

Abstract
IMPORTANCE
PLCG2-associated antibody deficiency and immune dysregulation (PLAID) is a newly characterized immunodeficiency syndrome associated with distinct cutaneous features. Awareness of the cutaneous skin findings associated with PLAID may facilitate diagnosis and improve patient care.
OBJECTIVES
To characterize the cutaneous manifestations of PLAID and identify potential cellular mechanisms of the disease.
DESIGN, SETTING, AND PARTICIPANTS
In this retrospective analysis of patients with PLAID and PLAID-like disease evaluated at the National Institutes of Health from January 1, 2005, through December 31, 2014, patients with deletions in PLCG2 leading to PLAID and patients with PLAID-like disease for whom a PLAID mutation was not identified were studied.
MAIN OUTCOMES AND MEASURES
Characterization of cutaneous manifestations of PLAID and PLAID-like disease and analysis of PLAID immune cell activation.
RESULTS
Among 36 patients with PLAID and PLAID-like phenotypes, all of whom had evaporative cold urticaria, 8 patients had a history of unique neonatal-onset ulcerative and cutaneous lesions in cold-sensitive regions of the body. Granulomatous skin lesions sparing warm regions (eg, flexural surfaces and skinfolds) were identified in 4 patients. Neutrophils and monocytes from patients with PLAID exhibited enhanced baseline activation in vitro, which was potentiated by ambient temperature exposure.
CONCLUSIONS AND RELEVANCE
Collectively, these findings suggest that early identification of neonatal lesions may help in the diagnosis of PLAID and that leukocyte hyperactivation may underlie cutaneous lesions in patients with PLAID. Further characterization of mechanisms underlying leukocyte hyperactivation may contribute to the fundamental understanding of granuloma formation.
Although the most common cutaneous manifestation of primary immunodeficiency (PID) is infection, a variety of noninfectious dermatologic findings may occur.1 Noninfectious cutaneous granulomas, for instance, are relatively frequent in patients with certain PIDs, including common variable immunodeficiency (CVID), severe combined immunodeficiency, chronic granulomatous disease (CGD), and ataxia-telangiectasia (A-T).2,3 Although not completely understood, cutaneous granulomas in PID may be a consequence of abnormal antigen processing, aberrant cytokine production, enhanced cell-mediated immune activity in the presence of a defective humoral system, abnormal neutrophil activation, or impaired macrophage regulation.2,4–6
Cutaneous granulomas are also a feature of a newly described PID, PLCG2-associated antibody deficiency and immune dysregulation (PLAID), caused by genomic deletions in PLCG2. PLAID is characterized by cold urticaria, autoimmunity, atopy, and humoral immune deficiency, resulting in recurrent sinopulmonary infections.7 PLAID-associated deletions of PLCG2 cause diminished receptor mediated activity at physiologic temperatures in B cells and natural killer cells with enhanced spontaneous signaling in mast cells and B cells at subphysiologic temperatures. The cold-induced signaling in mast cells leads to the characteristic cold urticaria observed in all patients with PLAID. Cutaneous granulomas have also been identified in 2 additional patients with missense mutations in PLCG2, leading to a related syndrome that lacks cold urticaria but has some other features of PLAID and striking autoinflammation termed APLAID.8 Studying these skin findings in the context of these molecular lesions can provide substantial insight into the pathogenesis of cutaneous granulomas.
We fully characterize the clinical and histologic spectrum of cutaneous disease in 27 patients with PLAID and 9 patients with a PLAID-like clinical phenotype who lack an identifiable PLCG2 mutation and contrast this cohort with patients with APLAID. In addition, we investigate an expanded subset of leukocytes and their responses to cold to gain insight into the pathologic mechanism underlying dermatologic disease in patients with PLAID and related disorders.
Methods
Study Participants
Thirty-six patients were referred to the National Institutes of Health for evaluation from January 1, 2005, through December 31, 2014. The National Institute of Allergy and Infectious Diseases institutional review board approved the clinical research protocol. The study was conducted according to the principles of the Declaration of Helsinki, and all patients provided written informed consent.
Patients included 27 individuals with PLAID from 3 different families and 9 individuals from 4 additional families with a similar clinical presentation to patients with PLAID (cold urticaria, immunoglobulin abnormalities, and recurrent infections) but in whom PLCG2 mutations were not identified (defined as a PLAID-like disease) (eMethods and eTable in the Supplement). Medical histories, physical examination findings, and laboratory and skin biopsy results that confirmed the presence of cutaneous disease were obtained. Cutaneous manifestations of the patients with PLAID were compared with those of 2 previously described patients with APLAID, a syndrome associated with a missense mutation in PLCG2 characterized by systemic autoinflammation, immune abnormalities, and bullous and granulomatous skin disease.
Effect of Temperature on Superoxide Production by Neutrophils
To promote neutrophil adherence, the wells of a 96-well plate (Primaria; BD Biosciences)were coated with fibrinogen by incubating 1 hour at 37°C with 32 μL of 2.5 mg/mL of fibrinogen (EMD Millipore). The wells were then washed 3 times with phosphate-buffered saline. Neutrophils (2 × 105/200 μL in 0.2% fetal calf serum in Hanks balanced salt solution with divalent cations, 10mM HEPES) and cytochrome C (100μM)were added to the wells in duplicate and preincubated at 20°C, 30°C, or 37°C for 5 minutes. An additional identical well that contained superoxide dismutase (100 μg/mL) served as a blank to validate superoxide production. The cells were then treated with buffer alone, tumor necrosis factor α (TNF-α) (50 ng/mL), or phorbol 12-myristate 13-acetate (100 ng/mL). The optical density at 549.5 nm(OD549.5 nm) of each well was determined at 5-minute intervals for 60 minutes using a thermally regulated Spectromax Plus 384 plate reader (Molecular Devices LLC) with shaking. The superoxide dismutase blank was subtracted from the duplicate experimental wells, and the relative change in OD549.5 nm vs time was plotted. Data were collected from 4 healthy controls and 8 patients with PLAID at 20°C; 11 controls, 8 patients with PLAID, and 2 patients with APLAID at 30°C; and 16 healthy controls, 13 patients with PLAID, and 2 patients with APLAID at 37°C. The variation in the number of healthy control and patient samples used in each experiment was a result of the limited availability of isolated cells on the day of analysis.
Surface Antigen Expression on Neutrophils, Monocytes, and Lymphocytes
Whole blood samples from 4 healthy controls and 8 patients with PLAID were collected into EDTA tubes (Vacutainer; BD Biosciences). One sample from each study participant was maintained at ambient temperature and the other maintained on warm sand (33°C–37°C during transport). Aliquots (100 μL) of whole blood were incubated for 15 minutes at ambient (room) temperature with the following antibodies: CD11b (eBioscience), CD16, CD18, CD45, or CD62L (BD Biosciences). The tubes were treated with OptiLyse C (Beckman Coulter Inc) for 10 minutes at room temperature to lyse the erythrocytes, washed twice, and then analyzed on the BD FACSCanto II cytometer (BD Biosciences). Expression, as measured by mean fluorescence intensity (MFI), was measured for each surface antigen.
Neutrophil Chemotaxis
Neutrophils isolated from 32 healthy controls or 7 patients with PLAID (1.0 μL of 5 × 106/mL)were added to the cell well of the EZ-TAXIS can (ECI Inc), and 1.0 μL of buffer or formyl-methionyl-leucyl phenylalanine (5 × 10−8M) was added to the opposing chemoattractant well. Images of cellular migration were captured every 30 seconds for 60 minutes at 37°C, and individual cells were tracked digitally. Images were converted to stacks (similar to video) using the ImageJ software, version 1.46r (National Institutes of Health). Ten randomly selected cells were electronically traced using the ImageJ plug-in MTrackJ. The paths of the migrating cells were plotted with the position of each cell at t = 0 anchored at the origin. With use of the coordinates obtained at each time point, the migratory path of each cell was resolved into a random migrational vector (orthogonal to the direction of the chemoattractant, along the x-axis) and a directed migrational vector (parallel to the direction of the chemoattractant, along the y-axis). In the absence of chemoattractant, the directed migrational vector should be equivalent to the random migrational vector.
Statistical Analysis
Differences in chemotaxis activity during 60 minutes and mean superoxide production by the samples from controls and patients with PLAID after 60 minutes were compared using unpaired 2-tailed t tests. The Wilcoxon signed rank test was applied to analyze mean MFI and temperature normalized MFI (MFIambient/MFIwarm) of surface antigens in patients compared with controls.
Results
Cold Urticaria and Neonatal Skin Lesions Associated With PLAID and PLAID-like Phenotypes
Cold urticaria manifested in early childhood among all 36 patients with PLAID and PLAID-like disease and many details regarding the manifestations have been previously reported.7,9 Briefly, in all patients, signs and symptoms of cold urticaria developed within the first year after birth. Urticarial reactions were noted as early as the first day after birth in some patients when warming of the neonate was delayed immediately after delivery. The rash that developed tended to lack the wheals observed in more typical, later-onset cold urticaria. Almost universally, evaporative cooling or immersion in coldwater led to the development of a blotchy, pruritic, erythematous rash, and in some cases cold ingested items led to an esophageal burning sensation. Of interest, contact with cold objects rarely prompted a rash. Syncopal episodes occurred only with failure to leave the coldwater or other prolonged generalized cold insult and initiate rewarming at the onset of symptoms. Antihistamines were effective in limiting the severity of the cold reactions; however, all patients reported that cold avoidance and/or rapid rewarming were the management solutions of choice. Although the severity of the cold urticaria reactions did not change as patients got older, the frequency of reactions was markedly reduced as patients learned to avoid cold triggers.
Eight patients (22%; 7 with PLAID and 1 with a PLAID-like phenotype) with a history of cold urticaria also had a history of cutaneous lesions during the neonatal period (Table). These patients reported spontaneous ulceration of the nasal tip at approximately the third day after birth. Nasal lesions had a hemorrhagic appearance with varying degrees of scale and crusting, ultimately producing a characteristic red-brown to black, escharlike plaque (Figure 1A). In 6 (75%) of 8 patients, nasal lesions resolved without intervention (range, 2–52 weeks) (Table and Figure 1B). The remaining 2 patients continued to have progressive, localized tissue destruction, resulting in erosion of the nasal cartilage during several years.
Table.
Cutaneous and Immunologic Features of Patients With PLAID and PLAID-Like Diseasea
| Patient No./Sex | PLCG2 Deletion | Cold Urticaria | Condition | Autoimmunity | Infectious History | Neonatal Lesion Distribution | Time to Lesion Resolution | Cutaneous Granuloma |
|---|---|---|---|---|---|---|---|---|
| 1/M | Yes | Yes | Food allergy (fish) | Seronegative inflammatory arthritis | Recurrent sinusitis and pneumonia, treated with IVIG | Nose, ears, toes, cheeks | NAb | Yes |
| 2/M | Yes | Yes | Asthma, AR, food allergy (fish) | None | Recurrent pneumonia, bronchitis | Nose | NAc | Yes |
| 3/F | Yes | Yes | AR, food allergy (shellfish and chocolate) | Positive ANA test result | Recurrent sinusitis | Nose | <1 y | Yes |
| 4/F | Yes | Yes | None | Hashimoto thyroiditis, vitiligo, positive ANA test result | Recurrent pneumonia in childhood | Nose | 6 mo | Yes |
| 5/M | Yes | Yes | Asthma, eczema | Positive ANA test result | None | Nose, fingers, toes | 2 wk | No |
| 6/M | Yes | Yes | None | Positive ANA test result | None | Nose, fingers, toes | 2 wk | No |
| 7/M | Yes | Yes | Asthma | Vitiligo, positive ANA test result | None | Nose | 1 wk | No |
| 8d/M | No | Yes | None | None | Recurrent sinopulmonary infections, treated with IVIG | Nose, fingers, toes | 6 wk | No |
Abbreviations: ANA, antinuclear antibody; AR, allergic rhinitis; IVIG, intravenous immunoglobulin; NA, not applicable; PLAID, PLCG2-associated antibody deficiency and immune dysregulation.
Genetic and immunologic data previously described by Ombrello et al.7
No resolution and complete nasal cartilage erosion.
Resolved with late-onset partial nasal cartilage erosion.
PLAID-like phenotype and no confirmed mutation.
Figure 1. Neonatal Lesions and Granulomatous Skin Disease in Patients With PLCG2-Associated Antibody Deficiency and Immune Dysregulation (PLAID) and PLAID-Like Phenotypes.
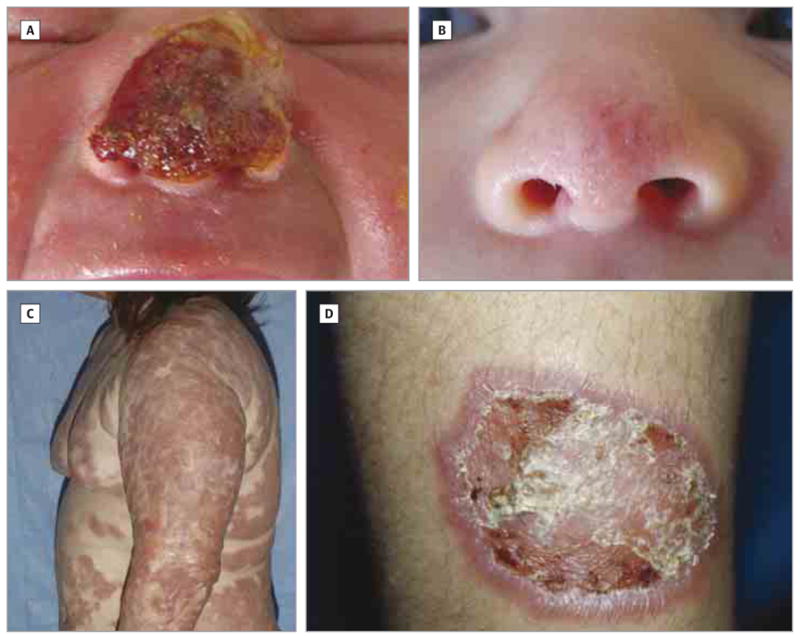
A, Neonatal nasal lesion showing hemorrhagic ulceration with crust involving the nasal tip and bridge in patient 8 during the first week after birth. B, Resolved nasal lesion in patient 8. C, Granulomatous skin disease in a patient with PLAID (patient 1). Confluent areas of tan to red plaques and nodules on the trunk and extremities with sparing of skinfolds and flexural surfaces. D, Well-demarcated plaque with thick scale and ulceration (patient 2).
Four patients with nasal ulceration also developed small papules and erosions on the fingers and toes in the neonatal period. Unlike the nasal lesions, there was no evidence of hemorrhage or ulceration, and acral lesions resolved without scar formation. Six of the 8 patients with neonatal skin lesions ultimately developed other cutaneous manifestations later in life. One patient developed vitiligo during adolescence, and one experienced recurrent red papules and patches on the head and upper trunk, which left residual brown patches during early childhood. Four patients developed granulomatous inflammation of the skin (Table).
Granulomatous Skin Disease in Patients With PLAID
The age at onset, distribution, and severity of cutaneous lesions differed among the 4 patients with biopsy-confirmed granulomatous dermatitis. Patient 1 had a history of neonatal nasal ulceration that ultimately led to complete erosion of his nasal cartilage. Unlike other patients, he developed additional facial lesions within the first few weeks of life. Granulomatous plaques first appeared on his cheeks, forehead, ears, and chin, sparing the scalp, nasolabial folds, and periorbital regions. These plaques began as firm, tender, red-brown plaques and nodules and evolved into scaly, red-brown and yellow- tan plaques with telangiectasia. Significant hypertrichosis was also noted during infancy. Throughout infancy and childhood, new plaques continued to emerge, eventually leading to widespread involvement of the face, trunk, and upper and lower extremities (Figure 1C). Areas of friction or pressure (eg, belt line), flexural surfaces (antecubital and popliteal fossae), skinfolds, and the skin overlying the vertebral column were spared. Progression of his disease resulted in extensive, disfiguring, coalescing, red-brown plaques and nodules and skin atrophy. In addition to complete erosion of the nasal cartilage, the patient had partial scarring of the tympanic membranes.
The results of skin biopsies of patient 1 performed at several time points revealed noninfectious, non–foreign-body granulomatous inflammation with nodular foci of CD68+ epithelial histiocytes and multinucleated giant cells surrounded by a mild CD4/CD8+ lymphocytic infiltrate and scattered eosinophils. Perineural granulomatous inflammation without nerve infiltration was also present. Findings of several biopsies revealed well-delineated, nonnecrotizing, sarcoid-type granulomas, whereas others revealed diffuse, poorly defined granulomatous inflammation, particularly in the superficial dermis (Figure 2A). Lymph node biopsy findings revealed well-circumscribed, sarcoid-type granulomatous inflammation with noncaseating necrosis and sclerotic stroma entirely replacing lymph node architecture. The results of stains for acid-fast organisms, bacteria, fungus, and spirochetes in all biopsy specimens were negative. Significant atypia or histologic features diagnostic of malignant tumors were not observed.
Figure 2. Histologic Characterization of PLCG2-Associated Antibody Deficiency and Immune Dysregulation Skin Granulomas.

A, Skin biopsy (middle back) showing fairly well-formed granuloma with several scattered eosinophils (patient 1). B, Skin biopsy (lower leg) revealing histiocytic infiltrate with multinucleated giant cells (patient 4) (hematoxylin-eosin, original magnification ×100).
Patient 2, a second-degree relative of patient 1, was born with a nasal lesion in infancy that led to partial erosion of his nasal cartilage. However, unlike patient 1, he did not develop granulomatous skin disease until he was in his 20s. At that time, numerous discrete, indurated, scaly, erythematous plaques developed on the trunk and extremities (Figure 1D). Spontaneous ulceration eventually led to hypopigmented, atrophic scarring. Results of a skin biopsy performed when the patient was in his 30s revealed sarcoid-type, granulomatous inflammation. According to the patient, new lesions stopped arising after he moved from the Northeastern United States to a warmer Southern climate.
Patients 3 and 4 developed granulomatous lesions limited to their lower extremities. Patient 3 developed a single small, tender, pruritic, and scaly plaque before the age of 10 years, which evolved into a chronic, nontender, scaly plaque. Histologic analysis revealed granulomatous dermatitis. Patient 4 developed 2 small, pruritic, pink papules on the medial aspect of each lower leg in her 50s. The lesions grew during several years, becoming indurated with raised borders, superficial ulceration, and an overlying layer of yellow fibrinoid material. The results of a skin biopsy revealed granulomatous inflammation, characterized by nonnecrotizing granulomas in the dermis. The granulomas were composed of a central core of histiocytes and multinucleated giant cells and were partially surrounded by a rim of lymphocytes. Scattered eosinophils were also present in the dermis (Figure 2B).
Treatment-Resistant Granulomatous Disease in Patients With PLAID
Patient 1 was treated with numerous topical and systemic therapies, including topical tacrolimus, topical nitrogen mustard, topical corticosteroids, methotrexate with mercaptopurine, cyclosporine, interferon alfa, thalidomide, etoposide phosphate, cis-retinoic acid, vinblastine sulfate, imatinib mesylate, and psoralen–UV-A(photochemotherapy);none of these altered the course of his skin disease. Systemic corticosteroid therapy was instituted in early infancy and remained the main-stay of treatment, often used in combination with other immunosuppressive agents. Although oral corticosteroids did not result in lesion resolution, withdrawal invariably led to disease exacerbation. Patient 2 was treated with phototherapy and topical and oral corticosteroids. Similar to patient 1, oral corticosteroids provided only short-term improvement. Patients 3 and 4 did not receive treatment for their limited skin involvement.
Autonomous and Cold-Induced Activation of PLAID Neutrophils and Monocytes
The skin granulomas in patients with PLAID and APLAID recapitulate other diseases of deficient immunity or increased inflammation, including CGD, severe combined immunodeficiency, CVID, A-T, and sarcoidosis.2,3 Proposed theories for granuloma formation in such diseases include increased or unregulated inflammatory activity of neutrophils and macrophages.2,5,6,10 Of note, 3 patients in the originally described PLAID cohort also had a concomitant diagnosis of CVID on the basis of present B cells, low IgG levels, and recurrent infections that required intravenous immunoglobulin replacement therapy. Given the association of PLAID with CVID and the presence of activated macrophages in PLAID granulomas, we hypothesized that alterations in PLCG2 result in abnormal neutrophil and monocyte and/or macrophage activation. Furthermore, a previous study11 suggested that recruitment of neutrophils (which attract macrophages) to granulomas is associated with mast cell activity. On the basis of the established in vitro findings of increased PLAID mast cell activity in cold temperatures, we also predicted that abnormal neutrophil or monocyte and/or macrophage activity associated with PLCG2 mutations may be enhanced in colder temperatures.
Expression of CD11b, an integrin that mediates leukocyte adhesion and migration, is increased in the setting of neutrophil or monocyte activation.12,13 Unstimulated PLAID neutrophils and monocytes maintained at ambient (subphysiologic) temperature consistently demonstrated a significant increase in spontaneous activation, as determined by CD11b MFI, compared with unstimulated PLAID cells maintained at warm (physiologic) temperature or unstimulated control cells maintained at both temperatures (Figure 3A–F). As expected, PLAID lymphocytes did not demonstrate increased expression of surface antigens. The increased expression of surface activation markers in subphysiologic conditions in the absence of chemical stimulation suggests that compared with healthy controls, PLAID neutrophils and monocytes are abnormally activated in subphysiologic temperature settings.
Figure 3. Expression of Surface Activation Markers in Different Temperature Settings.

A and B, Mean surface CD11b expression (mean fluorescence intensity [MFI]) in neutrophils (A) and monocytes (B) among healthy controls and patients with PLCG2-associated antibody deficiency and immune dysregulation (PLAID) at ambient temperatures. Error bars indicate SD. C and D, Mean surface CD11b expression (MFI) in neutrophils (C) and monocytes (D) among healthy controls and patients with PLAID at warm temperatures. Error bars indicate SD. E and F, Median relative expression of CD11b in ambient compared with warm temperatures in neutrophils (E) and monocytes (F) from patients with PLAID and healthy controls. Relative MFI of CD11b is calculated as (MFIambient/MFIwarm). Error bars indicate interquartile range. G, Basal superoxide production in neutrophils from patients with PLAID compared with healthy controls at 20°C, 30°C, and 37°C. H, Neutrophil superoxide production in response to tumor necrosis factor α in patients with PLAID.
aP < .05 (unpaired t test and Wilcoxon signed rank test).
bP < .01 (unpaired t test and Wilcoxon signed rank test).
cP < .001 (unpaired t test and Wilcoxon signed rank test).
dP < .33 (unpaired t test and Wilcoxon signed rank test).
eP = .78 (unpaired t test and Wilcoxon signed rank test).
Constitutive Production of Superoxide With Increased Activity in the Cold
Normal neutrophil activation results from a carefully coordinated sequence of events that involves phagocytosis and clearance of antigenic stimuli through an oxidative burst. Defects in this process, as observed in CGD, can result in granulomatous disease. Superoxide production in neutrophils from patients with PLAID and healthy controls under different stimulatory and temperature conditions were assayed. At all tested temperatures (20°C, 30°C, and 37°C), neutrophils from patients with PLAID produced significantly greater basal levels of superoxide than did neutrophils from healthy controls at 60 minutes (Figure 3G). Differences were most notable between the 2 groups at subphysiologic temperatures (20°C and 30°C). Although superoxide production in neutrophils from healthy controls was enhanced by TNF-α at both 30°C and 37°C, neutrophils from patients with PLAID failed to respond further to TNF-α activation at subphysiologic temperatures (Figure 3H; eFigure 1A in the Supplement).
Similar to cells from patients with PLAID, neutrophils from patients with APLAID produced significantly higher basal superoxide levels than neutrophils from healthy controls at 30°C and 37°C (eFigure 1B in the Supplement). However, in contrast to patients with PLAID, TNF-α stimulation of neutrophils from patients with APLAID at 37°C produced only a minimal increase in superoxide production, whereas stimulation at 30°C induced a significant increase in superoxide production (eFigure 1C in the Supplement).Taken together, these findings suggest that neutrophils from patients with PLAID and those with APLAID have increased basal activity that is further enhanced in cold settings, but neutrophils from patients with PLAID have a more significant defect in ligand-mediated superoxide production.
Neutrophils From Patients With PLAID and Impaired Chemotaxis In Vitro
Previous studies14–16 of neutrophils in the setting of granulomatous diseases, such as necrobiosis lipoidica, granuloma annulare, and sarcoidosis, found impaired neutrophil chemotaxis in vivo with normal migration in vitro, suggesting that neutrophil chemotactic defects may stimulate granuloma formation through over activation of macrophages. Compared with cells from healthy controls, neutrophils from patients with PLAID displayed reduced random migration under basal conditions and reduced directed migration to the chemoattractant formyl-methionyl-leucyl phenylalanine (eFigure 2 in the Supplement). The neutrophils from patients with PLAID had appropriate directionality but failed to attain the same increased velocity (distance divided by time) associated with directed migration in neutrophils from healthy controls (eFigure 2 in the Supplement).
Discussion
The present study highlights the unique cutaneous manifestations in patients with PLAID, APLAID, and PLAID-like phenotypes and temperature-sensitive cellular activation in patients with PLAID. Striking neonatal nasal and acral skin manifestations occurred in 22% of patients and provided the earliest signs of PLAID or PLAID-like disease. To our knowledge, the neonatal-onset ulcerative and self-resolving lesions in cold-sensitive regions of the skin have not been previously described as part of another syndrome. Although the histologic nature of the neonatal lesions remains unclear because of the lack of biopsy material, their self-limiting nature in most patients argues against an infectious process.
Given the associated observation that lower temperatures result in abnormal activation of PLAID monocytes and neutrophils under basal conditions, the acral location of neonatal lesions and sparing of warmer skin folds of granuloma is intriguing. It is possible that enhanced leukocyte activation caused by temperature variation in the skin (which can approach 26.8°C under normal conditions) may generate or perpetuate the cutaneous neonatal lesions and granulomata associated with PLAID and PLAID-like phenotypes.17 In our cohort, severity of granulomatous disease did not appear to correlate with genetic mutations, cellular responses, or familial clustering. Environmental factors and/or other background genetic differences may account for the heterogeneity.
In addition to cold-induced cellular activation, abnormal chemotaxis and impaired TNF-α–mediated activation observed in neutrophils from PLAID patients may also contribute to granuloma formation. Impaired neutrophil functioning could stimulate increased cell signaling and activation, potentially leading to the generation of granulomas. However, the TNF-α–mediated activation and chemotaxis data must be interpreted with caution because it is possible that cold induced activation of cells from patients with PLAID during processing (not performed at 37°C)may have led to abnormal activation of neutrophils before assay performance.
PLAID-associated granulomas may contribute to our understanding of granulomatous disease in other settings. Noncaseating cutaneous granulomas in patients with PLAID are histopathologically similar to those observed in patients with sarcoidosis and CVID. Like PLAID, granulomas in sarcoidosis and CVID may be caused by leukocyte hyperactivation. Inappropriate macrophage activation, similar to that seen in PLAID, has been proposed as a potential mechanism for granuloma formation in CVID and A-T.2,5 Tonic activation of neutrophils has also been implicated in the formation of CGD granulomas.6 Given that neutrophils and monocytes from patients with PLAID have spontaneous cold activation and that PLAID granulomas occur predominantly on cold skin surfaces, we hypothesize that innate immune leukocyte hyperactivation may underlie cutaneous granuloma formation in PLAID. This raises the possibility that leukocyte hyperactivation may provide a unique and direct pathway for cutaneous granuloma formation.
Conclusions
Testing for PLCG2 mutations should be considered in patients with a lifelong history of cold urticaria and a history of recurrent sinopulmonary infections or abnormalities on tests of immune cell functioning. A high index of suspicion should be maintained for patients with a strong family history of cold urticaria and recurrent infections in addition to their personal history. Patients with cold urticaria who have additional findings of granulomatous skin disease or a history of hemorrhagic lesions at acral sites in the early neonatal period are excellent candidates for genetic testing.
The identification of PLAID-like phenotypes among patients without identifiable PLCG2 mutations suggests that additional genetic defects may exist within this pathway, which also led to skin lesions and immune dysfunction. Future efforts will focus on identifying the genetic mutations responsible for PLAID-like phenotypes in these patients. Identification of patients using characteristic cutaneous manifestations that may precede immunodeficiency symptoms may help in early identification and improved care of patients with PLAID and PLAID-like phenotypes. Although direct correlations between patient phenotype and in vitro data have yet to be identified, the present study not only provides a laboratory and clinical model and an approach to study the basic mechanisms of PLAID, but also contributes to our understanding of the broader category of granulomatous disorders.
Supplementary Material
Acknowledgments
Funding/Support: This study was supported in part by the Intramural Research Program of the National Institutes of Health, the National Institute of Allergy and Infectious Diseases, and the National Institutes of Health Medical Research Scholars Program, a public-private partnership supported jointly by the National Institutes of Health and generous contributions to the Foundation for the National Institutes of Health from Pfizer Inc, The Doris Duke Charitable Foundation, The Alexandria Real Estate Equities Inc, Mr. and Mrs. Joel S. Marcus, and the Howard Hughes Medical Institute, as well as other private donors.
Footnotes
Supplemental content at jamadermatology.com
Conflict of Interest Disclosures: None reported.
Additional Information: Dr Cowen and Dr Milner are cosenior authors.
Additional Contributions: Danielle Fink, MS, Karen Lau, MS, Lauren Mendez, BS and Dara Riva, MS, conducted the chemotaxis, cytometry, and superoxide studies. We thank the patients and their families for participating in this study and the clinical staff of the Laboratory of Allergic Diseases for their efforts.
Author Contributions: Ms Aderibigbe and Dr Milner had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Study concept and design: Aderibigbe, Priel, Kuhns, Cowen, Milner.
Acquisition, analysis, or interpretation of data: All authors.
Drafting of the manuscript: Aderibigbe, Priel, Lyons, Kuhns, Cowen, Milner.
Critical revision of the manuscript for important intellectual content: All authors.
Statistical analysis: Aderibigbe, Priel, Ombrello, Kuhns, Milner.
Obtained funding: Ombrello, Lyons, Milner.
Administrative, technical, or material support: Ombrello, Lyons.
Study supervision: Prajapati, Liang, Lyons, Milner.
Role of the Funder/Sponsor: The funding sources had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and the decision to submit the manuscript for publication.
References
- 1.Al-Herz W, Nanda A. Skin manifestations in primary immunodeficient children. Pediatr Dermatol. 2011;28(5):494–501. doi: 10.1111/j.1525-1470.2011.01409.x. [DOI] [PubMed] [Google Scholar]
- 2.Chiam LY, Verhagen MM, Haraldsson A, et al. Cutaneous granulomas in ataxia telangiectasia and other primary immunodeficiencies: reflection of inappropriate immune regulation? Dermatology. 2011;223(1):13–19. doi: 10.1159/000330335. [DOI] [PubMed] [Google Scholar]
- 3.Sillevis Smitt JH, Kuijpers TW. Cutaneous manifestations of primary immunodeficiency. Curr Opin Pediatr. 2013;25(4):492–497. doi: 10.1097/MOP.0b013e3283623b9f. [DOI] [PubMed] [Google Scholar]
- 4.Keczkes K, Bilimoria S, Piercy DM. Pernicious anaemia and granulomatous skin lesions in a case of common variable hypogammaglobulinaemia. Br J Dermatol. 1979;101(2):211–217. doi: 10.1111/j.1365-2133.1979.tb05609.x. [DOI] [PubMed] [Google Scholar]
- 5.Mechanic LJ, Dikman S, Cunningham-Rundles C. Granulomatous disease in common variable immunodeficiency. Ann Intern Med. 1997;127(8 pt 1):613–617. doi: 10.7326/0003-4819-127-8_part_1-199710150-00005. [DOI] [PubMed] [Google Scholar]
- 6.Tintinger GR, Theron AJ, Steel HC, Anderson R. Accelerated calcium influx and hyperactivation of neutrophils in chronic granulomatous disease. Clin Exp Immunol. 2001;123(2):254–263. doi: 10.1046/j.1365-2249.2001.01447.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ombrello MJ, Remmers EF, Sun G, et al. Cold urticaria, immunodeficiency, and autoimmunity related to PLCG2 deletions. N Engl J Med. 2012;366(4):330–338. doi: 10.1056/NEJMoa1102140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou Q, Lee GS, Brady J, et al. A hypermorphic missense mutation in PLCG2, encoding phospholipase Cγ2, causes a dominantly inherited autoinflammatory disease with immunodeficiency. Am J Hum Genet. 2012;91(4):713–720. doi: 10.1016/j.ajhg.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gandhi C, Healy C, Wanderer AA, Hoffman HM. Familial atypical cold urticaria: description of a new hereditary disease. J Allergy Clin Immunol. 2009;124(6):1245–1250. doi: 10.1016/j.jaci.2009.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Petersen HJ, Smith AM. The role of the innate immune system in granulomatous disorders. Front Immunol. 2013;4:120. doi: 10.3389/fimmu.2013.00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.von Stebut E, Metz M, Milon G, Knop J, Maurer M. Early macrophage influx to sites of cutaneous granuloma formation is dependent on MIP-1α/β released from neutrophils recruited by mast cell-derived TNFα. Blood. 2003;101(1):210–215. doi: 10.1182/blood-2002-03-0921. [DOI] [PubMed] [Google Scholar]
- 12.Berger M, O’Shea J, Cross AS, et al. Human neutrophils increase expression of C3bi as well as C3b receptors upon activation. J Clin Invest. 1984;74(5):1566–1571. doi: 10.1172/JCI111572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller LJ, Bainton DF, Borregaard N, Springer TA. Stimulated mobilization of monocyte Mac-1 and p150,95 adhesion proteins from an intracellular vesicular compartment to the cell surface. J Clin Invest. 1987;80(2):535–544. doi: 10.1172/JCI113102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gange RW, Black MM, Carrington P. Defective neutrophil migration in granuloma annulare, necrobiosis lipoidica, and sarcoidosis. Arch Dermatol. 1979;115(1):32–35. [PubMed] [Google Scholar]
- 15.Gange RW, Black MM, Carrington P, McKerron R. Defective neutrophil migration in sarcoidosis. Lancet. 1977;2(8034):379–381. doi: 10.1016/s0140-6736(77)90306-3. [DOI] [PubMed] [Google Scholar]
- 16.Majewski BB, Rhodes EL, Watson B. Neutrophil mobility in granuloma annulare and necrobiosis lipoidica. Clin Exp Dermatol. 1981;6(6):583–590. doi: 10.1111/j.1365-2230.1981.tb02361.x. [DOI] [PubMed] [Google Scholar]
- 17.Zhu WP, Xin XR. Study on the distribution pattern of skin temperature in normal Chinese and detection of the depth of early burn wound by infrared thermography. Ann N Y Acad Sci. 1999;888:300–313. doi: 10.1111/j.1749-6632.1999.tb07964.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.