

Abstract
The amyloid hypothesis suggests that the progressive accumulation and deposition of central nervous system amyloid with aging is the proximate cause of Alzheimer’s disease (AD). Thus, targeting molecular mechanisms of aging may represent a viable treatment approach. Caloric restriction prevents diseases of aging, including AD, in animal models, perhaps by activation of sirtuins. The sirtuins (such as mammalian SIRT1) are deacetylases that link energy balance (NAD+/NADH) to regulation of gene transcription. Resveratrol is a potent activator of SIRT1, and thus may mimic caloric restriction to prevent diseases of aging. We conducted a randomized, double blind, placebo-controlled, phase II trial of resveratrol for individuals with mild to moderate AD. Resveratrol (1) is detectable in cerebrospinal fluid (at low nanomolar levels), (2) is safe and well tolerated, (3) alters AD biomarker trajectories, (4) preserves blood–brain barrier integrity, and (5) modulates the CNS immune response. Further studies are needed to determine the safety and efficacy of resveratrol and the validity of this approach in the treatment and prevention of AD and other diseases of aging.
Keywords: Alzheimer’s disease, resveratrol, sirtuin, polyphenol, amyloid
Introduction
Alzheimer’s disease (AD) is the most common cause of dementia and an increasingly common cause of morbidity and mortality in the elderly. The prevalence and incidence of AD increases as life expectancies advance around the globe. AD is not only a growing healthcare burden, but a growing social and economic burden as well. In 2016, an estimated 5.4 million people in the United States alone are living with AD, and this number is projected to increase to 14–20% of the population by 2030.1 AD is defined pathologically by the density and distribution of central nervous system (CNS) β-amyloid (Aβ) plaques and neurofibrillary tangles (composed of the microtubule-associated protein tau and hyperphosphorylated tau). According to the amyloid hypothesis, AD is initiated by the progressive accumulation and deposition of Aβ/amyloid—a fragment generated by proteolytic cleavage of the widely expressed transmembrane protein amyloid precursor protein (APP). Normal APP catabolism results in fragments (especially Aβ40 and Aβ42) with a propensity to alter conformation and aggregate into oligomers and amyloid fibrils that condense as parenchymal neuritic plaques and vascular amyloid.2 While neurofibrillary tangles also increase with aging, amyloid appears to accelerate their generation.3 Despite many other pathologies (synaptic and neuronal loss, gliosis, inflammation), a high-density and widespread distribution of plaques and tangles in CNS cortex remain the defining pathologic features of AD.
Non-modifiable risk factors
The major non-modifiable risk factors for late-onset (sporadic) AD are (1) older age,4 (2) a family history of AD,5 and (3) carrying the APOE4 allele,6 with aging being the most important. The great majority of individuals with AD are 65 years of age or older. Approximately 15% of affected individuals are between the ages of 65 and 74, while 44% are between 75 and 84.1 Having a first-degree relative with AD doubles the risk compared with those with no family history. Individuals with more than one first-degree relative are at an even higher risk. While increased genetic risk is only partially explained by APOE genotype, it is a proven inherited risk factor. APOE is expressed in the liver and encodes a lipoprotein that transports cholesterol and lipids in blood. APOE is also expressed in the brain, where it may play similar roles in CNS lipid and cholesterol transport. The three alleles E2, E3, and E4 influence AD risk—with E4 having the highest risk, E3 intermediate, and E2 the lowest risk. Individuals who inherit one copy of the APOE4 allele have a threefold higher risk of AD compared with those without, while individuals who inherit two copies of the APOE4 allele have an 8- to 12-fold higher risk.7 Approximately 40–65% of individuals with AD are APOE4+, while the general population frequency of APOE4+ in the United States is about 20–25%. Fortunately, the population frequency of the highest-risk group (APOE4/APOE4 or APOE4 homozygous) is only 1–2%.6 Inheriting the APOE4 allele does not guarantee that an individual will develop AD, and population studies cannot predict individual risk.
Modifiable risk factors
Aging and genetics/family history are classically considered to be non-modifiable risk factors for AD. Environmental risk factors, however, are modifiable. Regular physical activity and management of cardiovascular risk factors, such as diabetes, obesity, smoking, and hypertension, lower the risk of cognitive decline and dementia with aging.8,9 Additionally, a Mediterranean diet (fruits, vegetables, nuts, beans, fish, olive oil) and lifelong learning may also delay cognitive decline with aging.9 Accumulating evidence suggests that energy and glucose metabolism may be linked to APP and Aβ metabolism and thus influence AD risk. Caloric excess, insulin resistance, diabetes, obesity, and metabolic syndrome are likely the most important modifiable risk factors for AD. Conversely, exercise, physical activity, maintaining ideal body weight, and caloric restriction (or intermittent fasting) are preventive measures (Fig. 1).10 While genetics is classically considered non-modifiable, caloric excess (diabetes, obesity) versus caloric restriction and exercise regulate the repertoire of gene expression via epigenetic mechanisms (DNA methylation and histone acetylation). Similarly, new insights into molecular mechanisms of aging and their links to energy metabolism suggest that molecular pathways regulating aging may be potential therapeutic targets for AD. In other words, genetics and aging may also be modifiable risk factors for AD.
Figure 1.

Alzheimer’s disease (AD) as a metabolic disorder. Risk factors for mild cognitive impairment (MCI) and AD with aging include caloric excess and sedentary lifestyle, leading to insulin and glucose dysregulation. In contrast, exercise and caloric restriction (or intermittent fasting) may delay or prevent cognitive decline with aging. Prodromal AD refers to individuals who are cognitively intact but have a positive AD biomarker. Resveratrol may mimic effects of caloric restriction by activation of sirtuins—deacetylases that link energy balance (via NAD+/NADH regulation) to altered gene transcription (via epigenetics). Thus, although classically considered as non-modifiable risk factors for AD, genetics and aging may be modifiable.
Current treatment strategies
Current U.S. Food and Drug Administration (FDA)-approved drugs for dementia due to AD include the cholinesterase inhibitors donepezil, rivastigmine, and galantamine and the NMDA receptor antagonist memantine. These drugs support cholinergic neurotransmission or block excitotoxic neuronal injury and death. However, these drugs provide only modest, temporary, and palliative benefits. There is no evidence to suggest that they influence the underlying disease processes. Newer therapeutic strategies for AD are focused on the amyloid hypothesis of AD (Fig. 2A) but have not shown clinical efficacy to date.
Figure 2.

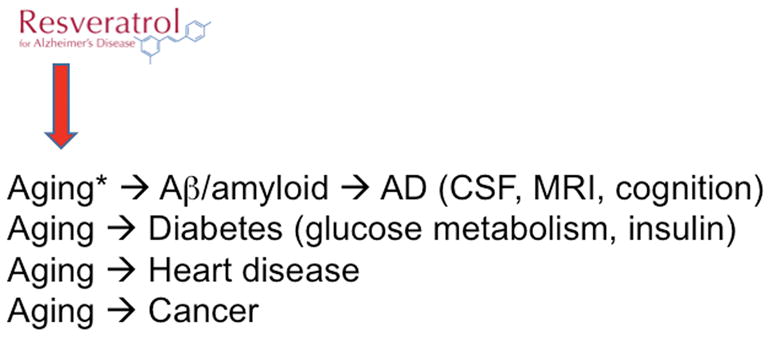
(A) Investigational agents for Alzheimer’s disease (AD) targeting amyloid. The majority of phase II/II trials in progress either promote Aβ clearance (active and passive immunotherapy) or inhibit Aβ generation (BACE1 inhibitors). Outcome measures for AD trials include cognitive, functional, and behavioral measures; MRI and PET neuroimaging; and cerebrospinal fluid (CSF) proteomics (especially Aβ, tau, and phospho-tau).
Following success with transgenic animal models of AD, passive immunization trials with the anti-Aβ monoclonal antibodies bapineuzumab, solanezumab, aducanumab, gantenerumab, and crenezumab have been completed or are in progress. These treatments consist of monthly intravenous administration of either humanized or human monoclonal antibodies targeting distinct Aβ epitopes and aggregates.11,12 However, phase III trials of bapineuzumab and solanezumab in individuals with mild-to-moderate AD failed to prove efficacy in pre-stated primary clinical outcomes. For example, a recent phase III trial of solanezumab in individuals with mild AD did not improve the primary cognitive outcome measure compared with placebo.13 The repeated failures of these and other large phase III trials of antiamyloid strategies raise serious questions regarding the amyloid hypothesis. Other investigators, however, suggest that treatment of individuals diagnosed with AD is “too little, too late” and that antiamyloid approaches may be successful if given in earlier disease stages. Utilizing recently developed amyloid positron emission tomography (PET) neuroimaging data, we now know that AD has a long subclinical phase—defined by progressive CNS amyloid deposition—10–20 years before cognitive decline commences.14 Thus, more recent clinical trials have focused on treatment of individuals with prodromal AD (mild cognitive impairment (MCI)) or older individuals who are cognitively normal but at risk (positive amyloid PET scan, or low Aβ and high tau in cerebrospinal fluid (CSF)).15–17 For example, in patients with prodromal or mild AD, 1-year treatment with aducanumab reduces brain Aβ in a dose- and time-dependent manner. This is accompanied by slowing of clinical decline. These promising results are supportive of the amyloid hypothesis of AD.18
A second major antiamyloid strategy focuses on inhibition of Aβ generation from APP rather than promoting amyloid clearance with antibodies. Inhibitors of β-amyloid cleaving enzyme 1 (BACE1) are now in phase II/III clinical trials for older individuals along the AD spectrum (at risk, MCI, or AD). BACE1 is the major CNS β-secretase required for the generation of Aβ from APP.19–23 Similar to the anti-Aβ antibody therapies, BACE1 inhibitor trials are also moving toward targeting prodromal and asymptomatic individuals instead of those with AD. A recent trial of the BACE1 inhibitor verubecestat in patients with mild-to-moderate AD was terminated owing to a lack of evident efficacy. However, a trial of verubecestat in patients with prodromal AD is now in progress.24 A new trial of a BACE1 inhibitor in healthy normal individuals at risk for AD will begin this year. The amyloid hypothesis will not be proven until or unless an antiamyloid treatment strategy shows clinical benefits in humans. New insights into the molecular mechanisms and pathways regulating aging, including in humans, suggest that these pathways may be exploited to develop novel therapeutics for AD, as well as other diseases of aging, such as cancer, diabetes, and heart disease (Fig. 2B).
Caloric restriction and Alzheimer’s disease
Many diseases leading to progressive disability and death occur later in life, implicating aging as a primary risk factor. Here, we define aging as Δx/Δt where t = time, but this definition does not mandate physiologic decline; some variables (x) may in fact improve (e.g., with a new treatment or by initiating an exercise program). Because of page limitations, we will not review molecular theories of aging here. For almost a century, however, we have known that the mild stressor caloric restriction (or consuming approximately two-thirds of normal daily calories) postpones and prevents diseases of aging in animal models, including nonhuman primates, and perhaps also in humans. While the molecular mechanism(s) remains unclear, activation of sirtuins (notably SIRT1) may be a critical pathway.25
Sirtuins
Molecular pathways regulating aging are now under intense scrutiny, and demonstrate that aging per se is genetically and pharmacologically modifiable in many laboratory animal species (typically with life span and/or health span as end points). Interestingly, many but not all of these genes and pathways implicate insulin signaling and glucose/energy metabolism. Included among these are the sirtuins (in mammals SIRT1–7), a family of NAD-dependent deacetylases. Sirtuins are regulated by the NAD+/NADH ratio (thus sensing cellular energy balance) and prolong the life span of model organisms. Sirtuins are essential in mediating the antiaging effects of caloric restriction. One of these, mammalian SIRT1, is a nuclear protein that deacetylates transcription factors governing central metabolic pathways. Targets of SIRT1 are important in energy metabolism, circadian rhythm, and aging.26,27 Additionally, SIRT1 is coupled to AMP kinase activity in a pathway that regulates cellular physiology during conditions of energy limitation.28
With regard to AD, SIRT1 overexpression in the brain reduces CNS AD pathologies via two putative mechanisms. First, SIRT1 directs the cleavage of APP away from the production of Aβ by activating α-secretase (cleaving APP within the Aβ sequence). Second, SIRT1 deacetylates tau, thus targeting its ubiquitination and proteasomal cleavage and reducing tangles.29 In support of this notion, mice overexpressing Aβ and SIRT1 demonstrate a decreased CNS amyloid burden.30
Jayasena et al. reviewed the potential roles and mechanisms of action of SIRT1–7 and the influence of polyphenols, particularly as they may relate to pathways involved in AD.31
Resveratrol
A screen of pharmacophore libraries for SIRT1 activators revealed resveratrol as the most potent molecule. Resveratrol, a polyphenol found in red grapes, red wine, and other plant foods, is receiving increasing attention owing to its medical potential. Accumulating evidence of the neuroprotective effects of red wine implicate several bioactive molecules, including quercetin, myricetin, catechins, tannins, anthocyanidins, ferulic acid, and resveratrol.32 Similar to caloric restriction,33,34 resveratrol treatment decreases age-dependent cognitive decline and AD-like pathologies in animal models.9,35 Preclinical studies with resveratrol suggest that it mimics the beneficial effects of caloric restriction via pharmacologic activation of SIRT1.36 These data support the notion that molecular mechanisms regulating aging may be exploited pharmacologically to delay or prevent diseases of aging, prolonging health span.
Preclinical studies indicate that resveratrol crosses the blood–brain barrier, resulting in detectable but low concentrations of the parent molecule in the brain, while much higher concentrations of resveratrol metabolites are found in blood. Thus, the bioavailability of oral resveratrol is poor.37,38 Since animals are unable to synthesize polyphenols, they must be ingested from a plant-rich diet to achieve pharmacologic effects.39 Polyphenols have potent anti-inflammatory and antioxidant properties that may affect brain function.40 Resveratrol increases cerebral arteriole dilation in rats, potentially via activation of NO-mediated vasodilation, thus improving postischemic cerebral perfusion.41 However, a similar study with human subjects failed to demonstrate improvement.42
Preclinical evidence supports the notion that resveratrol may play a role in the treatment and prevention of neurodegenerative diseases, such as Huntington’s disease, Parkinson’s disease, and AD.43 Through SIRT1 activation, resveratrol may protect neurons from reactive oxygen species (ROS), hydrogen peroxide free radicals, NO, Aβ, and other intra- and extracellular toxins and insults associated with neurodegenerative disorders.44–46 Resveratrol’s anti-inflammatory properties may also protect against Aβ-induced neuroinflammation and toxicity via inhibition of NF-κB signaling in microglia and astrocytes. Similarly, its antioxidant capabilities may decrease the generation of Aβ through inhibition of ROS-activated enzymes.45 In addition, resveratrol can dose-dependently inhibit Aβ42 fibril formation and cytotoxicity but does not prevent oligomer formation.47 The combination of well-established safety and promising preclinical data led to a growing number of clinical trials of resveratrol over the last decade.
Resveratrol for the treatment of Alzheimer’s disease
We conducted a randomized, double-blind, placebo-controlled trial of resveratrol for the treatment of mild-to-moderate dementia due to AD.10 This 52-week, phase II study assessed the safety and tolerability of resveratrol and effects on AD biomarkers (primary outcomes) as well as clinical outcomes (secondary). A total of 119 participants were randomized to placebo or pure synthetic resveratrol 500 mg orally once daily with a dose escalation by 500-mg increments every 13 weeks until a final dose of 1000 mg twice daily was reached for the final 13 weeks. Visits occurred at screening, baseline, and every 6 weeks with a total resveratrol exposure of 52 weeks. Compliance with resveratrol and with placebo was confirmed by mass spectrometry of blood samples. Despite high daily doses of oral resveratrol, we achieved only low nanomolar concentration of resveratrol in cerebrospinal fluid (and, by implication, in brain tissue). We confirmed the poor bioavailability of oral resveratrol, with much higher levels of glucuronidated and sulfated resveratrol metabolites detected in CSF and plasma. We cannot rule out the possibility, however, that these metabolites may also have bioactivity. At each visit, adverse events were assessed, along with clinical measures. MRI of the brain was performed at baseline, week 13, and week 52. CSF collections occurred at baseline and again at week 52. This study represented the largest (n = 119), longest (52 weeks), and highest-dose (ending with 2 g by mouth daily) human study of resveratrol to date.
A total of 104 participants completed the study, and 77 completed two CSF collections. Inclusion criteria included older individuals (> 49 years old) with a diagnosis of mild-to-moderate dementia due to AD, with stable medications and medically stable. No differences were found between the resveratrol- and placebo-treated groups in terms of safety and tolerability, with the only exception being weight loss in the resveratrol-treated group. The most common reported adverse events (AEs) were nausea and diarrhea, with 42% reported in the resveratrol group versus 33% in the placebo group. However, this was not a statistically significant difference. The high background of gastrointestinal AEs is likely due to concomitant treatment with a cholinesterase inhibitor approved for AD. Weight loss in the resveratrol-treated group may be due to enhanced mitochondrial biogenesis mediated by SIRT1 activation of PGC-1α25,36,48 We conclude that high-dose resveratrol is safe and well tolerated in individuals with mild-to-moderate AD.
We found that CSF Aβ40 and plasma Aβ40 levels declined significantly during the 52-week study in the placebo group, as expected with disease progression. In contrast, CSF Aβ40 and plasma Aβ40 levels were stabilized in the resveratrol-treated group, resulting in a significant difference at week 52 (n = 73, P = 0.002). Similar patterns were found for CSF Aβ42 and plasma Aβ42, although these results were not significant. However, in a post-hoc analysis of individuals with a biomarker-supported AD diagnosis (CSF Aβ42 < 600 ng/mL at baseline), we found a treatment effect on CSF Aβ42 as well (n = 70, P = 0.02). We have no direct evidence of sirtuin engagement from this study. While sirtuin activation is the presumed target, resveratrol may have many other molecular effects, including anti-inflammatory, antioxidant, and anti-Aβ aggregation. Other putative mechanisms of action include sirtuin activation with enhanced α-cleavage of APP49 and promotion of autophagy with removal of toxic intraneuronal proteinaceous aggregates.50
Volumetric MRI revealed that brain volumes (excluding CSF, brain stem, and cerebellum) declined significantly more (n = 96, P = 0.025) and ventricular volume increased more (n = 96, P = 0.05) in the resveratrol-treated group A subgroup analysis revealed that brain volume declined more with treatment in the APOE4 carriers compared with non-carriers. These findings persisted when participants with weight loss were excluded. The interpretation of greater brain volume loss with resveratrol treatment is unclear, but this finding was not associated with greater cognitive or functional decline. In fact, less decline (a clinical benefit) with treatment was detected in a functional measure—the Alzheimer’s Disease Cooperative Study-Activities of Daily Living (ADCS-ADL).
To test putative anti-inflammatory mechanisms mediating brain volume loss, we examined resveratrol’s ability to modulate neuroinflammation and induce adaptive immunity through further examination of CSF and plasma samples from the trial subjects.51 Since CSF tau and phospho-tau levels were unaffected (suggesting that neuronal loss was not an explanation for greater atrophy), we hypothesize that resveratrol has potent anti-inflammatory effects in the AD brain. Specifically, the etiology of greater brain volume loss with resveratrol may be due to decreased CNS edema, resulting in pseudoatrophy.52 This notion stems from the fact that similar effects are found with some active and passive antiamyloid immunotherapies for AD.53 In addition, effective drugs for the treatment of multiple sclerosis (another CNS inflammatory disorder) are also known to induce pseudoatrophy.54
We measured critical pro- and anti-inflammatory cytokines and chemokines, as well as metalloproteinases, in samples of CSF and plasma from a subset of individuals enrolled in the resveratrol for AD trial.10 In contrast to the parent study, we studied samples only from individuals with a biomarker-supported diagnosis of AD (CSF Aβ42 < 600 ng/mL at baseline), thus improving diagnostic accuracy. The most striking result is the ~ 50% decrease in CSF matrix metalloproteinase 9 (MMP-9) level with resveratrol—but not placebo—treatment. MMP-9 regulates blood–brain barrier permeability via release of cytokines and free radicals. Additionally, MMP-9 cleaves the vascular basal lamina and tight junctions in the neurovascular bed.55–57 MMP-9 has thus emerged as a major player in neurodegenerative and neuroinflammatory disorders, such as AD and multiple sclerosis.58 For example, MMP-9 gene knockout reduces neuroinflammation in animal models of experimental autoimmune encephalomyelitis.59 CSF MMP-9 is elevated in patients with bacterial meningitis and blood–brain barrier damage.60 Moreover, MMP-9 expression and activity is increased in serum, in CSF, and in demyelinating lesions in individuals with multiple sclerosis61 or ischemic stroke.62 In light of this evidence, decreased CSF MMP-9 levels suggest that resveratrol may reduce CNS permeability to limit the infiltration of leukocytes and other inflammatory mediators into the brain.
In addition to the effect seen on MMP-9, we also found that resveratrol may facilitate activation of microglia/macrophages, thereby inducing long-term adaptive immunity that may be clinically beneficial.51 Since chronic neuroinflammation likely contributes to the focal encephalopathy causing clinical dementia, activation of specific microglia/macrophages may be neuroprotective.63,64 As AD progresses, innate immune cells, such as resident microglia and macrophages, exhibit dysfunctional or senescent profiles characterized by impaired phagocytosis. As such, modulation of microglia/macrophages may have therapeutic benefits.65–67 Additionally, we found reduced plasma levels of the pro-inflammatory markers IL-1r4, IL-12P40, IL-12P70, and TNF-α in the treatment group, although plasma differences did not survive a statistical test of multiple comparisons.51
In summary, resveratrol robustly stabilizes the progressive decline in plasma Aβ40 and CSF Aβ40 level as dementia advances and is associated with brain pseudoatrophy. Additionally, despite the study being underpowered to detect clinical benefit, resveratrol attenuates decline in a functional measure— the ADCS-ADL. Although the numbers are small, we also found fewer cancers in the treatment group (one versus seven in six participants in the placebo group). Collectively, these data support the notion that targeting molecular mechanisms of aging may postpone or prevent several diseases of aging in parallel. With proven safety and suggestions of efficacy from the phase II trial of AD, the putative benefits of resveratrol and other sirtuin activators (STACs) must be further explored in well-powered studies of individuals with earlier disease stages.10,51,68
While these results are promising, there are several limitations. The phase II study was designed primarily to determine the safety and tolerability of high-dose oral resveratrol and to examine its pharmacokinetics. While certain AD biomarker trajectories were altered, there were no effects of treatment on other biomarkers or on secondary clinical outcomes, such as the Mini-Mental State Examination (MMSE) score, Alzheimer’s Disease Assessment Scale-cognitive (ASAS-cog), Clinical Dementia Rating-Sum of Boxes (CDR-SB), and Neuropsychiatric Inventory (NPI). There were also no significant effects of drug treatment on glucose or insulin metabolism—perhaps because we excluded individuals with known diabetes or treated for diabetes. Since a phase II study is inherently limited by small sample sizes, altered biomarker trajectories must be interpreted with caution. Although they may suggest CNS effects, they do not necessarily indicate clinical benefit.
Conclusions
Further studies are now needed to determine whether resveratrol and similar polyphenols may be beneficial for AD or other disorders of aging. Another phase II study is warranted to examine the safety and efficacy of higher doses and improved formulations with better pharmacokinetics and bioavailability. Similarly, as with the anti-Aβ antibody and BACE1 inhibitor studies, the next trial of resveratrol may enroll a different study population, such as those with prodromal AD or normal older individuals at risk (e.g., earlier along the AD spectrum). The working hypothesis is that resveratrol may delay or prevent AD onset when given to subjects with prodromal AD or cognitively normal individuals at risk. Alternatively, novel patentable molecules—sirtuin activators (STACs) and resveratrol mimetics—may be developed. Instead of approaching each disease of aging individually, with one or more drugs prescribed for each, targeting regulatory molecules of aging per se may target several diseases of aging simultaneously, thus minimizing polypharmacy and its associated risks and costs. While neurotransmitter-based therapies are the first wave of AD treatments, and amyloid agents the second wave (perhaps), the next wave may target aging mechanisms.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Alzheimer’s A. 2016 Alzheimer’s disease facts and figures. Alzheimers Dement. 2016;12(4):459–509. doi: 10.1016/j.jalz.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 2.Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256(5054):184–5. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 3.Wang X, et al. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim Biophys Acta. 2014;1842(8):1240–7. doi: 10.1016/j.bbadis.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hebert LE, et al. Change in risk of Alzheimer disease over time. Neurology. 2010;75(9):786–91. doi: 10.1212/WNL.0b013e3181f0754f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fratiglioni L, et al. Risk factors for late-onset Alzheimer’s disease: a population-based, case-control study. Ann Neurol. 1993;33(3):258–66. doi: 10.1002/ana.410330306. [DOI] [PubMed] [Google Scholar]
- 6.Saunders AM, et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology. 1993;43(8):1467–72. doi: 10.1212/wnl.43.8.1467. [DOI] [PubMed] [Google Scholar]
- 7.Loy CT, et al. Genetics of dementia. Lancet. 2014;383(9919):828–40. doi: 10.1016/S0140-6736(13)60630-3. [DOI] [PubMed] [Google Scholar]
- 8.Cognitive Aging: Progress in Understanding and Opportunities for Action. Mil Med. 2015;180(11):1111–3. doi: 10.7205/MILMED-D-15-00292. [DOI] [PubMed] [Google Scholar]
- 9.Baumgart M, et al. Summary of the evidence on modifiable risk factors for cognitive decline and dementia: A population-based perspective. Alzheimers Dement. 2015;11(6):718–26. doi: 10.1016/j.jalz.2015.05.016. [DOI] [PubMed] [Google Scholar]
- 10.Turner RS, et al. A randomized, double-blind, placebo-controlled trial of resveratrol for Alzheimer disease. Neurol. 2015;85:2382–1391. doi: 10.1212/WNL.0000000000002035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adolfsson O, et al. An effector-reduced anti-beta-amyloid (Abeta) antibody with unique abeta binding properties promotes neuroprotection and glial engulfment of Abeta. J Neurosci. 2012;32(28):9677–89. doi: 10.1523/JNEUROSCI.4742-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Legleiter J, et al. Effect of different anti-Abeta antibodies on Abeta fibrillogenesis as assessed by atomic force microscopy. J Mol Biol. 2004;335(4):997–1006. doi: 10.1016/j.jmb.2003.11.019. [DOI] [PubMed] [Google Scholar]
- 13.Lilly . Lilly Announces Top-Line Results of Solanezumab Phase 3 Clinical Trial. Eli Lilly and Company; Indianapolis: 2016. [Google Scholar]
- 14.Villemagne VL, et al. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: a prospective cohort study. Lancet Neurol. 2013;12(4):357–67. doi: 10.1016/S1474-4422(13)70044-9. [DOI] [PubMed] [Google Scholar]
- 15.Mullard A. Sting of Alzheimer’s failures offset by upcoming prevention trials. Nat Rev Drug Discov. 2012;11(9):657–60. doi: 10.1038/nrd3842. [DOI] [PubMed] [Google Scholar]
- 16.Shimada H. The DIAN study. Brain Nerve. 2013;65(10):1179–84. [PubMed] [Google Scholar]
- 17.Reiman EM, et al. Alzheimer’s Prevention Initiative: a plan to accelerate the evaluation of presymptomatic treatments. J Alzheimers Dis. 2011;26(Suppl 3):321–9. doi: 10.3233/JAD-2011-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sevigny J, et al. The antibody aducanumab reduces Ab plaques in Alzheimer’s disease. Nature. 2016;537:50–56. doi: 10.1038/nature19323. [DOI] [PubMed] [Google Scholar]
- 19.Hussain I, et al. Identification of a novel aspartic protease (Asp 2) as beta-secretase. Mol Cell Neurosci. 1999;14(6):419–27. doi: 10.1006/mcne.1999.0811. [DOI] [PubMed] [Google Scholar]
- 20.Lin X, et al. Human aspartic protease memapsin 2 cleaves the beta-secretase site of beta-amyloid precursor protein. Proc Natl Acad Sci U S A. 2000;97(4):1456–60. doi: 10.1073/pnas.97.4.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sinha S, et al. Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature. 1999;402(6761):537–40. doi: 10.1038/990114. [DOI] [PubMed] [Google Scholar]
- 22.Vassar R, et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286(5440):735–41. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- 23.Yan R, et al. Membrane-anchored aspartyl protease with Alzheimer’s disease beta-secretase activity. Nature. 1999;402(6761):533–7. doi: 10.1038/990107. [DOI] [PubMed] [Google Scholar]
- 24.Merck . APECS Study in People with Prodromal Alzheimer’s Disease to Continue. Business Wire; Kenilworth, NJ: 2017. Merck Announces EPOCH Study of Verubecestat for the Treatment of People with Mild to Moderate Alzheimer’s Disease to Stop for Lack of Efficacy. [Google Scholar]
- 25.Pasinetti GM, et al. Roles of resveratrol and other grape-derived polyphenols in Alzheimer’s disease prevention and treatment. Biochim Biophys Acta. 2015;1852(6):1202–8. doi: 10.1016/j.bbadis.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakahata Y, et al. The NAD+-dependent deacetylase SIRT1 modulates CLOCK-mediated chromatin remodeling and circadian control. Cell. 2008;134(2):329–40. doi: 10.1016/j.cell.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Asher G, et al. SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell. 2008;134(2):317–28. doi: 10.1016/j.cell.2008.06.050. [DOI] [PubMed] [Google Scholar]
- 28.Canto C, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458(7241):1056–60. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guarente L, Franklin H. Epstein Lecture: Sirtuins, aging, and medicine. N Engl J Med. 2011;364(23):2235–44. doi: 10.1056/NEJMra1100831. [DOI] [PubMed] [Google Scholar]
- 30.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8(2):101–12. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 31.Jayasena T, et al. The role of polyphenols in the modulation of sirtuins and other pathways involved in Alzheimer’s disease. Ageing Research Reviews. 2013;12:867–883. doi: 10.1016/j.arr.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 32.Caruana M, et al. Putative role of red wine polyphenols against brain pathology in Alzheimer’s and Parkinson’s disease. Frontiers in Nutrition. 2016;3:1–16. doi: 10.3389/fnut.2016.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang J, et al. Caloric restriction attenuates beta-amyloid neuropathology in a mouse model of Alzheimer’s disease. FASEB J. 2005;19(6):659–61. doi: 10.1096/fj.04-3182fje. [DOI] [PubMed] [Google Scholar]
- 34.Patel NV, et al. Caloric restriction attenuates Abeta-deposition in Alzheimer transgenic models. Neurobiol Aging. 2005;26(7):995–1000. doi: 10.1016/j.neurobiolaging.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 35.Marambaud P, Zhao H, Davies P. Resveratrol promotes clearance of Alzheimer’s disease amyloid-beta peptides. J Biol Chem. 2005;280(45):37377–82. doi: 10.1074/jbc.M508246200. [DOI] [PubMed] [Google Scholar]
- 36.Kulkarni SS, Canto C. The molecular targets of resveratrol. Biochim Biophys Acta. 2015;1852(6):1114–23. doi: 10.1016/j.bbadis.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 37.Juan ME, Maijo M, Planas JM. Quantification of trans-resveratrol and its metabolites in rat plasma and tissues by HPLC. J Pharm Biomed Anal. 2010;51(2):391–8. doi: 10.1016/j.jpba.2009.03.026. [DOI] [PubMed] [Google Scholar]
- 38.Wang Q, et al. Resveratrol protects against global cerebral ischemic injury in gerbils. Brain Res. 2002;958(2):439–47. doi: 10.1016/s0006-8993(02)03543-6. [DOI] [PubMed] [Google Scholar]
- 39.Ahmed T, et al. Resveratrol and Alzheimer’s Disease: Mechanistic Insights. Mol Neurobiol. 2016;54:2622–2635. doi: 10.1007/s12035-016-9839-9. [DOI] [PubMed] [Google Scholar]
- 40.Pathak L, Agrawal Y, Dhir A. Natural polyphenols in the management of major depression. Expert Opin Investig Drugs. 2013;22(7):863–80. doi: 10.1517/13543784.2013.794783. [DOI] [PubMed] [Google Scholar]
- 41.Ritz MF, et al. Chronic treatment with red wine polyphenol compounds mediates neuroprotection in a rat model of ischemic cerebral stroke. J Nutr. 2008;138(3):519–25. doi: 10.1093/jn/138.3.519. [DOI] [PubMed] [Google Scholar]
- 42.Wong RH, et al. Chronic resveratrol consumption improves brachial flow-mediated dilatation in healthy obese adults. J Hypertens. 2013;31(9):1819–27. doi: 10.1097/HJH.0b013e328362b9d6. [DOI] [PubMed] [Google Scholar]
- 43.Sun AY, et al. Resveratrol as a therapeutic agent for neurodegenerative diseases. Mol Neurobiol. 2010;41(2–3):375–83. doi: 10.1007/s12035-010-8111-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Albani D, Polito L, Forloni G. Sirtuins as novel targets for Alzheimer’s disease and other neurodegenerative disorders: experimental and genetic evidence. J Alzheimers Dis. 2010;19(1):11–26. [Google Scholar]
- 45.Anekonda TS. Resveratrol--a boon for treating Alzheimer’s disease? Brain Res Rev. 2006;52(2):316–26. doi: 10.1016/j.brainresrev.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 46.Graff J, et al. A dietary regimen of caloric restriction or pharmacological activation of SIRT1 to delay the onset of neurodegeneration. J Neurosci. 2013;33(21):8951–60. doi: 10.1523/JNEUROSCI.5657-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Feng Y, et al. Resveratrol inhibits beta-amyloid oligomeric cytotoxicity but does not prevent oligomer formation. Neurotoxicology. 2009;30:986–995. doi: 10.1016/j.neuro.2009.08.013. [DOI] [PubMed] [Google Scholar]
- 48.Bastianetto S, Menard C, Quirion R. Neuroprotective action of resveratrol. Biochim Biophys Acta. 2015;1852(6):1195–201. doi: 10.1016/j.bbadis.2014.09.011. [DOI] [PubMed] [Google Scholar]
- 49.Donmez G, et al. SIRT1 suppresses beta-amyloid production by activating the alpha-secretase gene ADAM10. Cell. 2010;142(2):320–32. doi: 10.1016/j.cell.2010.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 50.Pallauf K, Rimbach G. Autophagy, polyphenols and healthy ageing. Ageing Res Rev. 2013;12(1):237–52. doi: 10.1016/j.arr.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 51.Moussa C, et al. Resveratrol regulates neuro-inflammation and induces adaptive immunity in Alzheimer’s disease. J Neuroinflammation. 2017;14(1):1. doi: 10.1186/s12974-016-0779-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zivadinov R, et al. Mechanisms of action of disease-modifying agents and brain volume changes in multiple sclerosis. Neurology. 2008;71(2):136–44. doi: 10.1212/01.wnl.0000316810.01120.05. [DOI] [PubMed] [Google Scholar]
- 53.Fox NC, et al. Effects of Abeta immunization (AN1792) on MRI measures of cerebral volume in Alzheimer disease. Neurology. 2005;64(9):1563–72. doi: 10.1212/01.WNL.0000159743.08996.99. [DOI] [PubMed] [Google Scholar]
- 54.De Stefano N, et al. Clinical relevance of brain volume measures in multiple sclerosis. CNS Drugs. 2014;28(2):147–56. doi: 10.1007/s40263-014-0140-z. [DOI] [PubMed] [Google Scholar]
- 55.Candelario-Jalil E, Yang Y, Rosenberg GA. Diverse roles of matrix metalloproteinases and tissue inhibitors of metalloproteinases in neuroinflammation and cerebral ischemia. Neuroscience. 2009;158(3):983–94. doi: 10.1016/j.neuroscience.2008.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reijerkerk A, et al. Diapedesis of monocytes is associated with MMP-mediated occludin disappearance in brain endothelial cells. FASEB J. 2006;20(14):2550–2. doi: 10.1096/fj.06-6099fje. [DOI] [PubMed] [Google Scholar]
- 57.Verslegers M, et al. Matrix metalloproteinase-2 and -9 as promising benefactors in development, plasticity and repair of the nervous system. Prog Neurobiol. 2013;105:60–78. doi: 10.1016/j.pneurobio.2013.03.004. [DOI] [PubMed] [Google Scholar]
- 58.Vafadari B, Salamian A, Kaczmarek L. MMP-9 in translation: from molecule to brain physiology, pathology, and therapy. J Neurochem. 2016;139(Suppl 2):91–114. doi: 10.1111/jnc.13415. [DOI] [PubMed] [Google Scholar]
- 59.Dubois B, et al. Resistance of young gelatinase B-deficient mice to experimental autoimmune encephalomyelitis and necrotizing tail lesions. J Clin Invest. 1999;104(11):1507–15. doi: 10.1172/JCI6886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Leppert D, et al. Matrix metalloproteinase (MMP)-8 and MMP-9 in cerebrospinal fluid during bacterial meningitis: association with blood-brain barrier damage and neurological sequelae. Clin Infect Dis. 2000;31(1):80–4. doi: 10.1086/313922. [DOI] [PubMed] [Google Scholar]
- 61.Yong VW, et al. Elevation of matrix metalloproteinases (MMPs) in multiple sclerosis and impact of immunomodulators. J Neurol Sci. 2007;259(1–2):79–84. doi: 10.1016/j.jns.2006.11.021. [DOI] [PubMed] [Google Scholar]
- 62.Yang Y, Rosenberg GA. Matrix metalloproteinases as therapeutic targets for stroke. Brain Res. 2015;1623:30–8. doi: 10.1016/j.brainres.2015.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010;362(4):329–44. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- 64.Zuroff L, et al. Clearance of cerebral Abeta in Alzheimer’s disease: reassessing the role of microglia and monocytes. Cell Mol Life Sci. 2017;74:2167–2201. doi: 10.1007/s00018-017-2463-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chakrabarty P, et al. IL-10 alters immunoproteostasis in APP mice, increasing plaque burden and worsening cognitive behavior. Neuron. 2015;85(3):519–33. doi: 10.1016/j.neuron.2014.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Guillot-Sestier MV, et al. Il10 deficiency rebalances innate immunity to mitigate Alzheimer-like pathology. Neuron. 2015;85(3):534–48. doi: 10.1016/j.neuron.2014.12.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Michaud JP, Rivest S. Anti-inflammatory signaling in microglia exacerbates Alzheimer’s disease-related pathology. Neuron. 2015;85(3):450–2. doi: 10.1016/j.neuron.2015.01.021. [DOI] [PubMed] [Google Scholar]
- 68.Venigalla M, et al. Novel promising therapeutics against chronic neuroinflammation and neurodegeneration in Alzheimer’s disease. Neurochem Intl. 2016;95:63–74. doi: 10.1016/j.neuint.2015.10.011. [DOI] [PubMed] [Google Scholar]