-HCl, pH 8.0, 1× protease and phosphatase inhibitors, added fresh). Cells were incubated for 30 min on ice with regular vortexing to ensure complete lysis, then centrifuged to clear the lysate. Nascent proteins were labeled with biotin using the Click-iT Protein Reaction Buffer Kit according to manufacturer's instructions. Proteins were precipitated using methanol/chloroform, then resuspended in IP buffer (20 mm Tris, pH 7.5, 150 mm NaCl, 1 mm EDTA, 0.5% Nonidet P-40, 1× protease and phosphatase inhibitors (added fresh)). Samples were incubated 6 h with streptavidin-coated magnetic beads, then washed three times with IP buffer. Samples were then subjected to PAGE/immunoblot.
Nascent RNA labeling
Nascent RNA was pulse labeled using the Click-iT Nascent RNA Capture Kit (Thermo Scientific), then “chased” with media not containing ethynyl uridine. Total RNA was then isolated and biotin-labeled (nascent) RNA was “pulled down” with streptavidin beads according to manufacturer's instructions. These samples were then subjected to qRT-PCR.
Anoikis resistance assay
Cells were treated with the indicated siRNA, then trypsinized, washed, and prepared in media to 2.5 × 105 cells/ml. A total of 5 × 105 cells were added to each well of either normal or polyHEMA-coated 6-well tissue culture plates. The cells were incubated an additional 24 h, then collected for analysis via flow cytometry. Dual staining with annexin V conjugated to phycoerythrin and 7-aminoactinomycin D using the PE Annexin V Apoptosis Detection Kit (BD Biosciences) was performed according to the manufacturer's instruction. Cells were analyzed on a FACSCanto II flow cytometer and gating analysis determined using FACSDiva software.
Biostatistics
Biostatistical analyses were performed using either SPSS (Statistical Package for the Social Sciences) or R. Statistical tests used include Student's t test (in the case of only two groups), analysis of variance (ANOVA) (in the case of multiple groups), and a false discovery rate-adjusted p value (Benjamini-Hochberg correction) calculation (Cuffdiff program).
Animal declaration of approvals
Animal experiments were conducted at Virginia Commonwealth University. The research protocol was approved by the VCU Institutional Animal Care and Use Committee (IACUC) involving animal care in accordance with the United States Department of Agriculture Animal Welfare Regulations, the Public Health Service Policy on the Humane Care and Use of Laboratory Animals, and the United States Government Principles for the Utilization and Care of Vertebrate Animals Used in Testing, Research, and Training (AD10000529). VCU is in compliance with all provisions of the Animal Welfare Act and other federal statutes and regulations relating to animals. VCU is registered under the Animal Welfare Act as a class R research facility with the United States Department of Agriculture Animal and Plant Health Inspection Service (APHIS) Animal Welfare (registration number 52-R-0007). The Office of Laboratory Animal Welfare (OLAW) approved the VCU Animal Welfare Assurance in compliance with the Public Health Service Policy (assurance number A3281–01).
Author contributions
J. T. D., M. A. P., and G. L. contributed to the conception and design of experimental studies, development of novel methodologies, data acquisition, analysis and interpretation, and initial writing of the manuscript. M. A. P. contributed the acquisition and analysis of RNA-seq data, biostatistical analyses, and verification of all presented data, and technical support. M. A. P. and C. E. C. contributed to the funding support, technical support, and data analysis/interpretation as related to the use and cell signaling of CPEB2. C. E. C. and M. A. P. contributed to the overall study supervision, funding support, hypotheses generation, conception and design of study, and editing and review of the manuscript.
Acknowledgments
Services and products in support of the research project were generated by the VCU Massey Cancer Center Shared Resources supported, in part, with funding from NCI, National Institutes of Health, Cancer Center Support Grant P30 CA016059.
This work was supported by Veteran's Administration Research Grants VA Merit Review and BX001792 (to C.E.C.); Research Career Scientist Award 13F-RCS-002 (to C.E.C.); National Institutes of Health Grants TR000057-04 (to M.A.P.) and HL125353, HD087198, CA117950, CA154314, and RR031535 (to C.E.C.); and NH1C06-RR17393 (to Virginia Commonwealth University for renovation). This work was also supported by funds from the University of South Florida (USF) Foundation and the USF College of Arts and Sciences. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or reflect the position or policy of the Department of Veterans Affairs or the United States government.
J. T. DeLigio, G. Lin, C. E. Chalfant, and M. A. Park, unpublished results.
- AnR
- anoikis resistance
- AS
- alternative splicing
- CPEB2
- cytosolic polyadenylation element–binding protein 2
- EMT
- epithelial-to-mesenchymal transition
- TNBC
- triple negative breast cancer
- NGS
- next generation sequencing
- iNOS
- inducible nitric oxide synthase
- polyHEMA
- poly(2-hydroxyethylmethacrylate)
- RPKM
- reads per kilobase per million.
References
- 1. Dai X., Li T., Bai Z., Yang Y., Liu X., Zhan J., and Shi B. (2015) Breast cancer intrinsic subtype classification, clinical use and future trends. Am. J. Cancer Res. 5, 2929–2943 [PMC free article] [PubMed] [Google Scholar]
- 2. Haque R., Ahmed S. A., Inzhakova G., Shi J., Avila C., Polikoff J., Bernstein L., Enger S. M., and Press M. F. (2012) Impact of breast cancer subtypes and treatment on survival: An analysis spanning two decades. Cancer Epidemiol. Biomarkers Prev. 21, 1848–1855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cheang M. C., Chia S. K., Voduc D., Gao D., Leung S., Snider J., Watson M., Davies S., Bernard P. S., Parker J. S., Perou C. M., Ellis M. J., and Nielsen T. O. (2009) Ki67 index, HER2 status, and prognosis of patients with luminal B breast cancer. J. Natl. Cancer Inst. 101, 736–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vanharanta S., and Massagué J. (2013) Origins of metastatic traits. Cancer Cell 24, 410–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Samatov T. R., Tonevitsky A. G., and Schumacher U. (2013) Epithelial-mesenchymal transition: Focus on metastatic cascade, alternative splicing, non-coding RNAs and modulating compounds. Mol. Cancer 12, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Guadamillas M. C., Cerezo A., and Del Pozo M. A. (2011) Overcoming anoikis—pathways to anchorage-independent growth in cancer. J. Cell Sci. 124, 3189–3197 [DOI] [PubMed] [Google Scholar]
- 7. Johnson R. M., Vu N. T., Griffin B. P., Gentry A. E., Archer K. J., Chalfant C. E., and Park M. A. (2015) The alternative splicing of cytoplasmic polyadenylation element binding protein 2 drives anoikis resistance and the metastasis of triple negative breast cancer. J. Biol. Chem. 290, 25717–25727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. D'Ambrogio A., Nagaoka K., and Richter J. D. (2013) Translational control of cell growth and malignancy by the CPEBs. Nat. Rev. Cancer 13, 283–290 [DOI] [PubMed] [Google Scholar]
- 9. Nairismägi M. L., Vislovukh A., Meng Q., Kratassiouk G., Beldiman C., Petretich M., Groisman R., Füchtbauer E. M., Harel-Bellan A., and Groisman I. (2012) Translational control of TWIST1 expression in MCF-10A cell lines recapitulating breast cancer progression. Oncogene 31, 4960–4966 [DOI] [PubMed] [Google Scholar]
- 10. Nairismägi M. L., Füchtbauer A., Labouriau R., Bramsen J. B., and Füchtbauer E. M. (2013) The proto-oncogene TWIST1 is regulated by microRNAs. PLoS One 8, e66070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen P. J., and Huang Y. S. (2012) CPEB2-eEF2 interaction impedes HIF-1α RNA translation. EMBO J. 31, 959–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang R., Zhang P., Li J., Guan H., and Shi G. (2016) Ubiquitination is absolutely required for the degradation of hypoxia-inducible factor–1 alpha protein in hypoxic conditions. Biochem. Biophys. Res. Commun. 470, 117–122 [DOI] [PubMed] [Google Scholar]
- 13. Richter J. D. (2007) CPEB: A life in translation. Trends Biochem. Sci. 32, 279–285 [DOI] [PubMed] [Google Scholar]
- 14. Kim J. H., and Richter J. D. (2006) Opposing polymerase-deadenylase activities regulate cytoplasmic polyadenylation. Mol. Cell 24, 173–183 [DOI] [PubMed] [Google Scholar]
- 15. Kong X., Lin Z., Liang D., Fath D., Sang N., and Caro J. (2006) Histone deacetylase inhibitors induce VHL and ubiquitin-independent proteasomal degradation of hypoxia-inducible factor 1α. Mol. Cell Biol. 26, 2019–2028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hon W. C., Wilson M. I., Harlos K., Claridge T. D., Schofield C. J., Pugh C. W., Maxwell P. H., Ratcliffe P. J., Stuart D. I., and Jones E. Y. (2002) Structural basis for the recognition of hydroxyproline in HIF-1 α by pVHL. Nature 417, 975–978 [DOI] [PubMed] [Google Scholar]
- 17. Regan Anderson T. M., Ma S. H., Raj G. V., Cidlowski J. A., Helle T. M., Knutson T. P., Krutilina R. I., Seagroves T. N., and Lange C. A. (2016) Breast tumor kinase (Brk/PTK6) is induced by HIF, glucocorticoid receptor, and PELP1-mediated stress signaling in triple-negative breast cancer. Cancer Res. 76, 1653–1663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mahara S., Lee P. L., Feng M., Tergaonkar V., Chng W. J., and Yu Q. (2016) HIFI-α activation underlies a functional switch in the paradoxical role of Ezh2/PRC2 in breast cancer. Proc. Natl. Acad. Sci. U.S.A. 113, E3735–E3744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Granados-Principal S., Liu Y., Guevara M. L., Blanco E., Choi D. S., Qian W., Patel T., Rodriguez A. A., Cusimano J., Weiss H. L., Zhao H., Landis M. D., Dave B., Gross S. S., and Chang J. C. (2015) Inhibition of iNOS as a novel effective targeted therapy against triple-negative breast cancer. Breast Cancer Res. 17, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Finlay J., Roberts C. M., Lowe G., Loeza J., Rossi J. J., and Glackin C. A. (2015) RNA-based TWIST1 inhibition via dendrimer complex to reduce breast cancer cell metastasis. Biomed. Res. Int. 2015, 382745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rhodes L. V., Martin E. C., Segar H. C., Miller D. F., Buechlein A., Rusch D. B., Nephew K. P., Burow M. E., and Collins-Burow B. M. (2015) Dual regulation by microRNA-200b-3p and microRNA-200b-5p in the inhibition of epithelial-to-mesenchymal transition in triple-negative breast cancer. Oncotarget 6, 16638–16652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hsia T. C., Liu W. H., Qiu W. W., Luo J., and Yin M. C. (2014) Maslinic acid induces mitochondrial apoptosis and suppresses HIF-1α expression in A549 lung cancer cells under normoxic and hypoxic conditions. Molecules 19, 19892–19906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Goehe R. W., Shultz J. C., Murudkar C., Usanovic S., Lamour N. F., Massey D. H., Zhang L., Camidge D. R., Shay J. W., Minna J. D., and Chalfant C. E. (2010) hnRNP L regulates the tumorigenic capacity of lung cancer xenografts in mice via caspase-9 pre-mRNA processing. J. Clin. Invest. 120, 3923–3939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vu N. T., Park M. A., Shultz M. D., Bulut G. B., Ladd A. C., and Chalfant C. E. (2016) Caspase-9b interacts directly with cIAP1 to drive agonist-independent activation of NF-κB and lung tumorigenesis. Cancer Res. 76, 2977–2989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shapiro B. A., Vu N. T., Shultz M. D., Shultz J. C., Mietla J. A., Gouda M. M., Yacoub A., Dent P., Fisher P. B., Park M. A., and Chalfant C. E. (2016) Melanoma differentiation-associated gene 7/IL-24 exerts cytotoxic effects by altering the alternative splicing of Bcl-x pre-mRNA via the SRC/PKCδ signaling axis. J. Biol. Chem. 291, 21669–21681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vu N. T., Park M. A., Shultz J. C., Goehe R. W., Hoeferlin L. A., Shultz M. D., Smith S. A., Lynch K. W., and Chalfant C. E. (2013) hnRNP U enhances caspase-9 splicing and is modulated by AKT-dependent phosphorylation of hnRNP L. J. Biol. Chem. 288, 8575–8584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Simanshu D. K., Kamlekar R. K., Wijesinghe D. S., Zou X., Zhai X., Mishra S. K., Molotkovsky J. G., Malinina L., Hinchcliffe E. H., Chalfant C. E., Brown R. E., and Patel D. J. (2013) Non-vesicular trafficking by a ceramide-1-phosphate transfer protein regulates eicosanoids. Nature 500, 463–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Su Hui Teo C., Serwa R. A., and O'Hare P. (2016) Spatial and temporal resolution of global protein synthesis during HSV infection using bioorthogonal precursors and click chemistry. PLoS Pathog. 12, e1005927. [DOI] [PMC free article] [PubMed] [Google Scholar]