

Abstract
Many organisms and tissues display the ability to heal and regenerate as needed for normal physiology and with pathogenesis. However, these repair activities can also be observed at the single cell level. The physical and molecular mechanisms by which a cell can heal membrane ruptures and rebuild damaged or missing cellular structures remain poorly understood. This review presents current understanding in wound healing and regeneration as two distinct aspects of cellular self-repair by examining a few model organisms that have displayed robust repair capacity, including Xenopus oocytes, Chlamydomonas, and Stentor coeruleus. While many open questions remain, understanding how cells repair themselves is important for our mechanistic understanding of cell biology. It also holds the potential for new applications and therapeutic approaches for treating human disease.
Introduction
Cells are generally soft, squishy, and easily damaged. However, many can repair themselves after being punctured, torn, or even ripped in half when damaged due to the normal wear-and-tear of normal physiology or as a result of injury or pathology. A cell is like a spacecraft: when it is punctured, cytoplasm spills out like oxygen escaping from a damaged space module. Like Apollo 13, a damaged cell cannot rely on anyone to fix it. It must repair itself, first by stopping the loss of cytoplasm, and then regenerate by rebuilding structures that were damaged or lost. Understanding how they repair and regenerate themselves could guide treatments for conditions involving cellular damage.
A standard question we ask students is to define what it means to be alive. This is surprisingly hard to do in a precise way, but surely one of the remarkable features of living systems that distinguishes them from human-made machines is their ability to heal and repair themselves. At the multi-cellular level, repair and regeneration are effected by generating new cells to replace the ones that were lost. This type of repair thus ends up being a direct consequence of another basic feature of living systems – the ability of a cell to reproduce itself. No additional processes need to be invoked beyond cell division. At the single-cell level, it is much less obvious how self-repair is accomplished.
In this review, we will distinguish wound healing from regeneration as two aspects of self-repair which serve distinct purposes, and will discuss each aspect separately. Wound healing is the process that stops further loss of material, in much the same way a blood clot stops further loss of blood. Regeneration, on the other hand, is the process by which the cell specifically re-builds and replaces the missing components (organelles, plasma membrane, cytoplasm, etc.) after the wound has been stabilized. Some cells can heal wounds but cannot regenerate. For example, if the the giant unicellular ciliate Bursaria is cut in half, the halves heal their surfaces and live, but they lose all their cortical structures, become spherical, encyst, and then re-develop a new cortical pattern from scratch (1). We contrast such cases from cells which are able to detect missing structures and specifically regenerate the pieces that were missing or damaged.
Examples of self-repairing cells
Many cell types can heal wounds and regenerate missing structures (Figure 1). Neurons are sometimes able to repair and regenerate damaged axons (2, 3), which is important because they do not proliferate. Cardiac myocytes routinely suffer mechanical wounding as the heart beats, and are able to survive and heal membrane ruptures (4, 5).
Figure 1.

Examples of self-repairing cells. Healing of a punctured Xenopus oocyte, where the dark and light halves represent the animal and vegetal poles respectively. Regrowth of damaged axons in neurons. Regeneration of flagella in Chlamydomonas. In each case, regenerated components are highlighted in red. The dots represent the loss of cell content from damage sites.
Wound healing has been extensively studied in Xenopus oocytes (6, 7). The Xenopus oocyte, while produced by a multicellular animal, is a large, single cell easily obtained and manipulated. When punctured either with a glass needle or with laser ablation, the oocyte rapidly restores an intact plasma membrane.
Some unicellular model systems illustrate cellular regeneration. There is an advantage in studying free living single-celled organisms in that their normal behavior can be studied without the influence from any neighboring cells as would be the case for cells within a tissue. This is important because if a cell regenerates in a tissue context, one cannot be sure whether the necessary spatial information comes from the cell itself or from information supplied by neighboring cells.
Chlamydomonas is a unicellular green alga surrounded by a thick cell wall. Two motile flagella, approximately 10 microns long, protrude from holes in the cell wall, allowing the cell to swim. When the cell is stressed (for example by pH shock), it severs its own flagella. This process, known as flagellar autotomy (8), presumably helps the cell survive a hostile environment by eliminating the only point of vulnerability not surrounded by a cell wall. Once the stress is removed, flagella grow back within 90 minutes. Flagellar regeneration in Chlamydomonas provides a simple and highly reproducible paradigm for studying organelle regeneration.
Another classical system for studying regeneration in single cells is Stentor coeruleus (Figure 2). Stentor is a giant, blue-colored ciliate that can reach sizes of over a millimeter. A Stentor cell is cone-shaped, with a circular oral apparatus (OA), consisting of thousands of cilia at the anterior end of the cell, specialized for sucking in food particles. Stentor cells show an amazing ability to self-repair after wounding. When pieces of a Stentor cell such as the OA are removed using a glass needle, or when cells are cut in half, they can self-repair without losing cytoplasm (9). Instead, the cell continues to live and is able to regenerate the missing components in a matter of hours (10). In bisected cells, both halves can regenerate because Stentor contains a highly polyploid macronucleus such that even small cell fragments will contain enough genomic copies to survive. Regeneration of the OA in Stentor has been a major paradigm for studying cellular regeneration (Figure 2). Although the OA is located at the anterior end of the cell, it does not at first regenerate in its original location. Instead, the cell synthesizes all of the basal bodies necessary to assemble the OA at a site on the surface of the cell roughly halfway down the length of the cell body, where narrow and wide pigment stripes meet each other. These basal bodies organize into the complete structure of the OA, at which point it is known as an oral primordium. The primordium then migrates to the anterior end of the cell and twists into the correct orientation. This entire process sometimes occurs spontaneously in the presence of an existing OA, which is then resorbed and replaced by the new one. This replacement process is known as “reorganization”, which is hypothesized to help ensure proper scaling of OA size with cell size as the cell grows.
Figure 2.

The regeneration and reorganization of the oral apparatus (green) of Stentor coeruleus. Blue lines indicate surface pigment stripes, and the red region indicates the oral primordium. At the opposite end from the oral apparatus, Stentor possesses a posterior holdfast, which the cell uses to attach itself to a solid substrate. When an oral apparatus regenerates after removal, it first assembles as an oral primordium, which then migrates to the anterior end and becomes a new oral apparatus.
These examples illustrate the ability of cells to heal wounds and regenerate missing structures. The mechanisms used for self-repair in these cases remain unclear, and pose an interesting challenge for cell biology. Several questions apply to both wound-healing and regeneration. First, does repair reflect a constitutive behavior or does it need an active pathway to be triggered? For example, given that the Stentor OA is spontaneously replaced during reorganization, can the regeneration of the OA simply be considered as an accelerated form of this same turnover process by which an OA is occasionally replaced by a new one? Or is there a distinct signaling program that needs to be triggered to drive regeneration? Many self-organizing systems can restore patterning after perturbation. It is important to determine whether wound-healing and/or regeneration represents a “response” where pathways need to be turned on, or whether it simply reflects constitutive cellular activities. If repair turns out to be triggered, what is the stimulus? How does the cell detect that something is wrong? Finally, once a repair response is triggered, how is it actually carried out at the molecular level?
Wound healing
Before discussing how wounds are repaired, we first consider the size and time scales that characterize the wound-healing problem. Openings in cell membranes occur rather regularly, but not all openings are recognized as a wound by the cell to trigger a healing response. For example, small membrane pores form spontaneously from lipid motion. Based on studies on planar lipid bilayer membranes, it was found that when the pore is small (<~ 10s nm), the restoring force by the membrane tension will reseal these pores (11, 12). Under physiological conditions, the membrane can also be permeated in processes such as conjugation in bacteria, which utilize pili—tube-like conduits (~ 10 nm)—to transfer DNA from one cell to another (13). In vitro, cell membranes have been permeated during processes such as electroporation, micro-injection, and more recently “nanostraws” which are hollow metal-oxide nanotubes (~ 100 nm) for intracellular delivery of ions and molecules (14, 15). These processes do not typically damage the cell nor do they trigger a wound response. So then, when is an opening recognized as a wound?
One perspective is to consider the function of the membrane, which is to separate the intracellular environment from the external, and to maintain physiological concentrations of ions, proteins, and other macromolecules. Sudden change in the intracellular environment could thus serve as a proxy to alert the cell to a membrane rupture. Specifically, influx of calcium ions is often a key trigger for wound response (6, 16, 17). Physiologically, this makes sense given Ca2+ is an important intracellular messenger and underlies many important signaling pathways. Excessive intracellular Ca2+ levels are toxic and can lead to cell death (18, 19). While no work to our knowledge has explicitly described the minimum size of a membrane rupture to trigger wound response, one can expect that the opening should be big enough and last long enough to allow the influx of ions like Ca2+ to perturb the intracellular environment in order for it to be recognized as a wound.
Once a hole is recognized, how long does the cell have to fix it before irreversible damage is done? As with oxygen leaking from a spacecraft, loss can be tolerated, but only up to some limit. In the case of a cell, the wound must be sealed in time to prevent the excessive loss of cell mass, as well as the influx and accumulation of unwanted components from the external environment. In the absence of a wound response, one can apply the equation for the flow through a circular aperture to estimate the leakage rate Q of cell mass from a punctured cell (20). Assuming the cytoplasm is a simple fluid leaking out of a membrane wound, Q = r3ΔP/(3μi), where r is the radius of the wound, ΔP is the pressure difference across the membrane, and μi is the viscosity of the inner fluid. Assuming ΔP ~ 10 Pa, μi ~ 1 - 100 mPa-s (1 –100 times that of water), the leakage rate would be ~ 80% – 0.8% of cell volume per second for a wound size that is 10% of cell size. This simple approximation ignores the complexity of many aspects of the cell (e.g., viscoelastic properties of the cytoplasm). Nevertheless, purely from the physics and mass transport perspective, the approximation suggests that healing should take place on the order of seconds to tens of seconds, instead of days, to prevent the excessive loss of cell mass. If we consider the influx of unwanted components (e.g., Ca2+) by simple diffusion, we get a similar time scale t ~ x2/D ~ 1 – 10 seconds, for a distance x ~ 10 μm and a diffusivity D ~ 10−10 – 10−11 m2/s (21). Indeed, in studies across quite a wide range of cell types, including sea urchin eggs (22), fibroblasts (23), alveolar epithelial cells (24), the time scales of wound sealing were around seconds to tens of seconds.
However, some cells have been reported to take minutes to even hours to seal the wound. It is possible to rationalize such differences by considering the parameters in the mass transport approximations above. For example, a low extracellular Ca2+ level would slow down the diffusive influx and subsequent accumulation of Ca2+ to a level necessary to trigger wound response (17). Similarly, a high cellular viscosity would decrease the leakage rate of cell mass. In both cases, the cell would not need to heal as quickly. Relevant to the first case, it was found that in the absence of Ca2+, sea urchin eggs did not seal their wounds (22). Relevant to the second case, earthworm giant axons take minutes to heal. Coincidentally, these axons also have densely packed cytoskeleton which can slow down diffusion (25).
How do cells heal wounds on the necessary timescale? The mechanism of wound healing has been relatively well studied in a few model systems such as sea urchin eggs and Xenopus oocytes, and has been reviewed elsewhere (6, 17, 26, 27). Here we will briefly describe known mechanisms. It is generally recognized that Ca2+ is necessary to trigger a wound response. As early as 1930, Heilbrunn reported that Ca2+ is needed for wound healing (28), a requirement seen in many other cell types (22, 29). The use of Ca2+ as the trigger for wound response makes sense not just because of the physiological significance of Ca2+, but also from an engineering perspective. For many cell types, unbound Ca2+ has the steepest concentration gradient across the cell membrane (~104-fold difference for Ca2+; < 102-fold difference for other ions such as Na+, K+, and Mg2+) (30, 31). Using a species with a steep gradient as a trigger increases the sensitivity of the cell to wounding events. But what does calcium actually do to heal a wound?
In general, Ca2+ has been shown to trigger two complementary mechanisms based on prior studies in a few model organisms (Figure 3): 1) Patching of the membrane hole by inducing membrane fusion via a range of processes including exocytosis, endocytosis, and the more recently described “explodosis” which involves the fusion of intracellular compartments which then rupture outwards on the side facing the outside of the cell (17, 32). Extra membranes can be derived from intracellular vesicles and organelles. 2) Formation of an actomyosin purse string which contracts around the wound. This contraction brings intact membrane and underlying cortical cytoskeleton to close the wound (6, 17).
Figure 3.
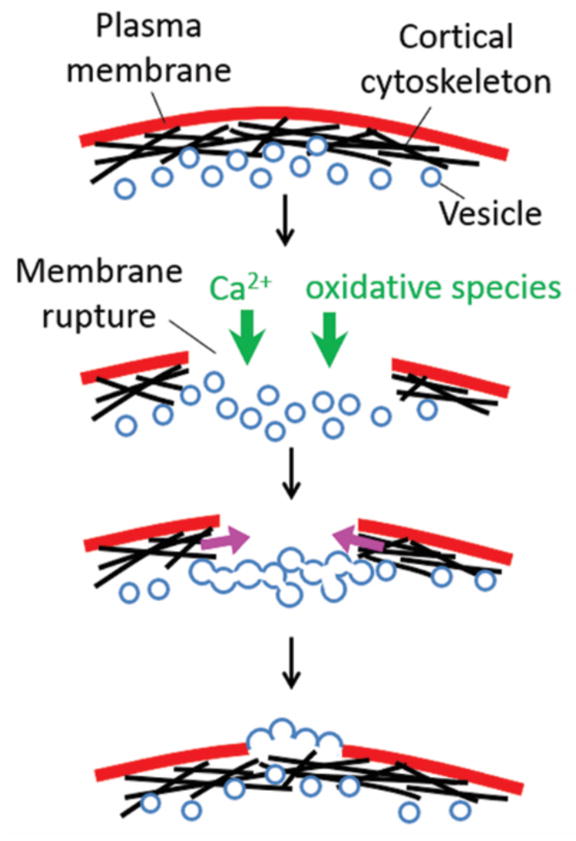
Wound healing studies in model cells such as Xenopus oocytes and muscle cells found that the process is triggered by the influx of Ca2+ and oxidative species. These species induce the fusion of membranes derived from intracellular vesicles and organelles, as well as the contraction of actomyosin purse string around the wound site.
Ca2+ may not be the only signal that triggers wound healing. Recently, oxidative species from the extracellular environment was found to play a role that is independent of the Ca2+-triggered wound response. Upon the wounding of striated muscles, oxidation was proposed to cause the oligomerization of MG53, a muscle-specific tripartite motif family protein, and could recruit MG53-containing vesicles to the wound site (33). The entry of Ca2+ then leads to the fusion of vesicles with the membrane to patch the wound. In a separate study on the transection of neurons, it was found that melatonin, an antioxidant, decreases healing rate, supporting oxidation as an additional trigger for wound response (2).
Regeneration
Once a wound has healed, the cell faces the problem of rebuilding damaged or lost structures. A population of dividing cells might not need an active regeneration mechanism, because as the cells proliferate they will be building new structures in the course of normal growth. In the absence of an active regeneration pathway, cellular structures would continue to grow in size and number, and then be partitioned between the daughters when the cell divides. For organelles that grow at constant rate and then partition between daughters, it has been shown that these two processes are sufficient to restore organelle size following a perturbation (34), but the restoration of size can take many cell generations. Other cells can rebuild lost or damaged structures within a single cell cycle. We will discuss two models systems – flagellar regeneration in Chlamydomonas and oral apparatus regeneration in Stentor.
Both flagellar regeneration in Chlamydomonas (35) and oral regeneration in Stentor (36) are accompanied by transcriptional activation and translation of proteins related to the structure being regenerated. Transcription is obligatory for Stentor OA regeneration but not for Chlamydomonas flagellar regeneration. If flagella are removed in the presence of translational inhibitors, flagella still regenerate, but only to half the normal length (37), suggesting that new protein synthesis is triggered during regeneration to provide enough material to reach the correct final length. How does a cell know that the flagellum or OA, respectively, has been removed, in order to trigger the transcriptional programs? In principle, a cell could know a structure is missing by sensing: 1) a loss of function of the structure; 2) removal of the structure leading to cellular stresses that occur when part of the cell is ripped out; or 3) absence of a signal molecule produced by the structure when present.
For flagella, whose function is to generate fluid flow, paralysis does not induce regeneration but severing does, suggesting the stimulus is not a loss of function. If only one flagellum is severed, it will regenerate with similar kinetics to when both flagella are severed. While this happens, the other flagellum shortens until both flagella reach the same length (38), presumably due to competition for precursor proteins (39). The two flagella then regrow to the normal length. If this experiment is repeated with translation inhibitors, the two flagella reach a shorter length (38), implying that gene expression can be triggered even by removal of just one flagellum. This result argues against a model in which the presence of flagella produces an inhibitory signal that keeps the genes turned off.
What is the trigger for regeneration in Stentor? If an OA is grafted onto a cell that is in the process of regenerating, regeneration immediately stops and the oral primordium is resorbed (40), suggesting the presence of an OA generates an inhibitory signal that prevents the formation of a new oral primordium. If a Stentor cell is scrambled with glass needles, it will typically form one or several oral primordia, but this is completely prevented if a new OA is grafted onto the minced cell (41). Conversely, if two cells are grafted together followed by the surgical removal of one OA, two oral primordia form, even though one intact OA remains (Figure 4) (42). This experiment suggests the stimulus triggering regeneration is not complete loss of all OAs but rather lack of an OA corresponding to a primordium site. Nevertheless, it has been reported that regeneration can be inhibited by insertion of an isolated OA randomly into the cytoplasm of a regenerating cell (43). The positional requirements for inhibition thus remain unclear.
Figure 4.

Evidence that oral regeneration in Stentor can be triggered by the loss of a single oral apparatus. One of the strengths of Stentor as a model system is the ability to graft cells and cell fragments together. In the experiment depicted here, two cells are grafted together to form a doublet cell, which has two oral apparatuses. If one OA is then surgically removed, both of the fused cells form oral primordia (red) such that one half of the doublet regenerates and the other reorganizes.
Opportunities and Applications
Although many studies on single-cell repair have been performed in lower organisms, the mechanisms identified have potential for new strategies for treating human disease. For example, metastasis can subject cancer cells to damaging mechanical forces. The ability for tumor cells to repair their nuclear envelope, which often ruptures during metastasis when they penetrate tissues, is necessary for cell survival (44). Inhibition of nuclear repair in such cells might reduce metastatic potential. In another example, cells can be punctured during attack by bacterial pathogens. For example, streptolysin-O (SLO) produced by Streptococcus pyogenes forms membrane pores with diameters up to 38 nm (45). If it was possible to make cells in a patient transiently more vulnerable to damage, it might induce a “scorched earth defense”, whereby infected cells would die, limiting further spread of infection. Conversely, understanding how cells repair and regenerate holds the potential for strategies in regenerative medicine in which damaged cells can be induced to regenerate in situ, rather than replacing them with new cells.
Pursuing these opportunities requires the development of tools and assays similar to those in tissue-level wounding studies. A reproducible, physiologically relevant, and high-throughput assay would permit identification of gene expression and molecular pathways involved. Wounding experiments in Stentor, for example, are still primarily based on manual surgery using glass needles (46). This method has not changed for over 100 years, and is slow and not easily reproducible. Recently, laser ablation is increasingly used for wounding. Laser wounding allows precise control on both the position and the size of the wound. The effect of laser on cells is still an active area of research, however (50, 51). The throughput may also be insufficient to generate enough cells in the same stage of their repair process for molecular studies such as RNA sequencing. In addition to the wounding method, methods are needed to quantify healing. Wound size measurement based on imaging is limited by optical resolution and the availability of membrane dyes. The alternative method – measuring rates of external dye uptake - can be misleading as uptake depends on not only the wound size but also the viscoelasticity of the cytoskeleton, which can change dynamically during the healing process.
Single-cell repair is increasingly recognized to be a conserved phenomenon across a wide range of biological systems. Clearly there are still many open questions, in particular about the molecular mechanisms of wound healing and regeneration. These phenomena also raise larger questions. For example, the question of the smallest cell fragment cells can heal from also leads one to think about the minimum set of components necessary for healing and subsequent survival. Such minimum set can perhaps be another approach to define a “minimum cell”. Understanding how cells repair themselves is thus important not only for our mechanistic understanding of cell biology, but also ultimately, for understanding what it means to be alive.
Acknowledgments
The authors thank members of their respective labs for many interesting conversations. Work in the authors’ labs on wound-healing and regeneration is supported by NSF Award #1517089 (ST and WM) and NIH grant GM113602 (WM), respectively. Both authors acknowledge support from NSF Award #1548297.
Footnotes
This manuscript has been accepted for publication in Science. This version has not undergone final editing. Please refer to the complete version of record at http://www.sciencemag.org/. The manuscript may not be reproduced or used in any manner that does not fall within the fair use provisions of the Copyright Act without the prior, written permission of AAAS.
Understanding how single cells repair and regenerate themselves underpins our mechanistic understanding of cell biology, and could guide treatments for conditions involving cellular damage.
References
- 1.Lund EJ. Reversibility of morphogenetic processes in Bursaria. J Exp Zool. 1917;24:1–33. [Google Scholar]
- 2.Spaeth CS, Boydston EA, Figard LR, Zuzek A, Bittner GD. A Model for Sealing Plasmalemmal Damage in Neurons and Other Eukaryotic Cells. J Neurosci. 2010;30:15790–15800. doi: 10.1523/JNEUROSCI.4155-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fishman HM, Bittner GD. Vesicle-mediated restoration of a plasmalemmal barrier in severed axons. News Physiol Sci. 2003;18:115–118. doi: 10.1152/nips.01429.2002. [DOI] [PubMed] [Google Scholar]
- 4.Han R, et al. Dysferlin-mediated membrane repair protects the heart from stress-induced left ventricular injury. J Clin Invest. 2007;117:1805–1813. doi: 10.1172/JCI30848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang XH, et al. Cardioprotection of Ischemia/Reperfusion Injury by Cholesterol-Dependent MG53-Mediated Membrane Repair. Circ Res. 2010;107:76–83. doi: 10.1161/CIRCRESAHA.109.215822. [DOI] [PubMed] [Google Scholar]
- 6.Sonnemann KJ, Bement WM. Wound Repair: Toward Understanding and Integration of Single-Cell and Multicellular Wound Responses. Annu Rev Cell Dev Biol. 2011;27:237–263. doi: 10.1146/annurev-cellbio-092910-154251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bement WM, Mandato CA, Kirsch MN. Wound-induced assembly and closure of an actomyosin purse string in Xenopus oocytes. Curr Biol. 1999;9:579–587. doi: 10.1016/s0960-9822(99)80261-9. [DOI] [PubMed] [Google Scholar]
- 8.Quarmby LM. Cellular deflagellation. Int Rev Cytol. 2004;233:47–91. doi: 10.1016/S0074-7696(04)33002-0. [DOI] [PubMed] [Google Scholar]
- 9.Slabodnick M, Prevo B, Gross P, Sheung J, Marshall W. Visualizing cytoplasmic flow during single-cell wound healing in Stentor coeruleus. J Vis Exp. 2013;82:e50848. doi: 10.3791/50848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morgan TH. Regeneration of proportionate structures in Stentor. Biol Bull. 1900;2:311–328. [Google Scholar]
- 11.Abidor IG, et al. Electric breakdown of bilayer lipid-membranes. 1. Main experimental facts and their qualitative discussion. Bioelectrochem Bioenerg. 1979;6:37–52. [Google Scholar]
- 12.Chernomordik LV, Melikyan GB, Chizmadzhev YA. Biomembrane fusion: a new concept derived from model studies using two interacting planar lipid bilayers. Biochim Biophys Acta. 1987;906:309–352. doi: 10.1016/0304-4157(87)90016-5. [DOI] [PubMed] [Google Scholar]
- 13.Dubey GP, Ben-Yehuda S. Intercellular Nanotubes Mediate Bacterial Communication. Cell. 2011;144:590–600. doi: 10.1016/j.cell.2011.01.015. [DOI] [PubMed] [Google Scholar]
- 14.VanDersarl JJ, Xu AM, Melosh NA. Nanostraws for Direct Fluidic Intracellular Access. Nano Lett. 2012;12:3881–3886. doi: 10.1021/nl204051v. [DOI] [PubMed] [Google Scholar]
- 15.Stephens DJ, Pepperkok R. The many ways to cross the plasma membrane. Proc Natl Acad Sci U S A. 2001;98:4295–4298. doi: 10.1073/pnas.081065198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McNeil PL, Terasaki M. Coping with the inevitable: how cells repair a torn surface membrane. Nat Cell Biol. 2001;3:E124–E129. doi: 10.1038/35074652. [DOI] [PubMed] [Google Scholar]
- 17.Moe AM, Golding AE, Bement WM. Cell healing: Calcium, repair and regeneration. Semin Cell Dev Biol. 2015;45:10–15. doi: 10.1016/j.semcdb.2015.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schanne FAX, Kane AB, Young EE, Farber JL. Calcium dependence of toxic cell death: a final common pathway. Science. 1979;206:700–702. doi: 10.1126/science.386513. [DOI] [PubMed] [Google Scholar]
- 19.Lemasters JJ, Diguiseppi J, Nieminen AL, Herman B. Blebbing, free Ca2+ and mitochondrial membrane potential preceding cell death in hepatocytes. Nature. 1987;325:78–81. doi: 10.1038/325078a0. [DOI] [PubMed] [Google Scholar]
- 20.Roscoe R. The flow of viscous fluids round plane obstacles. Philos Mag. 1949;40:338–351. [Google Scholar]
- 21.Allbritton NL, Meyer T, Stryer L. Range of messenger action of calcium-ion and inositol 1,4,5-trisphosphate. Science. 1992;258:1812–1815. doi: 10.1126/science.1465619. [DOI] [PubMed] [Google Scholar]
- 22.Terasaki M, Miyake K, McNeil PL. Large plasma membrane disruptions are rapidly resealed by Ca2+-dependent vesicle-vesicle fusion events. J Cell Biol. 1997;139:63–74. doi: 10.1083/jcb.139.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Togo T, Alderton JM, Bi GQ, Steinhardt RA. The mechanism of facilitated cell membrane resealing. J Cell Sci. 1999;112:719–731. doi: 10.1242/jcs.112.5.719. [DOI] [PubMed] [Google Scholar]
- 24.Godin LM, Vergen J, Prakash YS, Pagano RE, Hubmayr RD. Spatiotemporal dynamics of actin remodeling and endomembrane trafficking in alveolar epithelial type I cell wound healing. Am J Physiol Lung Cell Mol Physiol. 2011;300:L615–L623. doi: 10.1152/ajplung.00265.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krause TL, Fishman HM, Ballinger ML, Bittner GD. Extent and mechanism of sealing in transected giant axons of squid and earthworms. J Neurosci. 1994;14:6638–6651. doi: 10.1523/JNEUROSCI.14-11-06638.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Steinhardt RA. The mechanisms of cell membrane repair - A tutorial guide to key experiments. Ann N Y Acad Sci. 2005;1066:152–165. doi: 10.1196/annals.1363.017. [DOI] [PubMed] [Google Scholar]
- 27.McNeil PL, Steinhardt RA. Plasma membrane disruption: Repair, prevention, adaptation. Annu Rev Cell Dev Biol. 2003;19:697–731. doi: 10.1146/annurev.cellbio.19.111301.140101. [DOI] [PubMed] [Google Scholar]
- 28.Heilbrunn LV. The colloid chemistry of protoplasm. V A preliminary study of the surface precipitation reaction of living cells. Archiv Fur Experimentelle Zellforschung. 1927;4:246–263. [Google Scholar]
- 29.Chambers R, Kao CY. The effect of electrolytes on the physical state of the nerve axon of the squid and of stentor, a protozoon. Exp Cell Res. 1952;3:564–573. [Google Scholar]
- 30.Milo R, Phillips R. Cell Biology by the Numbers. Garland Science; New York, NY: 2015. [Google Scholar]
- 31.Bertini I, Gray HB, Lippard SJ, Valentine JS. Bioinorganic Chemistry. University Science Books; Mill Valley, CA: 1994. [Google Scholar]
- 32.Davenport NR, Sonnemann KJ, Eliceiri KW, Bement WM. Membrane dynamics during cellular wound repair. Mol Biol Cell. 2016;27:2272–2285. doi: 10.1091/mbc.E16-04-0223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cai C, et al. MG53 nucleates assembly of cell membrane repair machinery. Nat Cell Biol. 2009;11:56–U108. doi: 10.1038/ncb1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chan YHM, Reyes L, Sohail SM, Tran NK, Marshall WF. Organelle Size Scaling of the Budding Yeast Vacuole by Relative Growth and Inheritance. Curr Biol. 2016;26:1221–1228. doi: 10.1016/j.cub.2016.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stolc V, Samanta MP, Tongprasit W, Marshall WF. Genome-wide transcriptional analysis of flagellar regeneration in Chlamydomonas reinhardtii identifies orthologs of ciliary disease genes. Proc Natl Acad Sci USA. 2005;102:3703–3707. doi: 10.1073/pnas.0408358102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burchill BR. Synthesis of RNA and protein in relation to oral regeneration in the ciliate Stentor coeruleus. J Exp Zool. 1967;167:427–438. [Google Scholar]
- 37.Rosenbaum JL, Moulder JE, Ringo DL. Flagellar elongation and shortening in Chlamydomonas. The use of cycloheximide and colchicine to study the synthesis and assembly of flagellar proteins. J Cell Biol. 1969;41:600–619. doi: 10.1083/jcb.41.2.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Coyne B, Rosenbaum JL. Flagellar elongation and shortening in Chlamydomonas. II. Re-utilization of flagellar proteins. J Cell Biol. 1970;47:777–781. doi: 10.1083/jcb.47.3.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ludington WB, Shi LZ, Zhu Q, Berns MW, Marshall WF. Organelle Size Equalization by a Constitutive Process. Curr Biol. 2012;22:2173–2179. doi: 10.1016/j.cub.2012.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tartar V. Specific inhibition of the oral primordium by formed oral structures in Stentor coeruleus. J Exp Zool. 1958;139:479–505. [Google Scholar]
- 41.Tartar V. Cellular Mechanisms in Differentiation and Growth. Princeton University Press; Princeton, NJ: 1956. Pattern and substance in Stentor; pp. 73–100. [Google Scholar]
- 42.Tartar V. Reactions of Stentor coeruleus to homoplastic grafting. J Exp Zool. 1954;127:511–575. [Google Scholar]
- 43.Hyvert N, Pelvat B, de Haller G. Morphogenes experimentale chez les ciliates: IV. Sur le role de la Zon de Membranelles Adorales dans la regeneration chez Stentor coeruleus. Revue Suisse de Zoologie. 1972;79:1060–1068. [Google Scholar]
- 44.Denais CM, et al. Nuclear envelope rupture and repair during cancer cell migration. Science. 2016;352:353–358. doi: 10.1126/science.aad7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Duncan JL, Schlegel R. Effect of streptolysin O on erythrocyte membranes, liposomes, and lipid dispersions. A protein-cholesterol interaction. J Cell Biol. 1975;67:160–173. doi: 10.1083/jcb.67.1.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chambers R. The microvivisection method. Biol Bull. 1918;34:121–136. [Google Scholar]