

Abstract
Ctenophora, compromising approximately 200 described species, is an important lineage for understanding metazoan evolution and is of great ecological and economic importance. Ctenophore diversity includes species with unique colloblasts used for prey capture, smooth and striated muscles, benthic and pelagic lifestyles, and locomotion with ciliated paddles or muscular propulsion. However, ancestral states of traits are debated and relationships among many lineages are unresolved. Here, using 27 newly sequenced ctenophore transcriptomes, publicly available data, and methods to control systematic error we establish the placement of Ctenophora as the sister group to all other animals and refine phylogenetic relationships within ctenophores. Molecular clock analyses suggest modern ctenophore diversity originated approximately 350MYA ± 88 MY, conflicting with previous hypotheses of approximately 65 MYA. We recover Euplokamis dunlapae, a species with striated muscles, as the sister lineage to other sampled ctenophores. Ancestral state reconstruction shows the most recent common ancestor of extant ctenophores was pelagic, possessed tentacles, was bioluminescent, and did not have separate sexes. Our results imply at least two transitions from a pelagic to a benthic lifestyle within Ctenophora, suggesting such transitions were more common in animal diversification than appreciated.
Ctenophores, or comb jellies, have successfully colonized nearly every marine environment and can be key species in marine food webs1–6. For example, invasive ctenophores have caused dramatic fisheries collapses by voraciously preying on native fish larvae and their food, resulting in the economic loss of millions of US dollars to impacted areas4. Understanding morphological and life history diversity of ctenophores in a comparative context is essential for our knowledge of ctenophore and metazoan diversification as a whole7. Ctenophores have received considerable attention in regard to debate about whether they are the sister group to all other animals3,5,8–11, but relationships within Ctenophora has been the focus of only limited research3,12,13.
Putative ctenophore fossils date back to the Ediacaran period14 with substantial morphological diversity being present in the Cambrian15,16. All ctenophores possess smooth muscles, and at least one genus, Euplokamis, has striated muscles17. Most ctenophores possess tentacles (Fig. 1), but species in the genus Ocyropsis lose tentacles as adults18 and beroids lack them throughout their life cycle (Fig. 1)1,6. Many species are pelagic, but some are benthic or semi-benthic as adults and can have a relatively flattened body and lose the ciliary comb rose that otherwise characterize the phylum6,19 (Fig. 1). Relationships among ctenophore lineages remain poorly resolved as past phylogenetic analyses have either had too few taxa to recover broad evolutionary patterns3 or resulted in weak support for the deepest nodes, likely resulting from the use of only one or two genes12,13. Past researchers12,13 have also hypothesized that Ctenophora has undergone a bottleneck in species diversity, possibly as recently as 65 MYA. However, the age of crown group ctenophores has yet to be estimated with molecular dating methods. Here, we sequenced 27 transcriptomes from species across most of the known phylogenetic diversity of Ctenophora. New sequence data were combined with 10 ctenophore and 50 non-ctenophore publicly available transcriptomes (Supplementary Tables S1, S2) to clarify the phylogenetic placement of Ctenophora11,20–22. Thus, we performed analyses to determine appropriate outgroups and ctenophore placement among other metazoans using more ctenophore taxa than previous studies3,5,9–11,20 (Supplementary Table S2).
Fig. 1.
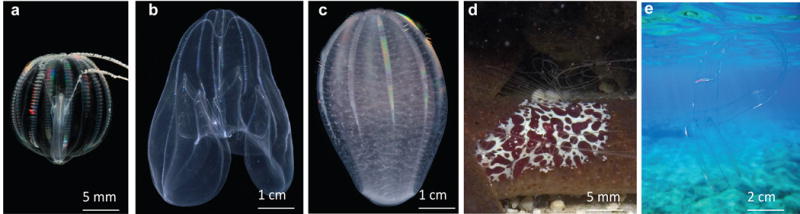
Exemplar morphological forms of Ctenophora. a) Cydippid morphology (ovate body, long tentacles); photograph taken by James Townsend. b) Lobate morphology (reduced tentacles, large lobes). c) Beroida morphology (lacking tentacles and lobes). d) Platyctenida morphology (flattened, long tentacles). e) Cestida morphology (ribbon-like); photograph taken by Roberto Pillon and contrast adjusted in Adobe Photoshop.
Results
Ctenophora is the sister lineage to all other extant metazoans
Using a variety of data filtering schemes and different substitution models to control for systematic error (Supplementary Table 2), we recovered ctenophores as the sister group to all other extant metazoans (1.00 Bayesian posterior probability (PP), 100% bootstrap support (BS); Fig. 2, Supplementary Figs. S1–S14). The percentage of individual genes favoring the hypothesis of ctenophores sister to all other animals was higher in every dataset (56.8%–75.4%; Table 1) than the percentage of genes favoring the hypothesis of sponges sister to all other animals (32.7%–43.2%; Table 1). Datasets that were trimmed of genes most likely to cause long-branch attraction had the highest percentage of genes supporting Ctenophora-sister, indicating that the Ctenophora-sister hypothesis is not a result of long-branch attraction. Our recovered placement of ctenophores does not change when concerns of Pisani et al.20 about outgroup choice and use of site-heterogeneous models are taken into account (see additional considerations in22,23).
Fig. 2.
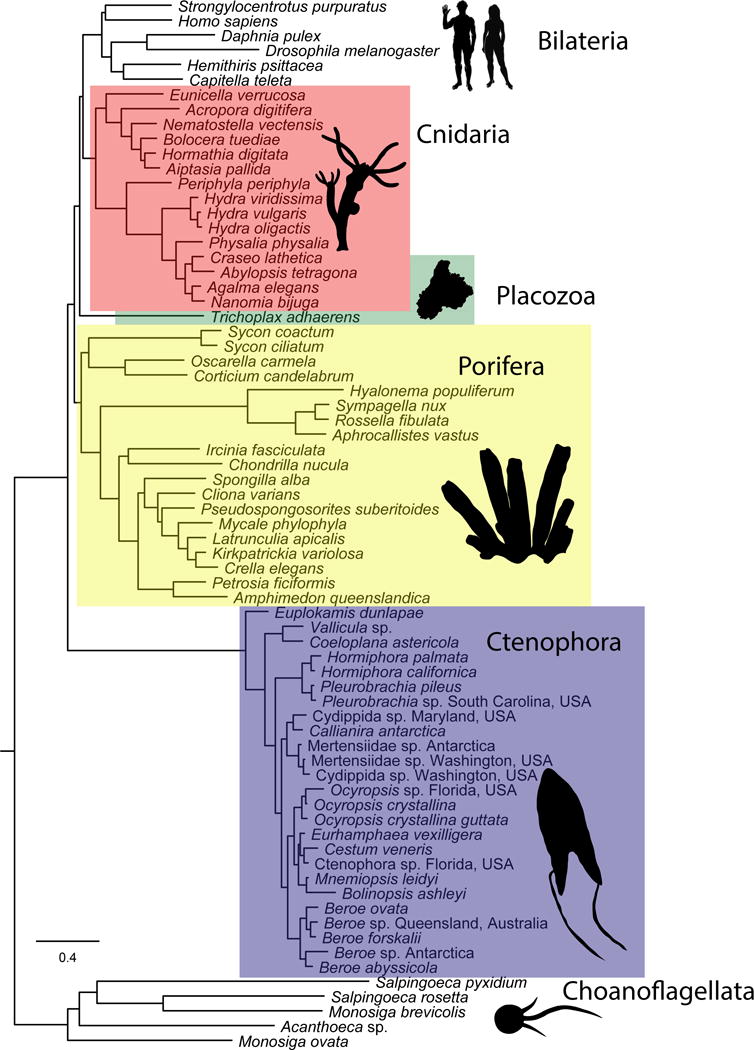
Relationships among metazoans inferred with the CAT-GTR substitution model and dataset Metazoa_Choano_RCFV_strict. All nodes have 100% PP. Inferred relationships among phyla are identical to those inferred with other models and datasets (Supplementary Figs. S1–S15; Supplementary Discussion). Scale bar in expected substitutions per site. Silhouette images downloaded from phylopic.org.
Table 1.
Number of genes and sites in each dataset supporting alternative hypotheses of the sister lineage to all other metazoans.
| Dataset1 | Genes supporting Ctenophora-sister |
Genes supporting Porifera-sister |
Sites supporting Ctenophora-sister |
Sites supporting Porifera-sister |
|---|---|---|---|---|
| Metazoa_full | 144 (64.3%) | 80 (35.7%) | 38,378 (56.4%) | 29,684 (43.6%) |
| Metazoa_RCFV_relaxed | 133 (64.8%) | 72 (35.2%) | 36,255 (55.5%) | 29,072 (44.5%) |
| Metazoa_RCFV_strict | 70 (60.3%) | 46 (39.7%) | 22,897 (52.9%) | 20,415 (47.1%) |
| Metazoa_LB_relaxed | 112 (68.3%) | 52 (31.7%) | 28,642 (55.9%) | 22,554 (44.1%) |
| Metazoa_LB_strict | 105 (69.5%) | 46 (30.5%) | 25,875 (55.1%) | 21,071 (44.9%) |
| Metazoa_RCFV_LB_relaxed | 97 (65.1%) | 52 (34.9%) | 26,647 (54.3%) | 22,389 (45.7%) |
| Metazoa_RCFV_LB_strict | 53 (71.6%) | 21 (28.4%9 | 15,194 (52.8%) | 13,558 (47.2%) |
| Metazoa_Choano | 144 (61.5%) | 90 (38.5%) | 41,971 (55.3%) | 33,850 (44.7%) |
| Metazoa_Choano_RCFV_relaxed | 111 (68.9%) | 50 (31.3%) | 32,434 (54.3%) | 27,247 (45.7%) |
| Metazoa_Choano_RCFV_strict | 87 (68.5%) | 40 (31.5%) | 27,257 (55.2%) | 22,131 (44.8%) |
| Metazoa_Choano_LB_relaxed | 104 (56.8%) | 79 (43.2%) | 33,268 (54.8%) | 27,417 (45.2%) |
| Metazoa_Choano_LB_strict | 156 (75.4%) | 51 (32.7%) | 29,875 (59.2%) | 20,595 (40.8%) |
| Metazoa_Choano_RCFV_LB_relaxed | 83 (63.8%) | 47 (36.2%) | 26,586 (54.2%) | 22,493 (45.8%) |
| Metazoa_Choano_RCFV_LB_strict | 56 (68.3%) | 26 (31.7%) | 17,873 (57.3%) | 13,334 (42.7%) |
See Supplementary Table S3 for more information on datasets.
A recent study by Simion et al.21 recovered sponges as the sister lineage to all other animals, but methodological problems in their analyses explain disagreement with our results. The placement of sponges as the sister lineage to all other animals was only recovered using the CAT-F81 substitution model (often referred to as “CAT”), which has been shown to sometimes result in less accurate phylogenetic hypotheses than models used here24. More problematically, not a single Bayesian analysis conducted by Simion et al.21 converged (Simion et al. pers. communication), rendering them statistically invalid. Use of other site-heterogeneous models that may not suffer from problems associated with CAT-F81 (see24 and Supplementary Discussion) resulted in Ctenophora sister to all other animals21, consistent with our findings (Fig. 2, Supplementary Fig. S1–S14) and those of two recent papers that employed novel methods25,26.
Wide consensus exists that Ctenophora is a hard lineage to place on the animal tree of life8,11,20,21, and increased taxon sampling is broadly accepted to aid in placement of difficult lineages27–29. Our datasets have greater ctenophore taxon sampling than past studies, including 27 novel ctenophore transcriptomes, and are arguably the most appropriate datasets, generated to date, for assessing the placement of Ctenophora. Using datasets with reasonably high ctenophore and other non-bilaterian taxon sampling, our results strongly reject the hypothesis that sponges are the sister lineage to all other extant metazoans.
Bayesian inference with a relaxed molecular clock also recovered ctenophores as the sister group to all other animals with maximum support (1.00 PP; Supplementary Figure S15, Supplementary Discussion). These analyses indicated that sampled ctenophores shared a common ancestor much more recently than either crown group sponges, cnidarians, or bilaterians (Supplementary Figure S15; Supplementary Discussion). Thus, our findings are consistent with the hypothesis that Ctenophora has undergone a species-diversity bottleneck, but we acknowledge uncertainty in our absolute diversification timing (350 MYA ± 88 MY, Supplementary Discussion). Nevertheless, this bottleneck appears to have occurred between 456-261 MYA (Supplementary Figure S15), much older than the 65 MYA previously hypothesized12,13. Given our results, ancestral ctenophores likely experienced a drastic decline before, or during, the Permian-Triassic (P-Tr) extinction (~250 MYA30). Early to mid Paleozoic Ctenophore fossils display substantially greater morphological diversity (e.g., more than eight comb rows) than seen today14–16, supporting the hypothesis that the phylum underwent a major diversity decline during the Paleozoic.
Evolution of Ctenophora
Relationships among ctenophores were assessed using a novel set of ctenophore-centric core orthologs. Orthology determination, subject to paralog and contamination screening, resulted in a primary dataset of 350 genes and 98,844 amino acid positions (Supplementary Table S3). Potential causes of systematic error were controlled for by creating additional datasets that removed potentially problematic genes (Supplementary Table S3)11. Phylogenetic analyses were conducted with data partitioning under maximum likelihood and with the CAT-GTR31 site-heterogeneous substitution model under Bayesian inference. All phylogenetic analyses focusing on intra-ctenophore relationships resulted in identical, highly-supported relationships (Fig. 3, Supplementary Figs. S16–S19).
Fig. 3.

Evolutionary relationships among Ctenophora and ancestral character state reconstruction of general body plan. Traditional orders labeled with colors matching corresponding body plan morphotype. Nodes are labelled with pie charts depicting posterior probability of character states. Phylogeny was inferred with dataset Ctenophore_RCFV_LB. Lines connect photographs of exemplars with species identity. Sponge and cnidarian outgroups that were used to root the tree were removed for illustrative purposes. Nodes have 100% BS or 1.00 PP support unless otherwise noted (BS/PP).
We found pervasive non-monophyly among currently recognized ctenophore higher taxonomic groups, including Tentaculata, Cydippida, and Lobata (Fig. 3, Supplementary Figs. S16–S19), corroborating previous analyses3,12,13. Other traditional groups based on morphology, like the benthic Platyctenida and atentaculate Beroida, were recovered monophyletic (Figs. 3, Supplementary Figs. S16–S19) congruent with past analyses3,12,13. Lobata was paraphyletic by inclusion of Cestida, represented by the ribbon-like Cestum veneris (Fig. 3, Supplementary Fig. S20). Ocyropsis species, which lose tentacles as adults, move by muscle propulsion, and are dioecious (Supplementary Figs. S21, S22), were monophyletic and sister to a clade with Cestida and all other lobates except the benthic Lobatolampea tetragona. These results indicate that the cydippid and lobate body plans are plesiomorphic (Fig. 3, Supplementary Fig. S20).
We recovered Euplokamis dunlapae as the sister lineage to all other sampled ctenophores with maximum support (Fig. 3, Supplementary Figs. S16–S19) consistent with initial genomic analyses3. Previous studies also recovered Mertensia ovum and Charistephane fugiens with Euplokamis dulapae as a united sister group to all other ctenophores13. Novel analyses based on 18S rRNA, which included many more taxa than our transcriptome-based analyses, recovered Mertensiidae as non-monophyletic and a clade including Mertensia ovum, Charistephane fugiens, and Euplokamis spp. sister to all other extant ctenophores (see Supplementary Discussion; Supplementary Fig. S23). As were unable to sample M. ovum and C. fugiens, we cannot reject that any three of these species, a clade of all three, or a yet to be discovered species could be the sister lineage to all other extant ctenophores. Euplokamis dunlapae is the only ctenophore species known to have striated muscles2, and Bayesian ancestral state reconstruction suggests that striated muscles likely evolved after the split between E. dunlapae and other ctenophores (PP = 0.90; Supplementary Fig. S24), rather than being present in the most recent common ancestor (MRCA) of extant ctenophores. Striated muscles have evolved at least three times: after the split of the Euplokamis dunlapae lineage from other ctenophores, in select Cnidaria32, and in bilaterians32 (Supplementary Fig. S25). Given that all extant ctenophores have smooth muscles, the MRCA of all extant ctenophores almost certainly possessed smooth muscles (PP = 1.0; Supplementary Fig. S24). Given our inferred relationships among ctenophores, sponges, placozoans, and cnidarians (Fig. 2), the MRCA to extant metazoans either possessed smooth muscles that were subsequently lost at least twice (in Porifera and in Placozoa), or, more parsimoniously, muscles evolved independently at least twice (in Ctenophora and the lineage leading to Cnidaria + Bilateria)3.
The MRCA of extant ctenophores was most likely pelagic (PP = 0.91; Fig. 4, Supplementary Fig. S24), with cydippid-like morphology (i.e., ovate body and branched tentacles; PP = 0.92; Fig. 3, Supplementary Fig. S20; Supplementary Discussion), and a simultaneous hermaphrodite (PP = 0.99; Supplementary Fig. S22). Ancestral state reconstruction suggests plesiomorphy of the cydippid body plan with most other morphotypes evolving from it (Fig. 3, Supplementary Fig. S20). The one exception appears to be the ribbon-like Cestida, which evolved from a lobate-like ancestor. Aside from beroids, which are atentaculate at all life stages, all ctenophores for which larval information is available have a free-swimming larval stage with cydippid-like morphology1,33. However, Platyctenids, and to a lesser extent lobates and cestids, undergo considerable morphological and functional changes during development33. Nevertheless, juvenile morphology among all ctenophores, except the derived beroids, resembles the inferred ancestral state of extant ctenophores (Fig. 3, Supplementary Fig. S20).
Fig. 4.

Evolutionary relationships of Ctenophora and ancestral character state reconstruction of benthic vs. pelagic lifestyle. Nodes (and unique taxa) are labelled with pie charts depicting posterior probability of character states. Traditional orders are labeled. a) Phylogeny was inferred with dataset Ctenophore_RCFV_LB. Sponge and cnidarian outgroups that were used to root the tree were removed for illustrative purposes. Nodes have 100% BS or 1.00 PP support unless otherwise noted (BS/PP). b) Benthic Platyctenida, Ceoloplana astericola on a seastar. c) Pelagic Pleurobrachia bachei. d) Benthic Lobata, Lobatolampea tetragona.
Ancestral state reconstruction indicates that ctenophores have transitioned from a pelagic to a benthic, or semi-benthic, adult lifestyle at least twice (Fig. 4, Supplementary Fig. S25). These two transitions occurred on the branches leading to Platyctenida and to Lobatolampea, but we cannot rule out additional transitions in undescribed benthic lineages. Interestingly, Lobatolampea was recovered as the sister lineage to a clade with all other lobates and Cestida, while Platyctenida was recovered as sister to all other ctenophores but Euplokamis. Thus, the two benthic lineages evolved separately. Transition between benthic and pelagic lifestyles has been studied in numerous invertebrate groups34, with most documented transitions occurring from a benthic to a pelagic existence. However, we found no evidence that any ancestrally benthic ctenophore lineage has evolved to occupy the water column (Fig. 4, Supplementary Fig. S25).
Pleurobrachiidae is one of the most common and well-studied groups of ctenophores and is often used as a reference for the phylum3,35. However, Pleurobrachiidae lacks bioluminescence35, and past uncertainty about the phylogenetic position of the family limited the ability to fully analyze evolution of bioluminescence in ctenophores12,13. We confidently recovered Pleurobrachiidae (i.e., Pleurobrachia and Hormiphora), plus Pukiidae, as a monophyletic lineage on a relatively long branch (Figs. 3, Supplementary Figs. S16–S19). Like Pleurobrachiidae, Pukiidae is incapable of bioluminescence. Ancestral state reconstruction suggests that the MRCA to extant ctenophores was bioluminescent (PP = 0.96; Supplementary Fig. S25; Supplementary Discussion), and this trait has likely been lost only once within Ctenophora. Bioluminescence is generally considered advantageous in deep water36, but most pleurobrachiids are found near-shore at shallow depths1,37,38, which may have relaxed selective pressures for maintaining bioluminescence.
The MRCA of extant ctenophores likely fed by capturing plankton with branched tentacles equipped with colloblasts, a unique synapomorphy of ctenophores. However, multiple transitions in adult feeding mode have occurred (Fig. 5, Supplementary Fig. S20; Supplementary Discussion). These transitions are associated with lineage-specific behavioral and morphological innovations38. For instance, the simplification of tentacles seen in Dryodora followed by the complete loss of tentacles in Beroe (Fig. 5, Supplementary Figs. S20, S21) is associated with engulfing larger prey items, rather than using tentacles and/or lobes for food capture as in other lineages (Fig. 5, Supplementary Fig. S20); although Dryodora has tentacles, they are likely used for sensing rather than capturing prey (Supplementary Discussion). The sister relationship between Dryodora and Beroe suggests a gradual transition from branched to reduced tentacles, followed by complete loss of tentacles. More broadly, ancestral state reconstruction of feeding behaviors produced three nodes where no character state had posterior probabilities of 90% or greater (Fig. 5, Supplementary Figs. S20). Ambiguity at these nodes is associated with a clear shift away from using primarily, or only, tentacles for prey capture as adults and dramatic morphological transitions.
Fig. 5.

Evolutionary relationships of Ctenophora and ancestral sate reconstruction of primary feeding mode. Traditional orders are labeled. Nodes are labelled with posterior probability of character states. Phylogeny was inferred with dataset Ctenophore_RCFV_LB. Sponge and cnidarian outgroups that were used to root the tree were removed for illustrative purposes. Nodes have 100% BS or 1.00 PP support unless otherwise noted (BS/PP).
Discussion
Using greater ctenophore taxon sampling than previous studies, data filtering schemes to remove potential causes of systematic error, and a variety of substitution models, we recovered Ctenophora as the sister lineage to all other animals. The debate surrounding the phylogenetic placement of Ctenophora has complicated studies on evolution of complex characters such as muscles and neurons. Genomic components of these features suggest extensive convergent and parallel evolution across Metazoa3, which is further supported by our phylogenetic results. However, events of independent origins of neural and muscular systems are not directly coupled with competing hypotheses of metazoan phylogeny3,39,40. Nevertheless, the placement of Ctenophora as the sister lineage to all other animals appears robust to error.
Our results suggest that Ctenophora has undergone a species-diversity bottleneck considerably farther in the past than previously hypothesized (Supplementary Fig. S15). Subsequent diversification resulted in numerous morphotypes evolving from a cydippid-like ancestor (Fig. 3). A benthic lifestyle has evolved convergently in at least two ctenophore lineages (Fig. 4), but evolution of striated muscles, loss of bioluminescence, and loss of tentacles throughout all life cycles appears to have only occurred once (Supplementary Figs. S20–S24). Ctenophora is in need of thorough taxonomic revision, and we expect progress to be made on that front in the coming years. Ctenophora is one of the most morphologically diverse and understudied metazoan groups, and our results provide a phylogenetic foundation for future studies on developmental, neuro-muscular, and tissue/organ evolution both within Ctenophora and among all metazoans.
Methods
Taxon sampling and sequencing
We sampled ctenophores from locations around the world (Table S1), mostly between 2013 and 2016. Ctenophore specimens were identified to as low of a taxonomic level as possible (Table S1). Many newly sequenced species, particularly those sampled from Antarctica, are undescribed species. Complementary DNA (cDNA) libraries for newly collected ctenophores were constructed using a template-switch method using the SMART™ cDNA library construction (Cat# 639537, Clontech). Full-length cDNA was amplified using the Advantage 2 PCR system (Cat# 639201, Clontech) and the minimum number of PCR cycles necessary for single-end sequencing for Ion Proton or 2 × 100 bp paired-end sequencing with Illumina. Illumina and Ion Proton sequencing libraries were subsequently prepared using NEBNext® Ultra™ DNA Library Prep Kit for Illumina® (Cat# E7645S, New England Biolabs Inc.) or NEBNext® Fast DNA Library Prep Set for Ion Torrent™ (Cat# E6270S, New England Biolabs Inc.). Each library was sequenced using either an Illumina NextSeq 500 or Ion Proton (see Table S1).
Publicly available ctenophore and non-ctenophore transcriptomes or gene models were retrieved from NCBI and other databases (Supplementary Tables S1, S2). Bolinopsis infundibulum from Moroz et al.3 was determined to be misidentified based on our sequencing of a novel B. infundibulum transcriptome. Thus, we now use name “Cydippida sp. Washington, USA” for the transcriptome labeled “Bolinopsis infundibulum” in Moroz et al.3.
We performed phylogenetic analyses at two scales to achieve different goals. First we inferred relationships among non-bilaterian metazoan phyla (with other opishtokonts as outgroups) to determine the sister lineage to Ctenophora. Second, we analyzed relationships and trait evolution within Ctenophora using appropriate outgroups as identified with the broader Metazoa analyses. Depending on the focal taxonomic scale, different taxon sampling schemes were used (see Supplementary Tables S1, S2). Datasets designed to examine relationships between metazoan phyla are named with the prefix “Metazoa_” followed by more specific information about the dataset as appropriate. For example, datasets with only choanoflagellates as outgroups are named “Metazoa_choano_”. Datasets designed to test relationships among ctenophores are named in a similar fashion except they have the prefix “Cteno_”. See Supplementary Table S3, and below, for additional information about dataset naming conventions.
When testing relationships among metazoan phyla, taxon sampling was similar to that of Whelan et al.11 with three exceptions. First, fewer bilaterians were included to decrease computational time. Second, a larger number of choanoflagellates were sampled, which we expected to result in more robust rooting of Metazoa than in past analyses3,5,8–11,24,41–43. Finally, more ctenophores were sampled than in previous studies3,5,8–11,24,41–43, which will likely increase the accuracy of ctenophore placement28,29,44.
For analyses that focused on relationships among metazoan phyla, we generated datasets that only had choanoflagellate outgroups and datasets that had Ichthyosporea, Filasterea, and choanoflagellate outgroups (Supplementary Tables S2, S3). These datasets had fewer ctenophores included than in datasets generated to test relationships within Ctenophora because we did not include individuals that were repetitive at or near the species level (e.g., only one individual identified as Pleurobrachia bachei was included in the broader analyses; Supplementary Tables S1, S2). This was done in order to decrease required computational time. Pukia falcata was also not included in the broad metazoan analyses, despite its inclusion in ctenophore-centric phylogenetic inference, because preliminary phylogenetic inference (not shown) revealed that its inclusion caused unstable relationships among metazoan phyla. Presumably, this was due to the comparably high amount of missing data in P. falcata. “Mertensiidae sp. (Antarctica)” was inadvertently not included in ctenophore specific dataset generation. However, inclusion of this species would likely not have affected overall conclusions about ctenophore evolution given its inferred placement from analyses with the metazoan datasets (Fig. 2, Supplementary Figures S1–S14).
Informatics and data matrix assembly
Prior to assembly, raw transcriptome reads were digitally normalized to a target of 30× coverage using normalize-by-median.py45 and assembled with Trinity 2014071746 using default parameters. After assembly, open reading frames and putative protein sequences were identified with TransDecoder46 using default parameters. We used HaMStR 13.247 and two core ortholog sets to recover orthologous groups (OGs) for phylogenomic analyses (Supplementary Table S3). The model organism core ortholog set packaged with HaMStR 13.2 was used for testing relationships among metazoan phyla because it was designed to be of broad taxonomic utility. For reconstructing ctenophore phylogeny, we designed a ctenophore-centric core ortholog set to increase the number of OGs in our datasets (Supplementary Table S3).
The ctenophore-centric core ortholog set was created by first performing an all-versus-all blastp search48 among transcriptomes of Beroe abyssicola, Coeloplana astericola, Euplokamis dunlapae, Mnemiopsis leidyi, Ocyropsis sp. from Florida, USA, and Pleurobrachia bachei. These species were chosen because they were hypothesized to represent a wide swath of ctenophore phylogeny and had relatively deeply sequenced transcriptomes. An e-value cutoff of 105 was used for blastp searches. Blastp results were used to perform Markov clustering with OrthoMCL 2.049 with an inflation parameter of 2.1 following Hejnol et al.10 and Kocot et al.50. Markov clustering resulted in 55,433 putative OGs. These OGs were further filtered to remove possible paralogs and low-quality OGs. First, any sequence that had less than 100 amino acids in length was removed. Each OG was then aligned with MAFFT51 using an automatically chosen alignment strategy and a “maxiterate” value of 1,000. After alignment, an approximately maximum likelihood tree was generated for each OG with FastTree 252 using “slow” and “gamma” options. Each tree and corresponding OG was processed with PhyloTreePruner53 to screen for paralogs; a bootstrap value of 90 was used for collapsing nodes. If more than one sequence for any of the six respective species was present after the paralog pruning step, then the longest sequence for that species was retained and others were discarded. Lastly, we removed OGs that had sequences for fewer than 4 species and any OG that did not have a Mnemiopsis leidyi sequence because it was chosen as the HaMStR primer taxon. The 2,354 remaining OG alignments were used to build protein hidden Markov models using HMMER tools hmmbuild and hmmcalibrate54. Our ctenophore core ortholog set has been deposited on figshare (doi:xxxx.xxx).
Transcriptomes and gene models were processed with HaMStR using one or both core ortholog sets (i.e., model organism or ctenophore) depending on which analyses each taxon was included in (Tables S1, S2). Post-HaMStR orthology filtering followed Whelan et al.11 with slight script modifications to increase speed and accuracy. For datasets generated to infer relationships among Bilateria and non-Bilateria phyla, OGs were discarded if they had less than 42 taxa present for datasets generated with all outgroups and less than 38 taxa present for datasets generated with only choanoflagellate outgroups (i.e., datasets Metazoa_full and Metazoa_Choano, respectively; Table S2). For datasets designed for testing relationships among ctenophores, OGs were discarded if they had less than 27 taxa present.
After orthology filtering of each dataset, single gene trees were generated with RAxML 8.2.455 using a gamma distribution to model rate heterogeneity and amino acid substitution models identified by model testing implemented in RAxML. We performed 100 fast bootstrap replicates for each gene tree to assess nodal support. Resulting gene trees were used with TreSpEx56 for more thorough screening of paralogs and contamination that may have passed through initial orthology determination. Briefly, we used the BLAST associated method in TreSpEx with the packaged Capitella teleta and Helobdella robusta blast databases following Struck56 and Whelan et al.11. All sequences identified as certain or uncertain paralogs by TreSpEx —such sequences may also be non-target sequence contamination—were removed from OGs. Subsequently, OGs that then had less than 42 taxa for dataset Metazoa_Full, 38 taxa for dataset Metazoa_Choano, and 27 taxa for dataset Ctenophore_full after paralog pruning with TreSpEx were also discarded to minimize missing data. For clarity, datasets Metazoa_full, Metazoa_Choano, and Ctenophore_full are herein referred to as “initial” datasets that were then filtered for OGs that had the highest potential for causing systematic error.
Systematic Error
To assess the effect of systematic error on phylogenetic inference we generated datasets with potential sources of systematic error removed from the initial datasets. Specifically, genes with the highest potential for causing long-branch attraction (LBA) or that had the highest levels of base compositional heterogeneity were removed. By creating nested datasets with different potential causes of systematic error removed, we were able to assess if inferred relationships were influenced by systematic error. Branch length heterogeneity scores (LB), which can be used to rank genes based on their possible contribution to LBA, were calculated using TreSpEx. This was done with individual trees for each OG in the three initial datasets; new trees for each paralog pruned OG were inferred with RAxML as described above. Density plots of LB score heterogeneity and upper quartile LB score for each OG and dataset were plotted using R57 (Supplementary Fig. S26). The two datasets designed to test relationships among metazoan phyla (i.e., Metazoan_full, Metazoa_Chaono) had fewer genes than the ctenophore-centric dataset. Thus, to strike a balance between removing OGs that may cause systematic error and not having enough phylogenetic signal (i.e., OGs) to accurately resolve relationships we identified a strict and a relaxed cutoff for removing genes with outlier LB scores (Supplementary Fig. S26). For the ctenophore-centric dataset, we only identified one set of genes as outliers (Supplementary Fig. S26). Using the initial datasets, nested datasets were generated by removing genes that were identified as having outlier LB scores (Supplementary Table S3). Relative Composition Frequency Variability (RCFV)58, which is a measure for how much base compositional heterogeneity is present in an OG, was calculated for each gene using BaCoCa59. A density plot of RCFV for each initial dataset was plotted in R (Supplementary Fig. S26). As with LB scores, for datasets Metazoa_full and Metazoa_Choano two sets of outliers were identified and removed to create datasets with all outlier RCFV genes removed (i.e., strict) and some outlier RCFV genes removed (i.e., relaxed; Supplementary Fig. S65, Table S3). Only a single set of RCFV outlier genes were identified for dataset Cteno_full (Supplementary Fig. S26, Table S3). We also created datasets that had both LB and RCFV outlier genes removed from the initial three datasets (Supplementary Fig. S26, Table S3). For the ctenophore-centric datasets, we created corresponding datasets with outgroups removed to test whether or not relationships among ctenophores were affected by relatively distantly related outgroups.
Phylogenetic reconstructions
Bayesian inference with the site-heterogeneous CAT-GTR substitution model was done with PhyloBayes MPI60. Analyses with CAT-GTR are notoriously time consuming24 so a number of steps were taken to facilitate convergence of independent Bayesian runs. First, only two datasets were analyzed with CAT-GTR: dataset Metazoa_Choano_RCFV_strict for testing relationships among metazoan phyla and dataset Cteno_RCFV_LB for determining relationships among ctenophore lineages. We removed three ctenophore taxa from dataset Metazoa_Choano_RCFV_strict to facilitate convergence; these three ctenophores were unstable in preliminary CAT-GTR analyses that failed to converge (see Supplementary Tables S1–S3). For CAT-GTR analyses on both datasets, two independent chains were sampled every generation. Trace plots of Markov chain Monte Carlo (MCMC) runs were visually inspected in Tracer 1.661 to assess stationarity and appropriate burn-in, which was determined to be 3,500 and 4,000 generations for datasets Metazoa_Choano_RCFV_strict and Cteno_RCFV_LB, respectively. Phylobayes runs were sampled for 18,436 generations on dataset Metazoa_Choano_RCFV_strict and for 23,947 generations on dataset Cteno_RCFV_LB. All parameters and tree shape reached convergence, which was considered to have occurred when the maxdiff value < 0.1 as measured by bpcomp60 and when rel_diff value < 0.3 and effective sample size > 50 as measured by tracecomp60. Although some have advocated for using CAT-F81 when CAT-GTR is deemed computationally prohibitive20,62, Whelan and Halanych24 recently showed the CAT-F81 can result in critically inaccurate trees. Thus, tree inference was not done with the CAT-F81 model on datasets which would have been too computationally demanding for analyses with CAT-GTR.
Maximum likelihood trees for each dataset were inferred with site-homogeneous amino acid substitution models coupled with data partitioning63. Best-fit partitions and amino acid substitution models for each dataset were inferred with PartitionFinder 2.064 using 20% relaxed clustering65, the rcluster_f command, and Bayesian information criteria. Maximum likelihood phylogenetic inference using best-fit partitions and amino acid substitution models was done with RAxML 8.2.455. A discrete gamma distribution with four categories was used on each partition for modeling rate heterogeneity. Nodal support was assessed with 100 fast bootstrap replicates. Files with best-fit partitions and models for each dataset have been deposited on FigShare (doi: XXXX).
Measuring support for competing hypotheses of non-bilaterian relationships
The number of genes and sites favoring each of the two competing hypotheses—sponges-sister to all other extant metazoans and ctenophores-sister to all other metazoans—was assessed under a maximum likelihood framework. For each metazoan dataset, site-wise likelihood scores were inferred for both hypotheses with RAxML 8.2.4 (option -f G). The same partitioning schemes and models utilized in the original tree inference were used. The two different phylogenetic hypotheses passed to RAxML (via -z) were the tree inferred with RAxML (i.e., the ctenophore-sister tree) and the corresponding tree that was modified to have sponges sister to all other metazoans; constraints were done by modifying the original tree in Mesquite 3.266. The number of genes and sites supporting each hypothesis were calculated with RAxML output and Perl scripts from Shen et al.26.
Molecular Clock Analyses
Past authors have hypothesized that Ctenophora underwent a species bottleneck, possibly as recently as 65 MYA12,13,67. However, the bottleneck hypothesis has not been tested with molecular clock methods. BEAST 268 is a well-tested and widely used program that implements molecular clock models, but analyses with amino acids can be prohibitively slow. Thus, for molecular clock analyses, we used our smallest dataset, Metazoa_Choano_RCFV_LB_strict. We also trimmed the same taxa that were deemed unstable for analyses with CAT-GTR (see above and Table S2). The same amino acid substitution models and best-fit partitions were inferred with PartitionFinder using 20% relaxed clustering with the rcluster_f command. The best-fit number of relaxed molecular clock models for use in BEAST 2 were inferred with ClockstaR70 using default parameters. One molecular clock was inferred to be most appropriate for this dataset. A relaxed molecular clock with a lognormal distribution71 and a Yule tree model was used. A calibration was placed on the node representing the most recent common ancestor (MRCA) of Metazoa using a normal distribution with a mean of 750 MYA and a standard deviation of 35 following the findings of dos Rios et al.72; monophyly of Metazoa was enforced. We only used one calibration point for the molecular clock analysis, even though this may result in inaccurate absolute branching time estimates. We attempted to perform analyses with a greater number of node-age calibrations (e.g., for sponges, cnidarians, and bilaterian lineages; see Supplementary Table S4)72, but Bayesian analyses failed to show evidence of convergence after over four months of run time. However, a single calibration point still allows for inference of relative timing of extant ctenophore diversification compared to better studied lineages and lineages with better fossil records. Thus, even if absolute timing of diversification events is imprecise in our molecular clock tree inference, we can analyze the inferred timing of ctenophore diversification relative to well-studied diversification events where timing of diversification is reasonably well known (e.g., Bilateria, protosomes) to estimate the age of the extant ctenophore MRCA.
Molecular clock analyses with BEAST 2 consisted of two independent runs with 27,246,750 MCMC generations sampled every 250 generations. Trace plots were viewed in tracer, burn-in was visually determined (12% for run 1 and 50% for run 2). Convergence was checked and confirmed by comparing trace plots in Tracer making sure the effective sample size of each parameter was greater than 50 and that stationarity appeared to have been achieved; most parameters has effective sample sizes well in excess of 200. A maximum clade credibility tree with median heights was calculated using TreeAnnotater68. Bayesian inference using a molecular clock resulted in identical branching patterns among phyla as analyses with RAxML and PhyloBayes (e.g., Ctenophora sister to all other animals PP = 1.00; Supplementary Figs. S1–S15).
Ancestral State Reconstruction
We performed ancestral state reconstruction for the following traits: 1) general body plan (i.e. “cydippid-like”, “lobata-like”, Platyctenida, Cestida; Fig. 3, Supplementary Fig. S20), 2) primary food capture mode (i.e., with tentacles, with body lobes, and engulfing prey with a comparatively large mouth; Supplementary Fig. S20), 3) presence/absence of tentacles as adults (Supplementary Fig. S21), 4) presence/absence of dioecy (Supplementary Fig. S22), 5) presence/absence of striated muscles (Supplementary Fig. S23), 6) presence/absence of smooth muscle (Supplementary Fig. S23), 7) pelagic vs. benthic/semi-benthic lifestyle (Supplementary Fig. S24), 8) ability to bioluminesce (Supplementary Fig. S24), 9) presence/absence of tentacles throughout life cycles (Supplementary Fig. S21). Characteristics were assigned using previous descriptive work2,6,17,19,37,38,73–79 and/or personal observations of individuals we collected (see Supplementary Table S5). Additional information about trait assignment can be found in Supplementary Discussion. Phylogenetic signal of each trait was measured with Blomberg’s K80, using the phytools 0.5–1081 package in R57; each trait had significant phylogentic signal (p <0.05).
Stochastic mapping of character evolution, a Bayesian method for ancestral state reconstruction82,83, was done to generate character state joint probabilities on the phylogeny inferred with dataset Cteno_RCFV_LB. This was done in R using phytools 0.5–10. Uncertainty in relationships was ignored because the only uncertain nodes were those at the tips among closely related taxa with identical character states (Figs. 3, Supplementary Figs. S16–S19; Table S5). Analyses that incorporated uncertainty in branch lengths were effectively the same as those that ignored uncertainty (Supplementary Discussion). For ancestral state reconstruction, Cydippida sp. from Friday Harbor was removed because this species was labeled as Bolinopsis infundibulum in Moroz et al.3, and we could not confidently assign character states given the misidentification. The larval ctenophore specimen (Ctenophora sp.) was also removed because many character states that would be present only in adults were undetermined. These tips were removed from trees using the R package Ape84. Outgroups were removed from all stochastic mapping analyses except presence/absence of striated and smooth muscle. The best-fit model of character evolution to be used for stochastic mapping was determined by fitting an equal rates model, a symmetrical model, and an all rates different model to each character state dataset using the R package Geiger85; corrected Akaike information criteria was used to determine the best-fit model for each respective character dataset. For each analysis, the prior probability of the root’s character state was estimated directly from the data and Bayesian MCMC was used to generate a posterior probability distribution for the character transition matrix. With these parameters, 1,000 stochastic maps were generated for each trait. Evolution of traits was visualized by displaying pie charts of posterior probabilities for each character state on every node.
Supplementary Material
Acknowledgments
This work was made possible in part by a grant of high-performance computing resources and technical support from the Alabama Supercomputer Authority and was supported by the US National Aeronautics and Space Administration (Grant NASA-NNX13AJ31G), the National Science Foundation (Grants ANT-1043670, ANT-1043745, 1557923, 1457162, 1548121, 1645219), and the Ministry of Education of the Russian Federation (#14W03.31.0015). We thank the International SeaKeeper Society, captains and crew of oceanic vessels Laurence M Gold, Capasetic, Penny Mae, Defiance, Basic Explorer, Harle of Fleet Miami, and Miss Phebe II as well as Mr. James Jacoby for their help in collection of ctenophores around the globe. Andrea Mills helped with lab work and data curation. Claudia Mills aided in species identifications. We thank three anonymous reviewers for their time and comments. The findings and conclusions in this article are those of the authors and do not necessarily represent the views of the U.S. Fish and Wildlife Service. This is Molette Biology Laboratory Contribution XXX and Auburn University Marine Biology Program Contribution XXX.
Footnotes
Author contributions: N.V.W, K.M.K, L.L.M, K.M.H. designed the study. K.M.K collected and identified australian speices. L.L.M, and G.P. collected and identified all other ctenophores. P.W., K.M., T.P.M., and L.L.M. sequenced and assembled ctenophore transcriptomes. N.V.W. and K.M.K. performed phylogenetic analyses and ancestral state reconstruction. N.V.W., K.M.K, L.L.M. and K.M.H wrote the manuscript. All authors edited manuscript versions and approved the final version.
Competing financial interests: We declare no competing financial interests
Code Data availability: Transcriptomes sequenced as a part of this study are available in NCBI Short Read Archive under BioProject PRJNA396415. 18S rRNA sequences have been uploaded to GenBank under accension numbers MF599304-MF599336. Datasets, model partitions, and tree files have been uploaded to FigShare (doi: XXX). Code used in this study is available from http://github.com/nathanwhelan.
References
- 1.Hyman L. The Invertebrates. Vol. 1. McGraw-Hill; 1940. [Google Scholar]
- 2.Mackie GO, Mills CE, Singla CL. Structure and function of the prehensile tentialla of Euplokamis (Ctenophora, Cydippida) Zoomorphology. 1988;107:319–337. [Google Scholar]
- 3.Moroz LL, et al. The ctenophore genome and the evolutionary origins of neural systems. Nature. 2014;510:109–114. doi: 10.1038/nature13400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roohi A, et al. Changes in biodiversity of phytoplanton, zooplankton, fishes and macrobenthos in the Southern Caspian Sea after the invasion of the ctenophore Mnemiopsis leidyi. Biol Invasions. 2010;12:2342–2361. [Google Scholar]
- 5.Ryan JF, et al. The genome of the ctenophore Mnemiopsis leidyi and its implications for cell type evolution. Science. 2013;342:1242592. doi: 10.1126/science.1242592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harbison GR. In: The origins and relationships of lower invertebrates. Morris SC, George JD, Gibson Ray, Platt HM, editors. Oxford University Press; 1985. pp. 78–100. [Google Scholar]
- 7.Dunn CW, Leys S, Haddock SHD. The hidden biology of sponges and ctenophores. Trends Ecol Evol. 2015;30:282–291. doi: 10.1016/j.tree.2015.03.003. [DOI] [PubMed] [Google Scholar]
- 8.Whelan NV, Kocot KM, Halanych KM. Employing phylogenomics to resolve the relationships among cnidarians, ctenophores, sponges, placozoans and bilaterians. Integr Comp Biol. 2015;55:1084–1095. doi: 10.1093/icb/icv037. [DOI] [PubMed] [Google Scholar]
- 9.Dunn CW, et al. Broad phylogenomic sampling improves resolution of the animal tree of life. Nature. 2008;452:745–749. doi: 10.1038/nature06614. [DOI] [PubMed] [Google Scholar]
- 10.Hejnol A, et al. Assessing the root of bilaterian animals with scalable phylogenomic models. Proc Biol Sci. 2009;276:4261–4270. doi: 10.1098/rspb.2009.0896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Whelan NV, Kocot KM, Moroz LL, Halanych KM. Error, signal, and the placement of Ctenophora sister to all other animals. Proc Natl Acad Sci USA. 2015;112:5773–2778. doi: 10.1073/pnas.1503453112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Podar M, Haddock SHD, Sogin ML, Harbison GR. A molecular phylogenetic framework for the phylum Ctenophora using 18S rRNA genes. Mol Phylogen Evol. 2001;21:218–230. doi: 10.1006/mpev.2001.1036. [DOI] [PubMed] [Google Scholar]
- 13.Simion P, Bekkouche N, Jager M, Quéinnec E, Manuel M. Exploring the potential of small RNA subunit and ITS sequences for resolving the phylogenetic relationships within the phylum Ctenophora. Zoology. 2015;118:102–114. doi: 10.1016/j.zool.2014.06.004. [DOI] [PubMed] [Google Scholar]
- 14.Tang F, Bengtson S, Wang Y, Wang X-l, Yin C-y. Eoandromeda and the origin of Ctenophora. Evol Dev. 2011;13:408–414. doi: 10.1111/j.1525-142X.2011.00499.x. [DOI] [PubMed] [Google Scholar]
- 15.Morris SC, Collins DH. Middle Cambiran ctenophores from Stephen Formation British Columbia, Canada. Philos Trans R Soc Lond, Ser B: Biol Sci. 1996;351:279–308. [Google Scholar]
- 16.Chen JY, et al. Raman spectra of a Lower Cambrian ctenophore embryo from southwestern Shaanxi, China. Proc Natl Acad Sci USA. 1997;104:6289–6292. doi: 10.1073/pnas.0701246104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mills CE. Revised classification of the genus Euplokamis Chun, 1880 (Ctenophora: Cydippida: Euplokamidae n. fam.) with a description of the new species Euplokamis dunlapae. Can J Zool. 1987;65:2661–2668. [Google Scholar]
- 18.Harbison GR, Miller RL. Not all ctenophores are hermaphrodites. Studies on the sytematics, distribution, sexuality and development of two species of Ocyropsis. Mar Biol. 1986;90:413–424. [Google Scholar]
- 19.Uyeno D, Lasley RM, Moore JM, Berumen ML. New records of Lobatolampea tetragona (Ctenophora: Lobata: Lobatolampeidae) from the Red Sea. Marine Biodiversity Records. 2015;8:e33. [Google Scholar]
- 20.Pisani D, et al. Genomic data do not support comb jellies as the sister group to all other animals. Proc Natl Acad Sci USA. 2015;112:15402–15407. doi: 10.1073/pnas.1518127112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Simion P, et al. A large and consistent phylogenomic dataset supports sponges as the sister group to all other animals. Curr Biol. 2017;27:958–967. doi: 10.1016/j.cub.2017.02.031. [DOI] [PubMed] [Google Scholar]
- 22.Halanych KM, Whelan NV, Kocot KM, Kohn AB, Moroz LL. Miscues misplace sponges. Proc Natl Acad Sci USA. 2016;113:E946–E949. doi: 10.1073/pnas.1525332113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moroz LL, Halanych KM. A sisterly dispute: methodological misconceptions. Nature. 2016;529:286–287. doi: 10.1038/529286a. [DOI] [PubMed] [Google Scholar]
- 24.Whelan NV, Halanych KM. Who let the CAT out of the bag? Accurately dealing with subtitutional heterogeneity in phylogenomics analyses. Syst Biol. 2017;66:232–255. doi: 10.1093/sysbio/syw084. [DOI] [PubMed] [Google Scholar]
- 25.Arcila D, et al. Genome-wide interrogation advances resolution of recalcitrant groups in the tree of life. Nature Ecology & Evolution. 2017;1:0020. doi: 10.1038/s41559-016-0020. [DOI] [PubMed] [Google Scholar]
- 26.Shen XX, Hittinger CT, Rokas A. Contentious relationships in phylogenomic studies can be driven by a handful of genes. Nature Ecology & Evolution. 2017;1:0126. doi: 10.1038/s41559-017-0126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heath TA, Hedtke SM, Hillis DM. Taxon sampling and the accuracy of phylogenetic analyses. Journal of Systematics and Evolution. 2008;46:239–257. [Google Scholar]
- 28.Hedtke SM, Townsend TM, Hillis DM. Resolution of phylogenetic conflict in large data sets by increased taxon sampling. Syst Biol. 2006;55:522–529. doi: 10.1080/10635150600697358. [DOI] [PubMed] [Google Scholar]
- 29.Zwickl DJ, Hillis DM. Increased taxon sampling greatly reduces phylogenetic error. Syst Biol. 2002;51:588–598. doi: 10.1080/10635150290102339. [DOI] [PubMed] [Google Scholar]
- 30.Benton MJ, Twitchett RJ. How to kill (almost all life): the end-Permian extinctoin event. Trends in Ecology and Evolution. 2003;18:358–365. [Google Scholar]
- 31.Lartillot N, Philippe H. A Bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Mol Biol Evol. 2004;21:1095–1109. doi: 10.1093/molbev/msh112. [DOI] [PubMed] [Google Scholar]
- 32.Steinmetz PRH, et al. Independent evolution of striated muscles in cnidarians and bilaterians. Nature. 2012;487:231–234. doi: 10.1038/nature11180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martindale MQ. In: Atlas of marine invertebrate larvae. Young CM, Sewell MA, Rice ME, editors. Academic Press; 2002. pp. 109–122. [Google Scholar]
- 34.Rigby S, Milsom C. Benthic origins of zooplankton: an environmentaly determined macroevolutionary effect. Geology. 1996;24:52–54. [Google Scholar]
- 35.Haddock SHD, Case JF. Not all ctenophores are bioluminescent: Pleurobrachia. Biol Bull. 1995;189:356–362. doi: 10.2307/1542153. [DOI] [PubMed] [Google Scholar]
- 36.Widder EA. Bioluminescence in the ocean: origins of biological, chemical, and ecological diversity. Science. 2010;328:704–708. doi: 10.1126/science.1174269. [DOI] [PubMed] [Google Scholar]
- 37.Gershwin L-a, Zeidler W, Davie PJF. Ctenophora of Australia. Mem Queensl Mus. 2010;54:1–45. [Google Scholar]
- 38.Haddock SHD. Comparative feeding behaviour of planktonic ctenophores. Integr Comp Biol. 2007;47:847–853. doi: 10.1093/icb/icm088. [DOI] [PubMed] [Google Scholar]
- 39.Moroz LL. Covergent evolution of neural systems in ctenophores. J Exp Biol. 2015;218:598–611. doi: 10.1242/jeb.110692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moroz LL, Kohn AB. Independent origins of neurons and synapses: insights from ctenophores. Philos Trans R Soc Lond, Ser B: Biol Sci. 2016;371:20150041. doi: 10.1098/rstb.2015.0041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Borowiec ML, Lee EK, Chiu JC, Plachetzki DC. Extracting phylogenetic signal and accounting for bias in whole-genome data sets supports the Ctenophora as sister to remaining Metazoa. BMC Genomics. 2015;16:987. doi: 10.1186/s12864-015-2146-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cannon JT, et al. Xenacoelomorpha is the sister group to Nephrozoa. Nature. 2016;530:89–93. doi: 10.1038/nature16520. [DOI] [PubMed] [Google Scholar]
- 43.Chang ES, et al. Genomic insights into the evolutionary origin of Myxozoa within Cnidaria. Proc Natl Acad Sci USA. 2015;112:14912–14917. doi: 10.1073/pnas.1511468112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nabhan AR, Sarkar IN. The impact of taxon sampling on phylogenetic inference: a review of two decades of controversy. Briefings in Bioinformatics. 2011;13:122–134. doi: 10.1093/bib/bbr014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brown T, Howe C, Zhang A, Pyrkosz Q, Brom AB. A reference-free algorithm for computational normalization of shotgun sequencing data. ArXiv e-prints. 2012;1203:4802. [Google Scholar]
- 46.Haas BJ, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. 2013;8:1494–1512. doi: 10.1038/nprot.2013.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ebersberger I, Strauss S, von Haeseler A. HaMStR: profile hidden markov model based search for orthologs in ESTs. BMC Evol Biol. 2009;9:157. doi: 10.1186/1471-2148-9-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li L, Stoeckert CJ, Roos DS. OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kocot KM, et al. Phylogenomics of Lophotrochozoa with consideration of systematic error. Syst Biol. 2017;66:256–282. doi: 10.1093/sysbio/syw079. [DOI] [PubMed] [Google Scholar]
- 51.Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Price MN, Dehal PS, Arkin AP. FastTree 2 – Approximately maximum-likelihood trees for large alignments. PLOS ONE. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kocot KM, Citarella MR, Moroz LL, Halanych KM. PhyloTreePruner: A Phylogenetic Tree-Based Approach for Selection of Orthologous Sequences for Phylogenomics. Evol Bioinform. 2013;9:429–435. doi: 10.4137/EBO.S12813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Finn RD, Clements J, Eddy SR. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011;39:W29–W37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Struck TH. TreSpEx—detection of misleading signal in phylogenetic reconstructions based on tree information. Evol Bioinform. 2014;10:51–67. doi: 10.4137/EBO.S14239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; Vienna, Austria: 2015. [Google Scholar]
- 58.Zhong M, et al. Detecting the symplesiomorphy trap: a multigene phylogenetic analysis of terebelliform annelids. BMC Evol Biol. 2011;11:369. doi: 10.1186/1471-2148-11-369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kück P, Struck TH. BaCoCa - A heuristic software tool for the parallel assessment of sequence biases in hundreds of gene and taxon partitions. Mol Phylogen Evol. 2014;70:94–98. doi: 10.1016/j.ympev.2013.09.011. [DOI] [PubMed] [Google Scholar]
- 60.Lartillot N, Rodrigue N, Stubbs D, Richer J. PhyloBayes MPI: phylogenetic reconstruction with infinite mixtures of profiles in a parallel environment. Syst Biol. 2013;62:611–615. doi: 10.1093/sysbio/syt022. [DOI] [PubMed] [Google Scholar]
- 61.Rambaut A, Drummond AJ. Tracer v 1.6. 2013 Available from http:/beast.bio.ed.ac.uk/Tracer.
- 62.Nosenko T, et al. Deep metazoan phylogeny: when different genes tell different stories. Mol Phylogen Evol. 2013;67:223–233. doi: 10.1016/j.ympev.2013.01.010. [DOI] [PubMed] [Google Scholar]
- 63.Brown JM, Lemmon AR. The importance of data partitioning and the utility of bayes factors in Bayesian phylogenetics. Syst Biol. 2007;56:643–655. doi: 10.1080/10635150701546249. [DOI] [PubMed] [Google Scholar]
- 64.Lanfear R, Frandsen PB, Wright AM, Senfeld T, Calcott B. PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol Biol Evol. 2016 doi: 10.1093/molbev/msw260. [DOI] [PubMed] [Google Scholar]
- 65.Lanfear R, Calcott B, Kainer D, Mayer C, Stamatakis A. Selecting optimal partitioning schemes for phylogenomic datasets. BMC Evol Biol. 2014;14:82. doi: 10.1186/1471-2148-14-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mesquite: a modular system for evolutionary analysis v. 2017 Version 3.2. http://mesquiteproject.org.
- 67.Jékely G, Paps J, Nielsen C. The phylogenetic position of ctenophores and the origin(s) of nervous systems. EvoDevo. 2015;6:1. doi: 10.1186/2041-9139-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bouckaert R, et al. BEAST 2: A Software Platform for Bayesian Evolutionary Analysis. PLoS Comput Biol. 2014;10:e1003537. doi: 10.1371/journal.pcbi.1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rice P, Longden I, Bleasby A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000;16:276–277. doi: 10.1016/s0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 70.Duchêne S, Molak M, Ho SYW. ClockstaR: choosing the number of relaxed-clock models in molecular phylogenetic analysis. Bioinformatics. 2014;30:1017–1019. doi: 10.1093/bioinformatics/btt665. [DOI] [PubMed] [Google Scholar]
- 71.Drummond AJ, Ho SYW, Phillips MJ, Rambaut A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006;4:e88. doi: 10.1371/journal.pbio.0040088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.dos Reis M, et al. Uncertainty in the timing of origin of animals and the limits of precision in molecular timescales. Curr Biol. 2015;25:29392950. doi: 10.1016/j.cub.2015.09.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Brusca RC, Moore W, Shuster SM. Invertebrates. Third. Vol. 1104. Sinauer Associates; 2016. [Google Scholar]
- 74.Eechkaut I, Flammang P, Bue CL, Jangoux M. Functional morphology of the tentacles and tentilla of Coeloplana bannworthi (Ctenophora, Platyctenida), and ectosymbiont of Diadema setosum (Echinodermata, Echinoida) Zoomorphology. 1997;117:165–174. [Google Scholar]
- 75.Harbison GR, Madin LP. In: Synopsis and classification of living organisms. Parker SP, editor. McGraw-Hill; 1982. pp. 707–715. [Google Scholar]
- 76.Horita T. An undescribed lobate ctenophore, Lobatolampea tetragona gen. nov. & spec. nov., representing a new family, from Japan. Zool Meded. 2000;73:457–464. [Google Scholar]
- 77.Matsumoto GI, Harbison GR. In situ observations of foraging, feeding, and escape behavior in three orders of oceanic ctenophores: Lobata, Cestida, and Beroida. Mar Biol. 1993;117:279–287. [Google Scholar]
- 78.Purcell JE, Sturdevant MV, Galt CP. In: Response of marine ecosystems to global change: Ecological impact of appendicularians. Gorsky G, Yongbluth MJ, Deibel D, editors. GB Scientific Publisher; 2004. [Google Scholar]
- 79.Stretch JJ. Observations on the abundance and feedign behavior of the cestid ctenophore, Velamen Parallelum. Bull Mar Sci. 1982;32:796–799. [Google Scholar]
- 80.Blomberg SP, Garland T, Jr, Ives AR. Testing for phylogenetic signal in comparative data: behavioral traits are more labile. Evolution. 2003;57:717–745. doi: 10.1111/j.0014-3820.2003.tb00285.x. [DOI] [PubMed] [Google Scholar]
- 81.Revell LJ. phytools: an R package for phylogenetic comparative biology (and other things) Methods Ecol Evol. 2012;3:217–223. [Google Scholar]
- 82.Huelsenbeck JP, Nielsen R, Bollback JP. Stochastic mapping of morphological characters. Syst Biol. 2003;52:131–158. doi: 10.1080/10635150390192780. [DOI] [PubMed] [Google Scholar]
- 83.Nielsen R. Mapping mutations on phylogenies. Syst Biol. 2002;51:729–739. doi: 10.1080/10635150290102393. [DOI] [PubMed] [Google Scholar]
- 84.Paradis E, Claude J, Strimmer K. APE: analysis of phylogenetics and evolution in R language. Bioinformatics. 2004;20 doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- 85.Harmon LJ, Weir JT, Brock CD, Glor RE, Challenger W. GEIGER: investigating evolutionary radiations. Bioinformatics. 2008;24:129–131. doi: 10.1093/bioinformatics/btm538. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.