

Abstract
Identification of the appropriate combination of radionuclide, target and targeting vehicle is critical for successful radioimmunotherapy. For the treatment of disseminated peritoneal diseases such as pancreatic or ovarian cancer, α-emitting radionuclides have been proposed for targeted radiation therapy. This laboratory has taken a systematic approach investigating targeted α-radiation therapy, allowing comparisons to now be made between 211At, 227Th, 213Bi and 212Pb. Herein, trastuzumab radiolabeled with 211At and 227Th was evaluated for therapeutic efficacy in the LS-174T i.p. tumor model. A dose escalation study was conducted with each radioimmunoconjugate (RIC). Therapeutic benefit was realized with 211At-trastuzumab with doses of 20, 30 and 40 μCi. At doses > 40 μCi, toxicity was observed with greater weight loss and 2-fold higher decrease in the platelet counts. Following a second study comparing the effect of 20, 30 and 40 μCi of 211At-trastuzumab, 30 μCi was selected as the dose for future studies. A parallel study was performed evaluating 0.25, 0.5, 1.0, 2.0 and 5.0 μCi of 227Th-trastuzumab. The 0.5 and 1.0 μCi injected dose resulted in a therapeutic response; a lower degree of weight loss was experienced by the mice in the 0.5 μCi cohort. When the data is normalized for comparing 211At, 227Th, 213Bi and 212Pb, the choice of radionuclide for RIT is perhaps not entirely based on simple therapeutic efficacy, other factors may play a role in choosing the “right” radionuclide.
Graphical Abstract
Choosing an appropriate α-emitter for targeted therapy resides in multiple variables beyond efficacy such as the economics of injected dose.
Introduction
To date, there are still only two radiolabeled mAb that have been approved by the Food and Drug Administration (FDA) for therapeutic applications (http://www.immunologylink.com/FDA-APP-Abs.html. Indicated for the treatment of relapsed Non-Hodgkin’s Lymphoma, both are anti-CD20 mAbs labeled with β-emitters. Zevalin, radiolabeled with 90Y, gained approval in 2002 while Bexxar, radiolabeled with 131I, was approved in 2003. β-Emitting radionuclides suitable for therapeutic applications have maximum energies of 0.3–2.3 MeV with ranges of ~2–11 mm in tissues.1 This translates to the β-particles being able to traverse 10–1000 cell diameters which then promotes the tailoring of a targeted radiation therapeutic to a specific presentation of disease. β-Radiation has low linear energy transfer (LET). The long path length means that energy deposition is dispersed, sparse, and occurs some distance from the actual initial decay event.2 Therapeutic benefits are derived from non-targeted tumor cells being in the crossfire of the decay event and the omnidirectional potential of the emission. The same crossfire distance and directionality can potentially impact surrounding normal tissues and cells, resulting in toxicity. For this reason, targeted β-radiation is considered more appropriate for the treatment of solid tumors that are > 1 cm in diameter.3 Patients with single cell diseases (e.g., leukemia), micrometastases, disseminated cancers (e.g., ovarian, pancreatic, gastric carcinomatosis) or post-surgical residual disease would not be appropriate candidates for β-radiation therapy. For these specific scenarios, a large proportion of the energy from the β-particles would be deposited beyond the tumor lesion or cell margins and wasted while at the same time normal tissue would be irradiated and again result in toxicity. 4–6 In contrast to targeted β-radiation therapy, the properties of α-emitting radionuclides appear to be ideally suited for the treatment of the above stated disease presentations when incorporated into targeted radiation.
There are >100 radionuclides that emit α-particles, however, only a short list of those that are considered suitable for therapeutic applications are currently under investigation for targeted radioimmunotherapy (RIT). Included in the list are 149Tb, 211At, 212Pb, 212Bi, 213Bi, 223Ra, 225Ac and 227Th and is defined by a combination of half-life, availability of chemistry and economics.7 Although the transition of 212Pb to 212Bi generates a β-particle, this serves as an in vivo generator of 212Bi which is a short-lived α-emitter.
The α-particle energies range from 5–9 MeV with a 50–90 μm (~2–10 cell diameters) range in tissue and have a high LET. α-Particles deposit ≥500 times more energy per unit of path length than β-particles.8 When treating single cell disease, it is estimated that to effect 99.99% cell kill, only a few hundred α-particle decays would be required with a much reduced risk of toxicity to adjacent normal tissues due to the short range of the particle. Targeted β-particle therapy would require hundreds of thousands of β-decays to attain a comparable level of cell kill.9 Targeted α-radiation therapy has been shown to effect cell death through production of double-stranded DNA breaks, DNA crosslinking, chromosomal rearrangements, the induction of apoptosis, perturbation of the cell cycle, and blockage or downregulation of the DNA damage repair mechanism specific to double-stranded DNA breaks.10–14
Clinical trials have been conducted with 211At, 225Ac, 213Bi, 212Pb, and 223Ra.7 The diseases treated include glioma, ovarian carcinoma, acute myeloid leukemia, B-cell malignancies, melanoma, advanced myeloid leukemia and castration resistant prostate cancer. These trials have demonstrated the safety of the radiopharmaceuticals, that toxicities have been limited and that administration of therapeutic doses are possible.15–19
Pre-clinical studies from this laboratory translated to a first in human phase 1 clinical trial with 212Pb-trastuzumab at the University of Alabama at Birmingham.20–22 All indications are that locoregional administration of 212Pb-RIT is safe. Eighteen patients with HER2 positive peritoneal malignancies that had failed standard therapies received 212Pb-trastuzumab. The single i.p. injection was well tolerated with grade 1 toxicities, mostly asymptomatic, reported.22 No late renal, hepatic, cardiac or other toxicity was found up to 1 year and no immune response to the RIC was detected. Furthermore, prior to the clinical trial, a toxicology study with cynomolgus monkeys injected i.p. with 212Pb-trastuzumab showed that ~90–100% of the 212Pb and 83–92% of the 212Bi daughter was retained within the peritoneum.23
The report presented herein describes studies evaluating the therapeutic efficacy of trastuzumab radiolabeled with 211At as well as parallel and comparable studies using 227Th. With a 7.2 h half-life, 211At is “intermediate” to 213Bi (45.7 min) and 212Pb (10.6 h) while 227Th is at the far end of the half-life spectrum at 18.7 d. The strategy for the application of 227Th is that it delivers a low dose rate that may overcome the difficulties of treating tumor cells that vary in the cell cycle phases, and therefore susceptibility to ionizing radiation, as well as capitalizing upon contributions from the daughter radionuclides. Both 211At and 227Th suffer their own specific limitations and complications (vide infra).
The impetus for these studies derives from the fundamental principle that the biological half-life of the targeting vector(s) should, as precisely as possible, match the radiological half-life of the radionuclide being employed as the therapeutic. From the plethora of published biodistribution studies pertaining to pre-clinical tumor targeting, one can easily deduce that matching these two half-lives is critical to efficacious therapeutic impact. Additionally, the radiological half-life can not only impact simple logistics of dose preparation, but also the specific disease presentation being treated. Studies with 211At and 227Th address half-life gaps around prior studies from this laboratory with 213Bi and 212Pb to treat animals bearing an intraperitoneal colorectal cancer xenograft. In doing so through directly comparable studies, the aim is to investigate if conclusions can be drawn regarding an optimal therapeutic radionuclide within the context of this disease presentation.
Materials and methods
Cell line and culture
In vivo studies were conducted using LS-174T cells, a human colon carcinoma cell line, grown inDulbecco’s minimum essential medium (DMEM). The DMEM was supplemented with 1 mM glutamine, 1 mM non-essential amino acids (NEAA) and 10% FetalPlex (Gemini Bioproducts, Inc., West Sacramento, Ca) as detailed elsewhere.24, 25 DMEM, glutamine and NEAA were obtained from Lonza, Walkersville, MD. Cells were harvested when 75–80% confluency was attained.
Conjugation and Radiolabeling
Trastuzumab (Herceptin; Genetech, Inc., South San Francisco, CA) and panitumumab (Vectibix, Amgen, Thousand Oaks, CA) were purchased through the National Institutes of Health (NIH), Division of Veterinary Resources Pharmacy. Polyclonal human immunoglobulin (HuIgG; MP Biochemicals), purified from human serum, has no known antigen with which it reacts. HuM195, an anti-CD33 antibody, was kindly provided by Dr. McDevitt at the Memorial Sloan-Kettering Cancer Center.
Conjugation of monoclonal antibodies (mAb) with the bifunctional ligands, 1, 4, 7, 10-tetraazacyclododecane-N, N′,N″,N‴-tetraacetic acid (DOTA) or with trans-cyclohexyl-diethylenetriamine-pentaacetic acid (CHX-A″), was performed at a 10-fold molar excess of ligand to mAb according to established methods.26–32 The final protein concentrations were determined by the Lowry method using a BSA standard.33 The number of DOTA and CHX-A″ molecules bound to mAb were quantitated using spectrophotometric assays based on the titration of lead-Arsenazo(III) and yttrium- Arsenazo(III) complex, respectively. 34, 35 The final chelate:protein ratios range from 2.6 to 4.0. HuIgG, or HuM195 similarly conjugated, served as negative controls in these studies.
Astatine-211 Labeling
Astatine-211 was produced by the 209Bi(α,2n)211At reaction using a disposable internal bismuth target with α-particles from a Cyclotron Corporation CS-30 cyclotron at the National Institutes of Health Cyclotron Facility. The 211At was recovered using dry-distillation as previously reported.36, 37 The 211At was recovered by elution with a 1:1 mixture of a solution of N-chlorosuccinimide in acetonitrile (0.4 mg/mL):a solution of SPEMS precursor (1 mg/mL) in acetonitrile containing acetic acid (0.4%).38 The mixture was allowed to stand at room temperature for 30 min and the astatinyl precursor (N-methyl-SAPS) was purified by normal phase HPLC.38 The fraction containing the N-methyl-SAPS was air dried and the mAb (10 mg/mL) in borate buffer (0.1 M, pH 8.5) was added directly to the residue, vigorously vortexed, and incubated for 15 min at room temperature. The resulting astatinated mAb were purified on a PD-10 column using PBS as eluate.
Thorium-227 Labeling
Thorium-227 was obtained from Oak Ridge National Laboratories (Oak Ridge, TN). The nuclide was freshly purified for each labeling using an AG MP-1M resin, 200–400 mesh (Bio-Rad, Hercules, CA). A suspension of the resin in H2O (0.5 – 0.6 mL column vol.) was loaded into a Poly-Prep Chromatography column (Bio-Rad), washed with HNO3 (10 mL, 8M), 0.1M HNO3 (10 mL) and then re-equilibrated in 8 M HNO3 (10 mL). 227Th and its daughters in HNO3 (100 μL, 8 M) were loaded unto the column followed by HNO3 (6 mL, 8 M). The 227Th was eluted into a vial with 1.5 ml 0.1M HNO3, the eluate evaporated to dryness and re-dissolved in 0.1M HNO3 for labeling. The 227Th was measured at 236 KeV using an ORTEC High Purity Ge detector (Model GEM10P4-70) system cooled by an ORTEC CryoSecure Power Controller and connected to an ORTEC DSPEC Jr 2.0 Spectrometer (Ortec, Oak Ridge, TN) using GammaVision-32 Version 7.02 software (ORTEC).
mAb-DOTA conjugates (trastuzumab, panitumumab, and HuM195) were labeled with 227Th by a modification of a previously published procedure.39 For labeling, ascorbic acid (25 μL, 150 mg/mL) was added to the 227Th contained in HNO3 (25 μL, 0.1M), and the mixture was neutralized to pH 5.5 with ammonium acetate buffer (5 M, 7.5 μL) before the addition of the immunoconjugate (400 – 500 μg) contained in ammonium acetate solution (0.15 M). The mixture was incubated at 42 °C for 2 h. At the end of incubation, saturated DTPA (10 μL) was added to quench the reaction and the products were purified on a disposable PD-10 column eluted with PBS.
Trastuzumab-DOTA was radiolabeled with 111In per previously published methodology.24
Radioimmunoassay
The immunoreactivity of the 211At- and 227Th-labeled trastuzumab preparations were assessed by a radioimmunoassay using purified recombinant human ERB2/Fc (rHuErb2/Fc; 50 ng per well, Sigma-Aldrich)as previously detailed.40 After adsorption of the rHuErb2/Fc to the wells of a 96-well plate, the rHuErb2/Fc was removed. Serial dilutions of radiolabeled trastuzumab (~200,000 cpm to 12,500 cpm in 50 μL of BSA/PBS) were added to the wells and incubated for 4 h at 37°C. The wells were washed, the radioactivity harvested and measured using a γ-scintillation counter. The percentage binding was calculated for each dilution and averaged. The specificity of the radiolabeled trastuzumab was confirmed by incubating one set of wells with radiolabeled trastuzumab and 10 μg of unlabeled trastuzumab.
The assay to determine the immunoreactivity of the 227Th-panitumumab was performed similarly using 100ng/well of hEGFR.41
In Vivo Studies
All in vivo studies were performed using 6–8 week old female athymic (NCr-nu/nu) mice (NCI-Frederick, Cat#01B70). The studies were conducted per protocols approved by the National Cancer Institute Animal Care and Use Committee.
Tumor Targeting
The in vivo behavior of labeled DOTA-trastuzumab was evaluated in athymic mice bearing s.c.. LS-174T tumor xenografts. Mice were injected s.c. in the right scapular region with 2×106 cells in 0.2 mL of DMEM as detailed elsewhere.42 After i.v. injection of the mice with 111In-trastuzumab (~5 μCi) or 227Th-trastuzumab (~0.5 μCi) when tumors were 0.2–0.4 mm in diameter, the mice were euthanized at 24, 48, 72, 96, and 168 h by CO2 inhalation. The blood, tumor, and major organs were collected, wet-weighed, and counted in a γ-scintillation counter. The percent injected dose per gram (%ID g−1) was determined for each tissue; the averages and standard deviations are presented. In the case of the 227Th-trastuzumab, the tissues were stored at −20 °C for 2 months for equilibrium to be reached; the samples were counted and calculations made accordingly.
Therapy
Athymic mice were injected i.p. with 1×108 cells in 0.5 mL of media; RIT studies detailed below were initiated 3 d thereafter. The radiolabeled mAbs were administered i.p. to mice in 0.5 mL PBS. The activity is indicated in each study description. Appropriately radiolabeled HuIgG or HuM195 served as non-specific controls. The mice were monitored 2–3 times per week; the body weight recorded 1–2 times per week for 4–6 weeks as a measure of toxicity due to therapy. Progression of disease was observed either as an extension of the abdomen, development of ascites or noticeable, palpable, nodules in the abdomen or, conversely, as weight loss. Mice were euthanized if found to be in distress, moribund, or cachectic. Euthanasia was also performed when disease progression was evident as cited above.
The ultimate objective of studies from this laboratory has been to develop a multi-modality treatment, i.e., combining RIT with chemotherapeutics. Towards this end, studies were conducted to determine the effective working dose of 211At-trastuzumab and 227Th-trastuzumab, not necessarily a maximum tolerated dose, that when combined with a chemotherapeutic would not result in unacceptable levels of toxicity. For the evaluation of 211At-trastuzumab, tumor-bearing mice (n=9–15) were administered 10, 20, 30, 40, 50, 60, 70 or 80 μCi of 211At-trastuzumab by i.p. injection. Additional sets of mice received 10, 20, 30, 40, 50 or 60 μCi of 211At-HuIgG. All treatment groups were compared to a set of mice that received no RIT.
A second study was performed with 211At-trastuzumab to confirm the final dose selection that could be used in future studies involving chemotherapeutics. Athymic mice were treated 3 d after i.p. injection of LS-174T cells with 20, 30 or 40 μCi of 211At-trastuzumab. Additional groups of mice were administered the same doses of 211At-HuIgG and one set of mice were left untreated.
With the same intent, a therapy study was conducted with 227Th-trastuzumab. Athymic mice (n=10) bearing 3d LS-174T i.p. tumor xenografts were injected i.p. with 0.25, 0.5, 1, 2 or 5 μCi (in 500 μL PBS) of 227Th-trastuzumab. Panitumumab has shown therapeutic efficacy has a single modality and in combination with chemotherapeutics. Thus, 227Th-panitumumab was included in this study for comparison with 227Th-trastuzumab
Blood Cell Analysis
Blood samples were collected from the mice receiving 211At-RIT for analysis of blood cell counts, platelets specifically. Fifty μL were collected from 5 mice in each group at the time points indicated. The blood samples were diluted 1:1 with potassium EDTA (1.5mg/mL). Cell counts performed within 24 h of collection on an Abaxis VetScan VM5.
Results and discussion
211At Studies
In vitro Analysis of 211At-trastuzumab
Radiolabeling of trastuzumab with 211At resulted in a 50 – 60% product yield with a specific activity of 6–8 mCi/mg. The 211At-trastuzumab retained reactivity for HER2 with 52.3% bound. Addition of excess trastuzumab to the reaction reduced the amount of 211At-trastuzumab bound to 4.9% thus demonstrating specificity. These results are consistent with previous 211At labelings of trastuzumab performed in this laboratory. The data is also in agreement with other groups investigating different 211At-labeled immuno-conjugates.38, 43–46
Tumor targeting
Studies from this laboratory demonstrating tumor targeting and normal organ distribution of 211At-trastuzumab in mice bearing s.c. LS-174T tumor xenografts have been published. 38, 43 In those studies, tumor targeting as well as the level of 211At in the intestines and stomach were comparable to what has been reported by others while the level of 211At in the lungs was considerably improved over what has been published. 45, 47–49 Since 211At-trastuzumab targeting of LS-174T tumor xenografts was established, the studies described herein began with a dose escalation RIT study to determine an effective working dose with the RIC.
Determination of effective therapeutic dose for 211At-trastuzumab
Injected doses (10, 20, 30, 40, 50, 60, and 80 μCi) of 211At-trastuzumab were administered i.p. to athymic mice (n=10–15 per group) bearing i.p. LS-174T tumor xenografts. The survival curves for these dose groups are presented in Figure 1. Therapeutic benefit was observed with each of the doses from 10–60 μCi. There was a higher median survival (MS) compared to the untreated group of mice (MS = 16 d) resulting in therapeutic indices (TI) ranging from 1.9 to 5.0. The 50 and 60 μCi doses resulted in the highest MS of 66 and 80 d, respectively (Table 1). The 80 μCi dose was not tolerated, evident with a MS of 13 d. Meanwhile, some therapeutic efficacy was also observed for the nonspecific control, 211At-HuIgG, with a MS of 23 d for the 40 and 50 μCi doses thereby demonstrating the necessity of including such controls. The highest TI was 1.4 which corroborates the specificity of the 211At-trastuzumab therapy. For the selection of the effective working dose, animal weights and platelet levels were also taken into consideration.
Fig. 1.

Effect of increasing 211At-trastuzumab doses (μCi) on animal survival. Groups of mice (n =6–15) bearing i.p. LS-174T tumor xenografts were injected i.p. with 10, 20, 30, 40, 50, 60, or 80 μCi of 211At-trastuzumab. Additional groups of mice were treated with 20, 30, 40, 50, or 60 μCi of the nonspecific control, 211At-HuIgG while one set of mice were left untreated.
Table 1.
Dose Escalation of i.p. Injected 211At-mAb: Median Survival of Athymic Mice Bearing i.p. LS-174T Tumor Xenografts
|
|
|||||
|---|---|---|---|---|---|
| Dose (μCi) | None | 211At-Trastuzumab | 211At-HuIgG | ||
|
| |||||
| Median Survival (d) | Therapeutic Index | Median Survival (d) | Therapeutic Index | ||
| 0 | 16 | ||||
| 10 | 30 | 1.9 | --- | --- | |
| 20 | 43 | 2.7 | 17 | 1.1 | |
| 30 | 36 | 2.3 | 17 | 1.1 | |
| 40 | 38 | 2.4 | 23 | 1.4 | |
| 50 | 66 | 4.1 | 23 | 1.4 | |
| 60 | 80 | 5.0 | 10 | 0.6 | |
| 80 | 13 | 0.8 | --- | --- | |
Therapuetic Index is the median survival of the treatment group divided by the median survival of the untreated group.
The degree of weight loss increased with the increasing dose of 211At-trastuzumab (Table S1) as early as 3 d post-administration. Beginning at the 50 μCi dose, weight loss was 14.5% and reached 21.7% with the 80 μCi dose. In conjunction with this data, platelet levels also show a dose-dependent effect compared to the untreated group (Figure S2). Ten days after 211At-RIT, there is a noticeable decrease in platelet levels at all doses. In the groups of mice that received 10, 20, 30 and 40 μCi, the decrease in platelet levels were 11.7, 30.2, 33.3 and 25.5%, respectively. Meanwhile at the higher doses of 50, 60 and 80 μCi, the platelet levels were diminished by 58.7, 64.7 and 67.4%, respectively, compared to the group of mice that did not receive any RIT.
Considering that there was no significant difference in the MS of the mice that received 20, 30 or 40 μCi of 211At-trastuzumab (p = 1) and that the decrease in platelet levels were comparable, these three doses were re-evaluated in a second study to make a final decision on which dose would be utilized in future therapy studies. This lack of distinction in the effectiveness of 211At-RIT at these dose levels has been noted before.50 Treatment of mice bearing i.p. ovarian tumor xenografts with ~11, 21 and 32 μCi of 211At-MX35 resulted in a tumor free fraction (TFF) of 80, 50 and 60%, respectively, at 60 d. The TFF did not correlate with the amount of injected activity. One hypothesis presented to explain this ambiguity was that the specific activity of the 211At-MX35 at 134±25 kBq/μg (3.6 μCi/μg) was too low. In the studies reported here, the specific activity was 6–8 μCi/μg. In fact, use of a higher specific activity product would most likely risk radiolysis and compromise the RIC.
To choose a dose for subsequent RIT studies with 211At with confidence, the second therapy study was conducted with larger groups of mice. Tumor bearing mice (n=30) per group were injected with 20, 30 or 40 μCi of 211At-trastuzumab. Additional groups of mice received 20 μCi (n=22), 30 μCi (n=25) or 40 μCi (n=15) of 211At-HuIgG or were untreated (n=30) (Figure 2). Therapeutic efficacy was observed with a MS of 24, 30 and 30 d for the mice treated with 20, 30 and 40 μCi, respectively, of 211At-trastuzumab (Table 2). Compared to the untreated mice, this translates to a TI of 1.8, 2.3 and 2.3, respectively. Once again, some therapeutic efficacy was observed in the 211At-HuIgG treated mice with MS of 20 d (20 μCi), 23 d (30 μCi) and 23 d (40 μCi). The therapeutic response was specific; differences between 211At-trastuzumab and 211At-HuIgG at each dose were significant (p ≤ 0.002) while there was no significance among the doses of 211At-trastuzumab (p ≥ 0.1).
Fig. 2.
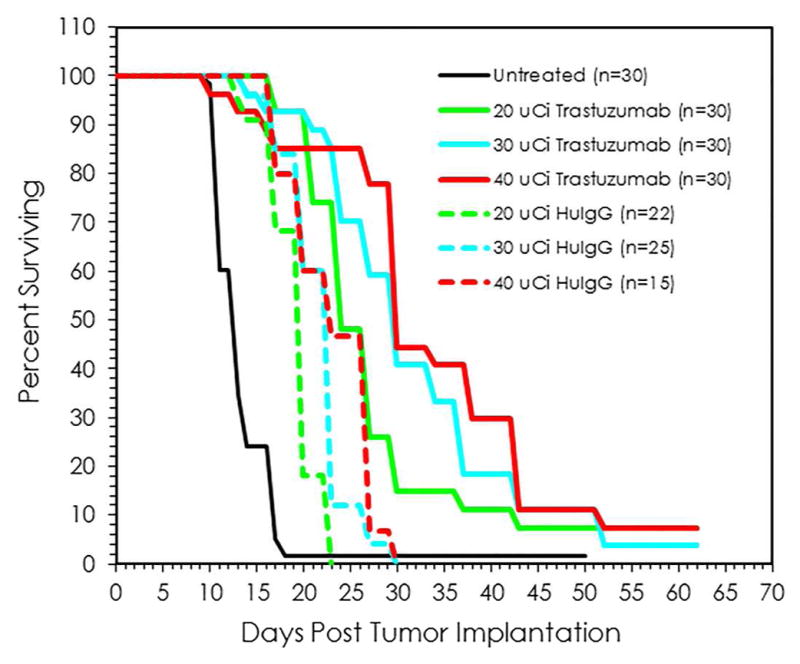
Validation of effective working dose for 211At-trastuzumab. Groups of mice were injected i.p. with 20, 30 or 40 μCi 211At-trastuzumab (n =30 per group) or 211At-HuIgG (n=15–22 per group). One group of mice were left untreated (n=30).
Table 2.
Dose Validation of Intraperitoneal 211At-RIT: Targeting HER2 Expressing LS-174 ip Xenografts with Trastuzumab
| Dose (μCi) | None | Trastuzumab | HuIgG | ||
|---|---|---|---|---|---|
|
| |||||
| Median Survival (d) | Therapeutic Index | Median Survival (d) | Therapeutic Index | ||
| 0 | 13 | ||||
| 20 | 24 | 1.8 | 20 | 1.5 | |
| 30 | 30 | 2.3 | 23 | 1.8 | |
| 40 | 30 | 2.3 | 23 | 1.8 | |
Therapuetic Index is the median survival of the treatment group divided by the median survival of the untreated group.
Of the groups injected with 211At-trastuzumab, the greatest weight loss was noted in those receiving 40 μCi (Table S2). At 7d post-RIT there was a loss of 12.5% which was never regained throughout the 32 d that the mice were weighed. Weight loss was also observed in the other two treatment groups which at 12 d were comparable, 10.4% loss in the 20 μCi group and 11.4% loss in the 30 μCi group. And as with the 40 μCi group, the mice even at these doses did not appear to completely recover from this weight loss.
Blood cell analysis (Figure 3) was consistent with previous RIT studies, there was the typical decrease in platelets 9 d after treatment at each of the 211At-trastuzumab doses. By the time of the next blood collection at 18 d, the platelet count had recovered with an apparent rebound effect (i.e., greater than standard deviations). By subsequent blood draws, platelet counts returned to pre-therapy levels. Interestingly, this initial drop in platelets and the subsequent recovery was not seen in the mice treated with 211At-HuIgG. One explanation for this may be the absence of an antigen target for the 211At-HuIgG that would shorten the retention time of the 211At-HuIgG in the peritoneum thereby decreasing toxicity. Based on the aggregate data, future studies were determined to be conducted with 30 μCi of 211At when the vector is an intact immunoglobulin.
Fig. 3.

Blood samples were collected from tumor bearing mice receiving 211At-RIT and platelet cell counts were performed. Tumor bearing mice received 20, 30 or 40 μCi of either 211At-trastuzumab or 211At-HuIgG. The platelets were quantitated for five mice in each group at the indicated days. The values are compared to the group of mice that did not receive 211At-RIT.
This dose does appear to agree with several reports in the literature, however, comparisons are difficult to make due to differences in the delivery vectors, the tumor model or the selection of the effective therapeutic dose. The highest dose that has been evaluated with 211At-MX35 has been ~33 μCi given i.p. in an ovarian cancer mouse model.51 Higher activities have not been evaluated to determine if this is an optimal effective therapeutic dose. In fact, this group has chosen 400 kBq (10.8 μCi) as their therapeutic dose for not only intact MX35 but also the F(ab′)2 fragment of MX35.51, 52 This dose selection is not based on direct empirical data but on data from a study evaluating the toxicity of i.v. and i.p. administered 211At-MX35 in nontumor bearing athymic Balb/c mice.50 Anti-HER2/neu diabodies radiolabeled with 211At have been found effective at 20 and 45 μCi in a breast cancer model.46 Achieving higher injected doses with a diabody is not surprising. At ~one-third the molecular weight and smaller in size than an intact IgG, diabodies should have greater tumor penetration and concomitantly a more rapid blood clearance. Perhaps more importantly, the study compared two diabodies in the same tumor model and observed differences in their therapeutic efficacy illustrating optimization is a requisite to appropriate application and to obtaining optimal benefit from each RIC. There is one report in which up to 92 μCi of 211At-trastuzumab was evaluated for therapeutic efficacy.53 The 211At-trastuzumab was administered locoregionally, via a surgically implanted intrathecal catheter, into the subarachnoid space along the brain and spine to treat carcinomatous meningitis in a athymic rat model. The investigators reported an increase in the MS at 33, 66 and 92 μCi in two experiments. Histopathological analysis of the neural axis revealed some toxicity that presented as edema, fibrosis, necrosis and demyelination that increased with dose. Tumor eradication was found to be most effective in the regions rostral to the injection site.
227Th Studies
In vitro analysis 227Th-labeled mAbs
The yield of radiolabeled product was 14% for panitumumab, 37% for trastuzumab and 39% for HuM195 with specific activities of 0.26 mCi/mg, 0.51– 0.68 mCi/mg and 0.72 mCi/mg, respectively. These specific activities are at least10-fold greater than what has been previously reported for 227Th-trastuzumab.39, 54–56 Meanwhile, the reported labeling yields for 227Th-trastuzumab range from 6% to 90%.55, 56 The percent bound to HER2 was 53.7% for 227Th-trastuzumab and 26.2% for 227Th-panitumumab to HER1. Addition of excess mAb to each of these RICs reduced the percent bound to 1.4% and 2.5%. The binding of 227Th-HuM195 to HER1 and HER2 was < 2%. The immunoreactivity of the 227Th-trastuzumab reported here is comparable to that obtained by other investigators.55 The low percent bound of the RICs with their respective antigens reflects the low specific activity of the products.
Tumor targeting and normal organ distribution of radiolabeled DOTA-trastuzumab
A study assessing and reconfirming tumor targeting and normal organ distribution of the DOTA-trastuzumab radiolabeled with 111In was performed in a s.c. tumor model prior to radiolabeling with 227Th. Tumor targeting of the 111In-DOTA-trastuzumab was lower compared to what has been published by this laboratory using the bifunctional chelate CHX-A″-DTPA (Table S3 and Figure S2).24 At 48 h post-injection of the RIC, the tumor percent injected per gram (%ID/g) peaked with a value of 13.20±1.91. In regards to normal organs, the distribution of radioactivity was comparable to previous studies and the %ID/g was <5.
A higher tumor %ID/g was observed in the mice injected i.v. with 227Th-trastuzumab (Table S4 and Figure S3), peaking at 22.99±8.09. The distribution of radioactivity in normal organs displayed a similar pattern and level as reported for 111In-CHX-A″-trastuzumab with the highest %ID/g found in the liver, followed by the spleen and kidneys.24 This data is consistent with a report by Abbas et al., in which 227Th-trastuzumab was evaluated in a murine model for ovarian cancer.54 Others have demonstrated tumor targeting with low uptake in normal organs following i.p. injection of 227Th-trastuzumab in mice bearing i.p. human ovarian (SKOV-3) and s.c. human breast cancer (SKBR-3) tumor xenografts.39, 55
Determination of effective therapeutic dose for 227Th-trastuzumab
A dose escalation study then followed to evaluate the therapeutic potential of a low dose rate α-particle emitting radionuclide. Athymic mice (n=10 per group) bearing 3 d i.p. LS-174T tumor xenografts were injected i.p. with 0.25, 0.5, 1.0, 2.0 or 5.0 μCi of 227Th-trastuzumab or the non-specific control, 227Th-HuM195. One set of 10 mice was left untreated. Some therapeutic benefit was evident with a MS of 30 d experienced by the mice that received either the 0.5 or the 1.0 μCi dose; the MS of the untreated group was 14 d (Table 3 and Fig. 4A). Apart from the 1.0 μCi dose, the MS of the mice receiving the 227Th-HuM195 was the same as that of the untreated group (Table 3 and Fig. 4C). The respective differences between 227Th-trastuzumab and 227Th-HuM195 at either the 0.5 or 1.0 μCi dose were not significant (p ≥ 0.64).
Table 3.
Dose Escalation of i.p. Injected 227Th-mAb: Median Survival of Athymic Mice Bearing i.p. LS-174T Tumor Xenografts
| Dose (μCi) | None | 227Th-Trastuzumab | 227Th-Panitumumab | 227Th-HUM195 | |||
|---|---|---|---|---|---|---|---|
| Median Survival (d) | Therapeutic Index | Median Survival (d) | Therapeutic Index | Median Survival (d) | Therapeutic Index | ||
| 0 | 14 | ||||||
| 0.25 | 17 | 1.2 | 21 | 1.5 | 14 | 1.0 | |
| 0.5 | 30 | 2.1 | 30 | 2.1 | 14 | 1.0 | |
| 1.0 | 30 | 2.1 | 24 | 1.7 | 11 | 0.8 | |
| 2.0 | 18 | 1.3 | 14 | 1.0 | 14 | 1.0 | |
| 5.0 | 14 | 1.0 | 11 | 0.8 | 14 | 1.0 | |
Therapuetic Index is the median survival of the treatment group divided by the median survival of the untreated group.
Fig. 4.

Effect of increasing 227Th-trastuzumab doses (μCi) on animal survival. Groups of mice (n =10) bearing i.p. LS-174T tumor xenografts were injected i.p. with 0.25, 0.5,1.0, 2.0 or 5.0 μCi of 227Th-trastuzumab (Panel A), 227Th-panitumumab or 227Th-HuM195 (Panel C). An additional group of mice was left untreated.
At these two doses, weight loss was greater in the mice treated with 1.0 μCi of 227Th-trastuzumab, 13.5% at 11 d, while the weight loss in the 0.5 μCi dose cohort was <10% (Table S5). Neither group recovered to their pre-therapy weight after 4 weeks of monitoring. In contrast, those mice that received the non-specific control, 227Th-HuM195, experienced a lower degree of weight loss. This may be the same phenomenon that was observed with the platelet counts of mice injected with 211At-HuIgG as explained above.
Panitumumab, targeting EGFR, has been radiolabeled with 212Pb and has demonstrated superior therapeutic efficacy compared to trastuzumab in the same intraperitoneal disseminated disease model.57 When evaluated in a 227Th-RIT setting, panitumumab as a delivery vector failed to provide any greater therapeutic benefit versus trastuzumab (Table 3 and Fig. 4B). No significant difference was found between the two mAbs at the 0.5 and 1.0 μCi doses (p ≥ 0.165). There was a difference at the 2.0 μCi dose with panitumumab resulting in a MS of 14 d vs. 18 d for trastuzumab (p = 0.002). The decrease in MS may be a function of the higher expression of EGFR on the LS-174T tumors.
Investigations from this laboratory have been systematic in identifying the effective working dose for each radionuclide that has been evaluated for targeted α-therapy. Holding as constants the tumor mouse model, the targeted molecule, and retaining the same delivery vehicle allows for very real comparisons to be made between the studies just described and the results that have been previously published for 213Bi and 212Pb-RIT. To compensate for the variance that can occur when working with in vivo model systems, and to facilitate comparisons of 211At, 227Th, 213Bi and 212Pb, the average median survivals along with the therapeutic indices of each of the RICs have been compiled in Table 4.
Table 4.
Comparison of Therapeutic Efficacy of Trastuzumab Radiolabeled with 212Pb, 213Bi, 211At and 227Th
| Radionuclide | Treatment | |||
|---|---|---|---|---|
|
| ||||
| Dose μCi | None | Trastuzumab | HuIgG | |
| 212Pb (n=7) | 10 | 18.6±3.4a | 48.0±11.5 | 29.1±6.5 |
| 213Bi n=7) | 500 | 20.2±5.4 | 46.9±13.7 | 34.0±15.5 |
| 211At (n=2) | 30 | 14.5±2.1 | 33.0±4.2 | 20.0±4.2 |
| 227Th (n=1) b | 0.5 | 14 | 30 | 14 |
| Ratios of Median Survival | ||||
|
|
||||
| Trastuzumab/None | HuIgG/None | Trastuzumab/HuIgG | ||
| 212Pb | 10 | 2.7±0.7 | 1.4±0.6 | 1.8±0.2 |
| 213Bi | 500 | 2.8±1.0 | 1.7±1.0 | 1.8±0.9 |
| 211At | 30 | 2.3±0.0 | 1.4±0.1 | 1.7±0.1 |
| 227Th | 0.5 | 2.1 | 1.0 | 2.1 |
Values are the median survival expressed in days.
The non-specific antibody used in the 227Th-RIT study was HuM195, an anti-CD33 mAb.
Statistical analysis (two-tailed, students t-test), comparing 212Pb-trastuzumab to 213Bi-trastuzumab, 212Pb-trastuzumab to 211At-trastuzumab and 213Bi-trastuzumab to 211At-trastuzumab reveals that there is no significant difference in the MS in any of these combinations (p > 0.05). The same is also true of these combinations for the radiolabeled negative control, HuIgG. The 227Th MS was not included in this analysis since only one RIT study was performed. This lack of a difference between the RICs can also be seen in the therapeutic indices (MS of RIC group/MS of untreated group). The TI of trastuzumab RICs range from 2.1–2.8 and the TI of the non-specific antibody ranges from1.0 – 1.7. Furthermore, the differences (ratios) between trastuzumab and the negative control not only demonstrates specificity of the RIT, but also remains constant.
In view of this last data analysis regarding all four radionuclides, the choice of radionuclide for α-RIT should perhaps not be entirely based on simple therapeutic efficacy and that other factors may play a very real role and ought to be taken into consideration in the making of such decisions. Variables such as half-life and tailoring that physical parameter to both accessibility of disease in combination with the kinetics of the targeting agents become important. Other variables that have very real impact are both local and general availability and production capability, as well as simple basic economics. A brief discussion of the impact of these variables within the context of the studied disease model system follows.
Bismuth-213, while readily available from 225Ac (10 d half-life) generators from at least two sources, and accompanied by established chemistry and a clinical trial track record, is (1) limited by its 47min half-life and (2) its cost per effective dose. The short half-life logistically limits clinical translation to the truly determined and innovative, an aspect that in part contributed to initial clinical trial translation.16 The disease presentation, AML, made accessibility by a full IgG a marginal issue, particularly with the target CD33 rapidly internalizing the RIC perhaps making this a convergence of positive factors. One might suggest that use of smaller targeting vectors would overcome half-life limits because of the expected improved target access, but then faster whole body clearance kinetics would require the injection of far higher activities to deliver equivalent therapeutic doses to tumor.58 This laboratory has reported on the treatment of a s.c. solid tumor with 213Bi targeted by a ΔCH2 engineered mAb which demonstrated that choosing an appropriate vector was a key variable to consider.58 A pre-targeting strategy may also prove suitable, but then very real economic considerations intrude again due to very efficient clearance kinetics of a small molecule.59 The data in the model studies here indicated that mouse doses of half a millicurie or higher are required for optimal survival; higher doses resulted in little improvement in efficacy and increased toxicity (weight loss), defining a point of diminishing returns. Thus, were one to attempt to scale such a single dose to human use, 213Bi becomes economically unfeasible and perhaps beyond the ability of most institutions to handle the amounts of the 225Ac required to generate a single dose.
For 211At, the half-life becomes considerably more reasonable in regards to biological targeting kinetics and handling logistics. Yet, the very limited number of cyclotron production sites, processing chemistry, transport and supply, and then melding in chemistry that remains not well understood complicates distribution. Notwithstanding these problems, clinical trials have been performed and/or are ongoing and have tended to also be restricted to cavitary contained disease presentation.19 These trials have also been tied tightly to active production sites of which there are a handful worldwide.7 A recent advance of an 211At generator system based on 211Rn may overcome some of these obstacles, but that system has yet to be fully validated, and scalability to clinical use is currently unknown.60 Evaluation of 211At in the LS-174T i.p. tumor model system was disappointing in long-term efficacy compared to prior 213Bi and 212Pb studies. And, here again, issues of general availability combined with scalability limits the general feasibility of this radionuclide.
With respect to 212Pb, this radionuclide is available from 224Ra (3. 6 d half-life) from at least two sources. The radionuclide is a 10.6 h β-emitter whose decay generates 212Bi and therefore acts as an in vivo or in situ generator. The chemistry is established, the generator system is well defined, and there has also been a clinical trial.20–22 The longer half-live is attractive as it begins to match up with the targeting and clearance kinetics of mAbs. The radionuclide is also perhaps limited to select disease presentations because of one of two complications associated with this radionuclide. The first complication is the loss of ~30% of the 212Bi formed in the decay process. This requires tailoring 212Pb to select applications, again cavitary contained disease. The second complication is a 2.6 meV γ-emission in the same abundance as the released 212Bi that requires appropriate shielding during radiopharmaceutical preparation to avoid excessive dose to personnel. In prior results of investigations from this laboratory, using the same model system, the injected activity of 212Pb to obtain an equivalent or even superior survival result was 20-fold less than that needed with 213Bi. On that basis, a significant advantage in economics was realized and by virtue of the generator production very large studies became feasible.
Lastly, 227Th has recently been pursued as another option for targeted α-therapy.39, 54, 55, 61, 62 Much of this pursuit is linked to 227Th being the parental radionuclide to 223Ra (Xofigo), recently approved for treating prostate cancer.63 While not being plagued by production, availability, chemistry, or actual dose economics issues, 227Th does however have two rather significant drawbacks. The first is that the half-life (18.7 d) is simply far too long for most biological targeting vectors without metabolic processing of the conjugate becoming a problem leading to toxicity. The second is that 227Th has a rather long decay chain with the first daughter being 223Ra, which with its long half-life will certainly traffic and be deposited in the bone, and may or not be another source of toxicity. This latter drawback also diminishes any thoughts that the α-decay cascade from the daughters of 227Th will contribute to therapy while the dose rate limits actual therapy from the parental radionuclide.
Conclusions
The development of α-emitting radionuclides for potential therapeutic applications currently has in place nearly all the requisite chemistry to permit their evaluation as targeted therapies in a host of possible disease models. One critical principle that remains applicable is the matching of radiological and biological half-lives to properly optimize such therapy with appropriate delivery vectors. Several of these radionuclides have been translated forward through to successful clinical trials all based in good decisions tailoring these variables appropriately either in cavitary contained or highly rapid targeting and internalization diseases.
The conclusion derived from the pre-clinical studies here along with prior studies that permit the direct comparison of 213Bi, 211At, 212Pb, and 227Th demonstrate that the matching principle noted above remains an important consideration. The analysis of the therapy results and comparisons of the therapeutic indices supports a contention that choosing an appropriate α-emitter for targeted therapy development may not reside in efficacy or even availability, but rather in very real variables as simple and overriding as economics of injected dose. Therefore, one can see that none of these radionuclides are perfect or without some compromise that requires very careful tailoring of its use to specific disease treatment scenarios. Some variables such as half-life and toxicity simply disqualify these radionuclides from use and others may be traversed with good decision making such as tailoring to specific disease scenarios, but there is no single best nor any one radionuclide that fits all situations.
Supplementary Material
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
References
- 1.Seidl C. Immunotherapy. 2014;6:431–458. doi: 10.2217/imt.14.16. [DOI] [PubMed] [Google Scholar]
- 2.Humm JL. J Nucl Med. 1986;27:1490–1497. [PubMed] [Google Scholar]
- 3.Larson SM. J Natl Cancer Inst. 1991;83:1602–1604. doi: 10.1093/jnci/83.22.1602. [DOI] [PubMed] [Google Scholar]
- 4.Azzam EI, de Toledo SM, Little JB. Curr Cancer Drug Targets. 2004;4:53–64. doi: 10.2174/1568009043481641. [DOI] [PubMed] [Google Scholar]
- 5.O’Donoghue JA, Bardies M, Wheldon TE. J Nucl Med. 1995;36:1902–1909. [PubMed] [Google Scholar]
- 6.Prise KM, Folkard M, Michael BD. Oncogene. 2003;22:7043–7049. doi: 10.1038/sj.onc.1206991. [DOI] [PubMed] [Google Scholar]
- 7.Kim YS, Brechbiel MW. Tumour Biol. 2012;33:573–590. doi: 10.1007/s13277-011-0286-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baidoo KE, Milenic DE, Brechbiel MW. Nucl Med Biol. 2013;40:592–599. doi: 10.1016/j.nucmedbio.2013.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Humm JL, Cobb LM. J Nucl Med. 1990;31:75–83. [PubMed] [Google Scholar]
- 10.Baidoo KE, Yong K, Brechbiel MW. Clin Cancer Res. 2013;19:530–537. doi: 10.1158/1078-0432.CCR-12-0298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seidl C, Port M, Apostolidis C, Bruchertseifer F, Schwaiger M, Senekowitsch-Schmidtke R, Abend M. Invest New Drugs. 2010;28:49–60. doi: 10.1007/s10637-008-9214-4. [DOI] [PubMed] [Google Scholar]
- 12.Seidl C, Schrock H, Seidenschwang S, Beck R, Schmid E, Abend M, Becker KF, Apostolidis C, Nikula TK, Kremmer E, Schwaiger M, Senekowitsch-Schmidtke R. Eur J Nucl Med Mol Imaging. 2005;32:274–285. doi: 10.1007/s00259-004-1653-3. [DOI] [PubMed] [Google Scholar]
- 13.Yong KJ, Milenic DE, Baidoo KE, Brechbiel MW. Mol Cancer Ther. 2012;11:639–648. doi: 10.1158/1535-7163.MCT-11-0671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yong KJ, Milenic DE, Baidoo KE, Kim YS, Brechbiel MW. Cancer Med. 2013;2:646–653. doi: 10.1002/cam4.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Andersson H, Cederkrantz E, Back T, Divgi C, Elgqvist J, Himmelman J, Horvath G, Jacobsson L, Jensen H, Lindegren S, Palm S, Hultborn R. J Nucl Med. 2009;50:1153–1160. doi: 10.2967/jnumed.109.062604. [DOI] [PubMed] [Google Scholar]
- 16.Jurcic JG, Larson SM, Sgouros G, McDevitt MR, Finn RD, Divgi CR, Ballangrud AM, Hamacher KA, Ma D, Humm JL, Brechbiel MW, Molinet R, Scheinberg DA. Blood. 2002;100:1233–1239. [PubMed] [Google Scholar]
- 17.Kratochwil C, Bruchertseifer F, Rathke H, Bronzel M, Apostolidis C, Weichert W, Haberkorn U, Giesel FL, Morgenstern A. J Nucl Med. 2017 doi: 10.2967/jnumed.117.191395. [DOI] [PubMed] [Google Scholar]
- 18.Parker C, Finkelstein SE, Michalski JM, O’Sullivan JM, Bruland O, Vogelzang NJ, Coleman RE, Nilsson S, Sartor O, Li R, Seger MA, Bottomley D. Eur Urol. 2016;70:875–883. doi: 10.1016/j.eururo.2016.06.002. [DOI] [PubMed] [Google Scholar]
- 19.Zalutsky MR, Reardon DA, Akabani G, Coleman RE, Friedman AH, Friedman HS, McLendon RE, Wong TZ, Bigner DD. J Nucl Med. 2008;49:30–38. doi: 10.2967/jnumed.107.046938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meredith R, Torgue J, Shen S, Fisher DR, Banaga E, Bunch P, Morgan D, Fan J, Straughn JM., Jr J Nucl Med. 2014;55:1636–1642. doi: 10.2967/jnumed.114.143842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meredith RF, Torgue J, Azure MT, Shen S, Saddekni S, Banaga E, Carlise R, Bunch P, Yoder D, Alvarez R. Cancer biotherapy & radiopharmaceuticals. 2014;29:12–17. doi: 10.1089/cbr.2013.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meredith RF, Torgue JJ, Rozgaja TA, Banaga EP, Bunch PW, Alvarez RD, Straughn JM, Jr, Dobelbower MC, Lowy AM. Am J Clin Oncol. 2016 doi: 10.1097/COC.0000000000000353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kasten BB, Azure MT, Schoeb TR, Fisher DR, Zinn KR. Nucl Med Biol. 2016;43:391–396. doi: 10.1016/j.nucmedbio.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 24.Milenic DE, Wong KJ, Baidoo KE, Nayak TK, Regino CA, Garmestani K, Brechbiel MW. mAbs. 2010;2:550–564. doi: 10.4161/mabs.2.5.13054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tom BH, Rutzky LH, Jakstys MH. In Vitro. 1976;12:180–191. doi: 10.1007/BF02796440. [DOI] [PubMed] [Google Scholar]
- 26.Chappell LL, Dadachova E, Milenic DE, Garmestani K, Wu C, Brechbiel MW. Nuclear medicine and biology. 2000;27:93–100. doi: 10.1016/s0969-8051(99)00086-4. [DOI] [PubMed] [Google Scholar]
- 27.Milenic DE, Garmestani K, Brady ED, Albert PS, Ma D, Abdulla A, Brechbiel MW. Cancer biotherapy & radiopharmaceuticals. 2005;20:557–568. doi: 10.1089/cbr.2005.20.557. [DOI] [PubMed] [Google Scholar]
- 28.Ray GL, Baidoo KE, Wong KJ, Williams M, Garmestani K, Brechbiel MW, Milenic DE. British journal of pharmacology. 2009;157:1541–1548. doi: 10.1111/j.1476-5381.2009.00327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu C, Kobayashi H, Sun B, Yoo TM, Paik CH, Gansow OA, Carrasquillo JA, Pastan I, Brechbiel MW. Bioorganic & medicinal chemistry. 1997;5:1925–1934. doi: 10.1016/s0968-0896(97)00130-2. [DOI] [PubMed] [Google Scholar]
- 30.Milenic DE, Garmestani K, Chappell LL, Dadachova E, Yordanov A, Ma D, Schlom J, Brechbiel MW. Nucl Med Biol. 2002;29:431–442. doi: 10.1016/s0969-8051(02)00294-9. [DOI] [PubMed] [Google Scholar]
- 31.Milenic DE, Wong KJ, Baidoo KE, Ray GL, Garmestani K, Williams M, Brechbiel MW. Cancer biotherapy & radiopharmaceuticals. 2008;23:619–631. doi: 10.1089/cbr.2008.0493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu C, Gansow OA, Brechbiel MW. Nucl Med Biol. 1999;26:339–342. doi: 10.1016/s0969-8051(98)00112-7. [DOI] [PubMed] [Google Scholar]
- 33.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 34.Dadachova E, Chappell LL, Brechbiel MW. Nucl Med Biol. 1999;26:977–982. doi: 10.1016/s0969-8051(99)00054-2. [DOI] [PubMed] [Google Scholar]
- 35.Pippin CG, Parker TA, McMurry TJ, Brechbiel MW. Bioconjugate chemistry. 1992;3:342–345. doi: 10.1021/bc00016a014. [DOI] [PubMed] [Google Scholar]
- 36.Pozzi OR, Zalutsky MR. J Nucl Med. 2007;48:1190–1196. doi: 10.2967/jnumed.106.038505. [DOI] [PubMed] [Google Scholar]
- 37.Schwarz UP, Plascjak P, Beitzel MP, Gansow OA, Eckelman WC, Waldmann TA. Nucl Med Biol. 1998;25:89–93. doi: 10.1016/s0969-8051(97)00165-0. [DOI] [PubMed] [Google Scholar]
- 38.Talanov VS, Garmestani K, Regino CA, Milenic DE, Plascjak PS, Waldmann TA, Brechbiel MW. Nucl Med Biol. 2006;33:469–480. doi: 10.1016/j.nucmedbio.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 39.Abbas N, Heyerdahl H, Bruland OS, Borrebaek J, Nesland J, Dahle J. EJNMMI research. 2011;1:18. doi: 10.1186/2191-219X-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Milenic DE, Baidoo KE, Brechbiel MW. Pharmaceuticals (Basel) 2015;8:435–454. doi: 10.3390/ph8030435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wong KJ, Baidoo KE, Nayak TK, Garmestani K, Brechbiel MW, Milenic DE. EJNMMI research. 2011:1. doi: 10.1186/2191-219X-1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chappell LL, Ma D, Milenic DE, Garmestani K, Venditto V, Beitzel MP, Brechbiel MW. Nucl Med Biol. 2003;30:581–595. doi: 10.1016/s0969-8051(03)00033-7. [DOI] [PubMed] [Google Scholar]
- 43.Talanov VS, Yordanov AT, Garmestani K, Milenic DE, Arora HC, Plascjak PS, Eckelman WC, Waldmann TA, Brechbiel MW. Nucl Med Biol. 2004;31:1061–1071. doi: 10.1016/j.nucmedbio.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 44.Garg PK, Harrison CL, Zalutsky MR. Cancer Res. 1990;50:3514–3520. [PubMed] [Google Scholar]
- 45.Gustafsson AM, Back T, Elgqvist J, Jacobsson L, Hultborn R, Albertsson P, Morgenstern A, Bruchertseifer F, Jensen H, Lindegren S. Nucl Med Biol. 2012;39:15–22. doi: 10.1016/j.nucmedbio.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 46.Robinson MK, Shaller C, Garmestani K, Plascjak PS, Hodge KM, Yuan QA, Marks JD, Waldmann TA, Brechbiel MW, Adams GP. Clin Cancer Res. 2008;14:875–882. doi: 10.1158/1078-0432.CCR-07-1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lindegren S, Andrade LN, Back T, Machado CM, Horta BB, Buchpiguel C, Moro AM, Okamoto OK, Jacobsson L, Cederkrantz E, Washiyama K, Aneheim E, Palm S, Jensen H, Tuma MC, Chammas R, Hultborn R, Albertsson P. PLoS One. 2015;10:e0126298. doi: 10.1371/journal.pone.0126298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Persson MI, Gedda L, Jensen HJ, Lundqvist H, Malmstrom PU, Tolmachev V. Oncol Rep. 2006;15:673–680. doi: 10.3892/or.15.3.673. [DOI] [PubMed] [Google Scholar]
- 49.Zalutsky MR, Stabin MG, Larsen RH, Bigner DD. Nucl Med Biol. 1997;24:255–261. doi: 10.1016/s0969-8051(97)00060-7. [DOI] [PubMed] [Google Scholar]
- 50.Elgqvist J, Bernhardt P, Hultborn R, Jensen H, Karlsson B, Lindegren S, Warnhammar E, Jacobsson L. J Nucl Med. 2005;46:464–471. [PubMed] [Google Scholar]
- 51.Elgqvist J, Andersson H, Back T, Hultborn R, Jensen H, Karlsson B, Lindegren S, Palm S, Warnhammar E, Jacobsson L. J Nucl Med. 2005;46:1907–1915. [PubMed] [Google Scholar]
- 52.Elgqvist J, Andersson H, Haglund E, Jensen H, Kahu H, Lindegren S, Warnhammar E, Hultborn R. Cancer biotherapy & radiopharmaceuticals. 2009;24:509–513. doi: 10.1089/cbr.2009.0618. [DOI] [PubMed] [Google Scholar]
- 53.Boskovitz A, McLendon RE, Okamura T, Sampson JH, Bigner DD, Zalutsky MR. Nucl Med Biol. 2009;36:659–669. doi: 10.1016/j.nucmedbio.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Abbas N, Bruland OS, Brevik EM, Dahle J. Nucl Med Commun. 2012;33:838–847. doi: 10.1097/MNM.0b013e328354df7c. [DOI] [PubMed] [Google Scholar]
- 55.Heyerdahl H, Abbas N, Sponheim K, Mollatt C, Bruland O, Dahle J. Curr Radiopharm. 2013;6:106–116. doi: 10.2174/18744710113069990018. [DOI] [PubMed] [Google Scholar]
- 56.Larsen RH, Borrebaek J, Dahle J, Melhus KB, Krogh C, Valan MH, Bruland OS. Cancer biotherapy & radiopharmaceuticals. 2007;22:431–437. doi: 10.1089/cbr.2006.321. [DOI] [PubMed] [Google Scholar]
- 57.Milenic DE, Baidoo KE, Kim YS, Barkley R, Brechbiel MW. Transl Oncol. 2017;10:535–545. doi: 10.1016/j.tranon.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Milenic D, Garmestani K, Dadachova E, Chappell L, Albert P, Hill D, Schlom J, Brechbiel M. Cancer biotherapy & radiopharmaceuticals. 2004;19:135–147. doi: 10.1089/108497804323071904. [DOI] [PubMed] [Google Scholar]
- 59.Frost SH, Back T, Chouin N, Hultborn R, Jacobsson L, Elgqvist J, Jensen H, Albertsson P, Lindegren S. Cancer biotherapy & radiopharmaceuticals. 2013;28:108–114. doi: 10.1089/cbr.2012.1281. [DOI] [PubMed] [Google Scholar]
- 60.Crawford JR, Yang H, Kunz P, Wilbur DS, Schaffer P, Ruth TJ. Nucl Med Biol. 2017;48:31–35. doi: 10.1016/j.nucmedbio.2017.01.011. [DOI] [PubMed] [Google Scholar]
- 61.Dahle J, Borrebaek J, Melhus KB, Bruland OS, Salberg G, Olsen DR, Larsen RH. Nucl Med Biol. 2006;33:271–279. doi: 10.1016/j.nucmedbio.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 62.Heyerdahl H, Abbas N, Brevik EM, Mollatt C, Dahle J. PLoS One. 2012;7:e42345. doi: 10.1371/journal.pone.0042345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wilson JM, Parker C. Expert Rev Anticancer Ther. 2016;16:911–918. doi: 10.1080/14737140.2016.1222273. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.