

Abstract
Objective:
To assess effects of caffeine on Parkinson disease (PD).
Methods:
In this multicenter parallel-group controlled trial, patients with PD with 1–8 years disease duration, Hoehn & Yahr stages I–III, on stable symptomatic therapy were randomized to caffeine 200 mg BID vs matching placebo capsules for 6–18 months. The primary research question was whether objective motor scores would differ at 6 months (Movement Disorder Society–sponsored Unified Parkinson's Disease Rating Scale [MDS-UPDRS]–III, Class I evidence). Secondary outcomes included safety and tolerability, motor symptoms (MDS-UPDRS-II), motor fluctuations, sleep, nonmotor symptoms (MDS-UPDRS-I), cognition (Montreal Cognitive Assessment), and quality of life.
Results:
Sixty patients received caffeine and 61 placebo. Caffeine was well-tolerated with similar prevalence of side effects as placebo. There was no improvement in motor parkinsonism (the primary outcome) with caffeine treatment compared to placebo (difference between groups −0.48 [95% confidence interval −3.21 to 2.25] points on MDS-UPDRS-III). Similarly, on secondary outcomes, there was no change in motor signs or motor symptoms (MDS-UPDRS-II) at any time point, and no difference on quality of life. There was a slight improvement in somnolence over the first 6 months, which attenuated over time. There was a slight increase in dyskinesia with caffeine (MDS-UPDRS-4.1+4.2 = 0.25 points higher), and caffeine was associated with worse cognitive testing scores (average Montreal Cognitive Assessment = 0.66 [0.01, 1.32] worse than placebo).
Conclusion:
Caffeine did not provide clinically important improvement of motor manifestations of PD (Class I evidence). Epidemiologic links between caffeine and lower PD risk do not appear to be explained by symptomatic effects.
Clinicaltrials.gov identifier:
Classification of evidence:
This study provides Class I evidence that for patients with PD, caffeine does not significantly improve motor manifestations.
Multiple studies have consistently linked the use of caffeine, an adenosine antagonist, to a lower risk of Parkinson disease (PD). The combined relative risk in a recent meta-analysis was 0.67 (95% confidence interval [CI] 0.58, 0.76).1 The mechanism for this robust finding is unclear. Although a neuroprotective effect is possible,2,3 there are other explanations, including reverse causality (prodromal PD reduces tolerability, benefit, or desire for caffeine), symptomatic benefit (caffeine treats motor symptoms, so delays diagnosis), or residual confounding by another factor (e.g., the Parkinson personality or changes in reward mechanisms).
Recently, we conducted a small randomized trial of caffeine in patients with PD with daytime somnolence.4,5 We observed a modest benefit on somnolence, but also found benefit on motor manifestations, with a 3.2-point improvement on the Unified Parkinson’s Disease Rating Scale part III. If caffeine could be used as a symptomatic agent for PD, it would have the notable advantage of being safe, well-tolerated, and inexpensive. However, the motor finding in our study was a secondary outcome. Also, the study was short duration (6 weeks), and given caffeine's well-known tachyphylactic properties for sleepiness,6,7 motor effects may be short-lived.
Therefore, we conducted a multicenter parallel-group randomized controlled trial to evaluate the symptomatic effects of caffeine in PD.
METHODS
Standard protocol approvals, registrations, and patient consents.
All patients provided written consent to participate, and research ethics boards of each center approved the study. The study is registered on clinicaltrials.gov (NCT01738178).
Patients.
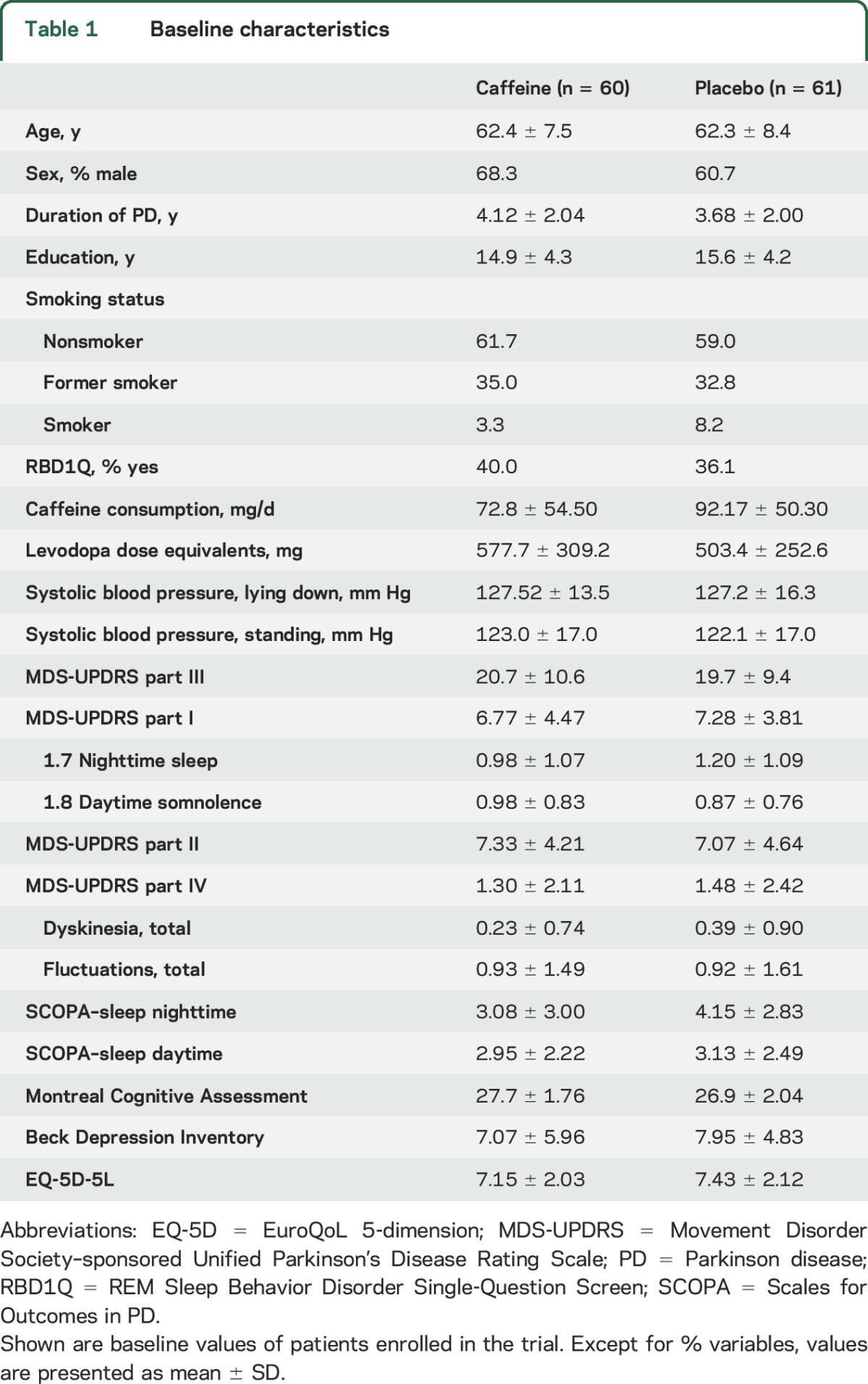
Patients were recruited from 7 sites at the McGill University Health Center; University of Manitoba; Toronto Western Hospital; Ottawa Civic Hospital; Pontifical Catholic University of Parana, Curitiba, Brazil; University of British Columbia; and University of Calgary between June 2014 and December 2015. All patients had idiopathic PD diagnosed as (1) parkinsonism according to UK brain bank criteria8 and (2) PD considered the likeliest cause according to the treating physician.9 Inclusion criteria were disease duration 6 months to 8 years, age 45–75 years, Hoehn & Yahr stage I–III, receiving symptomatic PD therapy for >6 months, and on stable dose for >3 months. Exclusion criteria included caffeine intake >150 mg per day (on standardized intake questionnaire), another adenosine antagonist, active peptic ulcers, supraventricular cardiac arrhythmia, uncontrolled hypertension, premenopausal women not using effective birth control, cognitive impairment (Montreal Cognitive Assessment [MoCA] <23/30),10 moderate to severe depression (Beck Depression Inventory [BDI] >19),11 anticipated need to change medications over the next 6 months, and current use of lithium or clozapine (pharmacokinetic interactions).
Intervention.
The treatment was caffeine-containing capsules 200 mg twice daily (or matching placebo in 1:1 ratio) in the morning and after lunch (this corresponding to approximately 3 daily cups of coffee, depending upon brewing technique). To improve tolerability and minimize unblinding, the dose was increased slowly (50 mg per week), with placebo for the first week, and full dose was reached at week 9.
Randomization and blinding.
Randomization was by block randomization (block size = 4), stratified to site, by a central statistician (L.J.) using PROC-PLAN (SAS Institute, Cary, NC). All patients, study investigators, and examiners were blinded to treatment assignment. Caffeine and placebo tablets were encapsulated as indistinguishable in appearance, prepared by a central pharmacy. To assess potential unblinding, patients and physician examiners were asked at study conclusion to guess each treatment allocation (forced choice of caffeine vs placebo).
Monitoring.
Compliance was assessed by patient report and pill counts. Dietary caffeine intake was assessed by standardized questionnaire12; all patients were instructed to avoid changing caffeine intake during the trial. For safety reasons, any patient who increased spontaneous caffeine intake to >300 mg would be withdrawn from the study. Adverse events were queried by semi-structured interview, screening for symptoms of gastrointestinal pain, other gastrointestinal problems, sleep changes, palpitations, sweating, and tremulousness, as well as open-ended adverse event reporting. Blood pressure was assessed to rule out new-onset hypertension.
Outcomes.
The primary outcome was change in objective motor parkinsonism severity over 6 months, rated by the Movement Disorder Society–sponsored Unified Parkinson's Disease Rating Scale (MDS-UPDRS) part III,13 assessed in the antiparkinson medication “on” state.
Secondary outcomes included all other components of the MDS-UPDRS (i.e., nonmotor symptoms, motor symptoms, fluctuations/dyskinesia). We also examined individual scores of each motor and nonmotor item from the MDS-UPDRS. Cognition was assessed with the MoCA,10 performed annually after 6 months using alternating versions 1–3 to prevent practice effects. Insomnia and somnolence were assessed with the Scales for Outcomes in PD (SCOPA) sleep scale (nighttime and daytime components),14 and REM sleep behavior disorder was screened with the REM Sleep Behavior Disorder Single-Question Screen.15 Both patient and examiner recorded a clinical global impression of change for overall PD severity. Quality of life was measured with the 5-item EuroQoL 5-dimension (EQ-5D).16 We also assessed antiparkinson medication doses, calculated as levodopa-dose equivalents.17
Study visits and study termination.
The study was divided into 3 stages. The first stage (the primary outcome measure of this publication) was designed to estimate motor benefits of caffeine while keeping symptomatic medications constant. Three visits were conducted, at baseline, 3 months, and 6 months of therapy. After 6 months, a second extension stage was started, in which patients continued caffeine/placebo with medication adjustments allowed as needed and followed every 6 months. Stage I was powered (80% power, α = 0.05) to find a change in MDS-UPDRS-III of 3 points; assuming SD for this change of 4.5 points, this required 38 patients per group. The study was also additionally powered for extension stages, for a planned total of 250 patients. Stage 2 was designed as a 4-year extension to assess whether symptomatic effects could be maintained, and how these effects might manifest on examination findings and medication utilization. Stage 3 was intended as a long-latency delayed start stage with all participants receiving caffeine, to be used as a pilot study for potential disease modification. However, following uncertain results of A2A antagonist programs (e.g., preladenant abandoned for failure at phase 3 trials18,19), and slower than expected recruitment, we elected to perform an interim analysis of data. At this stage, 121 patients had been recruited, ensuring sufficient accuracy to assess the primary stage 1 outcome. Based on results of this analysis, the study was stopped (because there was no evident symptomatic effect to be followed up in stage 2, and stage 3 was only an inadequately powered pilot stage). All patients were requested to complete stage 1, and patients in later stages were brought in for a final visit, after which intervention was terminated. As a secondary outcome, we analyzed results for all patients in the second stage, up until 18 months trial duration.
Analysis.
The primary analysis was intention-to-treat. Multiple imputation was planned for missing data; however, since only <1% of variables were missing, we omitted missing values. Adverse events and tolerability were reported descriptively, and the overall frequency of adverse events, serious adverse events, and dropout rates were compared by calculating 95% CIs for the between-group differences in proportions. We estimated treatment effects at each time point separately by creating baseline-to-time point differences within each patient, and comparing means across treatment groups with 95% CIs. For the analyses of other time points, we used 2 analyses; one was the same analysis as for the primary outcome, and the second considered all time points together. For the second, we adjusted for time by estimating a coefficient for each time point independently, that is, not assuming any shape or fixed slope for the outcome over time. A group measure then estimated the treatment effect, which assumed a constant difference between groups over time, after adjusting for baseline shape at each time point. We estimated treatment effects across these multiple time points using a Bayesian hierarchical random effects model, adjusting for time as described above, including age and sex as covariates.
RESULTS
Patient flow is illustrated in figure 1. A total of 121 patients were randomized: 60 to caffeine and 61 to placebo. Three patients withdrew and were lost to follow-up, all in the caffeine group. Therefore, 57 caffeine and 61 placebo patients were available for the primary analysis. Baseline variables are shown in table 1.
Figure 1. Patient flow throughout the course of the study.
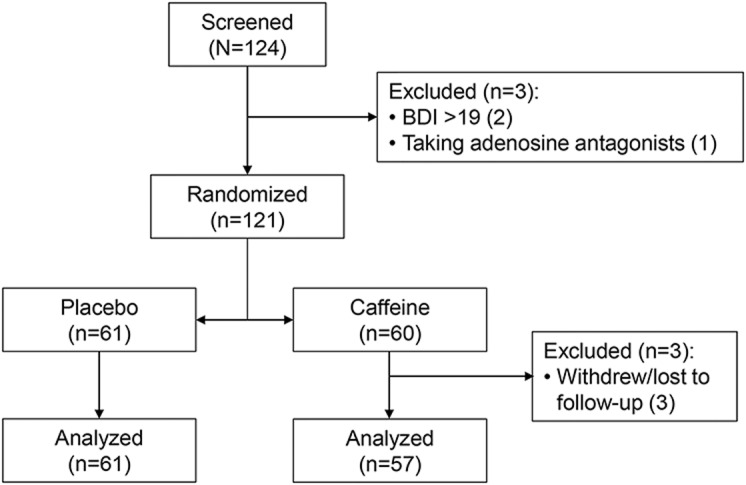
BDI = Beck Depression Inventory.
Table 1.
Baseline characteristics
Tolerability, adverse events, and blinding.
Over 6 months, 29 patients receiving caffeine and 31 patients receiving placebo reported adverse events (difference 2.5% [95% CI −15, 20]). No adverse event was significantly more common with caffeine (table e-1 at Neurology.org). One patient in each group had a serious adverse event, both deemed to be unrelated to study intervention. Of patients in the analysis, 7 receiving caffeine and 5 receiving placebo stopped the intervention because of side effects. There was no difference in blood pressure or caffeine consumption changes between groups. At the 6-month visit, 50% of patients guessed their treatment correctly (95% CI 41, 59), and examiners guessed correctly 57% (48%, 65%) of the time (chance 50%), indicating successful blinding.
Effect of caffeine at 6 months.
Over 6 months, the MDS-UPRDS III worsened by a mean of 0.64 (SD 7.25) points in the placebo group, compared to 0.16 (SD 7.68) points in the caffeine group (table 2, figure 2). There was no difference in decline between groups (0.48 points [−3.21, 2.25]). Removing protocol violations (including study medication termination) did not affect results (decline = 0.11 points less in placebo). Stratifying to baseline caffeine use also did not affect results (e.g., MDS-UPDRS III change in caffeine group = 0.10 ± 7.8 for those with baseline caffeine intake <80 mg/d vs 0.23 ± 7.0 for ≥80 mg/d). Caffeine response did not differ according to center (p = 0.64 for interaction between center and group on generalized linear modeling). Similarly, there was no difference in motor symptoms on the MDS-UPDRS part II total, or any of its individual components. The clinical global impression of change (rating overall PD severity) did not differ, either when assessed by patient or by examiner. There was no difference in depression or anxiety on the BDI or UPDRS part I, and no difference in quality of life on the EQ-5D.
Table 2.
Effect of caffeine at 6 months (primary outcome)
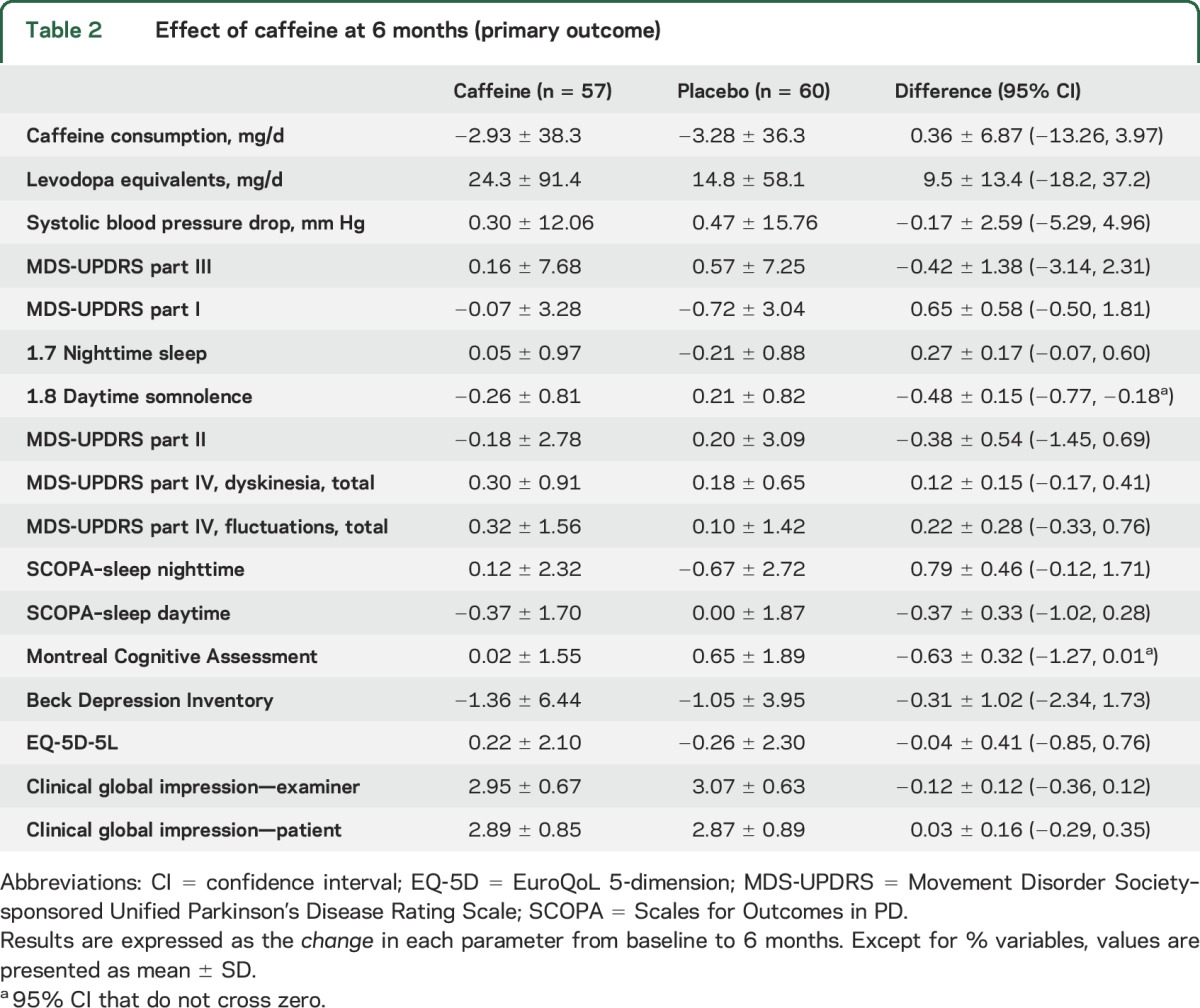
Figure 2. Change from baseline in selected Parkinson disease measures in caffeine vs placebo.

(A) Change in Movement Disorder Society–sponsored Unified Parkinson's Disease Rating Scale (MDS-UPDRS) III, (B) change in MDS-UPDRS 1.8 (daytime sleepiness), (C) change in MDS-UPDRS II, (D) change in Scales for Outcomes in PD (SCOPA) (daytime), (E) change in MDS-UPDRS IV (dyskinesia), (F) change in Montreal Cognitive Assessment. Note that the primary outcome is at 6 months; only a proportion of patients continued follow-up into 12 months (73%) and 18 months (55%). Error bars indicate standard error. The asterisk indicates a statistically significant difference between caffeine and placebo (p < 0.05).
Among the secondary outcomes, we noticed a small difference in the daytime somnolence score of the MDS-UPDRS in favor of caffeine (−0.49 [−0.79, −0.20] points). This was accompanied by an equivocal improvement in SCOPA–sleep daytime component (−0.37 [−1.02, 0.28] points), with 95% CI crossing zero. Nighttime sleep was slightly worse with caffeine, but 95% CIs crossed zero (0.28 [−0.06, 0.62] points on MDS-UPDRS and 0.81 [−0.11, 1.73] points on SCOPA–sleep nighttime). There was also a small difference in MoCA scores favoring placebo (difference = 0.65 points [−1.29, −0.01]).
Results at other time points.
At 3 months, results were broadly similar to 6 months (tables 3 and 4). Again there was no difference in MDS-UPDRS part III (0.85 points better in caffeine [−2.17, 1.46]). However, we noted a modest increase in dyskinesia severity (sum of MDS-UPDRS items 4.1 and 4.2) with caffeine compared to placebo (0.28 [0.01, 0.55]). This increase was also partially seen at 6 months, but CIs clearly crossed zero (0.12 [−0.17, 0.41]). SCOPA–sleep daytime scores showed improvement (−1.09 [−1.88, −0.30] points), whereas the MDS-UPDRS somnolence score had equivocal change (−0.18 [−0.49, 0.18] points).
Table 3.
Effects of caffeine at 3 months
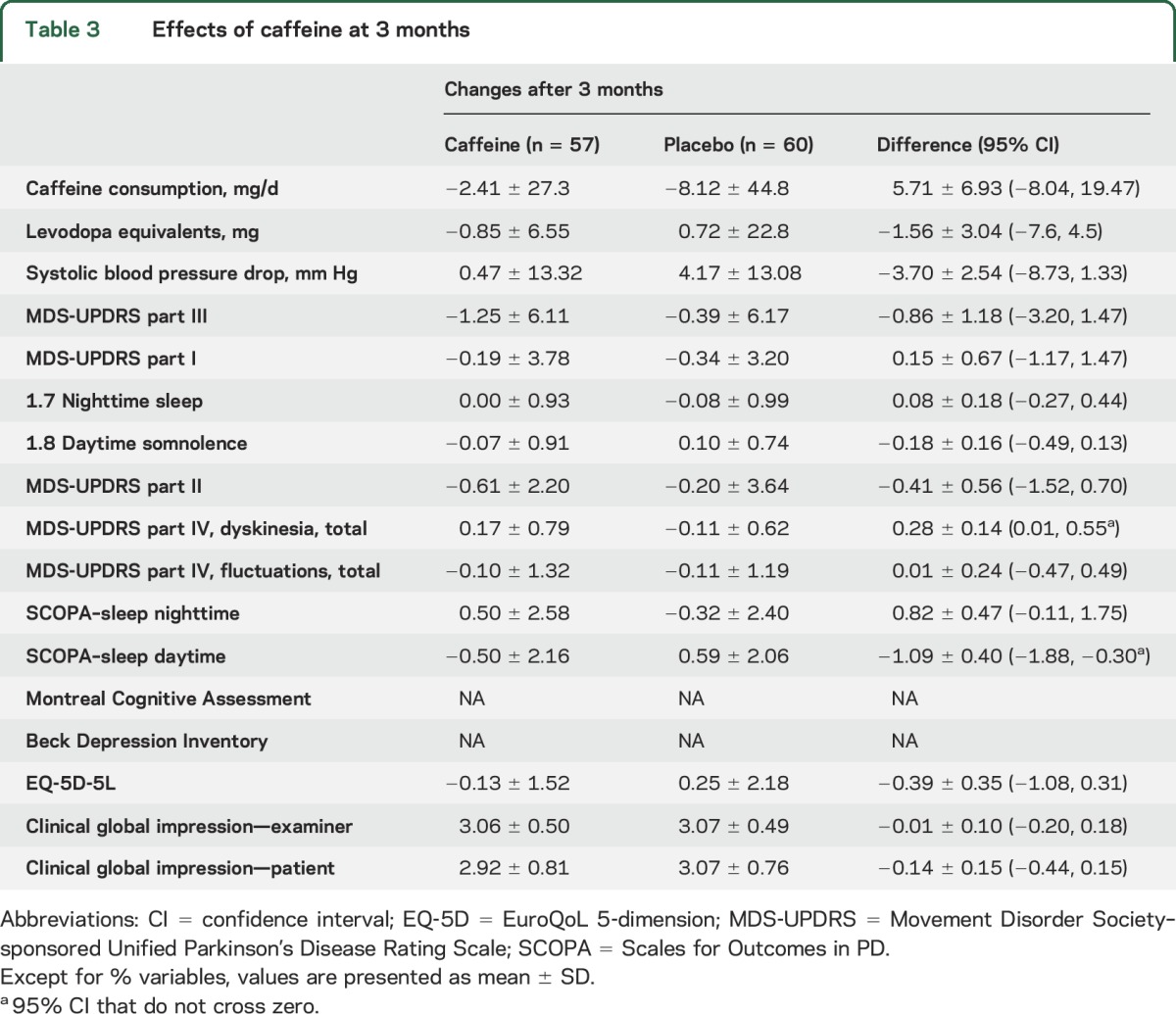
Table 4.
Effects of caffeine at 12 and 18 months
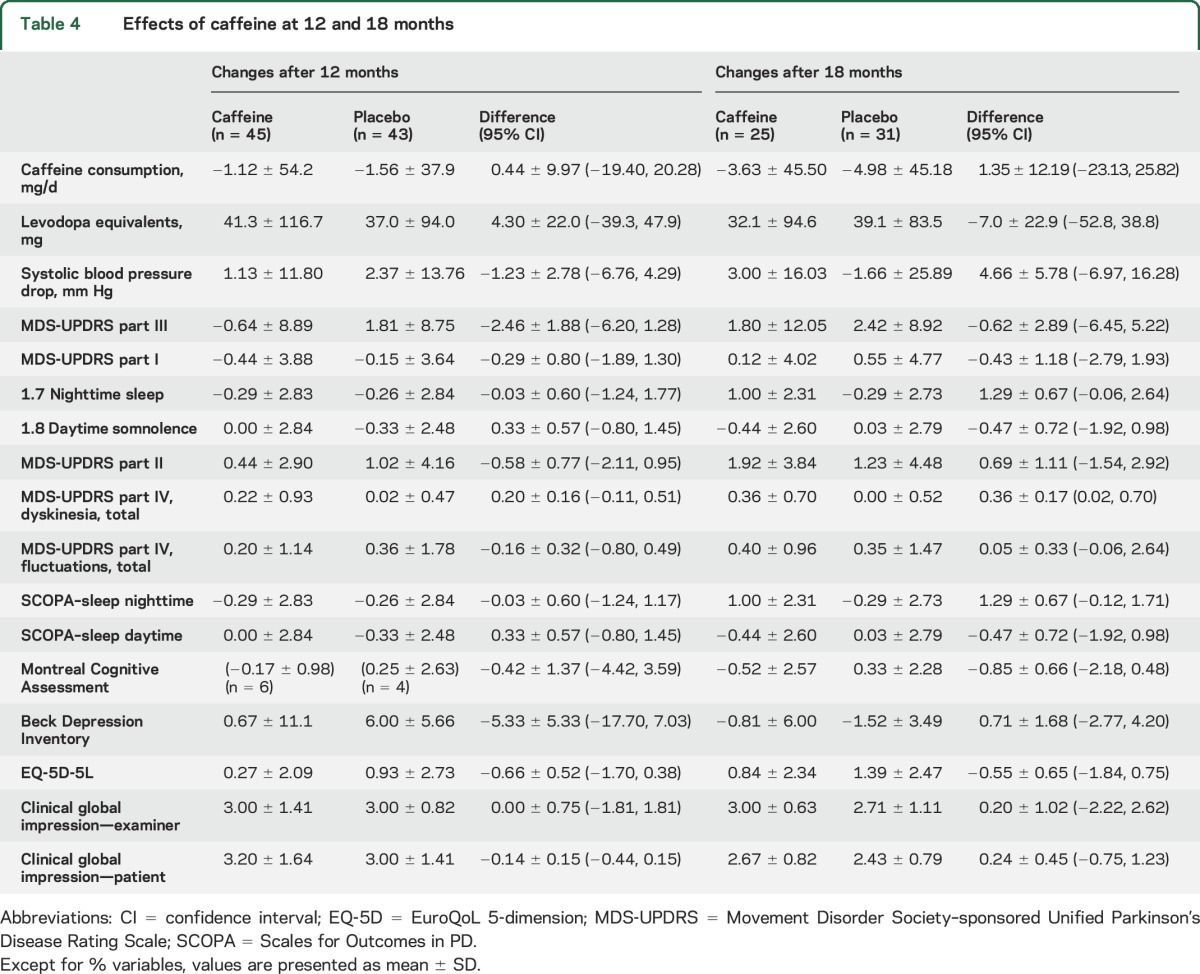
During stage II, a total of 88 patients were assessed at 12 months, and 66 at 18 months (tables 3 and 4). Again, results were broadly similar to 6 months. MDS-UPDRS III did not improve with caffeine at any time point (−2.5 [−6.2, 1.3] at 12 months, −0.62 [−6.5, 5.2] at 18 months). Dyskinesia scores were slightly higher at both 12 and 18 months; differences at 18 months were clearer [+0.36 (0.02–0.70] points). Point estimates of MoCA scores at 18 months suggested a similar possible worsening in the caffeine group (0.85 [−0.48, 2.18] points). However, improvement in somnolence measures seen at 3–6 months attenuated with time, with both the UPDRS–sleepiness and SCOPA–sleep daytime scores showing minimal difference between groups at later intervals (e.g., SCOPA–sleepiness = +0.33 [−0.9 to 1.5] at 12 months, −0.47 [−1.9 to 1.0] at 18 months).
Overall, on statistical analysis combining all time points, we again found no substantial change in MDS-UPDRS-III (−0.96 [−3.43, 1.54] points). MoCA worsened in the caffeine group by 0.66 (0.01, 1.32) points. Dyskinesia scores worsened by 0.25 (0.04, 0.45) points. Improvement on the SCOPA daytime sleepiness was equivocal (−0.42 [−1.09, 0.25]), but UPDRS sleepiness scores were reduced (−0.31 [−0.58, 0.05] points).
DISCUSSION
In this study, we found that caffeine does not produce sustained motor improvement in PD. On exploratory secondary outcomes, we found small improvements in measures of alertness, counterbalanced by an overall increase in dyskinesia and a slight lowering of cognitive test scores in those receiving caffeine.
The key finding of this study is that caffeine provided no improvement in motor PD. This is in contrast to our previous study, which found a 3.2-point improvement in UPDRS (1987 version). Based upon published conversion rates between the new and old UPDRS versions, the 3.2-point improvement corresponds to a 3.8-point change (3.2 × 1.2). In the current study, our 95% CI excludes this value (−3.2, 2.3 points). It also excludes the MDS-UPDRS-III estimated minimal clinically important change.20,21 There are many potential explanations for this difference, including study population differences (the previous trial included only somnolent patients who were older, more often male, and with longer disease duration) and a slower caffeine dose escalation. Perhaps the most likely explanation may be the different trial durations (6 weeks vs 6 months). Caffeine has well-known tachyphyllactic properties6,7; so caffeine may have a short-term benefit, which quickly dissipates. Regardless, our core finding is that caffeine cannot be recommended as symptomatic therapy for parkinsonism.
On secondary analysis, we again found some evidence that caffeine has temporary benefit on daytime alertness. There was some improvement on the MDS-UPDRS daytime sleepiness question at 6 months, with equivocal lowering of SCOPA–sleep. In our previous study, the Epworth Sleepiness Scale score (primary outcome) showed a nonsignificant improvement with caffeine (−1.71 points), with significantly improved clinical global impression of somnolence and significant improvement on per protocol analysis of the Epworth Sleepiness Scale (−1.97 points).5 Moreover, an open-label study of caffeine demonstrated reduced somnolence.4 This accords with the well-described effects of caffeine in the general population. Therefore, the balance of evidence suggests that caffeine probably has a modest effect on daytime somnolence or the sensation of alertness, which may wear off with prolonged exposure. Since caffeine is safe and generally well-tolerated, it seems reasonable to empirically try intermittent moderate doses of caffeine for somnolence, and repeat if improvement is seen.
There have been reports from observational studies that patients with PD who are caffeine users at baseline have less dyskinesia.22,23 Yet in this study, caffeine increased dyskinesia. This may be an effect of exposure time (i.e., caffeine prevents dyskinesia if provided before dopamine denervation or therapy, but increases dyskinesia if provided afterwards). Alternatively, this may be due to confounding in assessing markers of disease progression if those same markers also affect disease risk. For example, if a person develops PD despite years of exposure to a “protective” factor (e.g., caffeine), it may be that their disease is either (1) pathophysiologically distinct from those with a more typical risk profile (different underlying genetic profile, different reward mechanisms) or (2) anatomically distinct (if a “protective” factor works only upon the substantia nigra, leaving other PD-affected areas unchanged, patients exposed to this factor would have more advanced disease outside the substantia nigra by the time they present with measurable parkinsonism). The resulting differences in disease subtypes/stages could result in different PD manifestations like dyskinesia.
On secondary analysis, we also found small worsening of the MoCA with caffeine (mostly from unmatched improvement in the placebo group). Studies in the general population, including elderly persons, generally find small improvements in cognition with caffeine,6,12,24 unlike our results. Therefore, it is difficult to speculate a clear plausible mechanism for this finding. It should be emphasized that as a secondary outcome, this was an exploratory finding25; given multiple comparisons in the study, such findings may simply be chance.
Given the study results, what might be the implications for epidemiologic links between caffeine nonuse and PD? Some explanations have become less likely. Since caffeine is relatively well-tolerated in PD, reverse causality from side effects in prodromal disease becomes less probable. Similarly, since caffeine seems to have some alerting effects in PD, reverse causality related to loss of alerting benefit in prodromal stages becomes less probable. Also, given absent motor benefit, the lower risk cannot be simply explained as confounded by symptomatic treatment (caffeine, by treating motor PD, prevents diagnosis). Moreover, the absence of clinically meaningful symptomatic benefit documented in this trial is an important finding with respect to study design in any future disease-modifying trial. Our findings, however, do not necessarily imply that caffeine must be neuroprotective. First, although power is obviously limited, we saw no clear signal that motor measures were diverging between groups from 6 to 18 months. Second, and most critically, other plausible explanations remain, particularly residual confounding. For example, a similar inverse relationship exists between smoking and PD. A recent study found that patients developing PD find quitting smoking easier in the prodromal period,26 suggesting that systems of reward dependence/behavior reinforcement may differ in those at risk of PD, confounding the link.
Some limitations should be noted. All findings related to somnolence, cognition, and dyskinesia were secondary outcomes, and should be considered exploratory25; some may be chance findings. The study was not specifically designed to assess changes in dyskinesia, fluctuations, or dementia risk. Our SD of the estimated change in MDS-UPDRS-III was higher than expected (i.e., 6.2 and 7.3 vs the 4.5 observed in our original article). Therefore, we focused our result interpretation on the 95% CI, which excludes a >3.2-point improvement of MDS-UPDRS-III. We did not include biologic measures of caffeine or its metabolites; residual noncompliance could have affected results. The caffeine dose chosen was based upon previous studies and upon caffeine use patterns in the population (in which epidemiologic findings were observed)12; higher caffeine doses or other A2A antagonists may have different effects. Patients were assessed in the medication “on” state; using medication-naive patients or “off” evaluations might have different findings. The study was prematurely terminated, because analysis suggested that finding meaningful symptomatic benefit at longer time intervals was very unlikely. For example, the 18-month point estimate of difference for total MDS-UPDRS was 0.9 points. Even if this difference doubled over subsequent years, the study would have required approximately 1,000 patients for 80% power (our protocol had only 250). Note again that this was a trial of symptomatic therapy, and did not address potential disease modification.
Caffeine did not provide sustained symptomatic benefit on parkinsonism in PD. There may be temporary alerting effects, which need to be counterbalanced against potential increased dyskinesia and cognitive test worsening.
Supplementary Material
GLOSSARY
- BDI
Beck Depression Inventory
- CI
confidence interval
- EQ-5D
EuroQoL 5-dimension
- KVIQ
Kinesthetic and Visual Imagery Questionnaire
- MDS-UPDRS
Movement Disorder Society–sponsored Unified Parkinson's Disease Rating Scale
- MoCA
Montreal Cognitive Assessment
- PD
Parkinson disease
- SCOPA
Scales for Outcomes in PD
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
R.B.P. conceptualized the study, collected data, performed analysis, and drafted the initial version of the manuscript. J.A., M.M., D.G., S.F., R.M., S.C., A.M., A.B., and D.H. were responsible for generating data and for revision of the manuscript. A.P. was responsible for data collection, data analysis, and revising the manuscript. L.J. was responsible for data analysis and revision of the manuscript. A.E.L. was responsible for study conceptualization, generating data, and revision of the manuscript.
STUDY FUNDING
Supported by Canadian Institute of Health Research, Webster Foundation, Fonds de Recherche du Québec-Santé.
DISCLOSURE
R.B. Postuma received grant funding from Canadian Institute of Health Research, Webster Foundation, and Fonds de Recherche du Québec-Santé for this study. J. Anang, A. Pelletier, L. Joseph, M. Moscovich, D. Grimes, S. Furtado, R.P. Munhoz, S. Appel-Cresswell, A. Moro, A. Borys, D. Hobson, and A.E. Lang report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Noyce AJ, Bestwick JP, Silveira-Moriyama L, et al. Meta-analysis of early nonmotor features and risk factors for Parkinson disease. Ann Neurol 2012;72:893–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen X, Lan X, Roche I, Liu R, Geiger JD. Caffeine protects against MPTP-induced blood-brain barrier dysfunction in mouse striatum. J Neurochem 2008;107:1147–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schwarzschild MA, Chen JF, Ascherio A. Caffeinated clues and the promise of adenosine A(2A) antagonists in PD. Neurology 2002;58:1154–1160. [DOI] [PubMed] [Google Scholar]
- 4.Altman RD, Lang AE, Postuma RB. Caffeine in Parkinson's disease: a pilot open-label, dose-escalation study. Mov Disord 2011;26:2427–2431. [DOI] [PubMed] [Google Scholar]
- 5.Postuma RB, Lang AE, Munhoz RP, et al. Caffeine for treatment of Parkinson disease: a randomized controlled trial. Neurology 2012;79:651–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Higdon JV, Frei B. Coffee and health: a review of recent human research. Crit Rev Food Sci Nutr 2006;46:101–123. [DOI] [PubMed] [Google Scholar]
- 7.Harland BF. Caffeine and nutrition. Nutrition 2000;16:522–526. [DOI] [PubMed] [Google Scholar]
- 8.Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson's disease: a clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry 1992;55:181–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hughes AJ, Daniel SE, Lees AJ. Improved accuracy of clinical diagnosis of Lewy body Parkinson's disease. Neurology 2001;57:1497–1499. [DOI] [PubMed] [Google Scholar]
- 10.Nasreddine ZS, Phillips NA, Bedirian V, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc 2005;53:695–699. [DOI] [PubMed] [Google Scholar]
- 11.Beck AT, Ward CH, Mendelson M, Mock J, Erbaugh J. An inventory for measuring depression. Arch Gen Psychiatry 1961;4:561–571. [DOI] [PubMed] [Google Scholar]
- 12.Barone JJ, Roberts HR. Caffeine consumption. Food Chem Toxicol 1996;34:119–129. [DOI] [PubMed] [Google Scholar]
- 13.Goetz CG, Tilley BC, Shaftman SR, et al. Movement Disorder Society–sponsored revision of the Unified Parkinson's Disease Rating Scale (MDS-UPDRS): scale presentation and clinimetric testing results. Mov Disord 2008;23:2129–2170. [DOI] [PubMed] [Google Scholar]
- 14.Marinus J, Visser M, van Hilten JJ, Lammers GJ, Stiggelbout AM. Assessment of sleep and sleepiness in Parkinson disease. Sleep 2003;26:1049–1054. [DOI] [PubMed] [Google Scholar]
- 15.Postuma RB, Arnulf I, Hogl B, et al. A single-question screen for rapid eye movement sleep behavior disorder: a multicenter validation study. Mov Disord 2012;27:913–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.The EuroQol Group. EuroQol: a new facility for the measurement of health-related quality of life. Health Policy 1990;16:199–208. [DOI] [PubMed] [Google Scholar]
- 17.Hobson DE, Lang AE, Martin WR, Razmy A, Rivest J, Fleming J. Excessive daytime sleepiness and sudden-onset sleep in Parkinson disease: a survey by the Canadian Movement Disorders Group. JAMA 2002;287:455–463. [DOI] [PubMed] [Google Scholar]
- 18.Hauser RA, Stocchi F, Rascol O, et al. Preladenant as an adjunctive therapy with levodopa in Parkinson disease: two randomized clinical trials and lessons learned. JAMA Neurol 2015;72:1491–1500. [DOI] [PubMed] [Google Scholar]
- 19.Pinna A. Adenosine A2A receptor antagonists in Parkinson's disease: progress in clinical trials from the newly approved istradefylline to drugs in early development and those already discontinued. CNS Drugs 2014;28:455–474. [DOI] [PubMed] [Google Scholar]
- 20.Horvath K, Aschermann Z, Acs P, et al. Minimal clinically important difference on the motor examination part of MDS-UPDRS. Parkinsonism Relat Disord 2015;21:1421–1426. [DOI] [PubMed] [Google Scholar]
- 21.Schrag A, Sampaio C, Counsell N, Poewe W. Minimal clinically important change on the Unified Parkinson's Disease Rating Scale. Mov Disord 2006;21:1200–1207. [DOI] [PubMed] [Google Scholar]
- 22.Wills AM, Eberly S, Tennis M, et al. Caffeine consumption and risk of dyskinesia in CALM-PD. Mov Disord 2013;28:380–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scott NW, Macleod AD, Counsell CE. Motor complications in an incident Parkinson's disease cohort. Eur J Neurol 2016;23:304–312. [DOI] [PubMed] [Google Scholar]
- 24.Nehlig A. Is caffeine a cognitive enhancer? J Alzheimers Dis 2010;20(suppl 1):S85–S94. [DOI] [PubMed] [Google Scholar]
- 25.Bender R, Lange S. Adjusting for multiple testing: when and how? J Clin Epidemiol 2001;54:343–349. [DOI] [PubMed] [Google Scholar]
- 26.Ritz B, Lee PC, Lassen CF, Arah OA. Parkinson disease and smoking revisited: ease of quitting is an early sign of the disease. Neurology 2014;83:1396–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.