

Abstract
Using O-SP–core (O-SPcNH2) polysaccharide, isolated from Vibrio cholerae O1 lipopolysaccharide (LPS) and related synthetic substances, a detailed study of factors that affect conjugation of bacterial polysaccharides to protein carriers by squaric acid chemistry to form conjugate vaccines has been carried out. Several processes previously unrecognized, which take place during the squarate labelling of the O-SPcNH2 and subsequent conjugation of the squarate (O-SPcNH–SqOMe) formed, have been identified. The efficiency of conjugation at pH 8.5, 9.0, and 9.5 to each of bovine serum albumin (BSA) and the recombinant tetanus toxin fragment C (rTT-Hc) has been determined. The study led to a protocol for more efficient labeling of O-SPcNH2 antigen with the methyl squarate group, to yield a higher quality, more potent squarate conjugation reagent. Its use resulted in about two-fold increase of conjugation efficiency (from 23–26% on BSA to 51% on BSA and 55% on rTT-Hc). The spent conjugation reagent could be recovered and regenerated by treatment with MeI in the absence of additional base. The immunological properties of the experimental vaccine made from the regenerated conjugation reagent were comparable to those of the immunogen made from the parent O-SPcNH–SqOMe.
Keywords: O-specific polysaccharide, O-antigen, glycoconjugate vaccine, selective methylation, antigen recovery
Graphical Abstract
A detailed study of factors that affect conjugation of bacterial polysaccharides to protein carriers by squaric acid chemistry to form conjugate vaccines has been carried out. Several processes previously unrecognized, which take place during the squarate labelling of the O-SPcNH2 and subsequent conjugation of the squarate (O-SPcNH–SqOMe) formed, have been identified. The study led to a protocol whose use resulted in about two-fold increase of conjugation efficiency. The spent conjugation reagent could be recovered and regenerated. The immunological properties of the experimental vaccine made from the regenerated conjugation reagent were comparable to those of the immunogen made from the parent O-SPcNH–SqOMe.
Introduction
Because the squarate derivatives of carbohydrates are powerful tools for making neoglycoconjugates,1–6 squaric acid conjugation chemistry has a great potential in glycoconjugate vaccine development and manufacturing. Both synthetic7–10 and, as we more recently demonstrated,11,12 bacterial carbohydrate antigens can be chemically attached to protein carriers in this way to form potent immunogens.12–15 The method, which was originally designed for linker-equipped synthetic carbohydrates,2,16 is based (Scheme 1) on the reaction of squaric acid diesters (1) with amines at pH 7, to form squaric acid monoester monoamide (2). The latter can be made to react with another amine at higher pH, for example pH 92, to form squaric acid diamides (3). When the first amine is an amino group containing carbohydrate and the second amine is a protein, a glycoprotein (glycoconjugate) is formed.
Scheme 1.
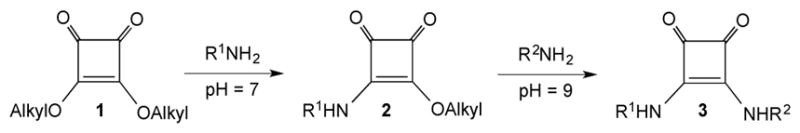
Conjugating amines through squaric acid diester chemistry.
These glycoconjugates, also series of conjugates in the one-pot manner,17 can be prepared to contain various, predetermined amounts of carbohydrate antigens, and used as experimental vaccines. Concerning vaccine development, the squaric acid chemistry method had been initially used only to prepare glycoconjugates from synthetic, linker-equipped carbohydrates. During our recent work toward a conjugate vaccine for cholera11, we conjugated for the first time bacterial polysaccharide using squaric acid chemistry. We used the O-SP–core polysaccharides from Vibrio cholerae O1, serotype Inaba and Ogawa (Fig. 1), known18 to contain a single amino group in the core region, and protein carrier BSA. The Inaba antigen had been conjugated to BSA before, also utilizing the free amino group in the core, but different conjugation chemistry was applied.19,20 The novelty of our approach was in the direct conjugation, namely without the introduction of linkers to either antigen or carrier. In this way, we avoided the demanding derivatization of the two components, and tedious purification of the linker-equipped intermediates to be conjugated. The efficiency of conjugation we initially achieved 11 (23–26%) was higher compared to the overall efficiency reported earlier but, clearly, preparation of vaccines by the squaric acid method would become more attractive to industry if the efficiency of conjugation could be further increased. Here, we report results of our efforts toward that goal, obtained within our work toward developing a conjugate vaccine for cholera caused by Vibrio cholerae O1. Our efforts led to a methodology with markedly increased efficiency of conjugation of bacterial carbohydrates to proteins by squaric acid chemistry, while maintaining the ability to prepare tailor-made conjugates3,17,21–23 with predictable/predetermined carbohydrate–protein ratios.
Figure 1.
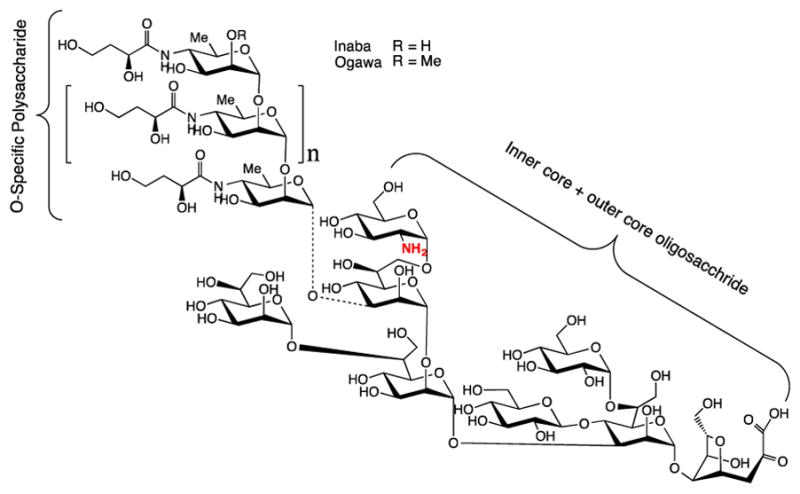
The amino group containing18 O-SP–core (O-SPcNH2) antigen isolated from Vibrio cholerae O:1; n = 7–22.19 The O-SP consists of (1→2)-α-linked D-perosamine (4-amino-4,6-dideoxy-D-mannopyranose) residues, which are N-acylated with 3-deoxy-L-glycero-tetronic acid;24 the two strains, Inaba and Ogawa differ in the absence or presence of methyl group in the terminal, upstream perosamine residue.25
Results and Discussion
To maintain a reasonable reaction rate, conjugations are normally conducted with excess of the squarate-labeled carbohydrate. The efficiency of the conjugation, i.e. the molar ratio of the squarate derivative attached to the carrier to the squarate derivative used at the onset of the conjugation, can potentially be increased by improving the overall conjugation process (labeling O-SPcNH2 followed by the actual conjugation), and by recovering unconjugated squarate-labeled antigen (which is normally discarded) and reusing it in another conjugation. Many variables affecting conjugation of synthetic oligosaccharides have already been optimized, and the original protocol1,2 for conjugation of such substances to proteins has been revised.3,26 These efforts led eventually to a simple and controllable protocol, which allows preparation of tailor-made conjugates with predetermined carbohydrate–protein ratios. In this context, it cannot be overemphasized that achieving the same in the case of bacterial polysaccharides presents a more formidable challenge because of their vastly different physico-chemical properties. Here we report our efforts toward increasing the efficiency of conjugation of bacterial polysaccharides using squaric acid chemistry.
1. Improving the conjugation protocol
1.1 Optimizing reaction conditions for O-SPcNH2→O-SPcNH–SqOMe conversion
When squaric acid chemistry was first used to conjugate bacterial polysaccharides,11 BSA was used as a model carrier protein. To establish conditions applicable for conjugation of O-SPcNH–SqOMe to a medically acceptable carrier, we used during the present work the recombinant tetanus toxin Hc fragment (rTT-Hc)27,28, in addition to BSA. Initially, we critically analyzed the original protocol.11 There, BSA was treated with O-SPcNH–SqOMe (Ogawa, 21.5 molar excess, antigen concentration ~5 mM) at pH 9 (0.5 M borate buffer). After 4 days, a conjugate showing antigen–protein ratio of 4.8 was formed (efficiency ~23%). Because of lack of suitable instrumentation, progress of the O-SPcNH2 →O-SPcNH–SqOMe conversion was not monitored, and the decision when the reaction should be terminated was based largely on chemical intuition. For obvious reasons, NMR and MS methods could not be used for monitoring the conversion of one mixture to another (c.f. Fig. 1 showing the structure of O-SPcNH2, which would be converted to the corresponding O-SPcNH–SqOMe; both starting and the target polysaccharides consist of a homologous series of species, namely the core oligosaccharide with O-SP consisting of ~7–22 N-acylated perosamine units attached). For the same reason, we did not find feasible monitoring this transformation by HPLC. Within attempts to find a reliable monitoring tool, we tried various HPLC columns, but satisfactory separation of the two polymolecular species could not be achieved. We considered using the conventional colorimetric fluorescamine assay29 to monitor the amine→squarate conversion by measuring the consumption of the free amino group-containing compounds in the mixture, but the sample amount required even by the micro plate assay would be too large, considering the scale of reactions we were working with (usually between ~10 and 200 μL). Therefore, in order to ensure a high degree of O-SPcNH2 → O-SPcNH–SqOMe conversion without monitoring the reaction progress, we used in the previous work11 a large excess of dimethyl squarate [3,4-dimethoxy-3-cyclobutene-1,2-dione, Sq(OMe)2, 4, ~22 molar eq.], in 0.05 M pH 7.0 phosphate buffer, and allowed for longer reaction time (48 h) than was necessary to complete similar conversions with oligosaccharides.3.
The remedy of the situation ensued when we acquired the NanoDrop 3300 spectrophotometer, which provided us with the ability to analyze sample volumes as small as 1 μL. With this technology at hand, we first re-examined our original squarate-forming reaction O-SPcNH2→O-SPcNH–SqOMe11 by monitoring its progress by fluorescamine microassay. (These experiments are not described in the Experimental). The protocol developed for primary amino group assays in proteins29 and recommended by the NanoDrop 3300 spectrophotometer supplier was applied. To briefly recapitulate, the reaction is run in the reaction buffer at pH 7 and samples are analyzed after dilution of an aliquot with the assay buffer pH ≥7. Measured is the amount of fluorescent dye formed from fluorescamine and free amine. The original O-SPcNH2→O-SPcNH–SqOMe experiment11 was repeated, and when the decrease of the relative fluorescent units leveled off the reaction mixture still contained ~7% of the unchanged O-SPcNH2. When the acidity of the reaction mixture was serendipitously checked at that stage of the transformation, but before dilution with the assay buffer, the pH was found to be 1.5. We reasoned that the very low pH must have been caused by the presence of the monoester acid [Sq(OMe)OH], formed by hydrolysis of Sq(OMe)2, which was used in large excess at the onset of the reaction. Evidently, the capacity of the reaction buffer used was insufficient to maintain the pH near neutral. When the pH was measured after the dilution with the assay buffer just before the fluorescamine analysis, pH measurement showed it to be 3.4, suggesting that the value “7%” (above) might not have been accurate because the reading was taken at pH much below 7. At such low pH (3.4), unchanged O-SPcNH2, if present, would largely be in the protonated form and escape the fluorescamine assay, making the assay inaccurate. Clearly, the protocol, which was not developed for monitoring conversions where acid could be generated during the reaction had to be modified to serve our purposes.
The dramatic drop of pH during O-SPcNH2→O-SPcNH–SqOMe conversion can be explained when we consider the three processes taking place simultaneously during the treatment of O-SPcNH2 with excess of Sq(OMe)2:
Amidation of O-SPcNH2 (O-SPcNH2→O-SPcNH–SqOMe);
Hydrolysis of the Sq(OMe)2 reagent (4, Scheme 2), which is known to be quite prone to hydrolysis,30 [Sq(OMe)2→ Sq(OMe)OH (5), and possibly30 also Sq(OMe)2→ Sq(OH)2 6]; and
Hydrolysis of the product squarate, O-SPcNH–SqOMe (O-SPcNH–SqOMe→O-SPcNH–SqOH).
Scheme 2.
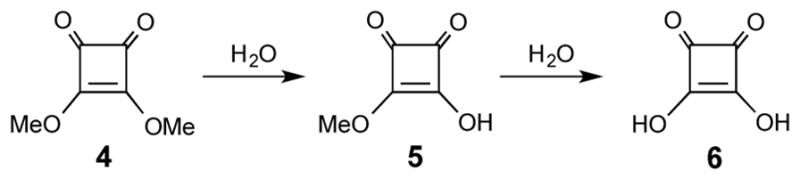
Hydrolysis of Sq(OMe)2.
During the O-SPcNH2→O-SPcNH–SqOMe conversion, while the amidation product O-SPcNH–SqOMe is being formed, the dimethyl squarate reagent 4 hydrolyzes at a considerable rate (half life in water, <3 hours 30). When the capacity of the reaction buffer is insufficient to keep pH from dropping substantially, variable amounts of the unchanged O-SPcNH2 become protonated and do not react with 4 to form the desired O-SPcNH–SqOMe. When pH of the reaction mixture drops too low, addition of the assay buffer (pH 7.2 PBS, 1×) to the sample analyzed does not bring pH back to neutral. The lower the pH of the analyzed sample, the higher percentage of unchanged O-SPcNH2 becomes protonated and escapes the fluorescamine assay. This makes the result inaccurate and misleading.
To verify the aforementioned reasoning and to gain better insight into the phenomena taking place during the amine to squarate conversion and sample preparation for the fluorescamine assay, we conducted the O-SPcNH2→O-SPcNH–SqOMe conversion using standard, commercially available low (0.05 M, pH 7) and high concentration (0.5 M pH 7) reaction buffers. For monitoring, two samples were taken from each of the reaction mixtures, and before fluorescamine assay one sample from each experiment was diluted with low concentration (pH 7.2 PBS, 1×) and the other with high concentration (pH 7.4 PBS, 10×) assay buffer. It must be kept in mind that the assay measures amine present in the samples diluted with the respective buffer and not in the whole reaction mixture.
As can be seen in Fig. 2A, when the reaction was run in 0.05M pH 7 phosphate buffer (pH of the reaction mixture after 24 h, 1.5; pH after dilution of the sample with the low concentration assay buffer after 24 h, 3.4) the fluorescamine assay showed virtually complete absence of free amine, suggesting that the PScNH2→O-SPcNH–SqOMe conversion was nearly complete after 24h. When the more concentrated pH 7.4 PBS, 10× assay buffer was used (Fig. 2B, pH of the reaction mixture after dilution of the sample with the high concentration buffer, 6.6), the result of the assay was quite different: ~55% of unchanged amine was still present after 24h. Obviously, because the samples were taken from the same mixture at the same time, only one set of these analyses, A or B, could be correct. Examination of the course of curves 2A and 2B, together with information about the pH provided clues for concluding that curve 2B reflects the true course of the reaction. The curve represents a situation when the amine protonated in the reaction mixture became deprotonated in the sample analyzed after the addition of pH 7.4 PBS 10× assay buffer. Knowing that fluorescamine assay measures the dye formed from free amine, only curve 2B provides a realistic picture about the state of the reaction, namely incomplete amine to squarate conversion.
Figure 2.
Monitoring the progress of the O-SPcNH2→O-SPcNH–SqOMe conversion by fluorescamine assay. A: Reaction bugger 0.05 M pH 7, assay buffer pH 7.2 PBS 1×; B: Reaction buffer 0.05 M pH 7, assay buffer pH 7.4 PBS 10×.
The foregoing rational is supported by results of an experiment where the stronger, higher capacity reaction buffer (0.5 M, pH 7 phosphate buffer) was used for the conversion. There, pH of the reaction mixture stayed near neutral throughout the conversion. At these conditions, the level of protonation of the amine was insignificant, and the fluorescamine assay indicated, independently of the assay buffer used, that the conversion of amine to squarate was virtually complete (Fig. 3A and 3B). Consequently, O-SPcNH2→O-SPcNH–SqOMe conversion in the protocol recommended (see Experimental) is conducted in 0.5 M pH 7 buffer.
Figure 3.
Monitoring the progress of the O-SPcNH2→O-SPcNH–SqOMe conversion by fluorescamine assay. A: Reaction bugger 0.5 M pH 7, assay buffer pH 7.2 PBS 1×; B: Reaction buffer 0.5 M pH 7, assay buffer pH 7.4 PBS 10×.
Having established that hydrolysis of 4 may affect quality of the conjugation reagent prepared from O-SPcNH2, next we determined the stability of 4 in the buffer that would be included in the revised protocol for O-SPcNH2→O-SPcNH–SqOMe conversion (0.5 M pH 7 reagent buffer). We used NMR spectroscopy to monitor composition of an 80 mM solution of Sq(OMe)2 in 0.5 M pD 7.0 buffer (this concentration is very close to that applied during conversion O-SPcNH2→O-SPcNH–SqOMe11 when 22 eq. excess of squarate reagent was used). The integration in the 1H spectrum of peaks for methyl groups at δ ~4.36 and 4.27 characteristic of Sq(OMe)2 and Sq(OMe)OD, respectively, and 3.32 for MeOD (formed by hydrolysis of the reagent) provided information about the processes taking place. The pD had not dropped below 6.45 during the reaction time of 24 h. It was found that after 200 min and 24 h, ~45% and 100%, respectively, of the Sq(OMe)2 reagent was hydrolyzed to form 5 (Fig. 4). This suggested that extending the reaction time11 of the squarate labeling of O-SPcNH2 beyond 24 h was not productive.
Figure 4.
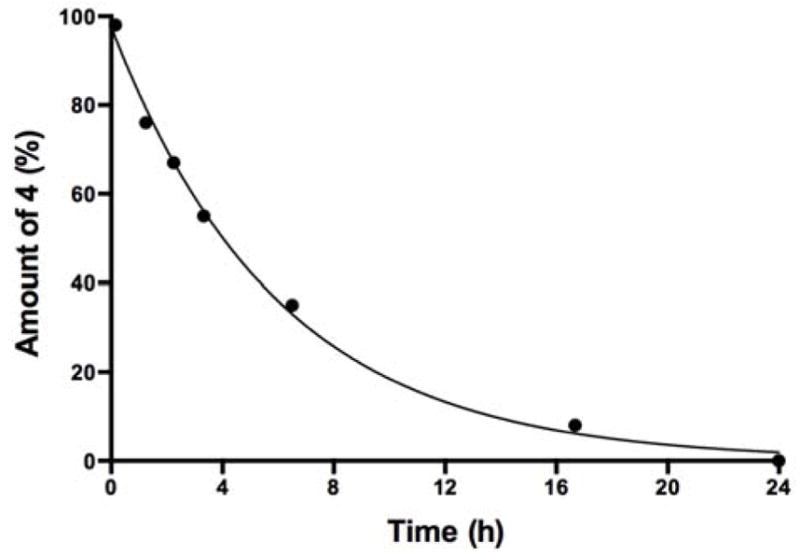
Hydrolysis of Sq(OMe)2 (4) in 0.5 M pD 7 buffer monitored by NMR spectroscopy.
Next, we deemed important to determine the effect of the amount of reagent 4 and the mode of addition of the reagent upon the squarate formation. For economical reasons, two amino dextrans, m.w. 3,000 and 10,000, each containing only one terminal amino group were used as model polysaccharides, instead of O-SPcNH2 that we had available in more limited quantity. Fig. 5 shows monitoring the reactions with amino dextran 10,000 by fluorescamine micro assay.
Figure 5.
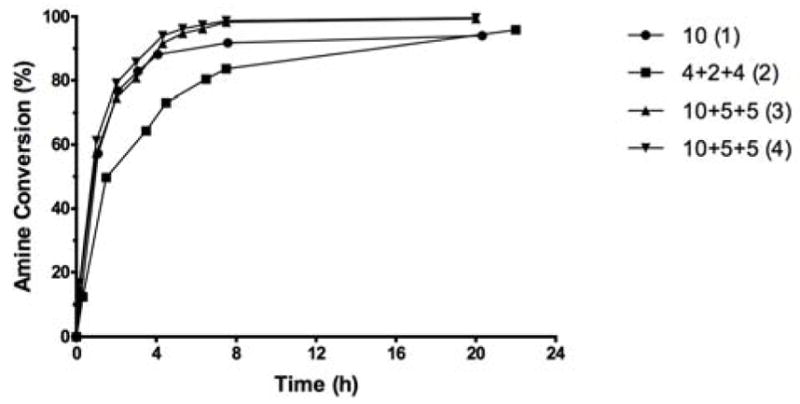
Squarate preparation from amino dextran 10,000 under different conditions. The reactions were conducted in 0.5 M pH 7 buffer at the dextran concentration of 4 mM; (1) single addition of 10 eq. of Sq(OMe)2; (2) portionwise addition of 4+2+4 eq. of Sq(OMe)2 at 0, 3 and 6 h; (3) and (4) parallel experiments to test reproducibility; portionwise addition of 10+5+5 eq. of Sq(OMe)2. The irregular course of the reaction reflect the stepwise addition of the reagent. For further details, see Experimental. The pH of the reaction mixtures, measured at the end of the reaction time, stayed above 6.4 in all cases.
When 10 eq. of Sq(OMe)2 was added in one portion, the fast initial reaction subsided gradually, and 94% conversion into the corresponding squarate was reached after ~21 h. As can be seen in Fig. 5 (2), portion-wise addition resulted in slightly more efficient utilization of the reagent and 96% conversion was achieved after ~22 h. Virtually complete conversion (~99.5%) could be achieved by portion-wise addition of 20 eq. of Sq(OMe)2 (10 + 5 + 5 eq. additions at 0, 3 and 6 h). The two parallel experiments, (3) and (4), showed excellent reproducibility.
The portion-wise addition of 20 eq. of reagent was then applied with the amino dextran 3,000. Two parallel experiments showed that after 22 h, more than 99.3% conversion was indicated by fluorescamine analysis (Fig. 6), and these are the conditions we recommend for conversion of polysaccharides to their corresponding methyl squarate derivatives.
Figure 6.
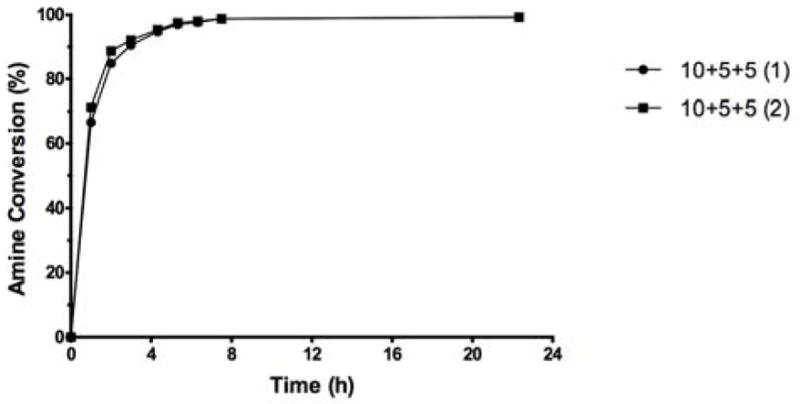
Reproducibility of the squarate preparation at optimized conditions using amino dextran, m.w. 3,000 as model compound; (1) and (2) represent two parallel experiments involving portionwise addition of 10+5+5 eq. of Sq(OMe)2.
When O-SPcNH–SqOMe was prepared at the optimized conditions [0.5 M pH 7 buffer, portion wise addition of 10+5+5 eq. of Sq(OMe)2 at 0, 3 and 6 h], the O-SPcNH2→O-SPcNH–SqOMe conversion was 98.5% after 24 h, as indicated by fluorescamine micro assay.
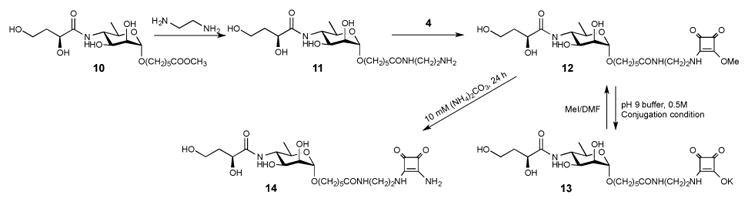
Another phenomenon that needs to be taken into account when examining preparation of the squarate derivatives from O-SPcNH2 is the possible hydrolysis of the formed O-SPcNH–SqOMe under conditions of its preparation to form the corresponding acid (PScNH–SqOMe→O-SPcNH–SqOH). Extent of such reaction was ascertained by subjecting model compound 12 (see Fig. 7) to hydrolysis experiments. When making squarate mono-amide mono-ester of low molecular mass substances, such as e.g. 12, the presence of products of their hydrolysis are easily detected by TLC and they are usually removed by chromatography. When high molecular mass O-SPcNH-SqOMe is being prepared, O-SPcNH–SqOH, if formed, remains undetected. The isolation of O-SPcNH-SqOMe is done with the aid of filtration devices equipped with appropriate cut-off membranes. This technique removes the buffer salts, excess of 4 and the product of its hydrolysis, but O-SPcNH-SqOH and O-SPcNH-SqOMe, the two groups of polysaccharides, are collected together. Thus, the conjugation reagent isolated after treatment of O-SPcNH2 with 4 contains always, in addition to O-SPcNH–SqOMe, a small but variable amount of the corresponding acid. Naturally, the more acid the conjugation reagent isolated contains, the less efficient of a conjugation reagent it becomes. We felt, therefore, that it was important to determine the stability of the O-SPcNH–SqOMe under conditions of their formation. For this purpose, model compound 12, (Scheme 4) was used (Fig. 7). When 12 was exposed to the original conditions11 for preparation of O-SPcNH–SqOMe [20 eq. of reagent, 0.05 M pH 7 buffer; Fig. 7 (1)], ~10% of the compound 12 hydrolyzed after 48 h. The rate of hydrolysis in 0.5 M buffer when 10 eq. of Sq(OMe)2 was added portion-wise or in one portion (Fig. 7) was similar. Under optimized conditions [(0.5 M pH 7 buffer, portion wise addition of 10+5+5 eq. of Sq(OMe)2, Fig. 7 (2)], only ~5% of 12 hydrolyzed after 48 h. Thus, albeit the hydrolysis of squarate monoesters, such as 12, takes place at conditions of their preparation, this side reaction is not extensive at optimized conditions (during the 24 h needed for virtually complete amine→squarate monoester conversion of high molecular mass substances).
Figure 7.
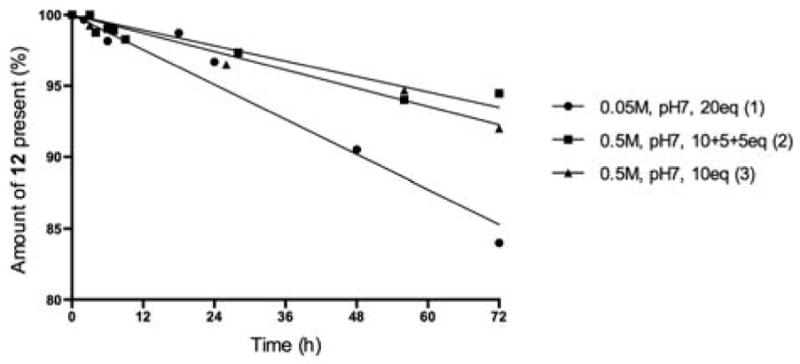
Stability of compound 12 under different conditions (concentration of 12, 4 mM).
Scheme 4.
Synthesis and derivatization of compound 12.
1.2 Effect of pH on conjugation efficiency
The benefit of conducting the conjugation of oligosaccharides at high concentration of hapten,3,26 has been established. However, problems due to the lower solubility of O-specific polysaccharides and higher viscosity of solutions of such substances at high concentration (20 mM and above3) could be anticipated. Therefore, we decided not to explore conjugation of polysaccharide antigens at higher concentrations and focused our efforts on optimizing conditions for conjugation at 5 mM antigen concentration.
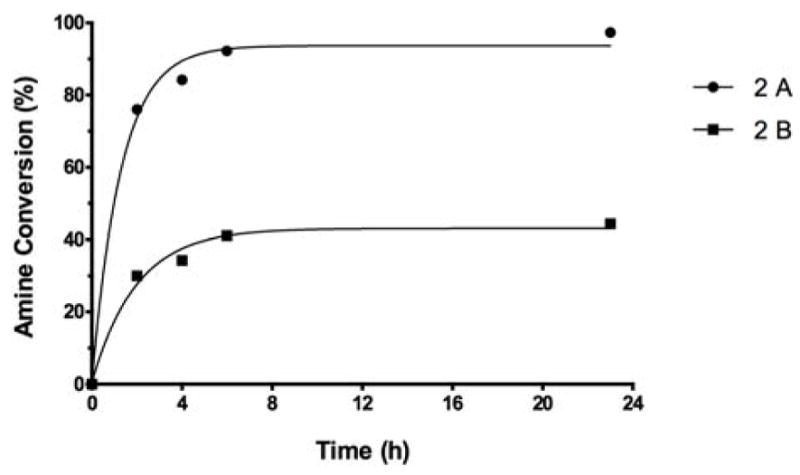
Most conjugations by squaric acid chemistry in aqueous media have been carried out at pH 9.0, which was suggested by Tietze et al. in their pioneering work.2 There are, however, examples in the literature of conjugations at higher pH.10,31–35 To examine the effect of pH on the efficiency of conjugation, we performed parallel conjugations with two different carrier proteins, BSA and rTT-Hc at pH 8.5, 9.0 and 9.5, while targeting molar carbohydrate–protein ratio (i.e. loading) 5:1. The conjugations to BSA were set up in 0.5 M borate buffer using 10 fold molar excess of O-SPcNH–SqOMe (Ogawa) at antigen concentration of 5 mM. The results are shown in Fig. 8. As can be seen, at the early stage of conjugation the reaction was much faster at pH 9.5 than at 9.0 or 8.5. After 24 h, the loading reached 4.0, 2.5 and 1.2, respectively. After 3 days, the loading in the reaction at 9.5 leveled off at ~5.0 while the other two reactions were still progressing. After 7 days, the loading in the reaction run at pH 9.0 reached 4.8 and the loading in the reaction run at pH 8.5 was still only ~3.9. The above observations show that higher pH made the conjugation faster, but the hydrolysis of the conjugation reagent (O-SPcNH–SqOMe) was also faster. That in the reaction run at pH 9.5 there was virtually no active squarate left after 3 days was indicated by no further increase of loading, while the loadings in the other two reactions were still slowly increasing.
Figure 8.
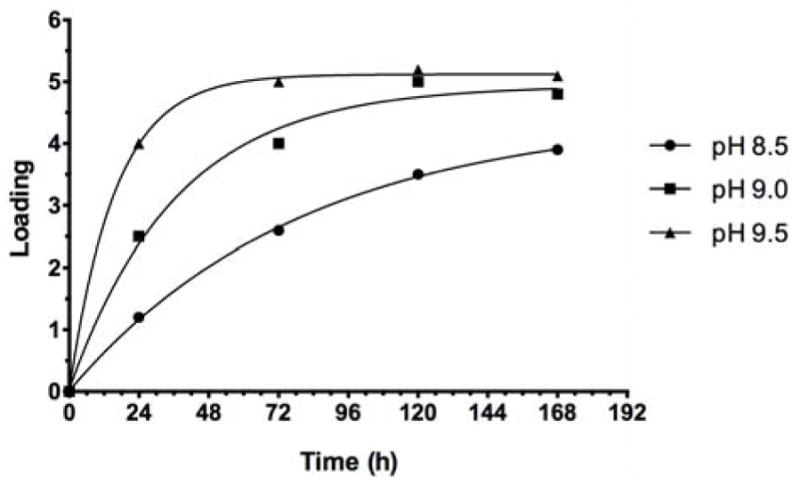
Conjugation of Ogawa O-SPcNH-SqOMe with BSA at pH 8.5, 9.0 and 9.5: Comparison of the reaction rate.
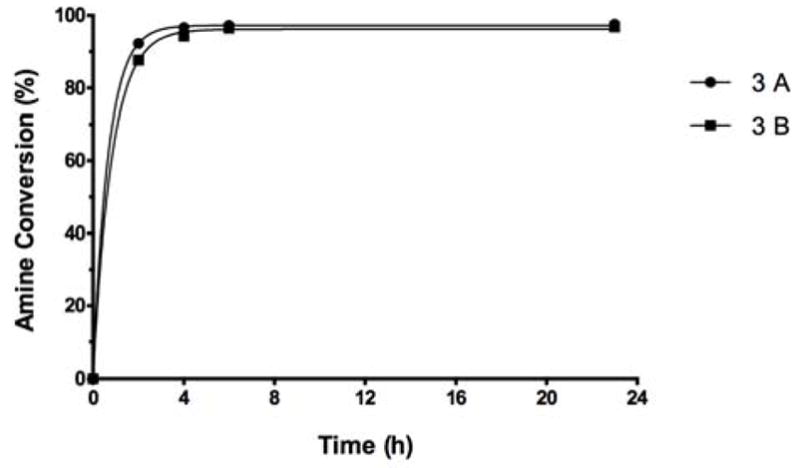
The same trend was observed when O-SPcNH-SqOMe (Inaba) was conjugated to rTT-Hc at pH 8.5, 9.0 and 9.5 (Fig. 9). The comparison of Fig. 8 and Fig. 9 shows that, generally, conjugation to rTT-Hc was faster than to BSA. The higher reactivity of rTT-Hc compared to that of BSA is likely due to structural differences of the two proteins.
Figure 9.
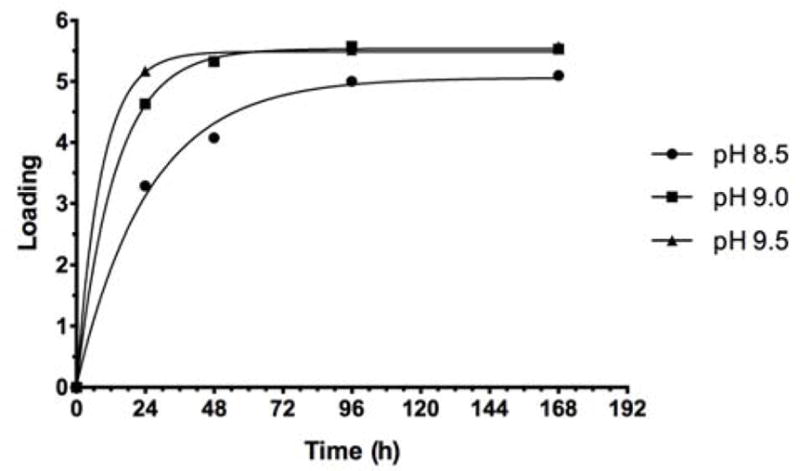
Conjugation of Inaba O-SPcNH–SqOMe with rTT-Hc at pH 8.5, 9.0 and 9.5: Comparison of the reaction rate.
The above results show the benefit of conducting conjugations by squaric acid chemistry at pH 9.5. Since the rate of the reaction may differ with different antigen/protein combinations, it is advisable to optimize reaction conditions to suit best any particular situation (reactivity and stability of antigens and proteins, etc.).
When O-SPcNH-SqOMe of Vibrio cholerae was conjugated at pH 9.5 to each of BSA and rTT-Hc (targeted carbohydrate–protein ratio ~5:1) with 10 eq. of antigen at 5 mM concentration, it yielded, after 96 hours of reaction time, conjugates with loading 5.1 (BSA, conjugation efficiency, 51%) and 5.5 (rTT-Hc, conjugation efficiency, 55%), respectively.
The increased overall efficiency of conjugation achieved, compared to the original protocol,11 lied mainly in the use of a better quality conjugation reagent. The latter resulted from the use of a higher capacity reaction buffer, which kept pH of the reaction environment for squarate labeling nearly neutral and minimized the protonation of O-SPcNH2. Also, monitoring by fluorescamine assay allowed shortening the reaction time for O-SPcNH2→O-SPcNH–SqOMe conversion. This, in turn, reduced the extent of hydrolysis of the squarate reagent formed. Consequently, the squarate labeled conjugation reagent contained a lesser amount of unconverted O-SPcNH2 and O-SPcNH–SqOH, making it a more potent conjugation reagent.
2. Recovery of spent squarate-labeled antigen and its regeneration by selective methylation at the squaric acid residue
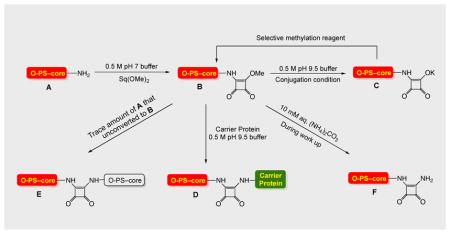
Conjugation by squaric acid chemistry is invariably accompanied by hydrolysis/saponification of the squaric acid amide ester reagent (e.g. O-SPcNH–SqOMe) to form the corresponding acid/salt.3 To compensate for losses of reagents brought about in this way, and to maintain an acceptable reaction rate, variable excess of squarate-labeled carbohydrate has been used. Because conjugation of oligosaccharides is highly efficient,3 recovery of unattached hapten is normally not an issue. Conjugation of bacterial polysaccharides is less efficient, and even after improvements achieved during this work approximately 50% of the labor-intensive polysaccharide antigen remained unconjugated. The unconjugated material can be presumed to be a multi-component mixture (see Fig. 11) of polymolecular materials, which is normally discarded, making it a rather expensive waste. The most valuable component in such mixture, O-SPcNH–SqOH(K), is the product of hydrolysis of the conjugation reagent, O-SPcNH–SqOMe. The recovery of the former, its conversion to the original ester O-SPcNH–SqOMe, and its use in another, similar conjugation would be desirable because it would increase the cost effectiveness of the conjugation process. This would make the squaric acid technology for production of conjugate vaccines from bacterial O-antigens more attractive to industrial application.
Figure 11.

Cartoon presentation of conversion of O-SPcNH2 (A) to O-SPcNH–SqOMe (B), conjugation of the latter to a carrier protein and recovery of the spent conjugation reagent.
During our study of the conjugation by squaric acid chemistry,3 we found methyl squarate derivatives of sugars very convenient conjugation reagents, and we use such reagents routinely for conjugation of both synthetic and bacterial carbohydrates. A prerequisite for successful regeneration of squarate-labeled carbohydrates, e.g. conversion of O-SPcNH–SqOH to O-SPcNH–SqOMe, is the availability of a methylation reagent which would methylate the squaric acid moiety and leave other groups amenable to methylation intact.
Because of lack of a method for monitoring the conversion O-SPcNH–SqOH→O-SPcNH–SqOMe, we examined the selectivity of methylation with various methylation reagents using suitable model compounds. This work was done within the research toward a vaccine for cholera caused by Vibrio cholerae O1 and, therefore, model compounds chosen contained structural motifs present in the O-SPcNH2 of that strain. Initially, we used the methyl glycoside of the upstream, terminal monosaccharide of the O-SP of Vibrio cholerae O1 (c.f. Fig. 1), serotype Ogawa (7)36 (Scheme 3), and treated it for 2 days with MeI and K2CO3 in DMF,37 which is a reagent known for high selectivity of methylation of carboxyl groups in the presence of hydroxyl and amide groups. The reagent would be a suitable candidate for inclusion in a future protocol for recovery of spent squarate-labeled antigen only if it left the model compound 7 intact.
Scheme 3.
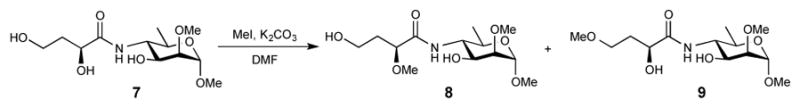
Products of treatment of 7 with MeI and K2CO3 in DMF for 48 h.
Two very minor, less polar products (Scheme 3, compounds 8 and 9) formed were isolated by chromatography and their structure was determined by spectral methods (see Experimental). Although the extent of methylation was negligible from the preparative point of view, this reagent would not be suitable for exclusive methylation of the squaric acid moiety in O-SPcNH–SqOH(K). Because products of our methylation were intended for use as antigenic components of conjugate vaccines, knowing that the byproducts of methylation would not be removed but would become part of the vaccine, even the slight modification of the antigenic determinants observed made this methylation method unacceptable. When a squaric acid derivative of a linker-equipped lactose3 was treated with diazomethane (this experiment is not described in the Experimental), it also led to various, very small amounts of less polar by-products.
Finally, we hypothesized that when O-SPcNH–SqOMe is hydrolyzed/saponified during conjugation at basic conditions, it is present in the conjugation mixture in form of its salt. If that salt could be rid of the excess base present (pH 9.5 buffer salts), the squarate moiety should hold its counter-ion because squaric acid is a very strong acid (pKl = 0.6, pK2 = 3.48).38 Subsequent treatment with MeI should then cause methylation exclusively at that site resulting in the conversion of the squaric acid residue into the corresponding methyl ester, and methylation at other sites should not occur. We tested the hypothesis with model compounds 13 (Scheme 4), 15 and 20 (Scheme 5), which are squarate derivatives of the fundamental fragments of O-SPcNH2 (c.f. Fig. 1). Compound 13 and 15 represent the upstream terminal mono- and hexasaccharide antigenic determinants of the O-SP of Vibrio cholerae O1 (Inaba and Ogawa, respectively), and compound 20 represents the carbohydrate moiety of the point of conjugation in O-SPcNH2 by squaric acid chemistry. To get the parent methyl esters 12, compound 1039 was treated first with ethylenediamine yielding 11 in 74% yield. Reaction of amine 11 with squarate dimethyl ester reagent 4 gave 12 as the exclusive product. To obtain the other parent methyl ester 19, monosaccharide 1740 was treated with hydrazine hydrate to remove the N-acetyl group. The formed amine 18, obtained in high yield, was allowed to react with dimethyl ester 4 yielding 19. Then, model compounds 13, 15 and 20 were prepared by subjecting the parent methyl esters 12 (Scheme 5), 1622, and 19 (Scheme 4) to the basic conjugation conditions to fully saponify the esters, followed by removal of excess of buffer salts by solid phase extraction. Treatment of a solution of the squarate salts 13, 15 and 20 in DMF with excess of MeI (5 eq.) each gave one largely predominating, faster moving product (>95%, TLC). A very minor, UV positive product (slower moving than 12, 16 and 19, respectively, TLC) was present in each of the three reactions. The major products were shown (TLC, MS, NMR) to be the expected esters 12, 16 and 19, respectively.
Scheme 5.
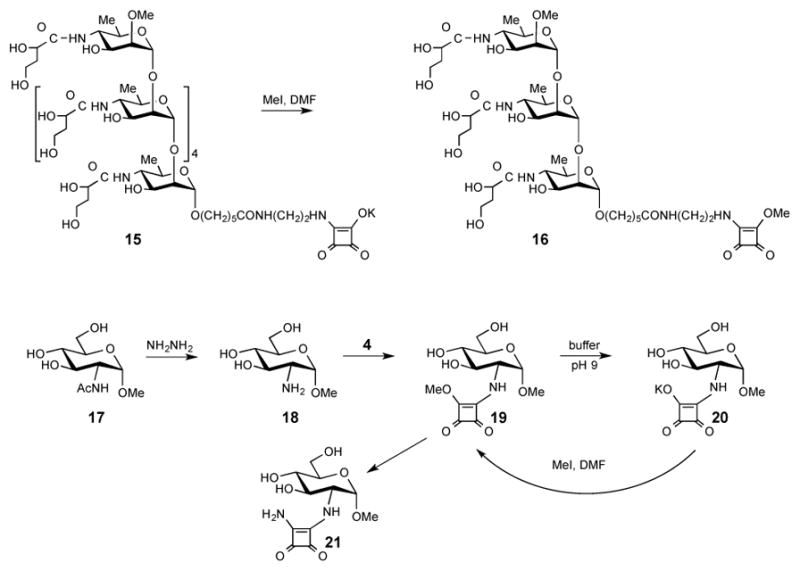
Synthesis/derivatization of model compounds 15 and 20.
In an attempt to elucidate the nature of the more polar, very minor by-products of methylation of 13, 15 and 20, the methylation of 13 was repeated on a larger scale and the by-product formed was isolated by chromatography. The compound turned out to be unstable in D2O, but its NMR spectra could be obtained for solution in CD3OD. Although the chromatographic mobility of this material was clearly different from that of 12 or 13, the HRMS of the compound agreed with that of 12. Its MS/MS showed loss of the same neutral fragments (m/z 102 and 247 Da) as those formed from 12 (Fig. 10), showing that the structure of the antigenic epitope was not altered during the methylation. Thus, while the definite structure of the very minute by-product of methylation of 13 has not been elucidated, it was clear from the above that the structural change during the treatment of 13 in DMF with MeI must have occurred within the squaric acid moiety.
Figure 10.
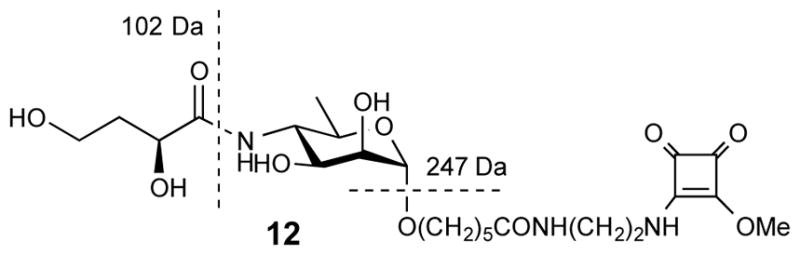
Structure of model compound 12 and loss of neutral fragments at conditions of MS/MS.
Thus, extrapolating to the situation involving a bacterial polysaccharide, if a compound analogous to the minor by product of methylation of 12 were to be formed along the regenerated O-SPcNH–SqOMe, it would either decompose during conjugation in the aqueous medium and not conjugate, or would conjugate and have the immunologically important carbohydrate epitope unchanged. Aided by the above reasoning, the methylation method used for 13 was deemed a suitable reagent to be included in the protocol for regeneration of spent squarate-labeled antigen, to effect the selective methylation of O-SPcNH–SqOK.
With the above results at hand, we recovered the spent conjugation reagent from one of our conjugations of O-SPcNH–SqOMe and subjected its solution in DMF to treatment with MeI. The product was isolated, conjugated to the medically acceptable protein, rTT-Hc, and the resulting glycoconjugate was tested for immunoreactivity in humans with cholera, and evaluated as a vaccine in mice (see below). The individual stages of the process just described were uneventful, except that the conjugation efficiency observed for the conjugation with the recovered and regenerated O-SPcNH–SqOMe was much lower than expected. A plausible explanation of the lower potency of the regenerated conjugation reagent is as follows. The likely composition of mixtures at the individual stages of the overall process, from labeling the O-SPcNH2 antigen with the squaric acid monoester residue to obtain O-SPcNH–SqOMe through the end of conjugation, until the recovered/regenerated conjugation reagent would be obtained is in the cartoon representation of the events involved (Fig. 11). Accordingly:
To convert O-SPcNH2 (A) to the conjugation reagent (B, O-SPcNH–SqOMe), dimethyl squarate 4 was added to a solution of O-SPcNH2 in 0.5 M pH 7 buffer. When the conversion was complete, as indicated by fluorescamine micro assay, the mixture was dialyzed against water through a 3 K cut-off membrane and the filtrate containing unchanged 4, products of its hydrolysis 5 and the buffer salts was discarded. The retentate containing the desired product B, perhaps some product of its hydrolysis present as salt C (because some hydrolysis of B is impossible to prevent), and, possibly, some unchanged A was freeze-dried, to give the product containing predominantly the conjugation reagent B.41
The conjugation reagent just obtained and carrier protein were allowed to react in pH 9.5 buffer to effect conjugation; only B present in the mixture reacts to form the conjugate D.42 Some B, still present because it was used in excess at the onset of the conjugation, is saponified during conjugation at pH 9.5 to form salt C. Theoretically, some amine A that remained unconverted in the squarate-forming reaction might react at pH 9.5 with squarate B to form dimer E (we have, however, determined experimentally that the extent of the dimer formation is minimal and can be neglected (see Experimental)). Nevertheless, if the recovery and regeneration is anticipated it is important to realize that the dimer, if formed, would be collected with the spent conjugation reagent. It would be present in the regenerated conjugation reagent as an inert impurity and decrease the efficiency of subsequent conjugation.
When the conjugate with a satisfactory antigen–carrier ratio had been formed, the mixture was dialyzed through 30 KDa cut-off membrane against 10 mM (NH4)2CO3.43 The contact of the unchanged methyl ester B44 with (NH4)2CO3 during work-up converts variable amounts of B (depending on factors such as time of interaction, concentration, temperature etc.) into the corresponding amide F.45 We have also established that, when targeted loading is ~5, our optimized conjugation protocol (See Experimental) requires the reaction time of conjugation to be ~3–4 days, as shown by monitoring the conjugation by SELDI-TOF mass spectrometry46,47. At that time, unchanged squarate B may still be present.42
When the exchange of pH 9.5 buffer for (NH4)2CO3 solution is deemed to be complete, the retentate containing the conjugate is freeze-dried, while the volatile salt (NH4)2CO3 is also removed. Because no other material whose molecular mass is higher than 30 KDa is present in the retentate, the freeze-dried material is the pure glycoconjugate D. It is obtained as a white fluffy solid, which can be readily reconstituted in aqueous media.
The filtrate, containing mainly the salt C, possibly some of each of amide F, ester B that was not destroyed during the work-up, unchanged O-SPcNH2 (A) that was carried over from squarate preparation step, the dimer (E) if any, and the buffer salts, is dialyzed against water using a 3 KDa cut-off membrane. The filtrate from this operation contains only buffer salts, and is discarded.
The retentate contains the same components as before dialysis except the buffer salts that have now been removed. In order to simplify the further discussion, this retentate (recovered material) will be denoted X (Fig. 11). Because squaric acid is a very strong acid30,38, the hydrolyzed squarate conjugation reagent is present in the form of salt C. When, after freeze-drying, a solution of X in DMF is treated with MeI in the absence of additional base, C is methylated at the squarate residue. The methylation mixture is dialyzed through a 3 KDa cut-off membrane against ultra pure water, and a solution of the retentate is freeze-dried to afford the recovered/regenerated conjugation reagent B-1 as a white solid. As already emphasized, the less unchanged O-SPcNH–SqOMe is present at the end of conjugation, the less amide F is formed during dialysis against the solution of (NH4)2CO3, resulting in higher quality of the recovered conjugation reagent B-1.
Comparison of NMR spectra of the foregoing product of methylation with those of the O-SPcNH–SqOMe made from the freshly prepared O-SPcNH2 was not very informative regarding the degree of methylation. However, when the regenerated methyl squarate B-1 was treated at pH 9.5 with rTT-Hc (10 eq., as in the experiment when fresh O-SPcNH–SqOMe was used, this experiment is not described in the Experimental), monitoring of the progress of conjugation by SELDI-TOF MS showed slow but steady increase of molecular mass of the formed glycoconjugate. The targeted antigen–rTT-Hc ratio ~5:1 was not reached after the expected reaction time of 4 d,11 but when 20 molar excess of the reagent prepared from the recovered material was used in another experiment, a conjugate (D-1) with antigen–rTT-Hc ratio 5.2:1 was obtained after 4 days. This showed that good quality conjugation reagent could be obtained by methylation of the spent antigen recovered from the conjugation reaction, although such conjugation reagent is less potent than the one prepared directly from O-SPcNH2. A plausible explanation for the lower potency of such conjugation reagent is as follows: firstly, one has to bear in mind that O-SPcNH2 is of bacterial origin, which is known to be a micro heterogeneous material. During isolation and purification of O-SPcNH2 from the bacteria, a small amount of material that does not contain the amino group may have been co-isolated and might be present in the O-SPcNH2 as a contaminant. Such material, if present, does not form the squarate derivative and, therefore, does not conjugate. After the conjugation cycle is complete, ~50% of the antigen used at the onset of conjugation attaches to the protein, and the material that did not conjugate owing to the lack of amino group is collected together with the material amenable to regeneration. Secondly, as already discussed, there would be also a certain amount of squarate dimer (E) and amide (F) carried over and not converted to methyl squarate. Lastly, experiments with model compounds have shown that the remethylation process is a high yielding reaction, but the same reaction with polysaccharide material may not be equally efficient. The extent of the side reactions just described may vary from batch to batch and is impossible to quantify at the present state of the art. Thus, variable amounts of different materials that cannot be converted into O-SPcNH–SqOMe are always present in the spent and recovered conjugation reagent as inert contaminants. Even though some, but not all undesired material could be removed from the crude conjugation reagent, not much would be accomplished by adding an additional operation to the protocol, which would result in manipulative losses of the active squarate reagent. Presence of material other than the active squarate in the crude conjugation reagent does not affect the quality of the resulting vaccine because all foreign material can be completely removed after conjugation. Thus, contaminants accumulate in the recovered/regenerated material and their contribution in weight-percentage increase after every regeneration cycle. Therefore, the recovery from the 2nd generation of spent conjugation reagent was not attempted.
In order to evaluate whether the recovered/regenerated methyl squarate of O-SPcNH2 from Vibrio cholerae O1 (Inaba) retained its immunoreactivity and immunogenicity, we used conjugate D-1 and (a) analyzed its immuno-recognition using plasma from humans with cholera and compared its immuno-recognition to that of OSP:rTTHc (D) made using “fresh” O-SPcNH2, and (b) compared the immunogenicity as a vaccine of the two preparations in mice. In brief, we found that immuno-recognition of the two preparations was the same in humans (Fig 12): humans recovering from cholera developed comparable immune responses by day 7 to both preparations, and this response was not present in humans recovering from typhoid fever. These results strongly suggest that the OSP-moiety of these two conjugates were equivalently displayed in a biologically relevant manner, and that the reprocessing involved in regenerating O-SPcNH–SqOMe did not affect immunoreactivity.
Figure 12.
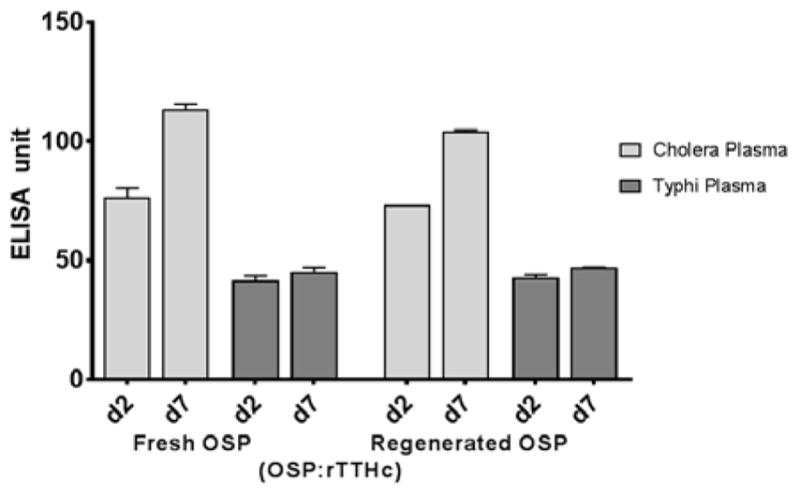
Equivalent and specific immunoreactivity of human plasma from patients with cholera or typhoid fever with OSP:rTTHc made from fresh or from regenerated V. cholerae O-SPcNH–SqOMe.
To further address the potential utility of conjugates made with fresh or regenerated O-SPcNH–SqOMe, we also immunized mice with OSP:rTTHc conjugates generated using one or the other O-SPcNH–SqOMe preparation. We found that immune responses induced by these two vaccine versions were equivalent (Fig 13). These results strongly support the utility of approaches incorporating regenerated O-SPcNH–SqOMe, and represent a further advantage of this conjugation technology based on squaric acid chemistry.
Figure 13.

Serum IgG responses to OSP:BSA in mice immunized intramuscularly with OSP:rTTHc generated using fresh or regenerated O-SPcNH–SqOMe. Mice were immunized on days 0, 14, and 28. Serum (day 0 and day 35) was analysed for IgG antibodies targeting V. cholerae O1 OSP. Immune responses were equivalent in mice receiving the two OSP:rTTHc vaccine preparations.
Conclusions
Processes that have not been previously recognized but take place during the conjugation of bacterial carbohydrates to proteins by squaric chemistry have been identified. Based on this new information, the original protocol11 for labeling bacterial O-SPcNH2 antigens with methyl squarate residue and conjugation of the resulting conjugation reagent has been revised. The main improvement resulted from conducting the squarate preparation in a higher capacity, higher concentration buffer. Beneficial was also conducting conjugation at pH 9.5, rather than the conventionally applied pH 9. Monitoring the O-SPcNH2→O-SPc-SqOMe conversion by fluorescamine micro assay allowed shortening of the reaction time during the conversion. Application of the revised protocol increased efficiency of conjugation of the amino group-containing fragment of the LPS from Vibrio cholerae O1 to BSA from ~25% to ~51% (the efficiency was 55% when rTT-Hc was the carrier). The overall efficiency of conjugation could be further increased somewhat by recovering the spent squarate reagent, and reactivating it by selective methylation at the squaric acid residue. The conjugate prepared from the recovered antigen was equally and specifically immunoreactive when analyzed with serum from humans with cholera, compared to the immunoreactivity of the conjugate made using fresh O-SPcNH2. Similarly, immunogenicity of the two conjugates made from the fresh and regenerated reagents was comparable when analyzed in vaccine studies in mice. The increased overall efficiency of conjugation and the ability to recover and regenerate bacterial polysaccharide-derived conjugation reagents improve the economy of the conjugate vaccine preparation, and make squaric acid technology more attractive for industrial preparation of conjugate vaccines. The conjugation conditions optimized here for the O-SP–core from Vibrio cholerae O1, may have to be adjusted for conjugation of other bacterial polysaccharides and/or carriers. The diverse processes involved in squarate labeling and conjugation identified here can aid in rational optimization of protocols when other bacterial carbohydrates are to be conjugated to carriers. Some objections48,49 to the use of squaric acid chemistry for preparation of immunogens were recently invalidated.15 Despite multi-facet processes taking place during conversion of amino group-containing polysaccharides to the corresponding glycoconjugates, the protocol suggested here for conjugation is simple and easy to perform. Thus, because of its efficiency and experimental simplicity, when polysaccharide antigens are amenable to conjugation by squaric acid chemistry, the latter should become a method of choice as a conjugation tool in conjugate vaccine development and manufacturing.
Experimental Section
General methods
Isolation and purification of the O-SPcNH2 fragment of the LPS of vibrio cholerae O1 has been described.11 The O-SPcNH2→O-SPcNH–SqOMe conversion was monitored by fluorescamine microassay29, conducted with a Thermo Scientific NanoDrop 3300 fluorospectrometer. The standard curve was constructed using the starting O-SPcNH2. The pH was measured with a Corning, Model 440 pH meter equipped with model MI-410 microelectrode (Microelectrodes, Inc. Bedford, NH, USA). Squarate labeling and conjugations were performed in WHEATON 0.3 or 1.0 mL V-vials, 13–425 S/T cap (Product No. W986273NG) equipped with Spin Vanes. Nuclear Magnetic Resonance (NMR) spectra were measured at 400 MHz, 500 MHz or 600 MHz for 1H and 100 MHz, 125 MHz or 150 MHz for 13C, with Bruker Avance spectrometers (Billerica, MA, USA). Solvent peaks were used as the internal reference relative to tetramethylsilane (0 ppm). Assignment of NMR signals were made by homonuclear and heteronuclear two-dimensional correlation spectroscopy, run with the software supplied with the spectrometers. Nuclei associated with the spacer are denoted with a prime in spectra assignments. The hydrolytic stability of compound 12 was monitored by HPLC using Agilent 1100 series chromatography system, and a 150 × 4.6 mm Phenomenex Luna 5 μ C18 column. Mobile phase A (0.1% TFA in MeCN) and B (0.1% TFA in H2O) were used for monitoring the course of the hydrolysis in different buffers. The elution was effected with a gradient from 0.5% of A to 60% of A over 30 min; flow rate 1 mL/min, injection volume 10 μL. Compounds were detected at 254 nm. Phenylmercuric acetate present in the pH 7 buffer as stabilizer was used as internal standard for quantitation. Buffers used for squarate labeling and conjugation were as follows. A, BuffAR pH 7.0 reference solution (0.05 M, Mallincrodt, Cat. No. 0031-04); B, 0.5 M borate buffer pH 9.0, made in house (1L) from boric acid (30.9 g), KCl (26.1 g), and KOH (8.42 g), and final adjustment to pH 9.5 by addition of solid KOH; C, pH 7.0 buffer, 0.5 M, was made by freeze-drying of 100 mL buffer A and re-dissolving the solid in 10 mL of ultra pure water. Solid Phase Extraction (SPE) cartridges Strata C18-E, 50 μm, 70A were purchased from Phenomenex. Amino dextrans m.w. 3,000 and 10,000 were purchased from Life Technologies. Solutions were concentrated at 25°C/~0.6 kPa. The approximate, average molecular mass of O-SPcNH2 (6,143 Da), was deduced from data extracted from Fig. 14. This value is close to what follows from independent determination by mass spectrometry.19 Fig. 14 shows a typical SELDI-TOF monitoring of a conjugation run at the optimized conditions. The peaks in the spectrum represent the most populated conjugate species. In this particular recording, knowing the molecular mass of the carrier protein (52,002 Da, based on the amino acid sequence50 using ExPASy Compute pl/Mw tool), the peaks at m/z 64969.47, 70841.62 and 76573.15 Da represent average loading of 2.0, 3.0 and 4.0 respectively. The average MW of the polymolecular antigen can be deduced by subtracting the molecular mass of the carrier from the m/z value at the center of a SELDI MS peak and dividing the difference by the respective anticipated loading. In this case, the average MW of the Inaba O-SPcNH2 is (76,573–52,002)/4 = 6,143 Da. Because the antigen is a biological, microheterogenic material, the average molecular mass of the antigen can vary with the source, and the mode of isolation. Based on our observation, the average MW of the Inaba O-SPcNH2 that we used ranges from ~6,000 to ~6,200 Da. The value 6,100 Da, which we use now routinely is a more realistic value for the average size of the Inaba antigen, compared to what we reported previously (5,100 Da).11. Fluorescamine was purchased from Sigma Life Science (F9015). A stock solution of fluorescamine in DMSO (3 mg/mL) was kept protected from light at room temperature. This solution is stable for at least 12 h.
Figure 14.

SELDI-TOF MS Monitoring of the conjugation of O-SPcNH–SqOMe to rTT-Hc after 24 h of reaction time.
Standard procedure for monitoring the conversion O-SPcNH2→O-SPcNH–SqOMe (fluorescamine micro assay)
A portion of the reaction mixture (1 μL) is transferred into a glass vial and diluted with pH 7.2 PBS (×1) assay buffer, to make the concentration of the sample 1 mg/mL. Fluorescamine stock solution (1/3 of the sample volume) is added, and the mixture is kept protected from light at room temperature for 15 min, whereupon it is analyzed with a NanoDrop 3300 fluorospectrometer, and the RFU (relative fluorescent units) value is recorded. For each fluorescamine assay, 2 μL of the analyzed solution is used and the average of five measurements is recorded. The initial reading should be taken before the reagent 4 is added.
Optimized O-SPcNH2→O-SPcNH–SqOMe conversion
O-SPcNH2 (11.9 mg, 0.00198 mmol, 1.0 eq.) is dissolved in 0.49 mL of pH 7.0 phosphate buffer (0.5 M) and 3,4-dimethoxy-3-cyclobutene-1,2-dione (4, 2.82 mg, 0.0198 mmol, 10.0 eq.) is added. A clear solution is formed in ~2 min. Every 3 hours, another portion of 4 (1.41 mg, 0.0099 mmol, 5.0 eq.) is added and the progress of the reaction is monitored by fluorescamine assay (see the Standard procedure above). When the total of 20 eq. of 4 had been added, the mixture is kept overnight at room temperature, when the fluorescamine assay shows that ~98.5% amine had been consumed. The mixture is filtered at 4°C through a Millipore Amicon Ultra-4 filtration device (3 KDa cut-off) and the retentate is washed with pure water (8 times). Freeze-drying of the retentate yields 10.3 mg (87%) of white, fluffy O-SPcNH–SqOMe.
Optimized conjugation protocol
Carrier protein (0.0002 mmol, 1.0 eq.) and O-SPcNH–SqOMe (0.002 mmol, 10.0 eq.) was placed into a 1 mL V vessel. 0.5 M pH 9.5 buffer (400 μL) was added and when a clear solution was formed (~5 min), it was kept at room temperature with intermittent monitoring of the progress of the conjugation by SELDI-TOF MS. When the carbohydrate/protein ratio leveled off (~3–4 d), the pH was ~9.3. The mixture was worked up as described below, to give the conjugate as white fluffy solid. In the case of Ogawa O-SPcNH–SqOMe/BSA conjugation, 5.1 loading was reached after 72 h of reaction time (yield 98%, efficiency 51%); when Inaba O-SPcNH–SqOMe was conjugated to rTT-Hc, after 96 h, 5.5 loading was reached (yield 88%, efficiency 55%). Unchanged/hydrolyzed conjugation reagent X (Fig. 11) was recovered (~35% in both cases) as white fluffy solid, as described below.
Standard work-up procedure for the conjugation reaction
When the desired antigen/protein ratio is reached, as shown by SELDI-TOF MS, the mixture is diluted with 10 mM aq. (NH4)2CO3 and passed, at 4°C, through Millipore Amicon Ultra 30 KDa cut-off ultrafiltration device (the cut-off varies depending on the mass of the conjugate; for speed, time and volume of the concentrate, manufacturer’s suggestions should be followed). The material that had passed through the membrane is collected separately. The retentate is ultrafiltered/washed 7 × with 10 mM aq. (NH4)2CO3, to ensure that all low-molecular mass material had been removed from the conjugate. The final retentate is transferred into a storage vial and lyophilized, yielding the conjugate as a white, fluffy solid.
For recovery and regeneration of the spent conjugation reagent, the material that had passed through the membrane, combined with the 7 washings is concentrated to a smaller volume and transferred into a Millipore Amicon Ultra 3k cut-off ultrafiltration devices and washed (8 ×) with pure water at 4°C as described above. The filtrate is discarded and the final retentate is transferred, with the aid of water, into a vial for freeze-drying, yielding the spent conjugation regent (X, Fig. 11) as white solid.
Regeneration of the spent conjugation reagent
In a 300 μL V vessel, the freeze-dried X (see the text above and Fig. 11) recovered from a conjugation reaction (2.85 mg, 0.000475 mmol, 1.0 eq.), is dissolved in anh. DMF (60 μL). A solution of MeI in DMF 1.1 μL (0.35 mg, 0.00246 μmol, 5.0 eq.) is added from a stock solution prepared using MeI (5.6 μL, 0.09 μmol) and DMF (34.4 μL). The reaction vessel is closed tightly and the mixture is kept in gentle motion at room temperature for 24 h. The mixture is diluted with water (follow the manufacturer’s guidelines for solvent–membrane compatibility), passed through a Millipore Amicon Ultra-4 (3 KDa cut-off membrane) device at 4°C, and the retentate is washed/ultrafiltered with pure water (7 ×). After freeze-drying, the regenerated O-SPcNH–SqOMe [B-1, 2.5 mg (88%)] is obtained as a white, fluffy solid.
Conjugation involving the regenerated O-SPcNH–SqOMe following the optimized conjugation protocol
Note: Preliminary experiments have shown that the amount of the regenerated conjugation reagent has to be doubled to achieve the same antigen–carrier ratio as with the reagent prepared from fresh O-SPcNH2.
The regenerated conjugation reagent (B-1, 4.0 mg, 0.000667 mmol, 20.0 eq.) and rTT-Hc (1.73 mg, 0.0000333 mmol, 1.0 eq.) were weighed into a 300 μL V vessel. pH 9.5 Buffer (133 μL) was added and a clear solution, which was formed within ~ 5 min, was kept at room temperature with gentle stirring, while the progress of the reaction was monitored by SELDI-TOF MS. After 4 days, loading ~5.2 was reached. The reaction was worked up following the standard work-up procedure, to give, after lyophilization, 2.2 mg (80%, efficiency 26%) of white fluffy solid conjugate D-1.
The overall conjugation efficiency is calculated from the combined amount of the antigen attached to the carrier in two consecutive conjugations, first using the conjugation reagent prepared from the fresh O-SPcNH2 (conjugation #1), and second from the recovered/regenerated conjugation reagent (conjugation #2). Because of the losses during recovery and regeneration, conjugation #2 starts with a much smaller amount of material than what would be expected based on the amount of antigen attached in conjugation #1. Accordingly, the increase of the conjugation efficiency, as a result of regeneration of the spent O-SPcNH2, amounts to ~8%.51 This raises the overall conjugation efficiency from 55% to 63%. The 8% increase of the overall efficiency might not seem very impressive, knowing that the efficiency of conjugation #1 was already 55%. However, when the efficiency of conjugation #1 with some less reactive antigens or proteins is low, it might be important to recover/regenerate the spent antigen and conduct #2 conjugation, to improve the overall conjugation efficiency.
Formation of the O-SPc dimer (E, Fig. 11)
pH 9.5 Buffer (33 μL) was added into a 300 μL V vessel containing O-SPcNH–SqOMe (1.0 mg, 0.000167 mmol, 1.5 eq.) and O-SPcNH2 (0.67 mg, 0.000111 mmol, 1.0 eq.), to form a clear solution whose concentration with respect to the squarate was 5 mM. The reaction was monitored by fluorescamine assay. The RFU reading changed after 53 h from the initial value of 3648 to 2759 (a 24% decrease). Considering that during O-SPcNH2®O-SPcNH–SqOMe conversion only a small amount (<2%) of O-SPcNH2 remains unchanged (Fig. 3) the formation of a dimer was not a significant side reaction during conjugation.
Treatment (Selective methylation) of 7 with MeI and K2CO3 in DMF and identification of products
A solution of compound 7 (250 mg, 0.85 mmol) in DMF (8 mL) was treated with MeI (605 mg, 4.26 mmol) and K2CO3 (589 mg, 4.26 mmol), and the mixture was stirred at room temperature for 48 h when TLC (15:1 CH2Cl2–MeOH) showed that most of 7 remained unchanged and that three minor, faster moving products were formed. The combined yield of the three faster moving products was less than ~5% (TLC). The mixture was filtered, concentrated and chromatography gave the two more abundant materials, sub milligram amount of each, identified by NMR and HRMS as methyl 4-(2-O-methyl-3-deoxy-L-glycero-tetronamido)-4,6-dideoxy-α-D-mannopyranoside (8) and methyl 4-(4-O-methyl-3-deoxy-L-glycero-tetronamido)-4,6-dideoxy-α-D-mannopyranoside (9). Compound 8: 1H NMR (500 MHz, CDCl3): δ 6.47 (d, 1H, J = 10.0 Hz, 4-NH), 4.79 (s, 1H, H-1), 3.95 (q, 1H, J = 10.0 Hz, H-4), 3.86 (t, 1H, J = 5.6 Hz, H-2′), 3.80-3.73 (m, 3H, H-4′, H-3), 3.68-3.62 (m, 1H, H-5), 3.50 (s, 3H, 2-OMe), 3.45 (dd, 1H, J = 1.5 Hz, 3.2 Hz, H-2), 3.43 (s, 3H, 2′-OMe), 3.38 (s, 3H, 1-OMe), 2.13-2.06 (m, 1H, H-3′a), 2.03-1.97 (m, 1H, H-3′b), 1.26 (d, 3H, J = 6.3 Hz, H-6); 13C NMR (125 MHz, MeOD): δ 97.5 (C-1), 80.7 (C-2′), 79.0 (C-2), 69.9 (C-3), 66.9 (C-5), 59.0 (2-OMe), 58.9 (C-4′), 58.2 (2′-OMe), 55.0 (1-OMe), 53.8 (C-4), 34.6 (C-3′), 18.0 (C-6); TOF-HRMS m/z: [M+H]+ calcd for C13H26NO7: 308.1704, Found 308.1703. Compound 9: 1H NMR (500 MHz, CDCl3): δ 6.80 (d, 1H, J = 9.5 Hz, 4-NH), 4.80 (s, 1H, H-1), 4.31 (br, 1H, H-2′), 3.93 (q, 1H, J = 10.0 Hz, H-4), 3.75-3.63 (m, 4H, H-4′, H-3, H-5), 3.50 (s, 3H, 2-OMe), 3.45 (dd, 1H, J = 1.5 Hz, 3.4 Hz, H-2), 3.39 (s, 3H, 1-OMe), 3.37 (s, 3H, 4′-OMe), 2.21-2.16 (m, 1H, H-3′a), 2.10-2.04 (m, 1H, H-3′b), 1.25 (d, 3H, J = 6.3 Hz, H-6); 13C NMR (125 MHz, MeOD): δ 97.4 (C-1), 79.2 (C-2), 73.4 (C-2′), 71.8 (C-4′), 70.0 (C-3), 67.0 (C-5), 59.1 (4′-OMe), 59.0 (2-OMe), 55.0 (1-OMe), 53.8 (C-4), 32.7 (C-3′), 17.9 (C-6); TOF-HRMS m/z: [M+H]+ calcd for C13H26NO7: 308.1704, Found 308.1703.
(2-Aminoethylamido)carbonylpentyl 4-(3-deoxy-L-glycero-tetronamido)-4,6-dideoxy-α-D-mannopyranoside (11)
Compound 1039 (1.7 g, 4.32 mmol) was dissolved in ethylenediamine (30 mL, 449 mmol) and stirred at 45°C for 18 h, when TLC showed that the reaction was complete. The mixture was concentrated and the residue was co-evaporated with water (× 2). The crude product was chromatographed (1:2:0.1 EtOAc–MeOH–30% NH4OH) to give compound 11 as colorless syrup (1.35 g, 74%). Compound 11: 1H NMR (600 MHz, MeOD): δ 4.72 (s, 1H, H-1), 4.18 (dd, 1H, J = 3.8 Hz, 8.6 Hz, H-2′), 3.83-3.81 (m, 2H, H-2, H-3), 3.77-3.75 (m, 2H, H-4, H-5), 3.63 (dd, 2H, J = 6.3 Hz, 7.0 Hz, H-4′), 3.59 (dt, 1H, J = 10.0 Hz, 6.7 Hz, H-1″a), 3.43 (dt, 1H, J = 10.0 Hz, 6.3 Hz, H-1″b), 3.19 (t, 2H, J = 6.2 Hz, H-7″), 2.71 (t, 2H, J = 6.2 Hz, H-8″), 2.17 (t, 2H, J = 7.5 Hz, H-5″), 1.96-1.90 (m, 1H, H-3′a), 1.78-1.72 (m, 1H, H-3′b), 1.54-1.49 (m, 4H, H-2″, H-4″), 1.29-1.23 (m, 2H, H-3″), 1.07 (d, 3H, J = 5.6 Hz, H-6); 13C NMR (150 MHz, MeOD): δ 177.2 (C-6″), 177.0 (C-1′), 99.5 (C-1), 69.1 (C-2), 68.7 (C-2′), 67.7 (C-3), 67.5 (C-1″), 67.1 (C-5), 57.6 (C-4′), 52.6 (C-4), 40.2 (C-7″), 39.4 (C-8″), 35.7 (C-3′), 35.4 (C-5″), 27.9 (C-2″), 24.7 (C-4″), 24.6 (C-3″), 16.5 (C-6); TOF-HRMS m/z: [M+H]+ calcd for C18H36N3O8: 422.2502, Found 422.2503.
1-[(2-Aminoethylamido)carbonylpentyl 4-(3-deoxy-L-glycero-tetronamido)-4,6-dideoxy-α-D-mannopyranoside]-2-methoxycyclobutene-3,4-dione (12)
3,4-Dimethoxy-3-cyclobutene-1,2-dione (600 mg, 4.22 mmol) was added to a solution of compound 11 (890 mg, 2.11 mmol) in MeOH (30 mL) and the mixture was stirred at room temperature overnight. The clear solution was concentrated, and chromatography (5:1→4:1 EtOAc–MeOH) gave compound 12 as white, amorphous solid (780 mg, 70%). Compound 12: 1H NMR (600 MHz, MeOD): δ 4.71 (d, 1H, J = 1.2 Hz, H-1), 4.37 (d, 3H, J = 15.0 Hz, -OCH3), 4.18 (dd, 1H, J = 4.0 Hz, 8.0 Hz, H-2′), 3.91 (t, 1H, J = 9.9 Hz, H-4), 3.85-3.77 (m, 3H, H-3, H-5, H-2), 3.73 (m, 2H, H-4′), 3.69-3.65 (m, 2H, H-1″a, H-8″a), 3.49 (t, 1H, J = 5.6 Hz, H-8″b), 3.42-3.37 (m, 3H, H-1″b, H-7″ab), 2.21-2.16 (m, 2H, H-5″), 2.03-1.98 (m, 1H, H-3′a), 1.86-1.80 (m, 1H, H-3′b), 1.64-1.59 (m, 4H, H-2″, H-4″), 1.42-1.38 (m, 2H, H-3″), 1.17 (d, 3H, J = 6.0 Hz, H-6); 13C NMR (150 MHz, MeOD): δ 190.0 (squarate-C), 189.8 (squarate-C), 185.1 (squarate-C), 185.0 (squarate-C), 178.7 (C-10″), 178.0 (C-10″), 178.0 (4-NHCO), 176.4 (C-6″), 175.0 (C-9″), 174.9 (C-9″), 101.6 (C-1), 71.5 (C-2), 70.8 (C-2′), 70.2 (C-3), 68.6 (C-5), 68.2 (C-1″), 61.2 (-OCH3), 61.1 (-OCH3), 59.4 (C-4′), 54.4 (C-4), 45.3 (C-8″), 45.1 (C-8″), 40.9 (C-7″), 40.6 (C-7″), 38.2 (C-3′), 36.9 (C-5″), 30.2 (C-2″), 26.9 (C-3″), 26.8 (C-3″), 26.6 (C-4″), 26.4 (C-4″), 18.3 (C-6); TOF-HRMS m/z: [M+Na]+ calcd for C23H37N3O11Na: 554.2326, Found 554.2322; Anal. Calcd for C23H37N3O11: C, 51.97; H, 7.02; N, 7.91. Found: C, 51.75; H, 6.96; N, 7.78.
Hydrolysis of compound 12 in different buffers monitored by HPLC
Fig. 7, condition (1): Compound 12 (1 mg, 1.88 μmol, 1.0 eq.) was dissolved in buffer A (470 μL) and 3,4-dimethoxy-3-cyclobutene-1,2-dione (5.36 mg, 37.6 μmol, 20.0 eq.) was added.
Fig. 7, condition (2): Compound 12 (1 mg, 1.88 μmol, 1.0 eq.) was dissolved in buffer B (470 μL) and 3,4-dimethoxy-3-cyclobutene-1,2-dione was added in portions at 0 h (2.67 mg, 18.8 μmol, 10.0 eq.), 3 h (1.33 mg, 9.4 μmol, 5 eq.) and 6 h (1.33 mg, 9.4 μmol, 5.0 eq.)
Fig. 7, condition (3): Compound 12 (1 mg, 1.88 μmol, 1.0 eq.) was dissolved in buffer B (470 μL) and 3,4-dimethoxy-3-cyclobutene-1,2-dione (2.67 mg, 18.8 μmol, 10.0 eq.) was added;
The progress of the reactions was monitored by HPLC at various time intervals, as described in General Methods. The results are shown in Fig. 7.
Saponification of squarates 12, 16 and 19
Each of compound 12, 16 and 19 was dissolved in pH 9 buffer (buffer B) to make a 20 mM solution, and the progress of the hydrolysis at room temperature was monitored by HPLC. After 14 days, more than 95% of the squarate was hydrolyzed. The reaction mixture was directly loaded on an SPE cartridge (loading should not exceed 1% of the cartridge packing) and eluted with pure water until the elution of the buffer salts was complete, as indicated by pH of the eluent (pH above 7 indicates presence of salt; basic eluate that chars has to be reprocessed). The elution was started with 2 column volumes of 100% water, and the concentration of MeOH was increased stepwise by 5% after every 2 column volumes, until 80:20 water–MeOH was reached. Because compound 20 was poorly retained, elution was conducted with 100% of water. Even then, several cartridges had to be used in order to effect purification. Compound 13: 1H NMR (600 MHz, MeOD): δ 4.69 (d, 1H, J = 1.4 Hz, H-1), 4.19 (dd, 1H, J = 4.0 Hz, 8.0 Hz, H-2′), 3.93 (t, 1H, J = 10.3 Hz, H-4), 3.84 (dd, 1H, J = 3.2 Hz, 10.3 Hz, H-3), 3.80-3.76 (m, 2H, H-2, H-5), 3.75-3.72 (m, 2H, H-4′), 3.68-3.64 (m, 3H, H-8″, H-1″a), 3.41-3.37 (m, 3H, H-7″, H-1″b), 2.19 (t, 2H, J = 7.3 Hz, H-5″), 2.04-1.98 (m, 1H, H-3′a), 1.87-1.81 (m, 1H, H-3′b), 1.64-1.55 (m, 4H, H-4″, H-2″), 1.41-1.36 (m, 2H, H-3″), 1.17 (d, 3H, J = 6.3 Hz, H-6); 13C NMR (150 MHz, MeOD): δ 195.8, 189.0, 181.3, 176.8 (C-1′), 175.2 (C-6″), 100.4 (C-1), 70.3 (C-2), 69.6 (C-2′), 69.0 (C-3), 67.4 (C-5), 67.1 (C-1″), 58.3 (C-4′), 53.1 (C-4), 42.8 (C-8″), 40.4 (C-7″), 37.0 (C-3′), 35.8 (C-5″), 29.0 (C-2″), 25.6 (C-3″), 25.3 (C-4″), 17.1 (C-6); TOF-HRMS m/z: [M+H]+ calcd for C22H35KN3O11: 556.1909, Found 556.1899. Compound 15: 1H NMR (600 MHz, D2O): δ 5.25 (s, 1H, H-1VI), 5.23 (s, 1H, H-1V), 5.22 and 5.21 (s, 3H, H-1II–IV), 4.92 (s, 1H, H-1I), 4.35-4.32 (m, 6H, H-2′I–VI), 4.22-4.19 (m, 8H, H-2II–V, H-3II–V), 4.12 (dd, 1H, J = 2.7 Hz, 10.8 Hz, H-3VI), 4.10 (dd, 1H, J = 2.7 Hz, 10.8 Hz, H-3I), 4.00 (m, 5H, H-2I, H-4II–V), 3.98 (m, 5H, H-5II–V, H-4I), 3.95 (m, 1H, H-5VI), 3.91 (m, 1H, H-5I), 3.88 (m, 1H, H-4VI), 3.82 (m, 1H, H-2VI), 3.80-3.77 (m, 12H, H-4′I–VI), 3.73 (t, J = 5.2 Hz, H-8″), 3.70-3.67 (m, 1H, H-1″a), 3.53 (s, 3H, 2VI-OMe), 3.54-3.51 (m, 1H, H-1″b), 3.45 (t, J = 5.2 Hz, H-7″), 2.25 (t, 2H, J = 7.1 Hz, H-5″), 2.11-2.06 (m, 6H, H-3′aI–VI), 1.93-1.87 (m, 6H, H-3′bI–VI), 1.61-1.55 (m, 4H, H-2″, H-4″), 1.35-1.29 (m, 2H, H-3″), 1.24-1.22 (m, 15H, H-6I–V), 1.19 (d, 3H, J = 6.0 Hz, H-6VI); 13C NMR (150 MHz, D2O): δ 194.1 (squarate), 181.3 (C-9″), 177.2 (C-1′I–V), 177.0 (C-1′VI), 176.9 (C-6″), 100.5 and 100.4 (C-1II–V), 98.7 (C-1VI), 98.1 (C-1I), 78.6 (C-2VI), 77.3 (C-2I), 77.2 (C-2V), 76.9 and 76.8 (C-2II–IV), 68.7 (C-2′I–VI), 68.0 (C-5II–V), 67.7 (C-5VI), 67.5 (C-1″), 67.4 (C-3I), 67.3 (C-5I), 67.2 (C-3VI), 67.2 and 67.1 (C-3II–V), 58.5 (2VI-OMe), 57.6 (C-4′I–VI), 52.9 (C-4VI), 52.8 (C-4I), 52.7 and 52.6 (C-4II–V), 43.0 (C-8″), 39.4 (C-7″), 35.7 and 35.6 (C-3′I–VI), 35.5 (C-5″), 27.9 (C-2″), 24.8 (C-4″), 24.5 (C-3″), 16.6 and 16.5 (C-6I–VI); TOF-HRMS m/z: [M+H]+ calcd for C73H122KN8O41: 1805.7345, Found 1805.7350. Compound 20: 1H NMR (600 MHz, D2O): δ 4.94 (d, 1H, J = 3.6 Hz, H-1), 4.12 (dd, 1H, J = 3.6 Hz, 10.2 Hz, H-2), 3.93 (d, 1H, J = 12.2 Hz, H-6a), 3.85-3.80 (m, 2H, H-6b, H-3), 3.76-3.74 (m, 1H, H-5), 3.55 (dd, 1H, J = 9.3 Hz, 9.9 Hz, H-4), 3.46 (s, 3H, 1-OMe); 13C NMR (150 MHz, D2O): δ 195.2 (squarate), 181.1 (squarate), 98.6 (C-1), 71.6 (C-3), 71.5 (C-5), 69.4 (C-4), 60.4 (C-6), 57.5 (C-2), 55.1 (1-OMe); TOF-HRMS m/z: [M+H]+ calcd for C11H15KNO8: 328.0435, Found 328.0433.
Methyl 2-amino-2-deoxy-α-D-glucopyranoside (18)
Compound 17 (1.0 g, 4.25 mmol) was dissolved in hydrazine hydrate (20 mL) and the solution was kept at 120°C for 36 h. TLC (3:1:0.1 CH2Cl2–MeOH–30% NH4OH) showed disappearance of the starting material and formation of a slower moving product. The mixture was concentrated at 60°C under high vacuum (<0.1 mmHg) to remove hydrazine. Chromatography of the residue (5:1:0.1→3:1:0.1 CH2Cl2–MeOH–30% NH4OH) gave 18 (705 mg, 3.65 mmol, 86%), m.p. 159–163°C (EtOH), [α]D 152.5° (c 0.84, H2O). Compound 18: 1H NMR (400 MHz, DMSO-d6): δ 4.51 (d, 1H, J = 3.6 Hz, H-1), 3.63 (dd, 1H, J = 1.8 Hz, 11.7 Hz, H-6a), 3.44 (dd, 1H, J = 5.7 Hz, 11.7 Hz, H-6b), 3.30 (m, 1H, H-5), 3.26 (s, 3H, 1-OMe), 3.16 (dd, 1H, J = 9.0 Hz, 9.6 Hz, H-3), 3.03 (dd, 1H, J = 9.0 Hz, 9.6 Hz, H-4), 2.36 (dd, 1H, J = 3.6 Hz, 9.8 Hz, H-2); 13C NMR (100 MHz, DMSO-d6): δ 100.1 (C-1), 75.2 (C-3), 73.0 (C-5), 70.4 (C-4), 61.0 (C-6), 56.3 (C-2), 54.3 (1-OMe); TOF-HRMS m/z: [M+H]+ calcd for C7H16NO5: 194.1028, Found 194.1025; Anal. Calcd for C7H15NO5: C, 43.52; H, 7.83; N, 7.25. Found: C, 43.56; H, 7.71; N, 7.33.
1-[2-N-(2-amino-2-deoxy-1-O-methyl-α-D-glucopyranose)-yl]-2-methoxycyclobutene-3,4-dione (19)
3,4-Dimethoxy-3-cyclobutene-1,2-dione (629 mg, 4.43 mmol) was added to a solution of 18 (570 mg, 2.95 mmol) in buffer A (10 mL, pH 7.0). The clear solution formed was kept at room temperature for 1 h, when TLC (5:1 EtOAc–MeOH) showed disappearance of the starting material. After concentration and chromatography (10:1→5:1 EtOAc-MeOH), compound 19 was obtained (800 mg, 89%), m.p. 168–169.5 °C (EtOH), [α]D -67.8° (c 1.0, MeOH). Compound 19: 1H NMR (400 MHz, MeOD): δ 4.77 (d, 1H, J = 3.6 Hz, H-1), 4.38, 4.37 (s, s, 3H, squarate-OMe), 4.14 (dd, J = 3.6 Hz, 10.4 Hz, H-2a), 3.84 (dd, 1H, J = 1.6 Hz, 12.0 Hz, H-6a), 3.75-3.68 (m, 2H, H-6b, H-3), 3.64 (dd, J = 3.6 Hz, 10.2 Hz, H-2b), 3.60-3.55 (m, 1H, H-5), 3.42, 3.41 (s, s, 3H, 1-OMe), 3.37 (m, 1H, H-4); 13C NMR (100 MHz, MeOD): δ 189.9, 189.6, 185.3, 178.5, 178.1, 175.0, 174.9 (squarate), 100.3 (C-1a), 100.1 (C-1b), 73.8 (C-5), 73.3 (C-3), 71.9 (C-4a), 71.8 (C-4b), 62.6 (C-6a), 62.5 (C-6b), 61.2 (squarate-OMe-a), 61.0 (squarate-OMe-b), 60.8 (C-2b), 59.9 (C-2a), 55.6 (1-OMe); TOF-HRMS m/z: [M+H]+ calcd for C12H18NO8: 304.1032, Found 304.1031; Anal. Calcd for C12H17NO8: C, 47.53; H, 5.65; N, 4.62. Found: C, 47.42; H, 5.71; N, 4.52.
1-[(2-Aminoethylamido)carbonylpentyl 4-(3-deoxy-L-glycero-tetronamido)-4,6-dideoxy-α-D-mannopyranoside]-2-aminocyclobutene-3,4-dione) 14
A solution of compound 12 (10.0 mg, 18.8 μmol) in 10 mM (NH4)2CO3 (100 mL) was kept at room temperature for 24 h. The solution was freeze-dried and the white fluffy solid residue was chromatographed (4:1 EtOAc–MeOH). Eluted first was product 14 (8.9 mg, 92%), which was freeze-dried and obtained as a fluffy amorphous solid. Eluted next was a small amount of the starting material 12 (0.8 mg, 8%). Compound 14: 1H NMR (600 MHz, D2O): δ 4.87 (d, 1H, J = 1.0 Hz, H-1), 4.34 (dd, 1H, J = 3.9 Hz, 8.7 Hz, H-2′), 3.99-3.97 (m, 2H, H-2, H-3), 3.93-3.89 (m, 2H, H-4, H-5), 3.80-3.77 (m, 4H, H-4′, H-8″), 3.70 (dt, 1H, J = 6.9 Hz, 9.8 Hz, H-1″a), 3.53 (dt, 1H, J = 6.3 Hz, 9.8 Hz, H-1″b), 3.48 (t, 2H, J = 5.4 Hz, H-7″), 2.26 (t, 2H, J = 7.2 Hz, H-5″), 2.11-2.06 (m, 1H, H-3′a), 1.93-1.87 (m, 1H, H-3′b), 1.65-1.56 (m, 4H, H-2″, H-4″), 1.36-1.32 (m, 2H, H-3″), 1.22 (d, 3H, J = 5.7 Hz, H-6); 13C NMR (150 MHz, D2O): δ 182.0, 177.0, 169.3, 99.5 (C-1), 69.1 (C-2), 68.7 (C-2′), 67.7 (C-3), 67.4 (C-1″), 67.1 (C-5), 57.6 (C-4′), 52.6 (C-4), 43.6 (C-8″), 39.3 (C-7″), 35.7 (C-3′), 35.5 (C-5″), 28.0 (C-2″), 24.8 (C-4″), 24.5 (C-3″), 16.5 (C-6); TOF-HRMS m/z: [M+Na]+ calcd for C22H36N4O10Na: 539.2329, Found 539.2333.
1-[2-N-(2-Amino-2-deoxy-1-O-methyl-α-D-glucopyranose)-yl]-2-aminocyclobutene-3,4-dione (21)
A solution of compound 19 (120 mg, 0.396 mmol) in 10 mM (NH4)2CO3 (60 mL) was kept at room temperature for 48 h. TLC (5:1 CH2Cl2–MeOH) showed that ~50% conversion was achieved. The solution was freeze-dried and the white fluffy residue was chromatographed (10:1→6:1 CH2Cl2–MeOH). Eluted first was starting material 19 (43 mg, 36%), which was freeze-dried and obtained as a fluffy amorphous solid. Eluted next was the title compound 21 (45 mg, 39%). After freeze-drying, 21 could be crystallized from EtOH as a slightly yellow crystals. Recrystallization from MeOH gave colorless crystals. Compound 21: [α]D +40° (c 0.62, MeOH); m.p. 303.5–305.5 °C (MeOH, dec.); 1H NMR (500 MHz, DMSO-d6): δ 7.41 (d, 1H, J = 9.5 Hz, 2-NH), 5.21 (d, 1H, J = 5.3 Hz, 3-OH), 5.14 (d, 1H, J = 5.3 Hz, 4-OH), 4.70 (d, 1H, J = 3.2 Hz, H-1), 4.61 (t, 1H, J = 5.8 Hz, 6-OH), 3.94 (m, 1H, H-2), 3.64 (m, 1H, H-6a), 3.48 (m, 1H, H-6b), 3.41 (m, 1H, H-3), 3.34 (m, 1H, H-5), 3.30 (s, 3H, 1-OMe), 3.15 (m, 1H, H-4); 13C NMR (125 MHz, DMSO-d6): δ 183.5, 169.7, 169.3 (squarate), 99.0 (C-1), 73.4 (C-5), 72.4 (C-3), 70.6 (C-4), 61.1 (C-6), 58.1 (C-2), 54.8 (1-OMe); TOF-HRMS m/z: [M+H]+ calcd for C11H17N2O7: 289.1036, Found 289.1035; Anal. Calcd for C11H16N2O7: C, 45.83; H, 5.59; N, 9.72. Found: C, 45.66; H, 5.53; N, 9.62.
Immunoreactivity study
Immunoreactivity of OSP:rTTHc made from “fresh” O-SPcNH–SqOMe was compared to that using regenerated O-SPcNH–SqOMe, as previously described.15 In brief, 96 well ELISA plates (Nunc) were coated with 100 ng/well of the OSP component of OSP:rTTHc made from fresh or regenerated O-SPcNH–SqOMe. Wells were probed with pooled sera (1:250 dilution 0.1% BSA in phosphate buffered saline-Tween) from 5 cholera patients (day 2 or day 7 of illness) or pooled sera of 5 humans with typhoid fever (day 2 or day 7).15 Samples were incubated at 37°C for 90 minutes and analyzed using horseradish peroxidase-labeled goat anti-human IgG (1:5000; Jackson ImmunoResearch) and developed as previously described.15 Samples from humans were collected under work approved by the Institutional Review Board of the Massachusetts General Hospital and the Ethical Review Committee of the International Centre for Diarrhoeal Disease Research in Dhaka, Bangladesh (icddr,b).
Vaccine analysis
Immunogenocity of OSP:rTTHc generated using either fresh or regenerated O-SPcNH–SqOMe was assessed in mice as previously described15. Briefly, cohorts of 10–12 Swiss Webster 3–5 week old female mice were immunized intramuscularly with 10 mcg of the OSP component of the vaccine preparations on days 0, 14, 28 and 42. We collected blood samples via tail-bleeds on day 0 (prior to vaccinating), and then on days 21, 35 and 49. We assessed OSP and TT-specific IgG responses as previously described15. Animal work met all governmental and institutional requirements, guidelines, and policies. This work was approved by the Massachusetts General Hospital Subcommittee on Research Animal Care (SRAC). The work adhered to the USDA Animal Welfare Act, PHS Policy on Humane Care and Use of Laboratory Animals, and the “ILAR Guide for the Care and Use of Laboratory Animals”.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH and NIDDK (PX and PK) and extramural support from NIAID (U01 AI106878 [ETR and FQ].
References
- 1.Kamath VP, Diedrich P, Hindsgaul O. Glycoconj J. 1996;13:315. doi: 10.1007/BF00731506. [DOI] [PubMed] [Google Scholar]
- 2.Tietze LF, Schröter C, Gabius S, Brinck U, Goerlach-Graw A, Gabius HJ. Bioconj Chem. 1991;2:148. doi: 10.1021/bc00009a003. [DOI] [PubMed] [Google Scholar]
- 3.Hou Sj, Saksena R, Kováč P. Carbohydr Res. 2008;343:196. doi: 10.1016/j.carres.2007.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wurm FR, Klok HA. Chem Soc Rev. 2013;42:8220. doi: 10.1039/c3cs60153f. [DOI] [PubMed] [Google Scholar]
- 5.Bundle DR. In: Vaccine Design: Innovative Approaches and Novel Strategies. Bagnoli F, Rappuoli R, editors. Caister Academic Press; 2011. p. 69. [Google Scholar]
- 6.Auzanneau FI, Borrelli S, Pinto BM. Bioorg Med Chem Lett. 2013;23:6038. doi: 10.1016/j.bmcl.2013.09.042. [DOI] [PubMed] [Google Scholar]
- 7.Chernyak A, Kondo S, Wade TK, Meeks MD, Alzari PM, Fournier JM, Taylor RK, Kováč P, Wade WF. J Infect Dis. 2002;185:950. doi: 10.1086/339583. [DOI] [PubMed] [Google Scholar]
- 8.Rollenhagen JE, Kalsy A, Saksena R, Quadri F, Caderwood SB, Kováč P, Wade WF, Ryan ET. Am J Trop Med Hyg Suppl. 2006;75:84. [Google Scholar]
- 9.Mawas F, Niggemann J, Jones C, Corbel MJ, Kamerling JP, Vliegenthart JFG. Infect Immun. 2002;70:5107. doi: 10.1128/IAI.70.9.5107-5114.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Safari D, Dekker HAT, Joosten JAF, Michalik D, de Souza AC, Adamo R, Lahmann M, Sundgren A, Oscarson S, Kamerling JP, Snippe H. Infect Immun. 2008;76:4615. doi: 10.1128/IAI.00472-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu P, Alam MM, Kalsy A, Charles RC, Calderwood SB, Qadri F, Ryan ET, Kováč P. Bioconj Chem. 2011;21:2179. doi: 10.1021/bc2001984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ryan ET, Kováč P, Qadri F, Xu P, Calderwood SB, Vann WF, Peterson DC. Conjugating Amines. 2013/009826. WO. 2013 Jan 17;
- 13.Ivers LC, Charles RC, Hilaire IJ, Mayo-Smith LM, Teng JE, Jerome JG, Rychert J, LaRocque RC, Xu P, Kováč P, Ryan ET, Qadri F, Almazor CP, Franke MF, Harris JB. Journal of Infectious Diseases. 2015;212:779. doi: 10.1093/infdis/jiv108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alam MM, Bufano MK, Xu P, Kalsy A, Yu Y, Freeman YW, Sultana T, Rashu MR, Desai I, Eckhoff G, Leung DT, Charles RC, LaRocque RC, Harris JB, Clements JD, Calderwood SB, Qadri Firdausi, Vann WF, Kováč P, Ryan ET. PLOS Neglected Tropical Diseases. 2014;8:e2683. doi: 10.1371/journal.pntd.0002683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sayeed MA, Bufano MK, Xu P, Eckhoff G, Charles RC, Alam MM, Kelly M, Sultana T, Rashu MR, Berger A, Gonzalez-Escobedo G, Mandlik A, Bhuiyan TR, Leung DT, LaRocque RC, Harris JB, Calderwood SB, Qadri FFVW, Kováč P, Ryan ET. PLOS Neglected Tropical Diseases. 2015;9:e3881. doi: 10.1371/journal.pntd.0003881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tietze LF, Arlt M, Beller M, Glüsenkamp KH, Jähde E, Rajewsky MF. Chem Ber. 1991;124:1215. [Google Scholar]
- 17.Saksena R, Ma X, Kováč P. Carbohydr Res. 2003;338:2591. doi: 10.1016/s0008-6215(03)00273-8. [DOI] [PubMed] [Google Scholar]
- 18.Vinogradov EV, Bock K, Holst O, Brade H. Eur J Biochem. 1995;233:152. doi: 10.1111/j.1432-1033.1995.152_1.x. [DOI] [PubMed] [Google Scholar]
- 19.Grandjean C, Boutonnier A, Dassy B, Fournier JM, Mulard AL. Glycocnjugate J. 2009;26:41. doi: 10.1007/s10719-008-9160-6. [DOI] [PubMed] [Google Scholar]
- 20.Gupta RK, Szu SC, Finkelstein RA, Robbins JB. Infect Immun. 1992;60:3201. doi: 10.1128/iai.60.8.3201-3208.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Saksena R, Adamo R, Kováč P. Bioorg Med Chem Lett. 2006;16:615. doi: 10.1016/j.bmcl.2005.10.056. [DOI] [PubMed] [Google Scholar]
- 22.Bongat AFG, Saksena R, Adamo R, Fujimoto YSZ, Peterson DC, Fukase K, Vann WF, Kováč P. Glycoconjugate J. 2010;27:69. doi: 10.1007/s10719-009-9259-4. [DOI] [PubMed] [Google Scholar]
- 23.Parameswar AR, Park IH, Saksena R, Kováč P, Nahm MH, Demchenko AV. ChemBioChem. 2009;10:2893. doi: 10.1002/cbic.200900587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kenne L, Lindberg B, Unger P, Gustafsson B, Holme T. Carbohydr Res. 1982;100:341. doi: 10.1016/s0008-6215(00)81047-2. [DOI] [PubMed] [Google Scholar]
- 25.Hisatsune K, Kondo S, Isshiki Y, Iguchi T, Haishima Y. Biochem Biophys Res Commun. 1993;190:302. doi: 10.1006/bbrc.1993.1046. [DOI] [PubMed] [Google Scholar]
- 26.Zhang J, Yergey A, Kowalak J, Kováč P. Carbohydr Res. 1998;313:15. doi: 10.1016/s0008-6215(98)00261-4. [DOI] [PubMed] [Google Scholar]
- 27.Makoff AJ, Ballantine SP, Smallwood AE, Fairweather NF. Bio/Technology. 1989;7:1043. [Google Scholar]
- 28.Louch HA, Buczko ES, Woody MA, Venable RM, Vann WF. Biochemistry. 2002;41:13644. doi: 10.1021/bi020291j. [DOI] [PubMed] [Google Scholar]
- 29.Udenfriend S, Stein S, Bohlen P, Dairman W, Leimgruber W, Weigele M. Science. 1972;178:881. doi: 10.1126/science.178.4063.871. [DOI] [PubMed] [Google Scholar]
- 30.Cohen S, Cohen SG. J Am Chem Soc. 1966;88:1533. [Google Scholar]
- 31.Benaissa-Trouw B, Lefeber DJ, Kamerling JP, Vliegenthart JFG, Kraaijeveld K, Snippe H. Infect Immun. 2001;69:4698. doi: 10.1128/IAI.69.7.4698-4701.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lefeber DJ, Kamerling JP, Vliegenthart JFG. Chem - Eur J. 2001;7:4411. doi: 10.1002/1521-3765(20011015)7:20<4411::aid-chem4411>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 33.Hossany RB, Johnson MA, Eniade AA, Pinto BM. Bioorg Med Chem. 2004;12:3743. doi: 10.1016/j.bmc.2004.03.075. [DOI] [PubMed] [Google Scholar]
- 34.Wang JW, Asnani A, Auzanneau FI. Bioorg Med Chem. 2010;18:7174. doi: 10.1016/j.bmc.2010.08.040. [DOI] [PubMed] [Google Scholar]
- 35.Chernyak A, Oscarson S, Turek D. Carbohydr Res. 2000;329:309. doi: 10.1016/s0008-6215(00)00189-0. [DOI] [PubMed] [Google Scholar]
- 36.Lei Ps, Ogawa Y, Flippen-Anderson JL, Kováč P. Carbohydr Res. 1995;275:117. doi: 10.1016/0008-6215(95)00147-l. [DOI] [PubMed] [Google Scholar]
- 37.Walvoort MTC, Sail D, van der Marel GA, Codée JDC. In: Carbohydrate Chemistry: Proven Synthetic Methods. Kováč P, editor. Vol. 1. CRC/Taylor and Francis; Boca Raton, FL: 2011. p. 99. [Google Scholar]
- 38.Schwartz LM, Howard LO. J Phys Chem. 1970;74:4374. [Google Scholar]
- 39.Xu P, Stevens ED, French AD, Kováč P. Molecules. 2015;20:2892. doi: 10.3390/molecules20022892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shashkov AS, Evtsigneev AJ, Derevitskaya VA. Carbohydr Res. 1979;72:215. [Google Scholar]
- 41.To minimize hydrolysis of B at conditions of its preparation, the conversion O-SPcNH2→O-SPcNH–SqOMe is conducted with appropriate excess of dimethyl squarate (determined experimentally with each antigen; the protocol described in the Experimental for O-SPcNH2 from Vibrio cholerae O1 should serve as a guideline) and without extending the reaction time unnecessarily (see text above and Fig.7). The capacity of the buffer has to be sufficient to maintain the pH near neutral. Using too large excess of dimethyl squarate may cause a cascade of unwanted events: hydrolysis of 4 (to give monoester acid, Sq(OMe)OH, 5) may generate enough acid to break the buffer, resulting in protonation of O-SPcNH2 and, in turn, termination of the squarate formation; more extensive hydrolysis of the squarate B might occur; that would result in free acid O-SPcNH–SqOH instead of salt C to be collected with squarate B, and the use of conjugation reagent containing a large amount of O-SPcNH–SqOH might, in turn, result in inefficient conjugation.
- 42.When recovery and reactivation of the conjugation reagent is anticipated, excess of the O-SPcNH–SqOMe reagent and the reaction time of the conjugation should be such (based on preliminary experiments) that, preferentially, no B remains unchanged when the desired carbohydrate loading onto protein is reached. This, because if a considerable amount of the unchanged B is present at the end of the conjugation, some of it will be irreversibly destroyed during work-up (dialysis against a solution of (NH4)2CO3, see later) to give O-SPcNH–SqNH2 (F).
- 43.The use of (NH4)2CO3, rather than water, is important to prevent losses of the conjugate due to its binding to microporous membranes through hydrophobic interactions.
- 44.It could be present in the conjugation mixture because it had been used in excess at the onset of conjugation and may not be fully saponified at the time the conjugation was deemed complete.
- 45.We had verified the formation of F by treating model compound 12 with 10 mM (NH4)2CO3 and identified the corresponding amide 14 (see Scheme 4, and Experimental Section for details).
- 46.Saksena R, Chernyak A, Karavanov A, Kováč P. In: Methods in Enzymology. Lee YC, Lee R, editors. Vol. 362. Academic Press; 2003. p. 125. [DOI] [PubMed] [Google Scholar]
- 47.Chernyak A, Karavanov A, Ogawa Y, Kováč P. Carbohydr Res. 2001;330:479. doi: 10.1016/s0008-6215(01)00018-0. [DOI] [PubMed] [Google Scholar]
- 48.Rich JR, Wakarchuk WW, Bundle DR. Chem - Eur J. 2006;12:845. doi: 10.1002/chem.200500518. [DOI] [PubMed] [Google Scholar]
- 49.Wu X, Ling CC, Bundle DR. Org Lett. 2004;6:4407. doi: 10.1021/ol048614m. [DOI] [PubMed] [Google Scholar]
- 50.McCarthy PC, Saksena R, Peterson DC, Lee CH, An Y, Cipollo JF, Vann WF. Glycoconj J. 2013;30:857. doi: 10.1007/s10719-013-9490-x. [DOI] [PubMed] [Google Scholar]
- 51.The contribution to overall efficiency of recovery = % of recovered spent conjugation reagent × yield of methylation (regeneration) × conjugation efficiency of conjugation #2, e.g (from above): 35% × 88% × 26% = 8%