

Abstract
The initial design, synthesis and validation of polymer-supported siloxane transfer agents have been achieved that permit the direct use of organolithium reagents in the palladium-catalyzed cross-coupling reactions. Through rational design, two generations of polymer support were developed that significantly simplify product purification and the transfer agent recycling.
Graphical abstract
Introduction
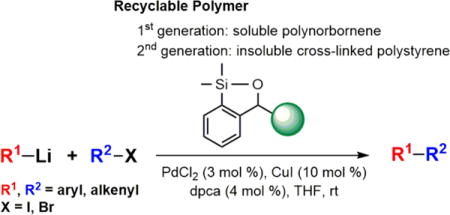
Over the past 50 years, palladium-catalyzed cross-coupling reactions (CCRs) of organometallic or main-group reagents with organic halides have revolutionized the method by which (sp2)carbon-(sp2)carbon bonds are constructed, permitting facile preparation of diverse, structurally complex molecules for the development of biologically active compounds as well as functional materials.1 Organolithium reagents are cheap and either commercially available or readily accessible through lithium-halogen exchange or via direct metalation. Not surprisingly, organolithium reagents are thus among the most commonly used highly reactive reagents in organic synthesis, and in turn comprised one of the earliest organometallic reagents to be exploited in cross-coupling reactions (i.e., the Murahashi2 CCR). Organolithiums in the Pd-catalyzed cross-coupling reactions however have some limitations, including in some cases low yields, narrow substrate scope due to the very high reactivity, and competitive formation of homo-coupled products. To overcome these issues, organolithium reagents are frequently converted to other less reactive organometallic reagents (e.g., organozincs,3 organoborons,4 organotins5 and organosilicons6) to be employed in CCR. A potentially concise and efficient method taking advantage of the direct use of organolithiums in CCR would be to eliminate the need for extra manipulations and purification steps required to generate the requisite organometallic reagents, as well as, to avoid the heavy metal and/or main group waste streams often observed in traditional cross-coupling reactions (Scheme 1).
Scheme 1.
The Waste Problem in Traditional Palladium-Catalyzed Cross-Coupling Reactions (CCR)
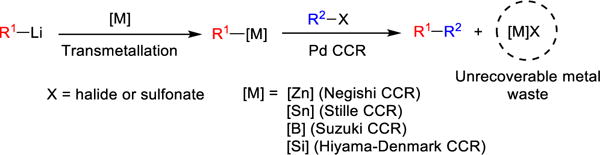
In connection with the evolution of Anion Relay Chemistry (ARC),7 we recently demonstrated that siloxane 1 and analogues8 thereof comprise competent recoverable silicon-based transfer agents for efficient palladium-catalyzed intermolecular cross-coupling reactions of aryl and alkenyl organolithium reagents at ambient temperature and pressure (Scheme 2). This tactic offers the solution to the intrinsic limitation of the Murahashi and importantly recent Feringa cross-coupling protocols2,9 of organolithiums, which are not compatible with substrates bearing sensitive functional groups such as nitriles or carbonyl functionalities (i.e., ketones, aldehyde and/or esters). Given that the siloxane-based transfer agents are fully regenerated following the CCRs, the siloxane-based method comprises an effective atom-economical approach for the construction of C-C bonds. The recovery of these transfer agents via either chromatographic separation or acid-base extraction, the latter by incorporation of a Brønsted base in the siloxane transfer structure,8b however in some cases has proven less than optimal. The development of solid-supported transfer agents hold the promise of significantly simplifying product purification, and thereby providing an attractive and practical method for recycling of transfer agents, further advancing the siloxane-based cross-coupling tactic (e.g., a polymer with attached siloxane transfer agents in flow reactors). We therefore recently undertook the incorporation of siloxane transfer agents onto readily recyclable polymer supports.10 Herein we record a full account on the rational design, synthesis, and validation of polymer-supported silicon reagents for palladium-catalyzed cross-coupling reactions of organolithiums.
Scheme 2.
Cross-Coupling Reactions of Organolithiums Mediated by Siloxane Transfer Agent
Results And Discussion
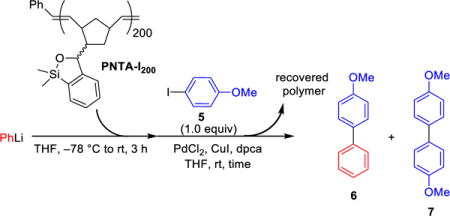
A. First-Generation Design, Synthesis and Validation of Polymer-Supported Siloxane Transfer Agents for Cross-Coupling Reactions
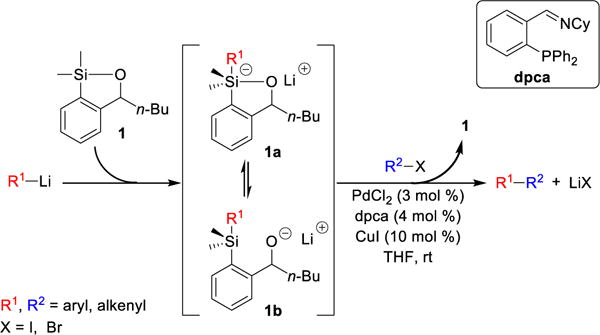
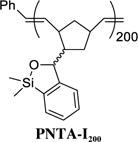
At the outset, we set two goals for the development of an effective polymer-supported siloxane transfer agents: (A) the synthesis of the polymer must be short, scalable, and inexpensive; and (B) the designed polymer preferably soluble in THF, the optimal reaction solvent for the cross-coupling reactions, and then upon addition of a more polar solvent become insoluble after completion of the cross-coupling reaction to permit facile removal of the polymer from the desired products. On the basis of these considerations, we chose to employ ring-opening metathesis polymerization (ROMP)11 to construct a polymeric siloxane transfer agent. To this end, the ROMP monomer was constructed via treatment of commercially available 5-norbornene-2-carboxaldehyde12 with PhMgCl to furnish benzylic alcohol 3, which upon ortho-lithiation with n-BuLi, followed by capture of the desired anion with Me2SiHCl, and ring-closure promoted by KOtBu13 leading to the slow evolution of hydrogen gas (Caution should be exercised!) led to siloxane 4 containing the desired norbornene moiety (Scheme 3). Ring-opening metathesis polymerization of 4 was then smoothly achieved with the first generation Grubbs catalyst14 to provide the polynorbornene-supported transfer agent PNTA-I200. Importantly, this short, efficient, and cost-effective route permits preparation of the polymer on multi-gram scale. The resulting polymer proved soluble in most organic solvents and insoluble in H2O and MeCN, which we took advantage of in the CCR purification process. Given that PNTA-I200 was produced in near quantitative yield employing the living polymerization technique, the siloxane loading was reasoned to be nearly identical to the molarity of the monomer (3.9 mmol/g); each polymer chains containing an average of 200 monomer units based on the ratio of monomer 4 to the Grubbs catalyst employed in the polymerization process.
Scheme 3.
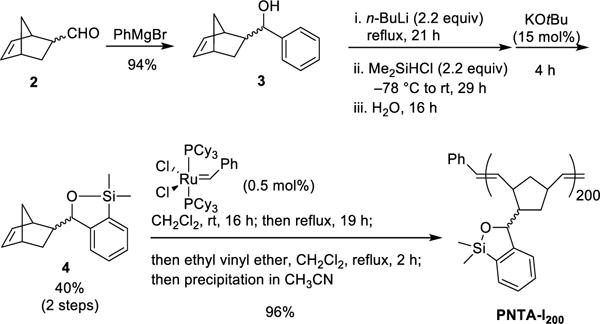
Synthesis of Polynorbornene-Supported Transfer Agent
Pleasingly, PNTA-I200 proved to be a viable transfer agent in cross-coupling reactions employing conditions similar to those reported for the small molecule transfer agents8 (Table 1). For example, use of 2.0 equiv of the siloxane polymer at a concentration of 15 mg/mL permitted cross-coupling between phenyllithium and 4-iodoanisole to furnish the desired product 6, albeit with the formation of a small amount of undesired homocoupled product 7 (entry 1). An increase in product yield was observed upon increasing the equivalence of PhLi and PNTA-I200 to 2.5 and 3.0, respectively (entry 2). Particularly pleasing, the cross-coupling efficiency was greatly improved by lowering the polymer concentration to 10 mg/mL, leading to complete conversion of aryl iodide 5 within 2 h at room temperature to furnish product 6 in 98% isolated yield (entry 3). Attempts to reduce the amount of PhLi or the siloxane loading, while maintaining the efficiency of the process, provided no enhancement (entries 4–6). As expected, and consistent with Murahashi’s observation,2 a control experiment in the absence of siloxane polymer resulted in inefficient cross-coupling in conjunction with significant formation of undesired homocoupled product (entry 7).
Table 1.
Optimization of CCR Employing PNTAa
| |||||
|---|---|---|---|---|---|
| entry | equiv PhLi |
equiv PNTA |
concd PNTA |
time |
1H-NMR resultsb (6 : 5 : 7) |
| 1c | 1.5 | 2.0 | 15 mg/mL | 25 h | 57 : 40 : 3 |
| 2d | 2.5 | 3.0 | 15 mg/mL | 15 h | 90 : 5 : 5 |
| 3e | 2.5 | 3.0 | 10 mg/mL | 2 h | 100 : nd : <1 |
| 4 | 1.5 | 3.0 | 10 mg/mL | 15 h | 84 : 11 : 5 |
| 5 | 2.0 | 3.0 | 10 mg/mL | 2 h | 72 : 18 : <1 |
| 6 | 2.0 | 2.5 | 10 mg/mL | 2 h | 88 : 9 : 3 |
| 7f | 2.5 | — | — | 2 h | 14 : 77 : 9 |
All reactions were performed on 0.3 mmol scale using 3 mol % PdCl2, 4 mol % dpca, 10 mol % CuI.
Determined by 1H-NMR analysis of the reaction mixture following an aqueous workup, extraction with Et2O and polymer removal via precipitation in CH3 CN.
Part of the recovered polymer remained insoluble in THF.
Small amount of the recovered polymer remained insoluble in THF; 82% yield of 6.
Polymer was recovered quantitatively and re-useable; 98% yield of 6.
Reaction was run in the absence of siloxane polymer.
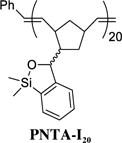
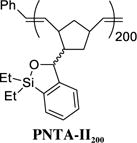
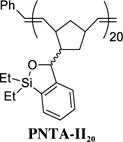
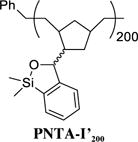
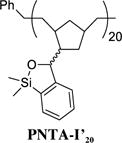
Having developed both an effective synthetic tactic for polymer construction and identification of optimal cross-coupling conditions employing the siloxane polymer PNTA-I200, we examined the effect of polymer structure on reactivity, vis-à-vis the ability to serve as a transfer agent. To this end, a series of polymer analogues were synthesized and employed in the cross-coupling reaction between PhLi and 4-iodoanisole (Table 2). Reducing the relative length of polymer chain to 20-mer units by employing a higher Grubbs catalyst loading in the ROMP process led to competing homocoupled product formation in the cross-coupling process (entry 2). The polymer’s ability to serve as a transfer agent also proved sensitive to the steric environment around the silicon center, as a small increase in the steric bulk by changing the substituents on the silicon atom on the transfer groups from methyl to ethyl resulted in an unproductive cross-coupling processes (entries 3 and 4). To explore the effect of the unsaturation present in the polymer chain, a tandem ROMP-hydrogenation protocol15 was developed to access polymers with a saturated backbone. Interestingly, while the saturated 20-mer displays physical and cross-coupling properties similar to those of the unsaturated analogue (i.e., complete conversion with significant homocoupling, entry 6), the saturated 200-mer proved insoluble in most organic solvents and yielded only a trace amount of desired product (entry 5). Best results in this series, with regard to the polymer physical properties (i.e., solubility), ease of handling, and cross-coupling efficiency were obtained with the unsaturated 200-mer possessing dimethyl substituents on silicon (i.e., PNTA-I200, entry 1).
Table 2.
Structure-Activity of Siloxane Polymer in Cross-Coupling Reactionsa
| |||
|---|---|---|---|
| entry | siloxane polymer | time |
1H-NMR resultsb (6 : 5 : 7) |
| 1 |
|
2 h | 100 : nd : <1c |
| 2 |
|
2 h | 80 : nd : 20 |
| 3 |
|
50 h | 1 : 99 : nd |
| 4 |
|
20 h | 42 : 33 : 25 |
| 5d |
|
20 h | 6 : 94 : nd |
| 6 |
|
2 h | 83 : nd : 17 |
All reactions were performed on 0.3 mmol scale using 2.5 equiv PhLi, 3.0 equiv siloxane polymer, 3 mol % PdCl2, 4 mol % dpca, 10 mol % CuI.
Determined by 1H-NMR analysis of the reaction mixture following an aqueous workup, extraction with Et2O and polymer removal via precipitation in CH3CN.
98% yield of 6.
The insoluble polymer was stirred heterogeneously in reaction mixture. nd = not detected.
To illustrate the utility and scope of the polynorbornene support, a series of cross-coupling reactions were carried out employing PNTA-I200 as the transfer agent (Table 3). Aryllithiums underwent effective coupling with various aryl and alkenyl iodides to provide the CCR products in excellent yield (entries 1,2,3, and 6). While the cross-coupling reaction was also effective with the electron-deficient p-cyanophenyl bromide, reaction employing the electron-rich p-methoxyphenyl bromide proved ineffective (entries 4 and 5). Alkenyl lithiums were also well tolerated in the cross-coupling reactions, proceeding in good yield with retention of the alkene geometry (entries 7–9). In all cases, the polymer was conveniently removed from the cross-coupling product mixture via precipitation in MeCN followed by filtration. Importantly, cross-coupling reactions employing PNTA-I200 are as rapid and high-yielding as those employing the small molecule transfer agents, with the added advantage of simple product isolation.
Table 3.
Validation of Polynorbornene Support across a Series of Cross-Coupling Reactionsa
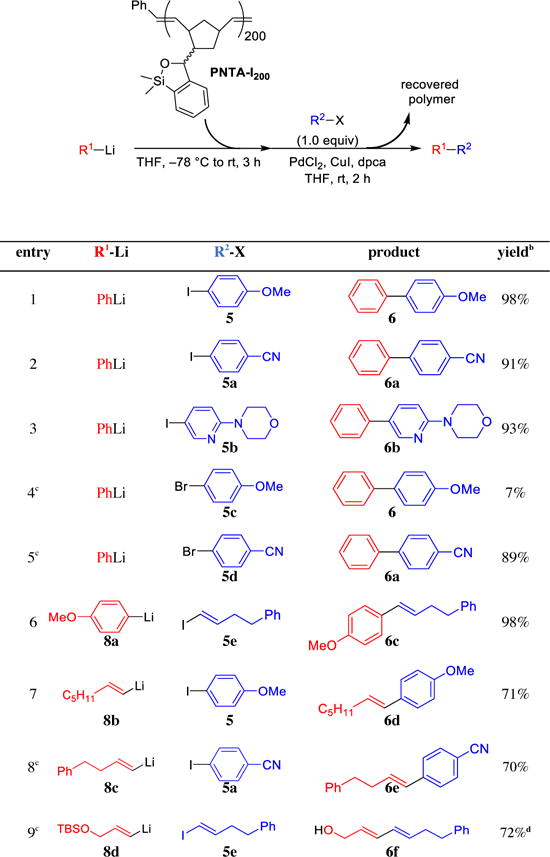
|
All reactions were run with 2.5 equiv R1Li, 3.0 equiv siloxane polymer, 3 mol % PdCl2, 4 mol % dpca, 10 mol % CuI.
Isolated yield.
12–24 h reaction time.
After polymer removal, the product mixture was treated with TBAF to remove the silyl group.
Attention was next directed toward the possibility of repeated recycling of the polynorbornene-supported transfer agent. We first examined the ability of PNTA-I200 to retain cross-coupling activity in multiple reactions employing the same coupling partners. As illustrated in Table 4, PhLi and 4-iodoanisole were employed for each experiment. The reaction was quenched after a 2 h reaction time to remove the excess lithium reagent. The siloxane polymer was then removed from the reaction products via precipitation in MeCN and employed in the subsequent run. Pleasingly, the polymer was recovered in near quantitative yield and reused for three cycles, furnishing the desired cross-coupling product in good to excellent yields. However, a small decrease in cross-coupling efficiency was observed after each cycle, in conjunction with an increase in the number average molecular weight (Mn) and polydispersity index (PDI) of the recovered polymer as determined by gel permeation chromatography (Figure 1). We reason that a change in the polymer structure after successive cross-coupling reactions was likely due to cross-linking of the polymers. As depicted in Scheme 4, addition of PhLi into the polymer solution in the siloxane activation step is envisioned to generate alkoxide intermediates. During the course of the reaction, an oxyanion could then attack a silicon atom of a nearby siloxane unit (either remaining intact after the activation step or regenerated after the cross-coupling step) residing on a different polymer chain, resulting in cross-linking of the polymer. The higher degree of cross-linking, the less efficient the recovered polymer would be in mediating the cross-coupling process. The observation that the recovered polymer appeared to have an increase in hardness, in conjunction with a small decrease in solubility in organic solvents after each cycle, supports this hypothesis. An increase in Mn was also observed in repeated cross-coupling reactions employing the saturated polymer (PNTA-I′20, Table 2), thus suggesting that the olefinic backbone is not responsible for the increase in molecular weight.
Table 4.
Recyclability of Polynorbornene-Supported Transfer Agent in Cross-Coupling Reactions Employing the Same Coupling Partnertsa
| |||
|---|---|---|---|
| cycle | isolated yield of 6 | polymer recoveredb |
1H-NMR Results (6 : 5 : 7) |
| 0 | (Mn = 63500, PDI = 1.2) | ||
| 1st | 98% | 98% (Mn = 73000, PDI = 1.6) | 100 : nd : <1 |
| 2nd | 91% | 99% (Mn = 104400, PDI = 2.3) | 97 : nd : 3 |
| 3rd | 81% | 99% (Mn = 94600, PDI = 2.6) | 86 : 11 : 3 |
All reactions were run with 2.5 equiv PhLi, 3.0 equiv siloxane polymer, 1.0 equiv 5, 3 mol % PdCl2, 4 mol % dpca, 10 mol % CuI.
Poly(methyl methacrylate) standards were used to determine Mn and PDI values. nd = not detected.
Figure 1.
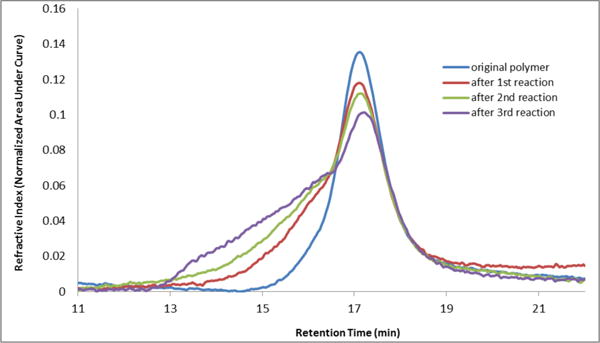
Gel Permeation Chromatogram of Recovered PNTA-I200 after Successive Cross-Coupling Reaction
Scheme 4.
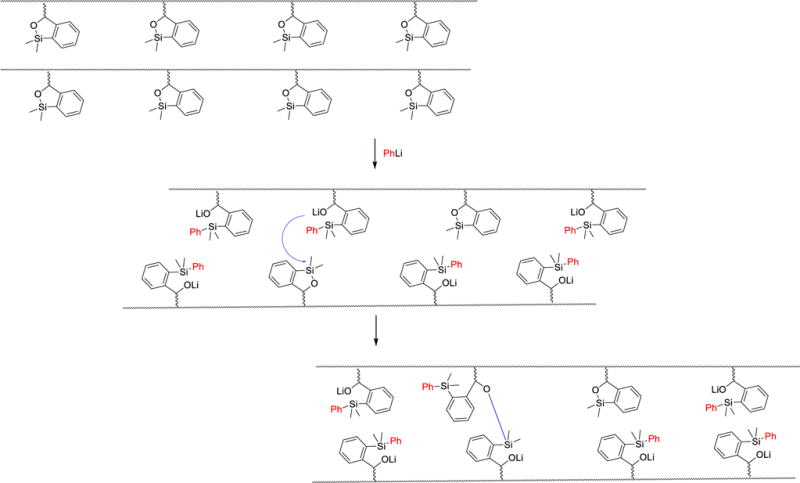
Polymer Cross-linking that Leads to Decrease in Reaction Efficiency after Repeated Cycles
We next performed a series of experiments to explore repeated recycling of the polynorbornene support in cross-coupling reactions employing different coupling partners, with particular attention paid to cross-contamination of the nucleophile in the subsequent cross-coupling products (Table 5). In the first experiment, two different nucleophilic coupling partners were employed in two successive cross-coupling reactions employing the same electrophilic coupling partner. Analysis of 1H-NMR of the product mixture revealed a small amount of the first cross-coupling product (4%) in the second cycle; this cross-over contaminant remained present in the third cycle, albeit in a minor amount (Table 5A). In the second experiment, the cross-over contamination of the nucleophile was also observed in repeated cross-coupling reactions employing different nucleophiles and electrophiles (Table 5B). These results clearly demonstrate nucleophile scrambling due to the incomplete reform of the siloxane moiety following quenching of the reaction mixture,16 leading to nucleophile carryover when the cross-coupling partners are changed in subsequent reactions.
Table 5.
Recyclability of Polynorbornene-Supported Transfer Agent in Cross-Coupling Reactions Employing Different Nucleophilic Coupling Partnertsa
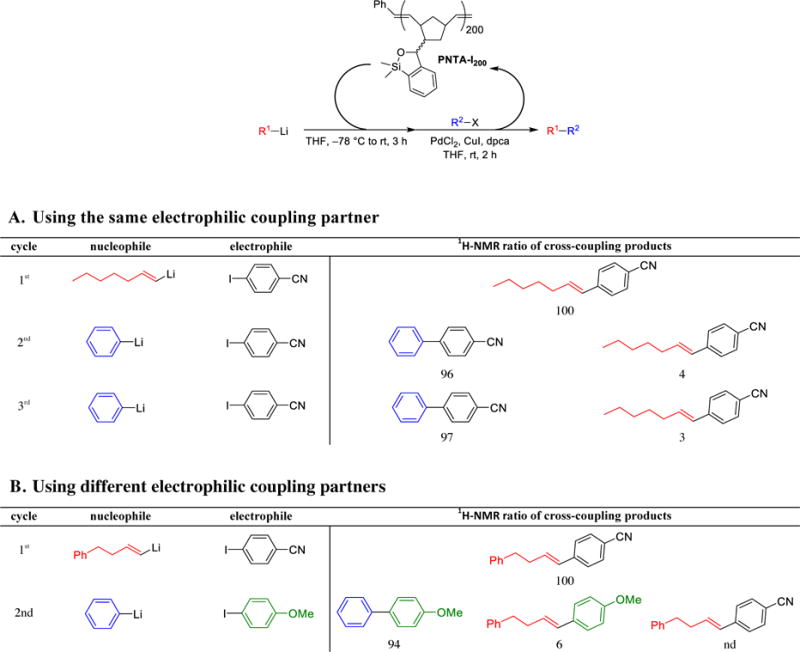
|
All reactions were run with 2.5 equiv R1Li, 3.0 equiv siloxane polymer, 1.0 equiv R2X, 3 mol % PdCl2, 4 mol % dpca, 10 mol % CuI. nd = not detected.
In summary, we successfully validated the use of ring-opening metathesis polymerization to incorporate an effective transfer agent onto a readily recoverable polynorbornene support that significantly simplifies product purification and transfer agent recycle. However, while this soluble linear polymer support proved effective in mediating the palladium-catalyzed cross-coupling reactions, we observed a decrease in reaction efficiency in conjunction with nucleophile cross-contamination after repeated reuse of the polymer-supported transfer agent.
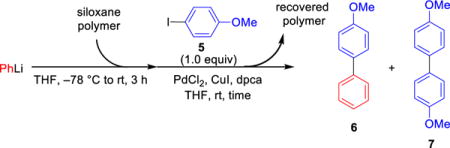
B. A Second-Generation Design, Synthesis and Validation of Polymer-Supported Siloxane Transfer Agents for Cross-Coupling Reactions
Attention was next directed toward the design of a more effective solid support with improved recycling properties (i.e., less cross-contamination). We began by addressing the issue of polymer cross-linking that we reasoned accounted for the drop in reaction efficiency. We hypothesized that we could suppress this process by restricting the polymer chain mobility and subsequently restrain the interaction of the reactive sites on the polymer chain via use of a more rigid, cross-linked, insoluble polymeric network. Towards this end, use of an inert co-monomer in the polymerization process would place the siloxane units further away from each other, minimizing the undesired cross-linking behavior. Another factor considered in the design was removal of the unsaturation element in the polymer chain, given the possible vulnerability of the olefinic backbone to chemical and thermal degradation. Based on our previous observation that steric hindrance at the silicon atom leads to inefficient reactivity, use of a spacer between the siloxane moiety and the polymer matrix was also considered to reduce any steric effect induced by a bulky polymer backbone (Scheme 5). On the basis of this logic, cross-linked polystyrene polymers emerged as suitable solid supports for the transfer agent.
Scheme 5.
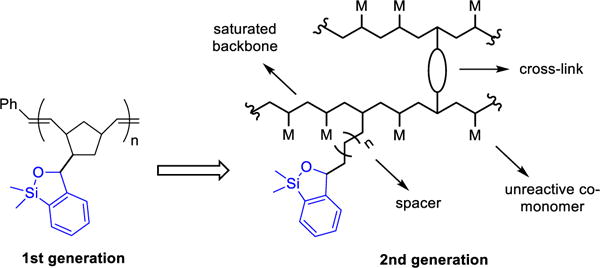
Design of 2nd Generation Polymer-Supported Transfer Agent
To test this hypothesis, we first directed our effort at strategies to attach the transfer agent onto commercially available cross-linked polystyrene resin beads. However, all attempts to install the siloxane moiety onto a polystyrene resin via nucleophilic substitution reactions proved unsuccessful, presumably due to undesired reactions at the silicon atom (Scheme 6). The use of a cross-coupling strategy employing 14 to functionalize the commercial resin beads also proved unsatisfactory as less than 10% of siloxane moiety was incorporated into the polystyrene resin 16.
Scheme 6.
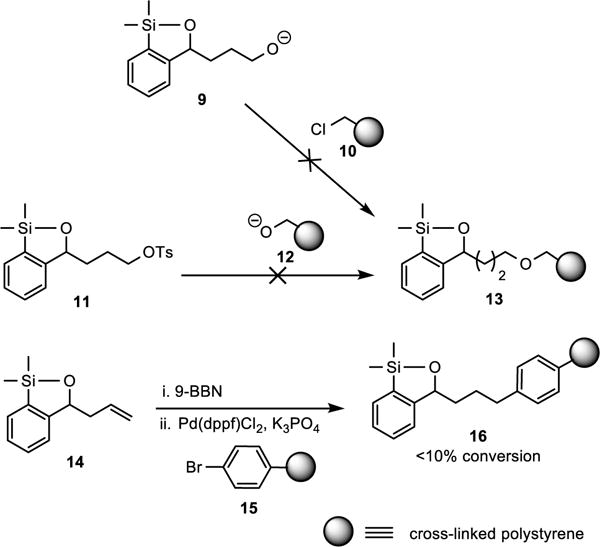
Unsuccessful Attempts to Immobilize Transfer Agent onto Commercially Available Resin Beads
We next turned to polymeric immobilization of the transfer agent, with a “bottom-up” tactic by first construction of a siloxane monomer (20, Scheme 7) having a styrene moiety which could then be subjected to co-polymerization with styrene and a suitable cross-linker to provide the desired cross-linked polystyrene-supported transfer agent. In this way we would have excellent control of both the siloxane loading and the swelling properties of the polymer, the latter simply a matter of mixing the monomer and the cross-linker of choice in the appropriate ratio. Again, we focused on simplicity for the polymer synthesis.
Scheme 7.
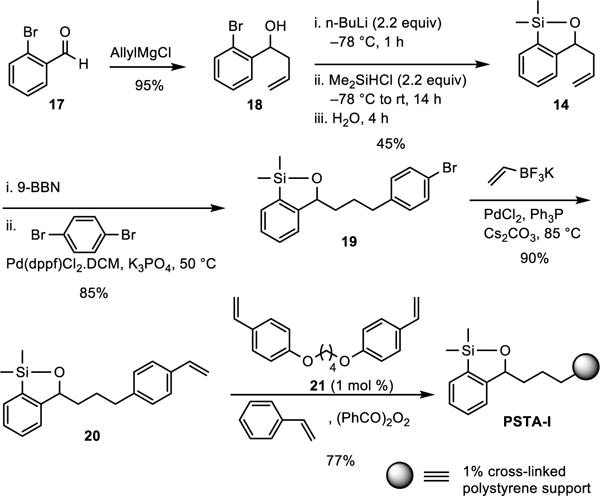
Synthesis of Polystyrene-Supported Siloxane Transfer Agent
We began with the synthesis of the siloxane monomer 20 via treatment of commercially available 2-bromobenzaldehyde 17 with allylmagnesium chloride to furnish benzylic alcohol 18, which was converted to siloxane 14 (Scheme 6) employing a one-pot, three step reaction sequence involving lithiation with n-BuLi, followed by anion capture with Me2SiHCl, and ring closure upon addition of H2O leading to the evolution of hydrogen gas (Caution should be exercised!). Hydroboration with 9-BBN followed by a Suzuki Pd-catalyzed cross-coupling reaction17 with p-dibromobenzene smoothly provided aryl bromide 19 in 85% yield. In turn, 19 was converted to siloxane monomer 20 via a second Suzuki cross-coupling reaction employing vinyltrifloroborate.18 Suspension co-polymerization19 of 20, with styrene and the tetrahydrofuran-derived cross-linker 21 (in a molar ratio of 12:87:1, respectively) was then smoothly achieved with benzoyl peroxide to provide the 1% cross-linked polystyrene-supported transfer agent PSTA-I as well-defined beads. Selection of the highly flexible cross-linker 14 in the polymerization process led to a gel-type resin that exhibits excellent swelling properties in THF,20 which again is the reaction solvent for many cross-coupling reactions. The siloxane loading of PSTA-I (1.5 mmol/g) was reasoned to be nearly identical to the silicon loading, the latter determined by elemental analysis.
We next evaluated PSTA-I as a viable transfer agent employing similar conditions as developed for the polynorbornene-supported transfer agent (Scheme 8). Pleasingly, PSTA-I efficiently mediated the palladium-catalyzed cross-coupling reaction between PhLi and 4-iodoanisole, furnishing the desired cross-coupling product 6 in 96% yield without observation of the homo-coupling product. Best results were again obtained employing 2.5 equiv of the lithium reagent and 3.0 equiv of the siloxane polymer. Importantly, the cross-coupling efficiency remained the same after six cross-coupling iterations employing the same batch of polymer, thereby demonstrating a significant improvement of the designed polystyrene support compared to the polynorbornene support. Also noteworthy was the ease of transfer agent recovery, as the polymer could be readily removed from the product mixture via simple filtration, followed by washing with organic solvents and drying under vacuum before being employed in the next cross-coupling cycle.
Scheme 8.
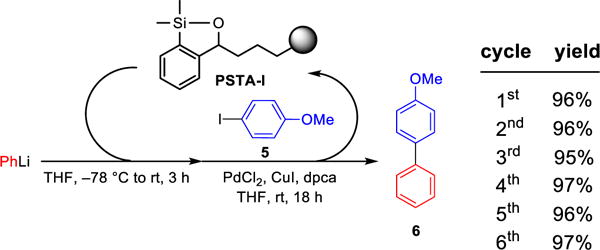
Recyclability of the Polystyrene Support in Cross-Coupling Reaction Employing the Same Nucleophile and Electrophilea
aAll reactions were run with 2.5 equiv PhLi, 3.0 equiv siloxane polymer, 1.0 equiv of 5, 3 mol % PdCl2, 4 mol % dpca, 10 mol % CuI. Siloxane loading was 1.5 mmol/g.
With PSTA in hand, we next explored the use of the new polymer support in repeated cross-coupling reactions employing different nucleophiles and electrophiles. As shown in Table 6, a single batch of siloxane polymer efficiently mediated a total of ten cross-coupling reactions, furnishing the desired cross-coupling products with no evidence of homocoupled products. Importantly, cross-coupling reactions employing recycled PSTA-I proved as high yielding as those employing the freshly made PNTA-I200. Equally significant, no nucleophile cross-contamination was detected across this series of reactions. Moreover, substrates containing highly sensitive functional groups such as nitriles and ketones were well tolerated in cross-coupling reactions mediated by PSTA-I,21 demonstrating the unique advantage of this protocol compared to other reported cross-coupling methods2,9 employing organolithium reagents as the nucleophilic coupling partners.
Table 6.
Recyclability of PSTA-I in Reactions Employing Multiple Cross-Coupling Partnersa
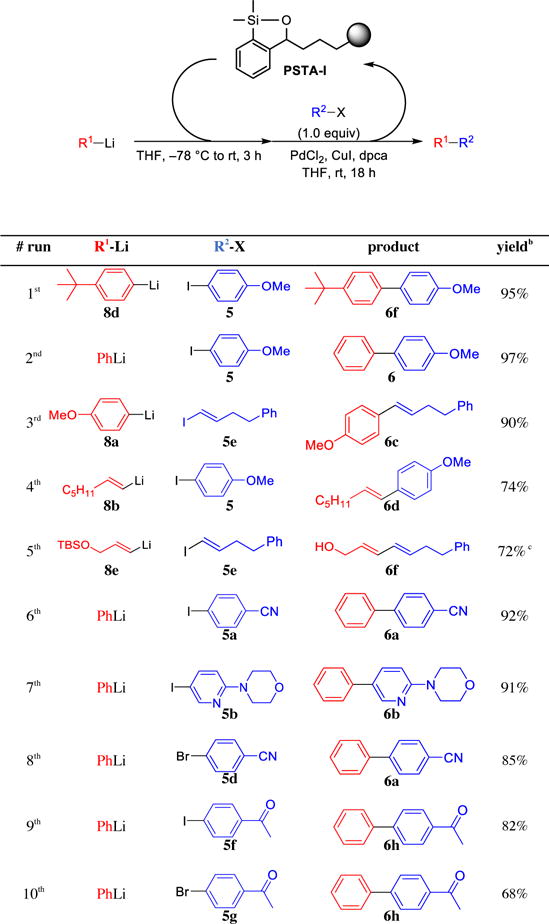
|
All reactions were run with 2.5 equiv R1Li, 3.0 equiv siloxane polymer, 3 mol % PdCl2, 4 mol % dpca, 10 mol % CuI. Siloxane loading was 0.74 mmol/g.
Isolated yield.
After polymer removal, the product mixture was treated with TBAF to remove the silyl group.
Having showcased the recyclability of PSTA-I, we next expanded the scope of this efficient siloxane polymer in the palladium-catalyzed cross-coupling reactions, with a focus on substrates containing heteroatoms (Table 7). To this end, electrophilic coupling partners comprising imidazole and all regioisomers of iodopyridine proceeded efficiently. Also noteworthy, trifluoromethyl groups were well tolerated both in electrophilic and nucleophilic coupling partners (6k and 6r). A variety of heteroaryl and hindered aryl lithiums were suitable to the cross-coupling conditions. We were particularly interested in cross-coupling reactions of 2-substituted heterocycles, given the inherently unstable and/or challenging-to-access 2-heterocyclic cross-coupling surrogates.22 Gratifyingly, use of PSTA-I permits direct cross-coupling of 2-lithiopyridine (8g), thus alleviating the need for preconstruction of 2-pyridyl N-methyliminodiacetic acid (MIDA) boronates.23 Furthermore, cross-couplings of 2-thienyllithium and 2-benzofuryllithium were successful, providing good yield of desired products (6n and 6q), with the latter accessed via a direct ortho-lithiation/cross-coupling sequence of benzofuran. Importantly, a single batch of polymer was employed and recycled in near quantitative yield following each reaction in this series of experiments.
Table 7.
Expanded Substrate-Scope of Pd-Catalyzed Cross-Coupling Reactions Employing Recycled PSTA-Ia
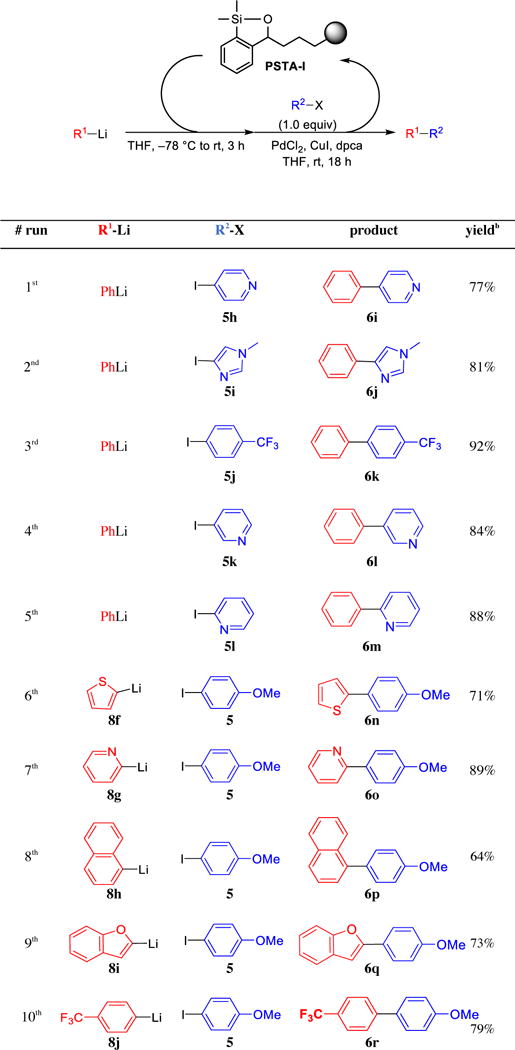
|
All reactions were run with 2.5 equiv R1Li, 3.0 equiv siloxane polymer, 3 mol % PdCl2, 4 mol % dpca, 10 mol % CuI. Siloxane loading was 0.57 mmol/g.
Isolated yield.
A proposed reaction mechanism for the siloxane-mediated cross-coupling reaction of organolithium reagents under catalytic amounts of Pd0 and CuI is illustrated in Scheme 9. Addition of organolithium to the transfer agent during the siloxane activation step generates an alkoxide intermediate, presumably in an equilibrium with a pentavalent siliconate complex.6 Transmetalation from silicon to copper24 furnishes the aryl-CuI species and in turn regenerates the transfer agent. A second transmetalation step from copper to palladium results in the ligated diarylpalladium(II) complex, which undergoes reductive elimination to provide the coupled products, and the resulting Pd0 then re-enters the catalytic cycle. A catalytic amount of CuI is required and serves as a molecular transport or shuttle between two transmetallation processes, i.e., first accepting nucleophile from activated transfer agent and then donating nucleophile to palladium center.
Scheme 9.
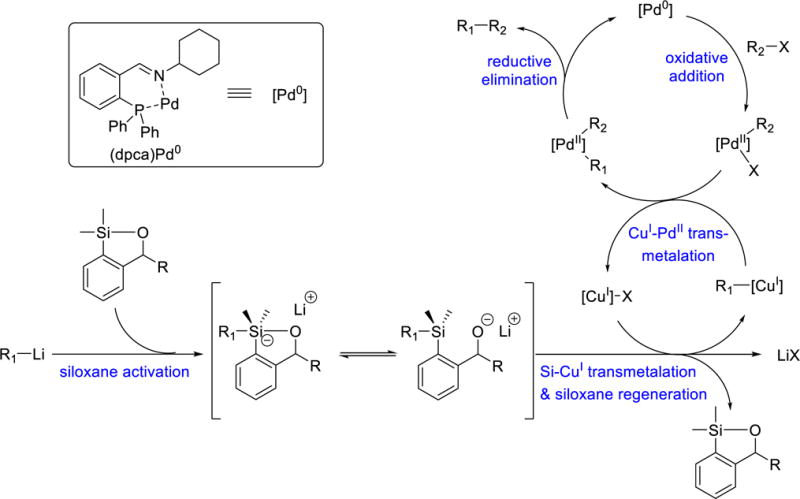
Proposed Reaction Mechanism
As part of an effort to expand the siloxane transfer agent tactic, we demonstrated that the polymer-supported transfer agent could be employed to mediate carbon-nitrogen bond formation, as recently demonstrated with monomeric siloxane transfer agents.25 A preliminary example is showed in Scheme 10 in which PNTA-I200 was used in the copper-catalyzed, electrophilic amination of PhLi to generate the desired aniline 16. This protocol featured a chromatography-free purification strategy in which the desired amination product was purified by acid-based extraction and the polymer was recovered via precipitation in MeCN and reused for two additional cycles.
Scheme 10.
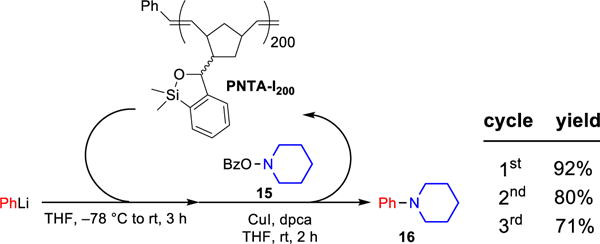
Electrophilic Amination of Organolithium Mediated by Polymer-Supported Transfer Agenta
aAll reactions were run with 2.5 equiv PhLi, 3.0 equiv siloxane polymer, 1.0 equiv 15, 10 mol % CuI, 10 mol % dpca.
Summary
The initial rational design, synthesis, and validation of two generations of polymer-supported transfer agents for use in the palladium-catalyzed cross-coupling reactions of organolithiums has been achieved. The polymer supports greatly simplify product purification, provide a practical method for transfer agent recovery, and maintain the efficiency of the cross-coupling process through multiple cycles. The mild reaction conditions and high functional group tolerance in conjunction with the operationally convenient protocol provided by the solid supports further advance the siloxane based transfer tactic in cross-coupling reactions of organolithiums. Studies to improve the design of polymer bound transfer agent to reduce the required equivalents of lithium reagents and to expand the concept of polymer-supported siloxane-transfer agent in other bond-forming processes as well as to explore the possibility of coating of siloxane polymer in flow micro-reactors continue in our laboratory.
Experimental Section
Materials and Methods
All moisture-sensitive reactions were performed using syringe-septum cap techniques under an inert atmosphere of N2. All glassware was flame dried or dried in an oven (140 °C) for at least 4 h prior to use. Reactions were magnetically stirred unless otherwise stated. Tetrahydrofuran (THF), dichloromethane (CH2Cl2) and diethyl ether (Et2O) were dried by passage through alumina in a solvent purification system. Unless otherwise stated, solvents and reagents were used as received. Analytical thin layer chromatography was performed on pre-coated silica gel plates (particle size 40–55 micron, 230–400 mesh). Column chromatography was performed using silica gel (40–63 micron particle size, 230–300 mesh) and compressed air pressure with commercial grade solvents. Yields refer to chromatographically and spectroscopically pure compounds, unless otherwise stated. NMR spectra were recorded at 500 MHz/125 MHz (1H NMR/ 13C NMR) on a 500 MHz spectrometer at 300 K. Chemical shifts are reported in parts per million with the residual solvent peak as an internal standard. 1H NMR spectra are tabulated as follows: chemical shift, multiplicity (s=singlet, d=doublet, t=triplet, q=quartet, dd=doublet of doublets, ddd= doublet of doublet of doublets, dddd= doublet of doublet of doublet of doublets, dt= doublet of triplets, m=multiplet, b=broad), coupling constant and integration. 13C NMR spectra are tabulated by observed peak. Melting points were determined using a capillar melting point apparatus and are uncorrected. Infrared spectra were measured on a FT/IR plus spectrometer. High-resolution mass spectra (HRMS) were measured on a LC-TOF mass spectrometer. GPC analysis of the polymer samples were done on a high-performance liquid chromatography (HPLC), equipped with a column oven (30 °C), an integration data station, a UV-vis detector (254 nm), a refractive index detector, and three gel columns (500 Å, 5 μm; 1000 Å, 5 μm; and 104 Å, 5 μm). THF (HPLC grade) was used as eluent at a flow rate of 1 mL/min. The number-average (Mn) and weight-average (Mw) molecular weights of polymer samples were determined with poly(methyl methacrylate) (PMMA) standards. Elemental analysis of Si was determined via inductively coupled plasma optical emission spectrometry (ICP-OES). Compound 5e26 and the corresponding vinyl iodides of 8b26 and 8d27 were prepared according to previously reported procedures. Caution: organolithium reagents, especially alkyl lithium reagents such as n-BuLi and t-BuLi are corrosive, flammable, and pyrophoric. Careful planning prior to execution of experiments and proper personal protective equipment are needed to safely handle organolithium reagents in the laboratory.
Preparation of water-washed silica gel for column chromatography (where specified)
Silica gel was suspended in deionized H2O and the slurry mixture was then packed into a prepared column. The obtained H2O-washed silica gel packed column was then rinsed with 2 column volumes of acetone, 1 column volume of EtOAc and 2 column volumes of hexanes, successively.
Synthesis and Cross-Coupling Reactions of Polynorbornene-supported Transfer Agents
Norborn-5-en-2-yl-phenol-methanol (3)
PhMgBr (3.0 M in Et2O, 17.55 mL, 52.64 mmol) was added slowly to a vigorously stirred solution of aldehyde 2 (95% purity, mixture of endo- and exo-isomers) (5.36 g, 41.68 mmol) in 200 mL Et2O at 0 °C. The obtained solution was stirred at rt for 15 h, and was quenched with saturated aqueous NH4Cl (50 mL). The organic layer was collected and the aqueous layer was extracted with Et2O (2 × 50 mL). The combined organic layers were washed with brine, dried with MgSO4, and concentrated in vacuo. The crude product was purified by flash chromatography on silica gel (5 – 30% Et2O/Hexanes) to afford the desired alcohol 3 as a mixture of diastereomers in a 1 : 1 : 3.7 : 3.7 ratio by 1H NMR spectroscopy (7.87 g, 39.3 mmol, 94%): colorless solid (M.P. = 50 – 53 °C); Rf 0.2 (20% Et2O in Hexanes); IR (neat, cm−1) 3376, 3060, 3029, 2967, 2939, 2869, 1603, 1494, 1454, 1335, 1068, 1015, 758, 719, 700; 1H NMR (500 MHz, CDCl3) δ 7.41 − 7.23 (m, 5.6 H), 6.26 − 6.23 (m, 0.77 H, vinyl proton), 6.20 − 6.15 (m, 0.50 H, vinyl proton), 6.10 − 6.05 (m, 0.22 H, vinyl proton), 6.00 − 5.96 (m, 0.50 H, vinyl proton), 4.41 (d, J = 9.5 Hz, 0.11 H, benzylic proton), 4.36 (d, J = 10.1 Hz, 0.11 H, benzylic proton), 4.03 (d, J = 10.1 Hz, 0.39 H, benzylic proton), 3.98 (d, J = 10.1 Hz, 0.39 H, benzylic proton), 3.16 (bs, 0.39 H, allylic proton), 3.10 (bs, 0.11 H, allylic proton), 2.92 (bs, 0.11 H, allylic proton), 2.88 (bs, 0.39 H, allylic proton), 2.77 (bs, 0.11 H, allylic proton), 2.75 (bs, 0.39 H, allylic proton), 2.51 – 2.43 (m, 0.80 H), 2.31 (bs, 0.39 H, allylic proton), 2.26 (bs, 0.11 H, allylic proton), 2.04 – 1.99 (m, 0.43 H), 1.85 – 0.89 (m, 5.73 H), 0.50 – 0.47 (m, 0.41 H); 13C NMR (125 MHz, CDCl3) δ 144.6, 144.5, 144.3, 144.2, 138.1, 138.1, 137.3, 137.1, 136.8, 136.8, 132.8, 132.4, 128.7, 128.6, 128.6, 128.3, 127.9, 127.9, 127.8, 127.0, 126.8, 126.5, 126.0, 80.0, 79.5, 79.3, 78.6, 49.9, 49.5, 48.1, 47.2, 46.9, 46.9, 45.7, 45.3, 44.9, 44.5, 44.4, 43.7, 43.0, 42.5, 42.3, 42.0, 30.9, 30.3, 30.1, 29.5; HRMS (CI+) m/z (M+H)+: Calcd for C14H17O: 201.1279, found: 201.1277.
3-(Bicyclo[2.2.1]hept-5-en-2-yl)-1,1-dimethyl-1,3-dihydrobenzo[c][1,2]oxasilole (4)
To a stirred solution of alcohol 3 (7.71 g, 38.55 mmol) in hexanes (125 mL, dried over MgSO4) and Et2O (100 mL) at 0 °C was added n-BuLi (2.3 M in hexanes, 36.9 mL, 84.87 mmol) dropwise. The solution was stirred at rt for 1 h, at which time reflux condenser was attached and the solution was heated to reflux at 70 °C for 21 h. The solution was then cooled to −78 °C and Me2SiHCl (9.22 ml, 84.87 mmol) was added dropwise. The solution was stirred and warmed to rt. After 29 h, the reaction mixture was quenched by addition of deionized H2O (100 mL) and stirred for 16 h. Note slow evolution of hydrogen gas (caution!). The organic layer was collected and the aqueous layer was extracted with hexanes (2 × 50 mL). The combined organic layers were washed with brine, dried with MgSO4, and concentrated in vacuo. The crude product was then taken up in THF (150 mL), and KOtBu (0.65 g, 5.79 mmol) was divided into 3 portions and added to the stirred solution sequentially, each portion after 1 h. The mixture was stirred for a total of 4 h, followed by addition of d.i. H2O (50 mL), and the resulting mixture was stirred for another 30 min. The organic layer was collected and the aqueous layer was extracted with Hexanes (2 × 50 mL). The combined organic layers were washed with brine, dried with MgSO4, and concentrated in vacuo. Kugelrohr distillation (110 – 160 °C, 0.025 mmHg), followed by flash chromatography on water-washed silica gel (0 – 0.5% Et2O/Hexanes) afforded the desired siloxane 4 as a mixture of diastereomers in a 1 : 1.5 : 3.4 : 4.9 ratio by 1H NMR spectroscopy (3.97 g, 15.51 mmol, 40%): colorless oil; Rf 0.5 (5% Et2O in Hexanes); IR (neat, cm−1) 3058, 2962, 1594, 1445, 1254, 1028, 873, 823, 787, 748; 1H NMR (500 MHz, CDCl3) δ 7.57 – 7.54 (m, 0.88 H), 7.46 – 7.22 (m, 5.14 H), 6.27 – 6.16 (m, 1.64 H, vinyl proton), 6.07 – 6.04 (m, 0.28 H, vinyl proton), 5.41 (d, J = 4.0 Hz, 0.14 H, benzylic proton), 5.25 (d, J = 5.5 Hz, 0.09 H, benzylic proton), 4.65 (d, J = 9.7 Hz, 0.45 H, benzylic proton), 4.55 (d, J = 10.3 Hz, 0.31 H, benzylic proton), 3.18 (bs, 0.35 H, allylic proton), 2.97 (bs, 0.48 H, allylic proton), 2.90 (bs, 0.21 H, allylic proton), 2.84 – 2.83 (m, 0.79 H, allylic proton), 2.76 (bs, 0.19 H, allylic proton), 2.58 (bs, 0.13 H, allylic proton), 2.13 – 2.07 (m, 0.80 H), 1.92 – 1.68 (m, 1.54 H), 1.48 – 1.13 (m, 3.26 H), 0.43 – 0.33 (m, 5.2 H); 13C NMR (125 MHz, CDCl3) δ 154.2, 153.5, 153.2, 138.0, 138.0, 137.8, 137.5, 137.4, 137.0, 136.1, 136.0, 135.9, 135.8, 133.5, 132.5, 131.2, 131.0, 130.9, 130.0, 129.6, 129.3, 129.2, 127.1, 127.0, 127.0, 126.9, 125.7, 123.2, 122.8, 122.7, 122.5, 85.9, 85.3, 84.3, 84.0, 49.7, 49.3, 49.0, 48.3, 47.0, 46.2, 46.2, 46.2, 46.0, 45.6, 44.7, 43.0, 42.5, 42.3, 42.2, 41.6, 30.0, 29.8, 29.8, 27.0, 1.8, 1.8, 1.6, 1.2, 1.0, 0.9, 0.8, 0.7; HRMS (CI+) m/z (M+H)+: Calcd for C16H21OSi: 257.1362, found: 257.1357.
3-(Bicyclo[2.2.1]hept-5-en-2-yl)-1,1-dimethyl-1,3-dihydrobenzo[c][1,2]oxasilole (4b)
To a stirred solution alcohol 3 (7.78 g, 38.89 mmol) in hexanes (125 mL, dried over MgSO4) and Et2O (100 mL) at 0 °C was added n-BuLi (2.3 M in hexanes, 37.2 mL, 85.56 mmol) dropwise. The solution was stirred at rt for 1 h, at which time reflux condenser was attached and the solution was heated to reflux at 70 °C for 21 h. The solution was then cooled to −78 °C and Et2SiCl2 (7.56 mL, 50.56 mmol) was added dropwise. The solution was stirred and warmed up to rt. After 21 h, the reaction mixture was quenched by addition of d.i. H2O (50 mL) and stirred for 15 min. The organic layer was collected and the aqueous layer was extracted with hexanes (2 × 50 mL). The combined organic layers were washed with brine, dried with MgSO4, and concentrated in vacuo. Kugelrohr distillation (120 – 150 °C, 0.025 mmHg), followed by flash chromatography on silica gel (0 – 1% Et2O/Hexanes) afforded the desired siloxane 4b as a mixture of diastereomers in a 1 : 1 : 1.8 : 4.8 ratio by 1H NMR spectroscopy (1.49 g, 5.25 mmol, 13%): colorless oil; Rf 0.55 (5% Et2O in Hexanes); IR (neat, cm−1) 3056, 2957, 1460, 1443, 1019, 962, 853, 741, 715; 1H NMR (500 MHz, CDCl3) δ 7.59 – 7.51 (m, 0.93 H), 7.44 – 7.34 (m, 1.36 H), 7.34 – 7.23 (m, 1.71 H), 6.31 – 6.15 (m, 1.67 H, vinyl proton), 6.09 – 6.03 (m, 0.33 H, vinyl proton), 5.37 (d, J = 4.4 Hz, 0.12 H, benzylic proton), 5.20 (d, J = 6.5 Hz, 0.12 H, benzylic proton), 4.63 (d, J = 10.1 Hz, 0.55 H, benzylic proton), 4.55 (d, J = 10.5 Hz, 0.21 H, benzylic proton), 3.20 (bs, 0.21 H, allylic proton), 3.00 (bs, 0.56 H, allylic proton), 2.92 (bs, 0.21 H, allylic proton), 2.87 – 2.81 (m, 0.77 H, allylic proton), 2.78 – 2.72 (m, 0.25 H, allylic proton), 2.15 – 2.05 (m, 0.79 H), 1.94 – 1.79 (m, 1.17 H), 1.52 – 0.71 (m, 13.04 H); 13C NMR (125 MHz, CDCl3) δ 154.9, 154.3, 153.9, 138.15, 137.8, 137.7, 137.5, 137.4, 136.9, 134.1, 134.1, 134.0, 133.9, 133.6, 132.5, 131.8, 131.8, 131.6, 131.5, 129.6, 129.3, 129.2, 126.9, 126.8, 126.8, 126.7, 123.1, 122.8, 122.7, 122.4, 86.1, 85.5, 84.7, 84.2, 49.7, 49.4, 48.9, 48.2, 47.4, 46.2, 46.1, 46.1, 46.0, 45.6, 44.6, 43.3, 42.6, 42.4, 42.1, 41.6, 30.1, 30.0, 30.0, 29.8, 27.0, 7.6, 7.4, 7.1, 7.1, 7.0, 6.9, 6.6, 6.5; HRMS (CI+) m/z (M+H)+: Calcd for C18H25OSi: 285.1675, found: 285.1664.
General procedure A: ring opening metathesis polymerization (ROMP)
Grubbs catalyst (1st generation) was dissolved in 0.5 mL DCM and stirred for 30 min. The catalyst solution was then cannulated into another flask containing siloxane monomer in 1 mL DCM (rinsed with 2 × 0.5 mL DCM). Under a flowing stream of N2, the obtained solution was stirred at rt for 16 h, at which time reflux condenser was attached, another 1 mL DCM was added and the solution was heated to reflux at 50 °C for 19 h. The obtained mixture was cooled to rt, diluted with 1 mL DCM, followed by addition of ethyl vinyl ether (2 mL). The solution was then heated to reflux for another 2 h. The solution was then cooled to rt, diluted with 1 mL DCM, followed by addition of a solution of tris(hydroxymethyl)phosphine in d.i. H2O (2 mL) and Et3N (freshly distilled, 0.1 mL). The obtained mixture was stirred vigorously for 30 min. The resultant organic layer was collected and the aqueous layer was extracted with DCM (2 × 5 mL). The combined organic layers were washed with brine, dried with MgSO4, and concentrated in vacuo. The obtained solid was then dissolved in 1 mL DCM and precipitated via dropwise addition into a vigorously stirred solution of CH3CN (250 mL). The precipitate was filtered and concentrated in vacuo to afford the desired polymer.
Siloxane Polymer PNTA-I200
Following general procedure A, ROMP of siloxane monomer 4 (0.256 g, 1.0 mmol) using first generation Grubbs catalyst (4.1 mg, 5.0 μmol, 0.5 mol %) and tris(hydroxymethyl)phosphine (120 mg, 1.0 mmol) afforded siloxane polymer PNTA-I200 (0.245 g, 96%) as a white solid: IR (CDCl3, cm−1) 3057, 2998, 2947, 2862, 1593, 1442, 1252, 1013, 910, 870, 823, 789, 740; 1H NMR (500 MHz, CDCl3) δ 7.60 – 7.41 (bs, 1H), 7.41 – 6.98 (bm, 3 H), 6.10 – 4.60 (bm, 3 H), 3.41 – 0.71 (bm, 7 H), 0.64 – 0.00 (bm, 6 H); Mn = 63500, PDI = 1.2. The transformation was successfully carried out on a multigram-scale.
Siloxane Polymer PNTA-I20
Following general procedure A, ROMP of siloxane monomer 4 (0.267 g, 1.0 mmol) using first generation Grubbs catalyst (42.9 mg, 0.052 mmol, 5.0 mol %) and tris(hydroxymethyl)phosphine (1.95 g, 15.7 mmol) afforded siloxane polymer PNTA-I20 (0.227 g, 85%) as a white solid: IR and 1H NMR identical to PNTA-I200; Mn = 7000, PDI = 1.5.
Siloxane Polymer PNTA-II200
Following general procedure A, ROMP of siloxane monomer 4b (0.270 g, 0.95 mmol) using first generation Grubbs catalyst (3.9 mg, 4.8 μmol, 0.5 mol %) and tris(hydroxymethyl)phosphine (46.2 mg, 0.37 mmol) afforded siloxane polymer PNTA-II200 (0.234 g, 87%) as a white solid: IR (CDCl3, cm−1) 3057, 2997, 2953, 2874, 1445, 1012, 737; 1H NMR (500 MHz, CDCl3) δ 7.63 – 7.39 (bs, 1H), 7.39 – 6.97 (bm, 3 H), 6.06 – 4.85 (bm, 3 H), 3.39 – 0.30 (bm, 17 H); Mn = 48600, PDI = 1.4.
Siloxane Polymer PNTA-II20
Following general procedure A, ROMP of siloxane monomer 4b (0.223 g, 0.78 mmol) using first generation Grubbs catalyst (32.3 mg, 0.039 mmol, 5.0 mol %) and tris(hydroxymethyl)phosphine (415 mg, 3.35 mmol) afforded siloxane polymer PNTA-II20 (0.220 g, 99%) as a pale yellow solid: IR and 1H NMR identical to PNTA-II200; Mn = 6500, PDI = 1.7.
Siloxane Polymer PNTA-I’200
First generation Grubbs catalyst (4.1 mg, 4.97 μmol) was dissolved in 0.5 mL DCM and stirred for 30 min. The catalyst solution was then cannulated into another flask containing siloxane monomer (0.255 g, 1.0 mmol) in 1 mL DCM (rinsed with 2 × 0.5 mL DCM). Under a flowing stream of N2, the obtained solution was stirred at rt for 16 h, at which time reflux condenser was attached, another 1 mL DCM was added and the solution was heated to reflux at 50 °C for 19 h. The obtained mixture was cooled to rt, diluted with 10 mL DCM, followed by addition of MeOH (5 mL) and Et3N (freshly distilled, 50 μL). The solution was then stirred and heated to 50 °C under an atmosphere of hydrogen (250 psi) in a Parr bomb for 19 h. The reaction mixture was then cooled to rt, removed from the Parr bomb, and the solvent was removed in vacuo. The obtained solid was washed with excess CH3CN and MeOH to afford the desired polymer (0.247 g, 97%) as a brown solid.
Siloxane Polymer PNTA-I’20
First generation Grubbs catalyst (31.2 mg, 0.038 mmol) was dissolved in 1.5 mL DCM and stirred for 30 min. The catalyst solution was then cannulated into another flask containing siloxane monomer (0.194 g, 0.759 mmol) in 1 mL DCM (rinsed with 2 × 0.5 mL DCM). Under a flowing stream of N2, the obtained solution was stirred at rt for 16 h, at which time reflux condenser was attached, another 1 mL DCM was added and the solution was heated to reflux at 50 °C for 19 h. The obtained mixture was cooled to rt, diluted with 5 mL DCM, followed by addition of MeOH (2 mL) and Et3N (freshly distilled, 50 μL). The solution was then stirred and heated to 50 °C under an atmosphere of hydrogen (250 psi) in a Parr bomb for 48 h. The solution was then cooled to rt, removed from the Parr bomb, followed by addition of a solution of tris(hydroxymethyl)phosphine (1.37 g, 11.05 mmol) in d.i. H2O (10 mL) and Et3N (freshly distilled, 1.0 mL). The obtained mixture was stirred vigorously for 30 min. The resultant organic layer was collected and the aqueous layer was extracted with DCM (2 × 5 mL). The combined organic layers were washed with brine, dried with MgSO4, and concentrated in vacuo. The obtained solid was then dissolved in 1 mL DCM and precipitated via dropwise addition into a vigorously stirred solution of CH3CN (250 mL). The precipitate was filtered and concentrated in vacuo to afford the desired polymer (0.186 g, 95%) as a gray solid: IR (CDCl3, cm−1) 3058, 2999, 2927, 2855, 1592, 1442, 1252, 908, 869, 822, 789, 741; 1H NMR (500 MHz, CDCl3) δ 7.60 – 7.43 (bs, 1H), 7.41 – 7.05 (bm, 3 H), 5.49 – 5.00 (bm, 1 H), 2.59 – 0.55 (bm, 11 H), 0.50 – 0.13 (bm, 6 H); Mn = 7200, PDI = 1.6.
General procedure B
To a cooled solution of siloxane polymer in THF (3.0 equiv, 10 mg/mL) at −78 °C was added PhLi in Bu2O (2.5 equiv) dropwise. The reaction mixture was allowed to warm to rt and was stirred for 3 h, and a white, cloudy solution developed. A solid mixture of PdCl2 (0.03 equiv), CuI (0.1 equiv), and dpca (0.04 equiv) was combined and added to the reaction flask, followed by addition of the aryl halide (1.0 equiv). The obtained reaction mixture was stirred vigorously at rt. Care should be taken so that all reactants are submerged in THF and no solid is deposited on the side of the flask. After 2 h, the reaction mixture was quenched with sat. aq. NH4Cl (5 mL), followed by addition of d.i. H2O (5 mL). The organic layer was collected and the aqueous layer was extracted with Et2O (2 × 10 mL). The combined organic layers were washed with brine, dried with MgSO4, and concentrated in vacuo to 1–2 mL in volume. The obtained concentrated solution was added dropwise into a vigorously stirred solution of CH3CN (250 mL). The precipitated polymer was filtered and the supernatant was concentrated in vacuo to provide the crude product. Following removal from the supernatant, the polymer was re-dissolved in DCM (20 mL) and filter through a glass fritted funnel to remove insoluble particles, if there is any. The obtained solution was then concentrated in vacuo to provide the recovered polymer.
General procedure C
To a solution of alkenyl or aryl iodide in THF (2.5 equiv, 0.1 M) at −78 °C was added t-BuLi in pentane (5.2 equiv), and a bright, yellow slurry developed. The reaction mixture was allowed to stirred at −78 °C for 1 h to ensure complete formation of the corresponding alkenyl or aryl lithium. A solution of siloxane polymer in THF (3.0 equiv) was prepared and added into the reaction flask drop-wise via syringe at −78 °C. Note that the final concentration of the siloxane polymer in the reaction flask should be 10 mg/mL. The reaction mixture was allowed to warm to rt and was stirred for 3 h, and a white, cloudy solution developed. A solid mixture of PdCl2 (0.03 equiv), CuI (0.1 equiv), and dpca (0.04 equiv) was combined and added to the reaction flask, followed by addition of the aryl halide (1.0 equiv). The obtained reaction mixture was stirred vigorously at rt. Care should be taken so that all reactants are submerged in THF and no solid is deposited on the side of the flask. After 2 h, the reaction mixture was quenched with sat. aq. NH4Cl (5 mL), followed by addition of d.i. H2O (5 mL). The organic layer was collected and the aqueous layer was extracted with Et2O (2 × 10 mL). The combined organic layers were washed with brine, dried with MgSO4, and concentrated in vacuo to 1–2 mL in volume. The obtained concentrated solution was added dropwise into a vigorously stirred solution of CH3CN (250 mL). The precipitated polymer was filtered and the supernatant was concentrated in vacuo to provide the crude product. Following removal from the supernatant, the polymer was re-dissolved in DCM (20 mL) and filter through a glass fritted funnel to remove insoluble particles, if there is any. The obtained solution was then concentrated in vacuo to provide the recovered polymer.
4-Phenylanisole (6)
Following general procedure B, using PSTA-I200 (0.213 g, 0.832 mmol), PhLi (384 μL, 1.8 M, 0.692 mmol), PdCl2 (1.5 mg, 8.3 μmol), dpca (4.1 mg, 0.011 mmol), CuI (5.3 mg, 0.028 mmol) and 5 (0.065 g, 0.277 mmol). The product was purified by chromatography on SiO2 (1% Et2O/ hexanes) to afford 6 (50.2 mg, 0.273 mmol, 98%) as a colorless solid. Analytical data matches that which has been previously reported for 6:28 1H NMR (500 MHz, CDCl3) δ 7.57–7.53 (m, 4 H), 7.42 (t, J = 7.6 Hz, 2 H), 7.31 (t, J = 7.4 Hz, 1 H), 6.99 (d, J = 8.7 Hz, 2 H), 3.86 (s, 3 H); 13C NMR (125 MHz, CDCl3) δ 159.3, 141.0, 133.9, 128.9, 128.3, 126.9, 126.8, 114.3, 55.5; HRMS (CI+) m/z (M+H)+: Calcd for C13H13O: 185.0966, found: 185.0963. Preparation of 6 from 5c was carried out in a similar fashion.
4-Cyanobiphenyl (6a)
Following general procedure B, using PSTA-I200 (0.193 g, 0.752 mmol), PhLi (348 μL, 1.8 M, 0.627 mmol), PdCl2 (1.3 mg, 7.5 μmol), dpca (3.7 mg, 0.01 mmol), CuI (4.8 mg, 0.025 mmol) and 5a (56.8 g, 0.248 mmol). The product was purified by chromatography on SiO2 (2% Et2O/ hexanes) to afford 6a (40.3 mg, 0.225 mmol, 91%) as a colorless solid. Analytical data matches that which has been previously reported for 6a:29 1H NMR (500 MHz, CDCl3) δ 7.73 (d, J = 8.3 Hz, 2 H), 7.69 (d, J = 8.4 Hz, 2 H), 7.59 (d, J = 7.4 Hz, 2 H), 7.49 (t, J = 7.2 Hz, 2 H), 7.43 (t, J = 7.3 Hz, 1 H); 13C NMR (125 MHz, CDCl3) δ 145.8, 139.3, 132.8, 129.3, 128.8, 127.9, 127.4, 119.1, 111.1; HRMS (CI+) m/z (M+H)+: Calcd for C13H10N: 180.0813, found: 180.0813. Preparation of 6a from 5d was carried out in a similar fashion.
4-(5-Phenylpyridin-2-yl)morpholine (6b)
Following general procedure B, using PSTA-I200 (0.238 g, 0.930 mmol), PhLi (431 μL, 1.8 M, 0.775 mmol), PdCl2 (1.7 mg, 9.3 μmol), dpca (4.6 mg, 0.012 mmol), CuI (5.9 mg, 0.031 mmol) and 5b (88.8 g, 0.306 mmol). The product was purified by chromatography on SiO2 (2 % Et2O/ hexanes) to afford 6b (68.4 mg, 0.285 mmol, 93 %) as a colorless solid. Analytical data matches that which has been previously reported for 6b:8a 1H NMR (500 MHz, CDCl3) δ 8.47 (d, J = 2.2 Hz, 1 H), 7.75 (dd, J = 2.5, 8.8 Hz, 1 H), 7.53 (d, J = 7.4 Hz, 2 H), 7.43 (t, J = 7.5 Hz, 2 H), 7.32 (t, J = 7.4 Hz, 1 H), 6.71 (d, J = 8.8 Hz, 1 H), 3.85 (app t, J = 4.9 Hz, 4 H), 3.56 (app t, J = 4.9 Hz, 4 H); 13C NMR (125 MHz, CDCl3) δ 158.9, 146.3, 138.4, 136.3, 129.1, 127.0, 126.9, 126.4, 106.8, 66.9, 45.8; HRMS (CI+) m/z (M+H)+: Calcd for C15H17N2O: 241.1341, found: 241.1344.
(E)-1-Methoxy-4-(4-phenylbut-1-en-1-yl)benzene (6c)
Following general procedure C, using PSTA-I200 (0.227 g, 0.887 mmol), 5 (0.173 g, 0.739 mmol), t-BuLi (1.06 mL μL, 1.45 M, 1.537 mmol), PdCl2 (1.6 mg, 8.9 μmol), dpca (4.4 mg, 0.012 mmol), CuI (5.6 mg, 0.030 mmol) and 5e (76.9 mg, 0.298 mmol, in 1 mL THF). The product was purified by chromatography on SiO2 (0.5 % Et2O/ hexanes) to afford 6c (69.5 mg, 0.292 mmol, 98%) as a colorless solid. Analytical data matches that which has been previously reported for 6c:30 1H NMR (500 MHz, CDCl3) δ 7.34 – 7.18 (m, 7 H), 6.85 (d, J = 8.5 Hz, 2 H), 6.38 (d, J = 15.9 Hz, 1 H), 6.13 (td, J = 6.9, 15.7 Hz, 1 H), 3.82 (s, 3 H), 2.79 (t, J = 7.8 Hz, 2 H), 2.52 (q, J = 7.2 Hz, 2 H); 13C NMR (125 MHz, CDCl3) δ 158.9, 142.3, 130.7, 129.9, 128.6, 128.5, 127.96, 127.21, 126.0, 114.1, 55.4, 36.2, 35.0; HRMS (CI+) m/z (M+H)+: Calcd for C17H19O: 239.1436, found: 239.1432.
(E)-1-(Hept-1-en-1-yl)-4-methoxybenzene (6d)
Following general procedure C, using PSTA-I200 (0.214 g, 0.836 mmol), (E)-1-iodohept-1-ene (the corresponding vinyl iodide of 8b) (0.156 g, 0.697 mmol), t-BuLi (1.01 mL, 1.43 M, 1.45 mmol), PdCl2 (1.5 mg, 8.4 μmol), dpca (4.1 mg, 0.011 mmol), CuI (5.3 mg, 0.028 mmol) and 5 (64.0 mg, 0.273 mmol). The product was purified by chromatography on SiO2 (0 – 0.5 % Et2O/ hexanes) to afford 6d (39.4 mg, 0.193 mmol, 71%) as a colorless oil. Analytical data matches that which has been previously reported for 6d:31 1H NMR (500 MHz, CDCl3) δ 7.28 (dd, J = 2.0, 6.5 Hz, 2 H), 6.84 (dd, J = 2.1, 6.6 Hz, 2 H), 6.32 (d, J = 15.7 Hz, 1 H), 6.09 (dt, J = 7.2, 15.8 Hz, 1 H), 3.80 (s, 3 H), 2.18 (m, 2 H), 1.46 (m, 2 H), 1.40–1.37 (m, 4 H), 0.90 (t, J = 7.1 Hz, 3 H); 13C NMR (125 MHz, CDCl3) δ 158.6, 131.2, 129.6, 129.5, 127.1, 114.2, 55.7, 33.5, 31.4, 29.5, 22.7, 14.1; HRMS (CI+) m/z (M+H)+: Calcd for C14H21O: 205.1592, found: 205.1621.
(E)-4-(4-Phenylbut-1-en-1-yl)benzonitrile (6e)
Following general procedure C, using PSTA-I200 (0.241 g, 0.942 mmol), (E)-(4-iodobut-3-en-1-yl)benzene (the corresponding vinyl iodide of 8c) (0.203 g, 0.785 mmol), t-BuLi (1.17 mL, 1.40 M, 1.63 mmol), PdCl2 (1.7 mg, 9.4 μmol), dpca (4.7 mg, 0.013 mmol), CuI (6.0 mg, 0.031 mmol) and 5a (71.6 mg, 0.313 mmol). The product was purified by chromatography on SiO2 (5 % Et2O/ hexanes) to afford 6e (51.3 mg, 0.220 mmol, 70%) as a colorless solid. Analytical data matches that which has been previously reported for 6e:32 1H NMR (500 MHz, CDCl3) δ 7.57 (d, J = 8.3 Hz, 2 H), 7.39 (d, J = 8.3 Hz, 2 H), 7.31 (t, J = 7.5 Hz, 2 H), 7.25 – 7.18 (m, 3 H), 6.46 – 6.35 (m, 2 H), 2.81 (t, J = 7.7 Hz, 2 H), 2.64 – 2.52 (m, 2 H); 13C NMR (125 MHz, CDCl3) δ 142.3, 141.4, 134.4, 132.5, 129.2, 128.6, 128.6, 126.6, 126.2, 119.2, 110.3, 35.6, 35.0; HRMS (CI+) m/z (M)+: Calcd for C17H15N: 233.1204, found: 233.1200.
(2E, 4E)-7-Phenylhepta-2,4-dien-1-ol (6f)
Following general procedure C, using PSTA-I200 (0.223 g, 0.871 mmol), (E)-t-butyl((3-iodoallyl)oxy)dimethylsilane (the corresponding vinyl iodide of 8d) (0.216 g, 0.726 mmol), t-BuLi (1.04 mL, 1.45 M, 1.51 mmol), PdCl2 (1.5 mg, 8.7 μmol), dpca (4.3 mg, 0.012 mmol), CuI (5.5 mg, 0.029 mmol) and 5e (74.8 mg, 0.290 mmol). The crude was taken up in THF (3 mL) and treated with TBAF (1.16 mmol, 4.0 equiv, 1.0 M in THF). The reaction mixture was stirred at rt for 4 h, quenched with sat. aq. NH4Cl (5 mL), followed by addition of d.i. H2O (5 mL). The organic layer was collected and the aqueous layer was extracted with Et2O (2 × 10 mL). The combined organic layers were washed with brine, dried with MgSO4, and concentrated in vacuo. The product was purified by chromatography on SiO2 (20 % Et2O/hexanes) to afford 6f (39.0 mg, 0.207 mmol, 72 %) as a colorless oil. Analytical data matches that which has been previously reported for 6f:8a 1H NMR (500 MHz, CDCl3) δ 7.31 – 7.26 (m, 2 H), 7.22 – 7.16 (m, 3 H), 6.22 (dd, J = 10.5, 15.1 Hz, 1 H), 6.09 (dd, J = 10.5, 15.1 Hz, 1 H), 5.79 – 5.70 (m, 2 H), 4.17 (d, J = 3.8 Hz, 2 H), 2.71 (t, J = 7.8 Hz, 2 H), 2.42 (q, J = 7.4 Hz, 2 H), 1.30 (bs, 1 H); 13C NMR (125 MHz, CDCl3) δ 141.8, 134.6, 132.0, 130.1, 130.0, 128.6, 128.5, 126.0, 63.7, 35.8, 34.6; HRMS (CI+) m/z (M-OH)+: Calcd for C13H15: 171.1174, found: 171.1182.
Synthesis and Cross-Coupling Reactions of Polystyrene-supported Transfer Agent
Preparation of Organolithium Reagents 8a, 8b, 8d and 8e
In a dried flask was added the corresponding aryl or vinyl iodide (2.0 mmol) in 1 mL THF and the solution was cooled to −78 °C. t-BuLi (2.0 equiv) was added dropwise and the obtained solution was stirred at −78 °C for 30 mins and then at room temperature for another 30 mins. The resulting organolithium solution was then titrated with diphenylacetic acid33 before use.
1-(2-Bromophenyl)but-3-en-1-ol (18)
Allylmagnesium chloride (2.0 M in THF, 52.20 mL, 104.47 mmol) was added slowly to a stirred solution of 2-bromobenzaldehyde (16.11 g, 87.05 mmol) in 250 mL THF at room temperature. The obtained solution was stirred for 15 h, and was quenched with saturated aqueous NH4Cl (50 mL). The organic layer was collected and the aqueous layer was extracted with Et2O (2 × 50 mL). The combined organic layers were washed with brine, dried with MgSO4, and concentrated in vacuo. The crude product was purified by flash chromatography on silica gel (10% EtOAc/Hexanes) to afford the desired alcohol 18 as a pale yellow oil (18.73 g, 82.88 mmol, 95%): IR (neat, cm−1) 3389, 3073, 2979, 2912, 1639, 1568, 1468, 1439, 1023, 917, 754; 1H NMR (500 MHz, CDCl3) δ 7.59–7.55 (m, 1 H), 7.52 (d, J = 7.9 Hz, 1 H), 7.34 (t, J = 7.5 Hz, 1 H), 7.16–7.11 (m, 1 H), 5.93–5.83 (m, 1 H), 5.24–5.16 (m, 2 H), 5.14–5.09 (m, 1 H), 2.69–2.62 (m, 1 H), 2.40–2.32 (m, 1 H), 2.14 (d, J = 3.4 Hz, 1 H); 13C NMR (125 MHz, CDCl3) δ 142.8, 134.4, 132.8, 129.0, 127.8, 127.5, 121.9, 118.9, 72.0, 42.3; HRMS (CI+) m/z (M-C3H5)+: Calcd for C7H6OBr: 184.9602, found: 184.9608.
3-Allyl-1,1-dimethyl-1,3-dihydrobenzo[c][1,2]oxasilole (14)
A solution of n-BuLi (2.55 M in hexanes, 62.10 mL, 158.30 mmol) was added dropwise to a stirred solution of benzyl alcohol 18 (16.33 g, 71.95 mmol) in 250 mL THF at −78 °C. The obtained solution was stirred for 1 h, followed by addition of Me2SiHCl (17.20 mL, 158.30 mmol) in one portion at −78 °C. The resulting reaction mixture was allowed to warm to rt and was stirred overnight. After 14 h, the reaction mixture was quenched by addition of d.i. H2O (100 mL) and stirred for 4 h. Note slow evolution of hydrogen gas (caution!). The organic layer was collected and the aqueous layer was extracted with hexanes (2 × 50 mL). The combined organic layers were washed with brine, dried with MgSO4, and concentrated in vacuo. Kugelrohr distillation (50 – 140 °C, 0.025 mmHg), followed by flash chromatography on water-washed silica gel (1% Et2O/Hexanes) afforded the desired siloxane 14 as a colorless oil (6.60 g, 32.35 mmol, 45%): IR (neat, cm−1) 3072, 3001, 2966, 2931, 2899, 2868, 1642, 1595, 1443, 1329, 1076, 1049, 988, 897, 825, 790; 1H NMR (500 MHz, CDCl3) δ 7.56 (d, J = 7.1 Hz, 1 H), 7.42–7.37 (m, 1 H), 7.31 (t, J = 7.1 Hz, 1 H), 7.24 (d, J = 7.7 Hz, 1 H), 5.87–5.76 (m, 1 H), 5.30 (dd, J = 3.9, 7.0 Hz, 1 H), 5.14–5.04 (m, 2 H), 2.73–2.65 (m, 1 H), 2.47–2.39 (m, 1 H), 0.38 (d, J = 15.5 Hz, 6 H); 13C NMR (125 MHz, CDCl3) δ 152.5, 135.9, 134.7, 131.0, 129.7, 127.2, 122.5, 117.7, 81.2, 43.5, 1.5, 0.7; HRMS (CI+) m/z (M-CH3)+: Calcd for C11H13OSi: 189.0736, found: 189.0733.
3-(3-(4-Bromophenyl)propyl)-1,1-dimethyl-1,3-dihydrobenzo[c][1,2]oxasilole (19)
A solution of 9-BBN (0.5 M in THF, 12.15 mL, 6.08 mmol) was added dropwise to a flask containing siloxane 14 (826.3 mg, 4.05 mmol) while stirring at room temperature. The obtained solution was stirred for 3 h, at which time TLC analysis indicated consumption of the starting material 14. A screw-cap vial was charged with 1,4-dibromobenzene (2.87 g, 12.15 mmol), Pd(dppf)Cl2.DCM (165.4 mg, 0.203 mmol), K3PO4 (1.72 g, 8.10 mmol), and the mixture was suspended in 10 mL DMF. The obtained mixture was stirred for 15 min at room temperature, followed by addition of the above solution containing siloxane 14/9-BBN adduct via cannula (rinsed with 0.5 mL DMF). The obtained vial was capped and heated to 50 °C for 16 h. The reaction mixture was then cooled to room temperature and quenched with d.i. H2O (10 mL). The resulting mixture was extracted with Et2O (2 × 25 mL). The combined organic layers were washed with brine, dried with MgSO4, and concentrated in vacuo. The crude oil was then taken up in THF (10 mL), followed by addition of solid NaBO3.4H2O (3.12 g, 20.25 mmol), and d.i. H2O (10 mL) for oxidation of borane byproducts to facilitate purification. The mixture was then stirred vigorously at room temperature, open to air, for 2 h. The aqueous layer was then extracted with hexanes (2 × 25 mL). The combined organic layers were washed with brine, dried with MgSO4, and concentrated in vacuo. Kugelrohr distillation (180 °C, 0.025 mmHg) afforded the desired aryl bromide 19 as pale yellow oil (1.25 g, 3.46 mmol, 85%): IR (neat, cm−1) 3058, 2942, 2860, 1593, 1487, 1252, 1077, 876, 818, 790; 1H NMR (500 MHz, CDCl3) δ 7.54 (d, J = 7.1 Hz, 1 H), 7.41–7.35 (m, 3 H), 7.29 (t, J = 7.1 Hz, 1 H), 7.15 (d, J = 7.7 Hz, 1 H), 7.03 (d, J = 8.3 Hz, 2 H), 5.25 (dd, J = 3.3, 7.4 Hz, 1 H), 2.68–2.53 (m, 2 H), 1.98–1.90 (m, 1 H), 1.82–1.59 (m, 3 H), 0.37 (d, J = 4.8 Hz, 6 H); 13C NMR (125 MHz, CDCl3) δ 153.0, 141.5, 135.7, 131.4, 131.0, 130.3, 129.8, 127.1, 122.3, 119.5, 81.5, 38.3, 35.3, 26.5, 1.5, 0.7; HRMS (CI+) m/z (M)+: Calcd for C18H21OBrSi: 360.0545, found: 360.0529.
1,1-dimethyl-3-(3-(4-vinylphenyl)propyl)-1,3-dihydrobenzo[c][1,2]oxasilole (20)
A solution of potassium vinyltrifluoroborate (336.6 mg, 2.51 mmol), PdCl2 (6.9 mg, 0.039 mmol), PPh3 (30.4 mg, 0.116 mmol), Cs2CO3 (1.89 g, 5.80 mmol), and aryl bromide 19 (698.5 mg, 1.93 mmol) in THF/H2O (9:1, 4.1 mL) was heated at 85 °C in a screw-cap vial. The reaction mixture was stirred at 85 °C for 19 h, then cooled to room temperature and diluted with H2O (5 mL), followed by extraction with dichloromethane (2 × 10 mL). The combined organic layers were washed with brine, dried with MgSO4, and concentrated in vacuo. Purification via flash chromatography on water-washed silica gel (1% Et2O/Hexanes) afforded the desired styrene 20 as a colorless oil (536.7 mg, 1.74 mmol, 90%): IR (neat, cm−1) 3055, 3001, 2941, 2858, 1629, 1511, 1442, 1252, 1085, 876, 823, 790; 1H NMR (500 MHz, CDCl3) δ 7.54 (d, J = 7.1 Hz, 1 H), 7.40–7.35 (m, 1 H), 7.33–7.26 (m, 3 H), 7.16 (d, J = 7.7 Hz, 1 H), 7.12 (d, J = 7.9 Hz, 2 H), 6.69 (dd, J = 10.9, 17.6 Hz, 1 H), 5.70 (d, J = 17.6 Hz, 1 H), 5.26 (dd, J = 3.2, 7.3 Hz, 1 H), 5.18 (d, J = 10.9 Hz, 1 H), 2.72–2.57 (m, 2 H), 2.01–1.90 (m, 1 H), 1.84–1.59 (m, 3 H), 0.38 (d, J = 7.1 Hz, 6 H); 13C NMR (125 MHz, CDCl3) δ 153.1, 142.3, 136.9, 135.7, 135.2, 131.0, 129.7, 128.7, 127.0, 126.3, 122.3, 113.0, 81.6, 38.4, 35.6, 26.5, 1.5, 0.7; HRMS (CI+) m/z (M)+: Calcd for C20H24OSi: 308.1596, found: 308.1581.
Polymer PSTA-I
A solution of acacia gum (3.2 g) and NaCl (2.0 g) in H2O (85 mL) was placed in a 100 mL- reaction flask equipped with a mechanical stirrer and deoxygenated by purging with N2 for 30 min. A solution of monomer 20 (433 mg, 1.40 mmol), styrene (1.20 mL, 10.47 mmol), cross-linker 21 (34.9 mg, 0.119 mmol), and benzoyl peroxide (20.0 mg, 0.083 mmol) in chlorobenzene (2.0 mL) was injected to the rapidly stirred aqueous solution. This mixture was heated at 85 °C for 17 h. The crude polymer was collected by filtration and washed sequentially with MeOH/H2O (3:1, 4 × 50 mL), MeOH (2 × 50 mL), THF (2 × 25 mL), Et2O (2 × 25 mL), hexanes (2 × 25 mL), followed by drying in vacuo to provide the desired polymer PSTA-I as white beads (724 mg) and a Si loading of 1.49 mmol/g was determined by inductively coupled plasma optical emission spectrometry (ICP-OES). The yield was 77% based on Si incorporation. IR (KBr, cm−1) 3026, 2914, 1943, 1721, 1492, 1448, 1372, 1329, 1249, 1082, 1026, 743, 698.
Procedure for Recyclability of PSTA-I Using the Same Nucleophile and Electrophile
To a cooled suspension of siloxane polymer PSTA-I (1.49 mmol/g, 500 mg, 0.745 mmol) swelling in THF (20 mL) at −78 °C was added PhLi in Bu2O (1.8 M, 345 μL, 0.621 mmol) dropwise. The reaction mixture was allowed to warm to room temperature and was stirred for 3 h. A solid mixture of PdCl2 (1.3 mg, 7.4 μmol), CuI (4.7 mg, 24.8 μmol), and dpca (3.7 mg, 9.9 μmol) was combined and added to the reaction flask, followed by addition of 4-iodoanisole (58.0 mg, 0.248 mmol). The obtained reaction mixture was stirred vigorously at room temperature for 18 h. The reaction mixture was then quenched with sat. aq. NH4Cl (5 mL), followed by addition of d.i. H2O (5 mL), and stirred for an hour at room temperature. The obtained mixture was filtered through a fritted filter to remove polymer. The filtered polymer was then washed with Et2O (4 × 50 mL). The combined filtrate was washed with brine, dried with MgSO4, and concentrated in vacuo, followed by purification via flash chromatography (1% Et2O/ hexanes) to provide the desired cross-coupling product 4-phenylanisole 6 as a white solid (43.8 mg, 0.238 mmol, 96%). Following filtration, the polymer was further washed sequentially with MeOH/H2O solution (1:1, 2 × 50 mL), MeOH (2 × 25 mL), CH2Cl2 (2 × 25 mL), Et2O (2 × 25 mL), hexanes (2 × 25 mL), and dried in vacuo to provide near quantitative recovery of siloxane polymer. The obtained polymer was re-used 5 more times employing the same procedure above and showed no loss in cross-coupling efficiency.
General Procedure for Recyclability of PSTA-I Using Multiple Nucleophiles and Electrophiles
To a cooled suspension of siloxane polymer PSTA-I (0.74 mmol/g, 3.0 equiv) swelling in THF (25 mL) at −78 °C was added the organolithium solution (2.5 equiv) dropwise. The reaction mixture was allowed to warm to room temperature and was stirred for 3 h. A solid mixture of PdCl2 (3 mol %), CuI (10 mol %), and dpca (4 mol %) was combined and added to the reaction flask, followed by addition of the aryl or alkenyl halide (1.0 equiv). The obtained reaction mixture was stirred vigorously at room temperature for 18 h. The reaction mixture was then quenched with sat. aq. NH4Cl (5 mL), followed by addition of d.i. H2O (5 mL), and stirred for an hour at room temperature. The obtained mixture was filtered through a fritted filter to remove polymer. The filtered polymer was then washed with Et2O (4 × 50 mL). The combined filtrate was washed with brine, dried with MgSO4, and concentrated in vacuo, followed by purification via flash chromatography to provide the desired cross-coupling product. Following filtration, the polymer was further washed sequentially with MeOH/H2O solution (1:1, 2 × 50 mL), MeOH (2 × 25 mL), CH2Cl2 (2 × 25 mL), Et2O (2 × 25 mL), hexanes (2 × 25 mL), and dried in vacuo to provide near quantitative recovery of siloxane polymer, which was employed in the next cross-coupling cycle.
4-(tert-Butyl)-4′-methoxy-1,1′-biphenyl (6g)
Following general procedure, using PSTA-I (750 mg, 0.555 mmol), 8e (926 μL, 0.50 M, 0.463 mmol), PdCl2 (1.0 mg, 5.6 μmol), dpca (2.7 mg, 7.4 μmol), CuI (3.5 mg, 18.5 μmol) and 5 (42.0 mg, 0.180 mmol). The product was purified by chromatography on SiO2 (1% Et2O/ hexanes) to afford 6g (41.2 mg, 0.172 mmol, 95%) as a colorless solid. Analytical data matches that which has been previously reported for 6g:34 1H NMR (500 MHz, CDCl3) δ 7.57–7.49 (m, 4 H), 7.48–7.44 (m, 2 H), 6.98 (d, J = 8.7 Hz, 2 H), 3.86 (s, 3 H), 1.38 (s, 9 H); 13C NMR (125 MHz, CDCl3) δ 159.1, 149.8, 138.1, 133.8, 128.1, 126.5, 125.8, 114.3, 55.5, 34.6, 31.5; HRMS (CI+) m/z (M+H)+: Calcd for C17H21O: 241.1592, found: 241.1603.
4-Phenylanisole (6)
Following general procedure, using PSTA-I (720 mg, 0.533 mmol), PhLi (234 μL, 1.9 M, 0.444 mmol), PdCl2 (0.9 mg, 5.3 μmol), dpca (2.6 mg, 7.1 μmol), CuI (3.4 mg, 17.8 μmol) and 5 (41.6 mg, 0.178 mmol). The product was purified by chromatography on SiO2 (1% Et2O/ hexanes) to afford 6 (31.8 mg, 0.173 mmol, 97%) as a colorless solid. Analytical data matches that which has been reported above.
(E)-1-Methoxy-4-(4-phenylbut-1-en-1-yl)benzene (6c)
Following general procedure, using PSTA-I (702 mg, 0.519 mmol), 8a (1.2 mL, 0.36 M, 0.433 mmol), PdCl2 (0.9 mg, 5.2 μmol), dpca (2.6 mg, 7.0 μmol), CuI (3.3 mg, 17.3 μmol) and 5e (44.7 mg, 0.173 mmol). The product was purified by chromatography on SiO2 (0.5 % Et2O/ hexanes) to afford 6c (37 mg, 0.155 mmol, 90%) as a colorless solid. Analytical data matches that which has been reported above
(E)-1-(Hept-1-en-1-yl)-4-methoxybenzene (6d)
Following general procedure, using PSTA-I (686 mg, 0.508 mmol), 8b (1.14 mL, 0.37 M, 0.423 mmol), PdCl2 (0.9 mg, 5.1 μmol), dpca (2.5 mg, 6.8 μmol), CuI (3.2 mg, 16.9 μmol) and 5 (39.6 mg, 0.169 mmol). The product was purified by chromatography on SiO2 (0–0.5 % Et2O/ hexanes) to afford 6d (25.5 mg, 0.125 mmol, 74%) as a colorless oil. Analytical data matches that which has been reported above.
(2E, 4E)-7-Phenylhepta-2,4-dien-1-ol (6f)
Following general procedure, using PSTA-I (662 mg, 0.490 mmol), 8d (1.4 mL, 0.30 M, 0.408 mmol), PdCl2 (0.9 mg, 4.9 μmol), dpca (2.4 mg, 6.5 μmol), CuI (3.1 mg, 16.3 μmol) and 5e (42.1 mg, 0.163 mmol). The crude was taken up in THF (3 mL) and treated with TBAF (0.65 mmol, 4.0 equiv, 1.0 M in THF). The reaction mixture was stirred at rt for 4 h, quenched with sat. aq. NH4Cl (5 mL), followed by addition of d.i. H2O (5 mL). The organic layer was collected and the aqueous layer was extracted with Et2O (2 × 10 mL). The combined organic layers were washed with brine, dried with MgSO4, and concentrated in vacuo. The product was purified by chromatography on SiO2 (20 % Et2O/ hexanes) to afford 6f (22.0 mg, 0.117 mmol, 72%) as a colorless oil. Analytical data matches that which has been reported above.
4-Cyanobiphenyl (6a)
Following general procedure, using PSTA-I (660 mg, 0.488 mmol), PhLi (214 μL, 1.9 M, 0.407 mmol), PdCl2 (0.9 mg, 4.9 μmol), dpca (2.4 mg, 6.5 μmol), CuI (3.1 mg, 16.3 μmol) and 5a (37.3 mg, 0.163 mmol). The product was purified by chromatography on SiO2 (2% Et2O/ hexanes) to afford 6a (26.8 mg, 0.150 mmol, 92%) as a colorless solid. Analytical data matches that which has been reported above. Preparation of 6a from 5d was carried out in a similar fashion.
4-(5-Phenylpyridin-2-yl)morpholine (6b)
Following general procedure, using PSTA-I (650 mg, 0.481 mmol), PhLi (211 μL, 1.9 M, 0.401 mmol), PdCl2 (0.9 mg, 4.8 μmol), dpca (2.4 mg, 6.4 μmol), CuI (3.0 mg, 16.0 μmol) and 5b (46.4 mg, 0.160 mmol). The product was purified by chromatography on SiO2 (20% Et2O/ hexanes) to afford 6b (34.9 mg, 0.145 mmol, 91%) as a colorless solid. Analytical data matches that which has been reported above.
4-Acetylbiphenyl (6h)
Following general procedure, using PSTA-I (620 mg, 0.459 mmol), PhLi (201 μL, 1.9 M, 0.382 mmol), PdCl2 (0.8 mg, 4.6 μmol), dpca (2.3 mg, 6.1 μmol), CuI (2.9 mg, 15.3 μmol) and 5f (36.5 mg, 0.148 mmol). The product was purified by chromatography on SiO2 (5% Et2O/ hexanes) to afford 6h (23.8 mg, 0.121 mmol, 82%) as a colorless solid. Analytical data matches that which has been previously reported for 6h:35 1H NMR (500 MHz, CDCl3) δ 8.04 (d, J = 8.3 Hz, 2 H), 7.69 (d, J = 8.1 Hz, 2 H), 7.63 (d, J = 7.3 Hz, 2 H), 7.48 (t, J = 7.5 Hz, 2 H), 7.41 (t, J = 7.3 Hz, 1 H), 2.64 (s, 3 H); 13C NMR (125 MHz, CDCl3) δ 197.9, 145.9, 140.0, 136.0, 129.1, 129.0, 128.4, 127.4, 127.4, 26.8; HRMS (CI+) m/z (M)+: Calcd for C14H12O: 196.0888, found: 196.0888. Preparation of 6h from 5g was carried out in a similar fashion.
4-phenylpyridine (6i)
Following general procedure, using PSTA-I (699 mg, 0.390 mmol), PhLi (176 μL, 1.85 M, 0.325 mmol), PdCl2 (0.7 mg, 4.0 μmol), dpca (2.0 mg, 5.2 μmol), CuI (2.6 mg, 13.0 μmol) and 5h (26.7 mg, 0.13 mmol). The product was purified by chromatography on SiO2 (40% EtOAc/ hexanes, Rf = 0.27) to afford 6i (15.6 mg, 0.100 mmol, 77%) as a colorless liquid. Analytical data matches that which has been previously reported for 6i:36 1H NMR (500 MHz, CDCl3) δ 8.67 (s, 2H), 7.65 (dd, J = 7.2, 1.8 Hz, 2H), 7.52 – 7.42 (m, 5H); 13C NMR (125 MHz, CDCl3) δ 150.2, 148.3, 138.2, 129.1, 129.0, 127.0, 121.7; HRMS (EI+): Calcd for C11H9N: 155.0734, found: 155.0735.
1-methyl-4-phenyl-1H-imidazole (6j)
Following general procedure, using PSTA-I (684 mg, 0.390 mmol), PhLi (176 μL, 1.85 M, 0.325 mmol), PdCl2 (0.7 mg, 4.0 μmol), dpca (2.0 mg, 5.2 μmol), CuI (2.6 mg, 13.0 μmol) and 5i (27.0 mg, 0.13 mmol). The product was purified by chromatography on SiO2 (40% EtOAc/ hexanes, Rf = 0.23) to afford 6j (16.6 mg, 0.105 mmol, 81%) as a colorless solid. Analytical data matches that which has been previously reported for 6j:37 1H NMR (500 MHz, CDCl3) δ 7.76 (s, 1H), 7.60 (s, 1H), 7.47 (d, J = 7.6 Hz, 2H), 7.36 (t, J = 7.6 Hz, 2H), 7.22 (t, J = 7.4 Hz, 1H), 3.94 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 136.7, 132.6, 128.8, 126.9, 126.3, 125.5, 123.2, 39.0; HRMS (EI+) m/z (M)+: Calcd for C10H10N2: 158.0844, found: 158.0833.
4-(trifluoromethyl)-1,1′-biphenyl (6k)
Following general procedure, using PSTA-I (652 mg, 0.371 mmol), PhLi (167 μL, 1.85 M, 0.309 mmol), PdCl2 (0.7 mg, 4.0 μmol), dpca (2.0 mg, 5.2 μmol), CuI (2.5 mg, 12.0 μmol) and 5j (33.7 mg, 0.124 mmol). The product was purified by chromatography on SiO2 (100% hexanes, Rf = 0.54) to afford 6k (25.1 mg, 0.114 mmol, 92%) as a colorless solid. Analytical data matches that which has been previously reported for 6k:36 1H NMR (500 MHz, CDCl3) δ 7.70 (s, 4H), 7.61 – 7.60 (m, 2H), 7.48 (t, J = 7.6 Hz, 2H), 7.41 (t, J = 7.3 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 144.7, 139.8, 129.5, 129.0, 128.2, 127.4, 127.3, 125.7 (q, JCF = 3.7 Hz), 123.2; HRMS (EI+) m/z (M)+: Calcd for C13H9F3: 222.0656, found: 222.0663.
3-phenylpyridine (6l)
Following general procedure, using PSTA-I (645 mg, 0.367 mmol), PhLi (167 μL, 1.85 M, 0.310 mmol), PdCl2 (0.6 mg, 3.6 μmol), dpca (1.8 mg, 4.8 μmol), CuI (2.3 mg, 12.0 μmol) and 5k (24.6 mg, 0.12 mmol). The product was purified by chromatography on SiO2 (40% EtOAc/ hexanes, Rf = 0.32) to afford 6l (15.6 mg, 0.101 mmol, 84%) as a colorless liquid. Analytical data matches that which has been previously reported for 6l:36 1H NMR (500 MHz, CDCl3) δ 8.86 (d, J = 2.3 Hz, 1H), 8.59 (dd, J = 4.7, 1.6 Hz, 1H), 7.88 (d, J = 8.0 Hz, 1H), 7.59 (d, J = 7.1 Hz, 2H), 7.48 (t, J = 7.6 Hz, 2H), 7.41 (t, J = 7.4 Hz, 1H), 7.37 (dd, J = 7.9, 4.8 Hz, 1H).; 13C NMR (125 MHz, CDCl3) δ 148.4, 148.3, 137.8, 136.6, 134.4, 129.1, 128.1, 127.1, 123.5; HRMS (EI+) m/z (M+H)+: Calcd for C11H9N: 155.0727, found: 155.0735.
2-phenylpyridine (6m)
Following general procedure, using PSTA-I (640 mg, 0.364 mmol), PhLi (169 μL, 1.80 M, 0.303 mmol), PdCl2 (0.6 mg, 3.6 μmol), dpca (1.8 mg, 4.8 μmol), CuI (2.3 mg, 12.0 μmol) and 5l (24.6 mg, 0.12 mmol). The product was purified by chromatography on SiO2 (40% EtOAc/ hexanes, Rf = 0.31) to afford 6m (16.3 mg, 0.106 mmol, 88%) as a colorless liquid. Analytical data matches that which has been previously reported for 6m:36 1H NMR (500 MHz, CDCl3) δ 8.70 (d, J = 4.5 Hz, 1H), 7.99 (d, J = 6.9 Hz, 2H), 7.77 – 7.72 (m, 2H), 7.48 (t, J = 7.3 Hz, 2H), 7.42 (t, J = 7.3 Hz, 1H), 7.23 (td, J = 4.8, 2.3 Hz, 1H).; 13C NMR (125 MHz, CDCl3) δ 157.5, 149.7, 139.4, 136.7, 128.9, 128.7, 126.9, 122.1, 120.5; HRMS (EI+) m/z (M)+: Calcd for C11H9N: 155.0735, found: 155.0717.
2-(4-methoxyphenyl)thiophene (6n)
Following general procedure, using PSTA-I (638 mg, 0.360 mmol), 8f (714 μL, 0.42 M, 0.30 mmol), PdCl2 (0.6 mg, 3.6 μmol), dpca (1.8 mg, 4.8 μmol), CuI (2.3 mg, 12.0 μmol) and 5 (27.5 mg, 0.12 mmol). The product was purified by chromatography on SiO2 (1% EtOAc/ hexanes, Rf = 0.31) to afford 6n (16.2 mg, 0.114 mmol, 71%) as a colorless solid. Analytical data matches that which has been previously reported for 6n:38 1H NMR (500 MHz, CDCl3) δ δ 7.54 (d, J = 8.7 Hz, 2H), 7.21 (d, J = 5.2 Hz, 1H), 7.20 (d, J = 3.5 Hz, 1H), 7.05 (dd, J = 5.1, 3.6 Hz, 1H), 6.92 (d, J = 8.7 Hz, 2H), 3.84 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 159.2, 144.3, 127.9, 127.3, 127.2, 123.8, 122.1, 114.3, 55.4; HRMS (EI+) m/z (M)+: Calcd for C11H10OS: 190.0452, found: 190.0436.
2-(4-methoxyphenyl)pyridine (6o)
Following general procedure, using PSTA-I (616 mg, 0.351 mmol), 8g (572 μL, 0.51 M, 0.292 mmol), PdCl2 (0.7 mg, 4.0 μmol), dpca (1.9 mg, 5.0 μmol), CuI (2.3 mg, 12.0 μmol) and 5 (26.8 mg, 0.117 mmol). The product was purified by chromatography on SiO2 (20% EtOAc/ hexanes, Rf = 0.37) to afford 6o (19.2 mg, 0.104 mmol, 89%) as a colorless solid. Analytical data matches that which has been previously reported for 6o:36 1H NMR (500 MHz, CDCl3) δ 8.65 (d, J = 3.9 Hz, 1H), 7.95 (d, J = 8.8 Hz, 2H), 7.71 (td, J = 7.7, 1.8 Hz, 1H), 7.67 (d, J = 8.0 Hz, 1H), 7.17 (t, J = 5.9 Hz, 2H), 7.00 (d, J = 8.8 Hz, 1H), 3.87 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 160.4, 157.1, 149.5, 136.7, 132.0, 128.1, 121.4, 119.8, 114.1, 55.3; HRMS (EI+) m/z (M)+: Calcd for C12H11NO: 185.0836, found: 185.0841.
1-(4-methoxyphenyl)naphthalene (6p)
Following general procedure, using PSTA-I (552 mg, 0.310 mmol), 8h (456 μL, 0.57 M, 0.260 mmol), PdCl2 (0.5 mg, 3.0 μmol), dpca (1.5 mg, 4.0 μmol), CuI (1.9 mg, 10.0 μmol) and 5 (22.9 mg, 0.10 mmol). The product was purified by chromatography on SiO2 (1% EtOAc/ hexanes, Rf = 0.25) to afford 6p (15.0 mg, 0.064 mmol, 64%) as a colorless solid. Analytical data matches that which has been previously reported for 6p:39 1H NMR (500 MHz, CDCl3) δ 7.94 – 7.90 (m, 2H), 7.84 (d, J = 8.2 Hz, 1H), 7.53 – 7.48 (m, 2H), 7.45 – 7.40 (m, 4H), 7.04 (d, J = 8.6 Hz, 2H), 3.90 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 158.9, 139.9, 133.8, 133.1, 131.8, 131.1, 128.2, 127.3, 126.9, 126.1, 125.9, 125.7, 125.4, 113.7, 55.4; HRMS (EI+) m/z (M)+: Calcd for C17H14O: 234.1048, found: 234.1045.
2-(4-methoxyphenyl)benzofuran (6q)
Following general procedure, using PSTA-I (524 mg, 0.300 mmol), 8i (694 μL, 0.36 M, 0.25 mmol), PdCl2 (0.5 mg, 3.0 μmol), dpca (1.5 mg, 4.0 μmol), CuI (1.9 mg, 10.0 μmol) and 5 (22.9 mg, 0.10 mmol). The product was purified by chromatography on SiO2 (1% EtOAc/ hexanes, Rf = 0.18) to afford 6q (16.4 mg, 0.073 mmol, 73%) as a colorless solid. Analytical data matches that which has been previously reported for 6q:40 1H NMR (500 MHz, CDCl3) δ 7.80 (d, J = 8.8 Hz, 2H), 7.56 (d, J = 7.4 Hz, 1H), 7.50 (d, J = 7.6 Hz, 1H), 7.30 – 7.19 (m, 2H), 6.98 (d, J = 8.8 Hz, 2H), 6.89 (s, 1H), 3.87 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 159.9, 156.0, 154.7, 129.5, 125.4, 123.7, 123.3, 122.8, 120.5, 114.2, 111.0, 99.6, 55.3; HRMS (EI+) m/z (M)+: Calcd for C15H12O2: 224.0837, found: 224.0834.
4-methoxy-4′-(trifluoromethyl)-1,1′-biphenyl (6r)
Following general procedure, using PSTA-I (480 mg, 0.270 mmol), 8j (437 μL, 0.53 M, 0.230 mmol), PdCl2 (0.5 mg, 3.0 μmol), dpca (1.5 mg, 4.0 μmol), CuI (1.7 mg, 9.0 μmol) and 5 (20.6 mg, 0.09 mmol). The product was purified by chromatography on SiO2 (1% EtOAc/ hexanes, Rf = 0.23) to afford 6r (17.9 mg, 0.071 mmol, 79%) as a colorless solid. Analytical data matches that which has been previously reported for 6r:36 1H NMR (500 MHz, CDCl3) δ 7.68–7.64 (m, 4H), 7.55 (d, J = 8.7 Hz, 2H), 7.01 (d, J = 8.7 Hz, 2H), 3.87 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 159.8, 144.3, 132.2, 128.5, 126.9, 125.6 (q, J = 3.9 Hz), 114.4, 55.4; HRMS (EI+) m/z (M)+: Calcd for C14H11F3O: 252.0762, found: 252.0767.
Supplementary Material
Acknowledgments
Financial support was provided by the NIH through Grant GM-29028. We thank Professor Virgil Percec and Dr. Nga H. Nguyen for helpful discussions and assistance with GPC, Dr. Bruno Melillo and Dr. Stephen P. Brown for helpful suggestions on polymer selection and handling, and Dr. Rakesh Kohli and Dr. Charles W. Ross, III for assistance with HRMS.
Footnotes
Supporting Information
1H-NMR and 13C-NMR spectral data of new compounds and cross-coupling products.
References
- 1.(a) de Meijere A, Diederich F, editors. Metal-catalyzed Cross-coupling Reactions. 2nd. Wiley-VCH; Weinheim: 2004. [Google Scholar]; (b) Seechurn CCJ, Kitching MO, Colacot TJ, Snieckus V. Angew Chem, Int Ed. 2012;51:5062. doi: 10.1002/anie.201107017. [DOI] [PubMed] [Google Scholar]
- 2.(a) Murahashi SI, Tanba Y, Yamamura M, Moritani I. Tetrahedron Lett. 1974;15:3749. [Google Scholar]; (b) Murahashi SI, Tanba Y, Yamamura M, Yoshimura N. J Org Chem. 1978;43:4099. [Google Scholar]; (c) Murahashi SI. J Organomet Chem. 2002;653:27. [Google Scholar]; For recent examples of cross-coupling of organolithiums employing Ni and Fe catalysis, see:; (d) Tao J, Wang Z. Asian J Org Chem. 2016;5:521. [Google Scholar]; (e) Heijnen D, Gualtierotti J, Hornillos V, Feringa BL. Chem Eur J. 2016;22:3991. doi: 10.1002/chem.201505106. [DOI] [PubMed] [Google Scholar]; (f) Jia Z, Liu Q, Peng X, Wong HNC. Nat Commun. 2016;7:10614. doi: 10.1038/ncomms10614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Negishi E, Baba S. Chem Commun. 1976;596 [Google Scholar]; (b) Baba S, Negishi E. J Am Chem Soc. 1976;98:6729. [Google Scholar]; Reviews:; (c) Negishi E. Acc Chem Res. 1982;15:340. [Google Scholar]; (d) Negishi E, Hu Q, Huang Z, Qian M, Wang G. Aldrichimica Acta. 2005;38:71. [Google Scholar]
- 4.(a) Miyaura N, Yamada K, Suzuki A. Tetrahedron Lett. 1979;20:3437. [Google Scholar]; (b) Miyaura N, Suzuki A. J Chem Soc Chem Commun. 1979:866. [Google Scholar]; Reviews:; (c) Miyaura N, Suzuki A. Chem Rev. 1995;95:2457. [Google Scholar]; (d) Suzuki A, Yamamoto Y. Chem Lett. 2011;40:894. [Google Scholar]
- 5.(a) Milstein D, Stille JK. J Am Chem Soc. 1978;100:3636. [Google Scholar]; (b) Milstein D, Stille JK. J Am Chem Soc. 1979;101:4992. [Google Scholar]; Reviews:; (c) Stille JK. Angew Chem Int Ed Engl. 1986;25:508. [Google Scholar]; (d) Mitchell TN. Synthesis. 1992;803 [Google Scholar]
- 6.(a) Nakao Y, Imanaka H, Sahoo AK, Yada A, Hiyama T. J Am Chem Soc. 2005;127:6952. doi: 10.1021/ja051281j. [DOI] [PubMed] [Google Scholar]; (b) Nakao Y, Takeda M, Matsumoto T, Hiyama T. Angew Chem Int Ed. 2010;49:4447. doi: 10.1002/anie.201000816. [DOI] [PubMed] [Google Scholar]; (c) Nakao Y, Hiyama T. Chem Soc Rev. 2011;40:4893. doi: 10.1039/c1cs15122c. [DOI] [PubMed] [Google Scholar]; (d) Denmark SE, Choi JY. J Am Chem Soc. 1999;121:5821. [Google Scholar]; (e) Denmark SE, Sweis RF. J Am Chem Soc. 2004;126:4876. doi: 10.1021/ja0372356. [DOI] [PubMed] [Google Scholar]; (f) Denmark SE, Regens CS. Acc Chem Res. 2008;41:1486. doi: 10.1021/ar800037p. and references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Ai Y, Kozytska MV, Zou Y, Khartulyari AS, Smith AB., III J Am Chem Soc. 2015;137:15426. doi: 10.1021/jacs.5b11540. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nguyen MH, Imanishi M, Kurogi T, Smith AB., III J Am Chem Soc. 2016;138:3675. doi: 10.1021/jacs.6b01731. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Liu Q, Chen Y, Zhang X, Houk KN, Liang Y, Smith AB., III J Am Chem Soc. 2017;139:8710. doi: 10.1021/jacs.7b04149. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Deng Y, Liu Q, Smith AB., III J Am Chem Soc. 2017;139:9487. doi: 10.1021/jacs.7b05165. [DOI] [PMC free article] [PubMed] [Google Scholar]; For reviews, see:; (e) Smith AB, III, Adams CM. Acc Chem Res. 2004;37:365. doi: 10.1021/ar030245r. [DOI] [PubMed] [Google Scholar]; (f) Smith AB, III, Wuest WM. Chem Commun. 2008:5883. doi: 10.1039/b810394a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Smith AB, III, Hoye AT, Martinez-Solorio D, Kim W-S, Tong R. J Am Chem Soc. 2012;134:4533. doi: 10.1021/ja2120103. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Martinez-Solorio D, Hoye AT, Nguyen MH, Smith AB., III Org Lett. 2013;15:2454. doi: 10.1021/ol400922j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Martinez-Solorio D, Melillo B, Sanchez L, Liang Y, Lam E, Houk KN, Smith AB., III J Am Chem Soc. 2016;138:1836. doi: 10.1021/jacs.5b13260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Giannerini M, Fañanás-Mastral M, Feringa BL. Nature Chem. 2013;5:667. doi: 10.1038/nchem.1678. [DOI] [PubMed] [Google Scholar]; (b) Hornillos V, Giannerini M, Vila C, Fañanás-Mastral M, Feringa BL. Org Lett. 2013;15:5114. doi: 10.1021/ol402408v. [DOI] [PubMed] [Google Scholar]; (c) Giannerini M, Hornillos V, Vila C, Fananas-Mastral M, Feringa BL. Angew Chem Int Ed. 2013;52:13329. doi: 10.1002/anie.201306427. [DOI] [PubMed] [Google Scholar]; (d) Vila C, Giannerini M, Hornillos V, Fañanás-Mastral M, Feringa BL. Chem Sci. 2014;5:1361. doi: 10.1039/c4sc03117b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Hornillos V, Giannerini M, Vila C, Fañanás-Mastral M, Feringa BL. Chem Sci. 2015;6:1394. doi: 10.1039/c4sc03117b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Heijnen D, Hornillos V, Corbet BP, Giannerini M, Feringa BL. Org Lett. 2015;17:2262. doi: 10.1021/acs.orglett.5b00905. [DOI] [PubMed] [Google Scholar]; (g) Pinxterhuis EB, Giannerini M, Hornillos V, Feringa BL. Nat Commun. 2016;7:11698. doi: 10.1038/ncomms11698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Nguyen MH, Smith AB., III Org Lett. 2013;15:4258. doi: 10.1021/ol402047d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nguyen MH, Smith AB., III Org Lett. 2014;16:2070. doi: 10.1021/ol5007086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Herisson JL, Chauvin Y. Makromol Chem. 1971;141:161. [Google Scholar]; (b) Murdzek JS, Shrock RR. Macromolecules. 1987;20:2640. [Google Scholar]; (c) Grubbs RH, Tumas W. Science. 1989;243:4893. doi: 10.1126/science.2645643. [DOI] [PubMed] [Google Scholar]; Reviews:; (d) Bielawski CW, Grubbs RH. Prog Polym Sci. 2007;32:1. [Google Scholar]; (e) Hilf S, Kilbinger AFM. Nat Chem. 2009;1:537. doi: 10.1038/nchem.347. [DOI] [PubMed] [Google Scholar]
- 12.Starting material was employed as a mixture of endo- and exo- isomers.
- 13.Weickgenannt A, Oestreich M. Chem Asian J. 2009;4:406. doi: 10.1002/asia.200800426. [DOI] [PubMed] [Google Scholar]
- 14.Schwab P, France MB, Ziller JW, Grubbs RH. Angew Chem Int Ed Engl. 1995;34:2039. [Google Scholar]
- 15.Drouin SD, Zamanian F, Fogg DE. Organometallics. 2001;20:24. [Google Scholar]
- 16.An intriguing phenomenon was observed with the small molecule siloxane transfer agents in which reclosure of intermediate alkoxides occurred following the cross-coupling reaction and aqueous work-up.
- 17.Miyaura N, Ishiyama T, Sasaki H, Ishikawa M, Satoh M, Suzuki A. J Am Chem Soc. 1989;111:314. [Google Scholar]
- 18.Molander GA, Brown AR. J Org Chem. 2006;71:9681. doi: 10.1021/jo0617013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilson ME, Paech K, Zhou WJ, Kurth MJ. J Org Chem. 1998;63:5094. [Google Scholar]
- 20.(a) Toy PH, Janda KD. Tetrahedron Lett. 1999;40:6329. [Google Scholar]; (b) Toy PH, Reger TS, Garibay P, Garno JC, Malikayil JA, Liu G, Janda KD. J Comb Chem. 2001;3:117. doi: 10.1021/cc000083f. [DOI] [PubMed] [Google Scholar]
- 21.The inertness of lithium reagents to sensitive functional groups suggests that the excess organolithium reagent is attached to the siloxane polymer in the form of alkoxides, which then became protonated following aqueous work-up to furnish reclosure of the siloxane.
- 22.(a) Tyrrell E, Brookes P. Synthesis. 2003;469 [Google Scholar]; (b) Molander GA, Ellis N. Acc Chem Res. 2007;40:275. doi: 10.1021/ar050199q. [DOI] [PubMed] [Google Scholar]; (c) Billingsley KL, Buchwald SL. Angew Chem, Int Ed. 2008;47:4695. doi: 10.1002/anie.200801465. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Gros P, Doudouh A, Fort Y. Tetrahedron Lett. 2004;45:6239. [Google Scholar]; (e) Yang DX, Colletti SL, Wu K, Song M, Li GY, Shen HC. Org Lett. 2009;11:381. doi: 10.1021/ol802642g. [DOI] [PubMed] [Google Scholar]; (f) Perkins JR, Carter RG. J Am Chem Soc. 2008;130:3290. doi: 10.1021/ja7113486. [DOI] [PubMed] [Google Scholar]; (g) Denmark SE, Smith RC, Chang WT, Muhuhi JM. J Am Chem Soc. 2009;131:3104. doi: 10.1021/ja8091449. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Bailey TR. Tetrahedron Lett. 1986;27:4407. [Google Scholar]
- 23.(a) Knapp DM, Gillis EP, Burke MD. J Am Chem Soc. 2009;131:6961. doi: 10.1021/ja901416p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dick GR, Knapp DM, Gillis EP, Burke MD. Org Lett. 2010;12:2314. doi: 10.1021/ol100671v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Dick GR, Woerly EM, Burke MD. Angew Chem, Int Ed. 2012;51:2667. doi: 10.1002/anie.201108608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.For the transmetalation of an aryl or alkenyl group from silicon to copper, see refs 6a–c and; (a) Ito H, Sensui H, Arimoto K, Miura K, Hosomi A. Chem Lett. 1997;639 [Google Scholar]; (b) Pierrat P, Gros P, Fort Y. Org Lett. 2005;7:697. doi: 10.1021/ol047482u. [DOI] [PubMed] [Google Scholar]; (c) Taguchi H, Ghoroku K, Tadaki M, Tsubouchi A, Takeda T. J Org Chem. 2002;67:8450. doi: 10.1021/jo025973r. [DOI] [PubMed] [Google Scholar]; (d) Taguchi H, Takami K, Tsubouchi A, Takeda T. Tetrahedron Lett. 2004;45:429–432. [Google Scholar]; (e) Suginome M, Kinugasa H, Ito Y. Tetrahedron Lett. 1994;35:8635. [Google Scholar]; (f) Takeda T, Uruga T, Gohroku K, Fujiwara T. Chem Lett. 1999;821 [Google Scholar]; (g) Taguchi H, Gohroku K, Tadaki M, Tsubouchi A, Takeda T. Org Lett. 2001;3:3811. doi: 10.1021/ol016837w. [DOI] [PubMed] [Google Scholar]; (h) Taguchi H, Miyashita H, Tsubouchi A, Takeda T. Chem Commun. 2002;2218 doi: 10.1039/b207325k. [DOI] [PubMed] [Google Scholar]
- 25.Nguyen MH, Smith AB., III Org Lett. 2013;15:4872. doi: 10.1021/ol4023084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stille JK, Simpson JH. J Am Chem Soc. 1987;109:2138. [Google Scholar]
- 27.Huang Z, Negishi E. Org Lett. 2006;8:3675. doi: 10.1021/ol061202o. [DOI] [PubMed] [Google Scholar]
- 28.Manolikakes G, Knochel P. Angew Chem Int Ed. 2009;48:205. doi: 10.1002/anie.200803730. [DOI] [PubMed] [Google Scholar]
- 29.Kobayashi O, Uraguchi D, Yamakawa T. Org Lett. 2009;11:2679. doi: 10.1021/ol900778m. [DOI] [PubMed] [Google Scholar]
- 30.Satoh T, Hanaki N, Yamada N, Asano T. Tetrahedron. 2000;56:6223. [Google Scholar]
- 31.Denmark SE, Sweis RF. J Am Chem Soc. 2001;123:6439. doi: 10.1021/ja016021q. [DOI] [PubMed] [Google Scholar]
- 32.Tseng C, Li M, Mo B, Warren SA, Spivey AC. Chem Lett. 2011;40:995. [Google Scholar]
- 33.Kofron WG, Baclawski LM. J Org Chem. 1976;41:1879. [Google Scholar]
- 34.Zhang L, Qing J, Yang P, Wu J. Org Lett. 2008;10:4971. doi: 10.1021/ol802049t. [DOI] [PubMed] [Google Scholar]
- 35.Metallinos C, Zaifman J, Dodge L. Org Lett. 2008;10:3527. doi: 10.1021/ol8013182. [DOI] [PubMed] [Google Scholar]
- 36.Gurung S, Thapa S, Vangala A, Giri R. Org Lett. 2013;15:5378. doi: 10.1021/ol402701x. [DOI] [PubMed] [Google Scholar]
- 37.Pavlik J, Kebede N. J Org Chem. 1997;62:8325. doi: 10.1021/jo970897r. [DOI] [PubMed] [Google Scholar]
- 38.Reddy V, Qiu R, Iwasaki T, Kambe N. Org Lett. 2013;15:1290. doi: 10.1021/ol400230y. [DOI] [PubMed] [Google Scholar]
- 39.Li X, Zhang J, Geng Y, Jin Z. J Org Chem. 2013;78:5078. doi: 10.1021/jo4005537. [DOI] [PubMed] [Google Scholar]
- 40.Alcaide B, Almendros P, Busto E, Lazaro-Milla C. J Org Chem. 2017;82:2177. doi: 10.1021/acs.joc.6b03006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.