

Abstract
Objective:
To identify the gene defect in patients with hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC) who are negative for TUBB4A mutations.
Methods:
We performed homozygosity mapping and whole exome sequencing (WES) to detect the disease-causing variant. We used a Taqman assay for population screening. We developed a luciferase reporter construct to investigate the effect of the promoter mutation on expression.
Results:
Sixteen patients from 14 families from different countries fulfilling the MRI criteria for H-ABC exhibited a similar, severe clinical phenotype, including lack of development and a severe epileptic encephalopathy. The majority of patients had a known Roma ethnic background. Single nucleotide polymorphism array analysis in 5 patients identified one large overlapping homozygous region on chromosome 13. WES in 2 patients revealed a homozygous deletion in the promoter region of UFM1. Sanger sequencing confirmed homozygosity for this variant in all 16 patients. All patients shared a common haplotype, indicative of a founder effect. Screening of 1,000 controls from different European Roma panels demonstrated an overall carrier rate of the mutation of 3%–25%. Transfection assays showed that the deletion significantly reduced expression in specific CNS cell lines.
Conclusions:
UFM1 encodes ubiquitin-fold modifier 1 (UFM1), a member of the ubiquitin-like family involved in posttranslational modification of proteins. Its exact biological role is unclear. This study associates a UFM1 gene defect with a disease and sheds new light on possible UFM1 functional networks.
Hypomyelination with atrophy of the basal ganglia and cerebellum (H-ABC) (MIM 612438) is a rare leukodystrophy that was identified by MRI pattern analysis.1 The 2 most important MRI features are hypomyelination and a very small or absent putamen. The disease is associated with dominant de novo mutations in the TUBB4A gene (MIM 602662), encoding tubulin β-4A.2,3 In the Amsterdam Database of Leukoencephalopathies, a small number of patients fulfilling the MRI criteria of H-ABC did not harbor a pathogenic TUBB4A mutation, suggesting that mutations in at least one other gene are involved in the disease. In this study, we aimed at identifying the causal genetic defect in this group of unsolved H-ABC cases.
METHODS
Standard protocol approvals, registrations, and patient consents.
We received approval from the ethical standards committee for gene identification research on patients with unclassified leukoencephalopathies at the VU University Medical Center Amsterdam. Written informed consent was obtained from the guardians of the patients participating in this study.
Patients.
Sixteen patients from 14 families fulfilled the following MRI criteria for H-ABC: (1) hypomyelination, defined as a mildly elevated T2 signal intensity of most cerebral white matter in combination with mild T1 hypointensity, T1 isointensity, or mild T1 hyperintensity relative to the cortex4; and (2) very small or absent putamen without signal abnormality indicating lesion or scarring in the region where the putamen should be. We obtained DNA from all patients, parents, and unaffected siblings and obtained clinical data by a standardized form for physicians. We performed Kaplan-Meier analysis in SPSS version 22 (SPSS Inc., Chicago, IL) to estimate the median survival. For each patient, at least one MRI was available. Two investigators evaluated MRIs by visual assessment, as previously described.3
Analysis of the TUBB4A gene.
Sanger sequencing of the TUBB4A gene was performed in 16 patients as previously described.3 In addition, we performed multiplex ligation-dependent probe amplification (MLPA) analysis of the TUBB4A gene in 9 patients according to manufacturer's instructions (MRC-Holland, Amsterdam, the Netherlands; complete description of the MLPA probe mix is available upon request).
Single nucleotide polymorphism (SNP) array analysis.
We executed SNP array analysis (CytoScan HD array; Affymetrix, Santa Clara, CA) according to the manufacturer's protocol in 5 patients to identify runs of homozygosity larger than 1 Mb and overlapping regions (Nexus version 7 [BioDiscovery, Hawthorne, CA]). SNP array-based genotypes were created using Chromosome Analysis Suite 2.1.0.16 (Affymetrix).
Whole exome sequencing (WES).
We performed WES on genomic DNA from 2 patients and analyzed data as previously described.5 Based on family data indicating consanguinity and shared Roma (Gypsy) ethnic background for several families, variant filtering was executed under the hypothesis of a homozygous recessive inheritance model. We focused on rare variants, exonic as well as intronic, located in the identified overlapping homozygous region, filtering for a minor allele frequency of less than 1% in public databases (dbSNP, 1000 Genomes Project, Exome Variant Server, and NHLBI Exome Sequencing Project) as well as an occurrence below 1% in the heterozygous state and absence in the homozygous state in our in-house WES control database.
Segregation of the UFM1 variant and exclusion of other mutations in the region of interest.
After identification of the Chr13 (GRCh38): g.38349765_38349767del UFM1 variant by WES, hereafter called c.-273_-271delTCA (NM_001286704.1), we performed segregation analysis in all patients, parents, and healthy siblings by Sanger sequencing. In 3 patients, we investigated all exons and intron-exon boundaries of the protein-encoding genes present in the overlapping homozygous region that was identified by SNP array analysis by Sanger sequencing. Primers were designed using Primer 3, V.0.4.0 (tables e-1 and e-2 at Neurology.org).6
Microsatellite marker haplotype analysis.
To analyze a possible founder effect in all patients and see if the shared haplotype identified by SNP array analysis could be further narrowed down, we genotyped microsatellite and SNP markers spanning 2.4 Mb around UFM1 (table e-3) in 16 patients, 24 parents, and 6 siblings. For the microsatellite marker analysis, we analyzed PCR products with an Applied Biosystems (Mulgrave, Australia) Genetic Analyzer 3730 with GS-500 Liz as a size standard. For the SNP marker analysis, we performed Sanger sequencing. We used control DNA from CEPH individual 1347-02 as a reference and analyzed the data with GeneMapper v3.7 software.
Carrier frequency rate analysis.
We tested a panel of 670 Roma controls from a range of subisolates collected for population genetic and genetic epidemiology studies7 for the c.-273_-271delTCA UFM1 mutation using custom-designed TaqMan SNP Genotyping Assays (Applied Biosystems; primers in table e-4). Because one individual originating from a community in Eastern Slovakia was homozygous for the variant, we subsequently screened an additional panel of 273 samples from Roma adults from Eastern Slovakia8 and a panel of 57 samples from inhabitants of the community in question.
Logarithm of the odds (LOD) score.
Upon identification of the candidate variant, we calculated a LOD score on the basis of the following assumptions: autosomal recessive pattern of inheritance, complete penetrance, and equal distribution between male and female participants. We applied 2 calculations, one based on all 12 families in which DNA of both parents was available, and one excluding the families that were used for the original candidate region and candidate variant selection to avoid possible ascertainment bias. Unaffected siblings were also included.
Construction of UFM1 promoter reporters and transfections.
We cloned the wild-type and mutant UFM1 promoter (c.-1889 to c.-1 of NM_001286704.1) into the pNL1.1 reporter (Promega, Madison, WI) using the infusion protocol (Clontech, Mountain View, CA; oligonucleotide primers in table e-5). The pNL1.1 plasmid encodes nanoluciferase, a sensitive reporter protein for chemiluminescence-based assays (Promega). We transfected the UFM1 promoter reporters into HeLa (cervix carcinoma), SY-5Y (neuroblastoma), H02-F2 (oligodendrocytoma), and U373 (astroglioma) cell lines. The pNL1.1 empty vector was included as negative control. To normalize for transfection differences, we cotransfected the pGL3 plasmid (Promega) that expresses firefly luciferase driven by the SV40 promoter (Promega). All cells were grown in DMEM/F-12 with 10% fetal bovine serum at 37°C under 5% CO2. We performed the transfections in white, half area 96-well plates with clear bottom. One day before transfection, we seeded 3,000 cells per well. We transfected 5 ng pNL1.1-based vector and 75 ng pGL3 vector using Fugene 6 (1:3 ratio of Fugene 6:DNA; Promega). We measured firefly luciferase and nanoluciferase activity with a microplate reader (Victor2; Perkin-Elmer Life Sciences, Waltham, MA) 40 hours post-transfection according to the manufacturer's protocol. Nanoluciferase activity was normalized to firefly luciferase activity. Results were expressed as mean of 2 independent experiments performed in duplicate. Factor correction was applied to eliminate between-session variation.9 Differences were analyzed by Student t test.
RESULTS
Sixteen patients from 14 families were included in the study; 3 patients had similarly affected deceased siblings, for whom we did not have DNA. Parental consanguinity was reported in 7 families and 2 families were related. All but 2 families were known to originate from the Roma population, which led us to suspect a common founder effect. In our search strategies, we therefore chose to focus on a rare variant with homozygous autosomal recessive inheritance.
The clinical phenotype consists of a severe encephalopathy with early death.
Detailed clinical characteristics of all patients are described in table e-6. In table 1, we compared the present cohort with our published cohort of 41 patients with H-ABC with TUBB4A mutations, separately considering patients with the common TUBB4A mutation and patients with a different mutation, who exhibited a more severe phenotype.3 All current patients demonstrated severe developmental delay, typically without intentional movements and language development. Almost all patients exhibited spasticity and extrapyramidal movement abnormalities, mostly dystonia. Seizures were frequent and present in all patients 18 months and older. The epilepsy was often severe and drug-resistant, including West syndrome. Stunted growth was frequent and all patients had microcephaly. Six patients underwent tracheostomy between 6 and 17 months, 4 of whom were on intermittent or permanent ventilation. Median survival was 2 years. Nine patients died at ages between 7 months and 7 years, most often due to respiratory insufficiency. Compared to the cohort of TUBB4A-mutated patients, the present patients were all at the most severe end of the H-ABC spectrum.
Table 1.
Clinical data on TUBB4A- and UFM1-mutated patients with hypomyelination with atrophy of the basal ganglia and cerebellum
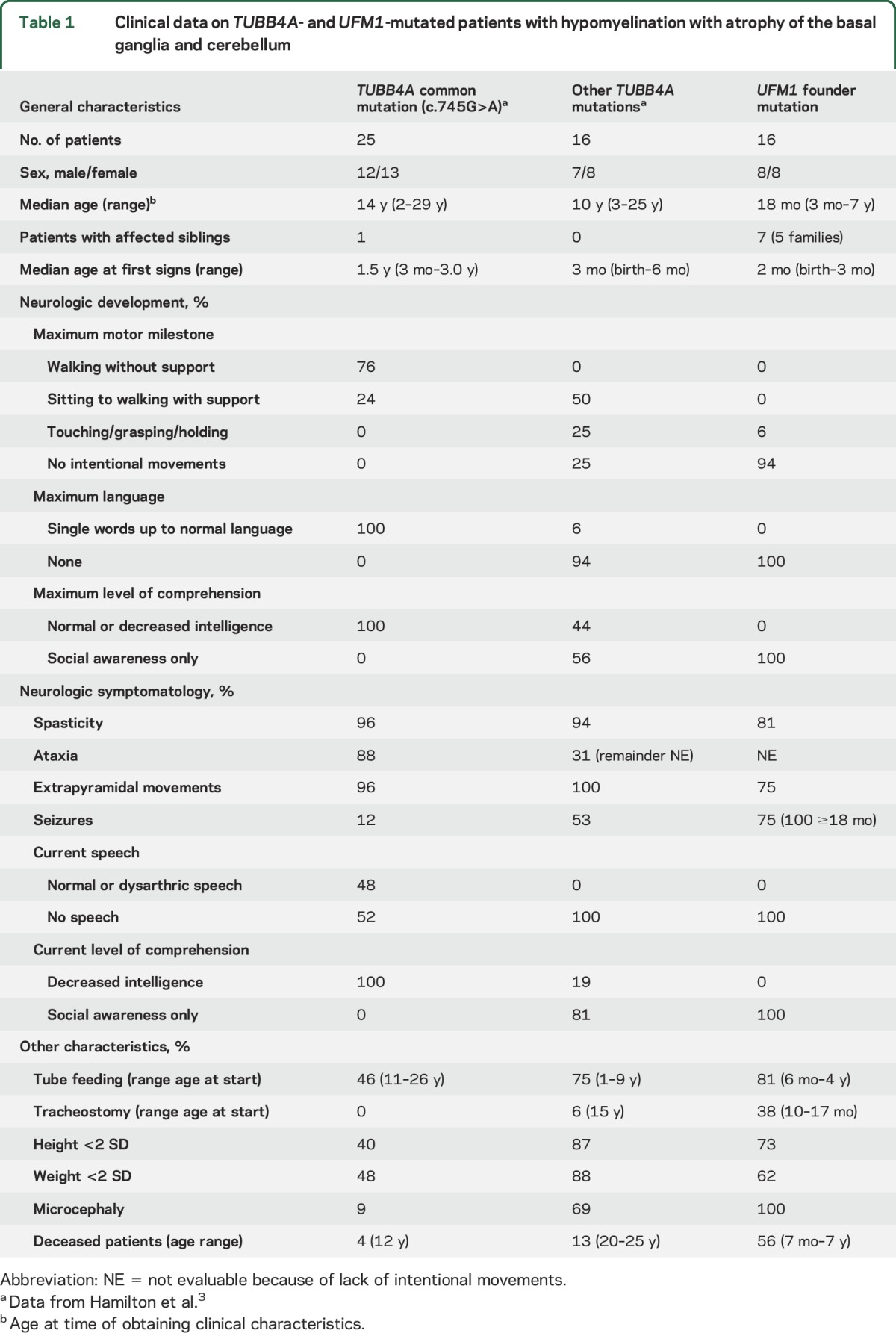
MRI shows severe hypomyelination, putamen atrophy, and distinctive caudate nucleus abnormalities.
Detailed MRI findings are outlined in table e-7, and a summary is presented in table 2, comparing the MRI characteristics of this cohort of patients to the early MRI characteristics in TUBB4A-mutated patients with H-ABC.3
Table 2.
Early MRI findings in TUBB4A- and UFM1-mutated patients
The first MRIs invariably revealed severe lack of myelin (figure 1) suggesting hypomyelination, although a single MRI does not allow an MRI-based diagnosis of hypomyelination in infants. None of the patients showed a normal putamen and in all patients the caudate nucleus was small. In all, the lateral part of the head of the caudate nucleus showed an area of abnormally high signal on T2-weighted images, resulting in a signal intensity similar to the hypomyelinated white matter. By contrast, in patients with TUBB4A mutations, the caudate nucleus signal was normal (74%) or hyperintense throughout (26%).3 Atrophy of the caudate nucleus led to widening of the anterior horns of the lateral ventricles. In addition, in 50% of patients there was a moderate dilation of lateral and third ventricles. On the first MRI, 56% of patients had mild cerebellar atrophy, restricted to the vermis.
Figure 1. MRI findings in TUBB4A (left) and UFM1 (right) mutated patient with hypomyelination with atrophy of the basal ganglia and cerebellum.

Sagittal T1-weighted (A–D) and axial T2-weighted (E, F) and T1-weighted (G, H) images in a patient with the common dominant c.745 G>A TUBB4A mutation at age 3 years (A, C, E, G) and a patient with the homozygous recessive UFM1 mutation at age 13 months (B, D, F, H). The patient with the common TUBB4A mutation shows a mildly hyperintense white matter signal on T1-weighted (C, G) and T2-weighted (E) images, indicating a moderate lack of myelin. In the UFM1-mutated patient, the white matter T1 signal is hypointense, indicating a profound lack of myelin (D, H). This patient also shows mild cerebral atrophy (F, H). Both patients have a mild cerebellar atrophy (A–D), most notable at the vermis (A, B). In both patients, there is no putamen visible and there is no visible lesion in this region (arrowheads in E–H). In the TUBB4A-mutated patient, the caudate nucleus has a normal signal (arrow in E). In the UFM1-mutated patient, the caudate nucleus is atrophic and the lateral part of the head has an abnormal hyperintense T2 signal (arrow in F) and hypointense T1 signal (arrow in H).
The follow-up MRIs showed slight progress of myelination after 3–13 months in 3 out of 5 patients with a follow-up scan, but myelination remained severely deficient, confirming hypomyelination. In 2 patients, progression of the cerebral atrophy was observed. In 4 patients who initially lacked cerebellar atrophy, follow-up MRIs revealed cerebellar atrophy after 3–15 months.
Genetic analysis identifies UFM1 as the only candidate gene.
Sanger sequencing was negative for TUBB4A mutations in 16 patients, and MLPA analysis revealed neither TUBB4A deletions nor duplications in the 9 patients who were investigated.
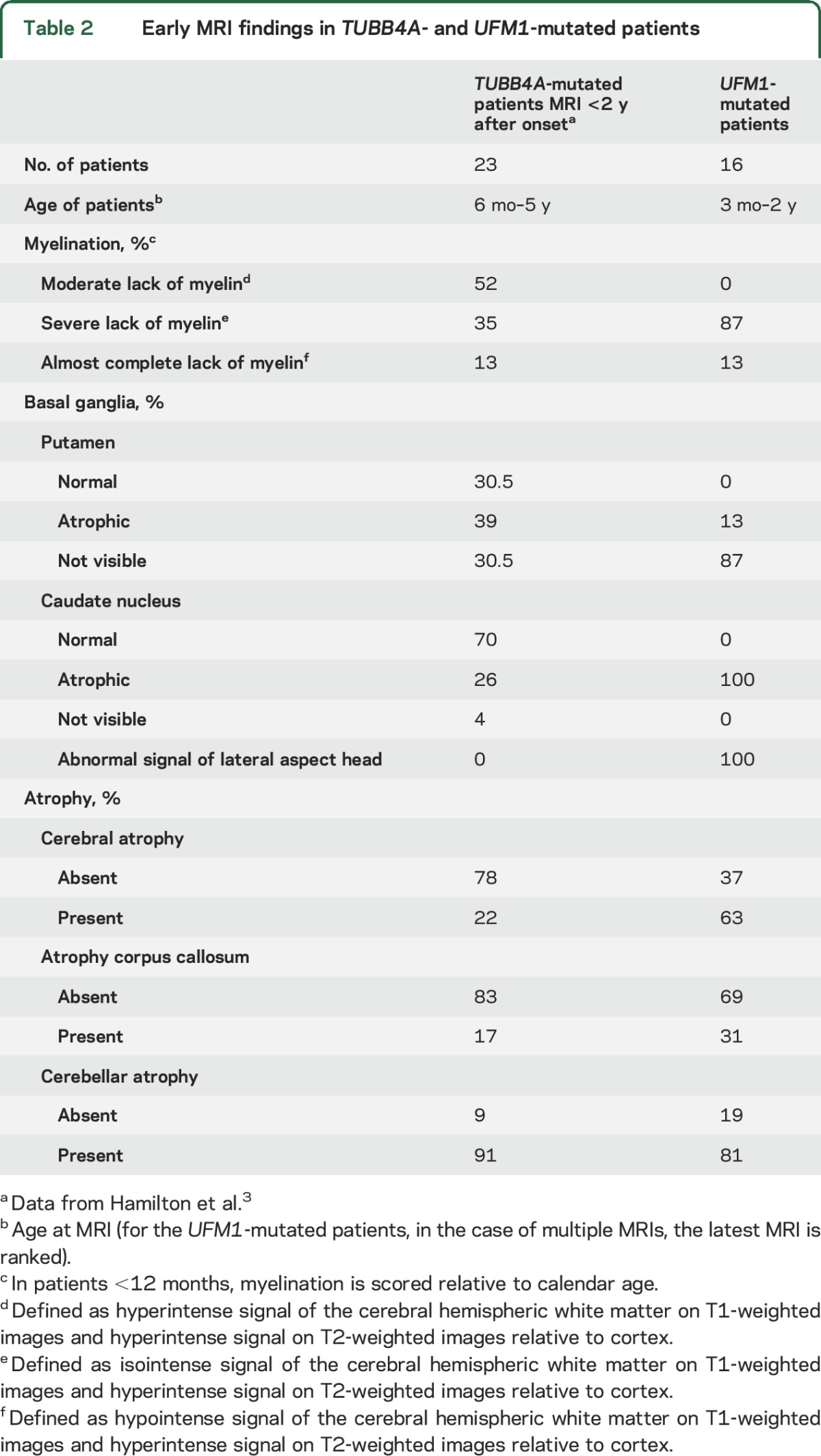
An SNP array identified a 0.8-Mb overlapping homozygous region on chromosome 13q13 (genomic coordinates 37,940,556–38,786,096 GRCh38) shared by all 5 patients in whom the SNP array was performed (figure 2A). This region encompasses long intergenic noncoding RNAs, the UFM1 gene and the first 6 exons of the FREM2 gene (NM_207361.5). UFM1 encodes ubiquitin fold modifier 1. FREM2 encodes FRAS1-related extracellular matrix protein 2 and is associated with Fraser syndrome, a developmental and malformative disorder involving multiple organs.
Figure 2. Founder mutation haplotype analysis.

Schematic representation of the candidate region on chromosome 13q13 containing UFM1 and part of FREM2. The area of overlapping haplotype is highlighted in light blue: section A depicts the results of the single nucleotide polymorphism (SNP) array in 5 patients: 238 overlapping homozygous SNPs starting with rs17068530 and ending with rs248423 (shown in black), flanked by rs9532135 and rs9566363 (shown in red). Section B depicts the microsatellite and SNP markers analyzed in the candidate region, which confirmed the presence of a common haplotype in all patients (markers depicted in black), flanked by microsatellite markers D13S219 and D13S1288 (shown in red).
WES analysis of 2 patients revealed a single homozygous 3 bp deletion in the promoter of UFM1 (MIM 610553) as candidate causal mutation: c.-273_-271delTCA (NM_001286704.1, dbSNP rs747359907). No other rare variants were detected in the candidate region. The variant has been reported to dbSNP in heterozygous state by 2 independent submitters with unknown allele frequency. UFM1 mutations have not been associated with a disease phenotype. The deleted nucleotides are moderately to highly conserved based on PhyloP conservation scores ranging from 0.32 to 2.09 and Phast conservation scores from 0.89 to 0.98.
Sanger sequencing confirmed that segregation of the UFM1 deletion was in perfect agreement with inheritance of the disease in all families.
Sanger sequencing of all exons and intron–exon boundaries in the shared haplotype in 3 patients revealed no other possible pathogenic variants.
LOD score calculation confirms linkage and haplotype analysis reveals a founder effect.
LOD score calculations for the UFM1 variant showed a maximum LOD score of 9.18 when all 12 families were included in the calculation, and a maximum LOD score of 5.32 when the families used for identification of the UFM1 variant were excluded.
Haplotype analysis showed that all patients were homozygous for an identical haplotype containing the UFM1 mutation, indicative of a founder effect (table e-3). The smallest putative shared region was flanked by microsatellite markers D13S219 and D13S1288 (figure 2B).
Population screening reveals high carrier rates in Roma subisolates.
An important step for validation was to prove absence of homozygosity for the UFM1 deletion among healthy Roma individuals. Screening of 670 Roma controls revealed 30 carriers with an overall carrier rate of 4.5% among different Roma communities across Europe and one individual who was homozygous for the deletion. Retrospective review of this case revealed that this sample was derived from a boy with severe encephalopathy with spasticity, who had died at age 2.5 years and had erroneously been included among the controls. He had a similarly affected sibling, who had also died. No imaging had been performed, but the severe phenotype was similar to that of the patients in this study. The child lived in an endogamous community in Eastern Slovakia with a high rate of consanguinity. An additional panel of Eastern Slovak Roma samples revealed a carrier frequency of 3.3% (9 out of 273). Subsequent investigation of the carrier rate in the specific community where the homozygous individual came from revealed a carrier rate of approximately 25% (14 out of 57 individuals).
The promoter mutation reduces reporter gene expression in specific CNS cell lines.
To study the effect of the deletion on promoter activity in different cell types, the candidate promoter sequence with or without the deletion was cloned into luciferase vectors and transfected into different cell lines to test its activity. The deletion significantly reduced promoter activity in SY-5Y and U373 but not in HeLa and HOG-F2 cell lines (figure 3).
Figure 3. c.-271_-271delTCA reduces UFM1 promoter activity in SY-5Y neuroblastoma and U373 astroglioma cell lines.

(A–D) c.-273_-271delTCA in human UFM1 (hUFM1) promoter significantly reduces reporter gene expression in SY5Y (p = 0.001) and U373 (p = 0.014) but not in HeLA (cervix carcinoma) or HOG-F2 (oligodendrocytoma) cell lines. Promoter activity was measured with dual luciferase reporter assays. The nanoluciferase/firefly luciferase ratio measured in cells transfected with wildtype hUFM1 was arbitrarily set at 1 on the y-axis. delTCA indicates cells transfected with human UFM1 promoter harboring c.-273_-271delTCA. Data represent mean ± SD values obtained from 2 independent experiments testing the luciferase activity in duplo and were analyzed by 2-tailed t test.
DISCUSSION
This study, focused on identifying the mutated gene in patients with H-ABC without TUBB4A mutations, revealed a homozygous 3-bp deletion in the UFM1 promoter area, which perfectly segregates with the disease. Most patients were known to have a Roma background, and haplotype analysis indicated that the shared UFM1 deletion originates from a common ancestor. Within the candidate region, the UFM1 promoter deletion was the only candidate. The Taqman screening assay confirmed a high carrier frequency and absence of homozygosity for the mutation among healthy Roma controls. Worldwide, our centers in Amsterdam and Washington know by far the highest number of patients with H-ABC and up until now we have not identified other UFM1 mutations in unrelated families. The genetically isolated Roma population harbors several unique autosomal recessive disorders caused by “private” founder mutations in genes, in which no other mutations have been found until now.10,11 In a luciferase assay, we showed that the UFM1 promoter deletion results in a significantly reduced transcription activity only in selected neural but not in other cell lines. This finding supports the pathogenicity of the mutation and suggests a cell-specific effect.
UFM1 encodes ubiquitin-fold modifier 1 (UFM1), a ubiquitin-like (UBL) protein, which is ubiquitously expressed, including in brain.12 UFM1 is hypothesized to post-translationally modify (“ufmylate”) proteins in a manner analogous to ubiquitination.12,13 Ubiquitin and UBL pathways are involved in control of numerous functions, including signal transduction, transcriptional regulation, and stress response.14 Several studies indicate an association between the UFM1 pathway and both neurodevelopment and neurodegeneration.15,16 Its exact biological role and working mechanism are poorly understood. Most evidence points to a role of UFM1 in endoplasmic reticulum homeostasis and protection against apoptosis.17–19
Genetic defects in the ubiquitin-proteasome system have been associated with several neurologic disorders, particularly cerebellar ataxias.20,21 Recently, various recessive mutations in UBA5, encoding UFM1 activating enzyme 5 (UBA5), were associated with a progressive, childhood-onset cerebellar ataxia.19,22,23 The most severe variants presented with infantile onset encephalopathy with hypotonia, spasticity, movement abnormalities, refractory epilepsy, microcephaly, failure to thrive, and cerebellar atrophy,19,22 similar to the encephalopathy in our patients. Strikingly, biochemical and experimental findings indicated that the described UBA5 mutations result in UFM1 system impairment.19,22,23 Drosophila models with knockdown of UBA5 and UFM1 homologues exhibited a neurologic phenotype with reduced motor activity and shortened lifespan, with UFM1 knockdown resulting in the most severe phenotype.23 CNS-specific knockout of Ufm1 in mice caused neonatal death with microcephaly and apoptosis of neurons in specific brain regions.22
Altogether, existing data suggest that the UFM1 system is crucial for neuronal development, function, and protection against apoptosis.14–19,22,23 This concept is substantiated by the current study, showing association of UFM1 mutations with an infantile-onset, severe epileptic encephalopathy and failure of development. Intriguingly, while the Ufm1 knockout mouse showed apoptosis in restricted brain areas, MRI also suggests apoptosis of selected neuronal cell populations in both TUBB4A- and UFM1-related H-ABC, substantiated by histopathology in the first.1,24 Muona et al.22 suggest that ufmylation may be spatiotemporally regulated and cell type–specific. Specificity is reinforced by the results of our transfection studies showing that only selective neural cell lines are vulnerable for the promoter mutation.
The relationship between H-ABC caused by monoallelic TUBB4A mutations and H-ABC caused by biallelic UFM1 mutations is unclear. TUBB4A mutations are associated with a disease spectrum ranging from early infantile, severe encephalopathy to adult-onset dystonia type 4, with a strong genotype–phenotype correlation.3 All patients with the c.-273_-271delTCA UFM1 promoter mutation have an early infantile, severe encephalopathy with early death. Although the UFM1-mutated patients fulfill the MRI criteria of H-ABC, they consistently have an additional feature: signal abnormality of the lateral part of the head of the caudate nucleus suggestive of local apoptosis. This feature has not been observed in TUBB4A-related H-ABC and may be pathognomonic for UFM1-related H-ABC. Whether UFM1-related disease and TUBB4A-related disease are basically variants of one disease or unrelated phenocopies is still to be clarified. The striking similarity between the 2 suggests that the encoded proteins may be at least partially involved in the same processes. A possible link could be that ufmylation is involved in the regulation of microtubule dynamics, which has been shown for other UBLs.25 Recently, recessive ZNF335 mutations were described in a single family, in which patients displayed features compatible with early-onset severe H-ABC.26 Further confirmation and characterization are necessary, but if the finding is confirmed in other families, studies focused on possible crossroads of tubulin β-4A, ubiquitin-fold modifier 1, and zinc finger protein 335 are warranted. Considering that a few patients with H-ABC are presently still genetically unclassified despite targeted Sanger sequencing and whole exome and genome sequencing, some more genes and proteins may be added.
The current study sheds new light on possible UFM1 functional networks and directly benefits patients and families. The identification of the disease-causing variant enables better clinical and genetic counseling, the possibility of prenatal testing, and the option of carrier testing in populations with a high carrier frequency.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the patients and families for their cooperation and contributions; Carola G. van Berkel, Emiel Polder, and Nienke L. Postma, technicians, Department of Child Neurology, Marjan M. Weiss, molecular geneticist, Department of Clinical Genetics, and Robbert Zalm, technician, Department of Functional Genomics, VU University Medical Center, Amsterdam, the Netherlands, for laboratory assistance; Matthijs Verhage, head of the Department of Functional Genomics, VU University Medical Center, for the use of his laboratory; Zoltán Bochdanovits, Department of Clinical Genetics, VU University Medical Center, for statistics advice; Marzia Pollazzon, Clinical Genetics Unit, Arcispedale S. Maria Nuova–Reggio Emilia, Italy, and Francisco Menor, Radiología Infantil Hospital Universitari i Politecnic La Fe, Valencia, Spain, for contribution to the acquisition of clinical data; and Chiara Aiello and Lorena Travaglini, Unit of Neuromuscular and Neurodegenerative Disorders, Laboratory of Molecular Medicine, “Bambino Gesù” Children's Hospital, Rome, Italy, for their contribution to the genetic testing.
GLOSSARY
- H-ABC
hypomyelination with atrophy of the basal ganglia and cerebellum
- LOD
logarithm of the odds
- MLPA
multiplex ligation-dependent probe amplification
- SNP
single nucleotide polymorphism
- UBA5
UFM1 activating enzyme 5
- UBL
ubiquitin-like
- UFM1
ubiquitin-fold modifier 1
- WES
whole exome sequencing
Footnotes
Supplemental data at Neurology.org
Contributor Information
Collaborators: Recessive H-ABC Research Group, Adele D'Amico, Ana Boban Raguž, Paola la Boria, Graziella Cefalo, Argirios Dinopoulos, Sira Domènech, Maria A. Donati, Daniele Frattini, Serena Gasperini, Lucio Giordano, Elena Procopio, Anita Rauch, Agusti Rodriguez-Palmero, Komudi Siriwardena, and Miguel Tomás-Vila
AUTHOR CONTRIBUTIONS
Eline M.C. Hamilton collected and analyzed patient and imaging data, analyzed the genetic data, and wrote the manuscript. Enrico Bertini collected and analyzed patient and imaging data and identified the disease in Italy. Luba Kalaydjieva performed carrier frequency analysis. Bharti Morar performed carrier frequency analysis. Dana Dojčáková performed carrier frequency analysis. Judy Liu performed laboratory experiments to substantiate the pathogenicity of the mutation. Adeline Vanderver performed laboratory experiments to substantiate the pathogenicity of the mutation. Julian Curiel performed laboratory experiments to substantiate the pathogenicity of the mutation. Claudia M. Persoon performed laboratory experiments to substantiate the pathogenicity of the mutation. Daria Diodato collected and analyzed patient and imaging data and contributed to the genetic studies. Lorenzo Pinelli collected and analyzed patient and imaging data and contributed to the genetic studies. Nathalie L. van der Meij performed genetic analyses. Barbara Plecko collected and analyzed patient and imaging data and contributed to the genetic studies. Susan Blaser collected and analyzed patient and imaging data and contributed to the genetic studies. Nicole I. Wolf contributed to disease concept formation and collected and analyzed patient and imaging data. Quinten Waisfisz performed genetic analyses and analyzed the genetic data. Truus E.M. Abbink performed genetic analyses, performed laboratory experiments to substantiate the pathogenicity of the mutation, and wrote the manuscript. Marjo S. van der Knaap conceptualized and coordinated the project, collected and analyzed patient and imaging data, and wrote the manuscript.
STUDY FUNDING
This study received financial support from the ZonMw TOP grant 91211005 (M.S.v.d.K.), the Optimix Foundation for Scientific Research (E.M.C.H., M.S.v.d.K.), and the Italian Ministry of Health (E.B.).
DISCLOSURE
E. Hamilton, E. Bertini, L. Kalaydjieva, B. Morar, D. Dojčáková, J. Liu, A. Vanderver, J. Curiel, C. Persoon, D. Diodato, and L. Pinelli report no disclosures relevant to the manuscript. N. van der Meij is an employee of MRC-Holland, provider of commercially available MLPA assays. B. Plecko, S. Blaser, N. Wolf, Q. Waisfisz, T. Abbink, and M. van der Knaap report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.van der Knaap MS, Naidu S, Pouwels PJ, et al. New syndrome characterized by hypomyelination with atrophy of the basal ganglia and cerebellum. AJNR Am J Neuroradiol 2002;23:1466–1474. [PMC free article] [PubMed] [Google Scholar]
- 2.Simons C, Wolf NI, McNeil N, et al. A de novo mutation in the beta-tubulin gene TUBB4A results in the leukoencephalopathy hypomyelination with atrophy of the basal ganglia and cerebellum. Am J Hum Genet 2013;92:767–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hamilton EM, Polder E, Vanderver A, et al. Hypomyelination with atrophy of the basal ganglia and cerebellum: further delineation of the phenotype and genotype-phenotype correlation. Brain 2014;137:1921–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schiffmann R, van der Knaap MS. Invited article: an MRI-based approach to the diagnosis of white matter disorders. Neurology 2009;72:750–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wolf NI, Salomons GS, Rodenburg RJ, et al. Mutations in RARS cause hypomyelination. Ann Neurol 2014;76:134–139. [DOI] [PubMed] [Google Scholar]
- 6.Koressaar T, Remm M. Enhancements and modifications of primer design program Primer3. Bioinformatics 2007;23:1289–1291. [DOI] [PubMed] [Google Scholar]
- 7.Kalaydjieva L, Morar B, Chaix R, Tang H. A newly discovered founder population: the Roma/Gypsies. Bioessays 2005;27:1084–1094. [DOI] [PubMed] [Google Scholar]
- 8.Bozikova A, Gabrikova D, Sovicova A, et al. The frequency of factor V Leiden and prothrombin G20210A mutations in Slovak and Roma (Gypsy) ethnic group of Eastern Slovakia. J Thromb Thrombolysis 2012;34:406–409. [DOI] [PubMed] [Google Scholar]
- 9.Ruijter JM, Thygesen HH, Schoneveld OJ, Das AT, Berkhout B, Lamers WH. Factor correction as a tool to eliminate between-session variation in replicate experiments: application to molecular biology and retrovirology. Retrovirology 2006;3:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Varon R, Gooding R, Steglich C, et al. Partial deficiency of the C-terminal-domain phosphatase of RNA polymerase II is associated with congenital cataracts facial dysmorphism neuropathy syndrome. Nat Genet 2003;35:185–189. [DOI] [PubMed] [Google Scholar]
- 11.Hantke J, Chandler D, King R, et al. A mutation in an alternative untranslated exon of hexokinase 1 associated with hereditary motor and sensory neuropathy: Russe (HMSNR). Eur J Hum Genet 2009;17:1606–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Komatsu M, Chiba T, Tatsumi K, et al. A novel protein-conjugating system for Ufm1, a ubiquitin-fold modifier. EMBO J 2004;23:1977–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tatsumi K, Sou YS, Tada N, et al. A novel type of E3 ligase for the Ufm1 conjugation system. J Biol Chem 2010;285:5417–5427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem 1998;67:425–479. [DOI] [PubMed] [Google Scholar]
- 15.Homrich M, Wobst H, Laurini C, Sabrowski J, Schmitz B, Diestel S. Cytoplasmic domain of NCAM140 interacts with ubiquitin-fold modifier-conjugating enzyme-1 (Ufc1). Exp Cell Res 2014;324:192–199. [DOI] [PubMed] [Google Scholar]
- 16.Martin DD, Ladha S, Ehrnhoefer DE, Hayden MR. Autophagy in Huntington disease and huntingtin in autophagy. Trends Neurosci 2015;38:26–35. [DOI] [PubMed] [Google Scholar]
- 17.Lemaire K, Moura RF, Granvik M, et al. Ubiquitin fold modifier 1 (UFM1) and its target UFBP1 protect pancreatic beta cells from ER stress-induced apoptosis. PLoS One 2011;6:e18517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Y, Zhang M, Wu J, Lei G, Li H. Transcriptional regulation of the Ufm1 conjugation system in response to disturbance of the endoplasmic reticulum homeostasis and inhibition of vesicle trafficking. PLoS One 2012;7:e48587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Colin E, Daniel J, Ziegler A, et al. Biallelic variants in UBA5 reveal that disruption of the UFM1 cascade can result in early-onset encephalopathy. Am J Hum Genet 2016;99:695–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ciechanover A, Brundin P. The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron 2003;40:427–446. [DOI] [PubMed] [Google Scholar]
- 21.Ronnebaum SM, Patterson C, Schisler JC. Emerging evidence of coding mutations in the ubiquitin-proteasome system associated with cerebellar ataxias. Hum Genome Var 2014;1:14018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muona M, Ishimura R, Laari A, et al. Biallelic variants in UBA5 link dysfunctional UFM1 ubiquitin-like modifier pathway to severe infantile-onset encephalopathy. Am J Hum Genet 2016;99:683–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duan R, Shi Y, Yu L, et al. UBA5 mutations cause a new form of autosomal recessive cerebellar ataxia. PLoS One 2016;11:e0149039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van der Knaap MS, Linnankivi T, Paetau A, et al. Hypomyelination with atrophy of the basal ganglia and cerebellum: follow-up and pathology. Neurology 2007;69:166–171. [DOI] [PubMed] [Google Scholar]
- 25.Lytle BL, Peterson FC, Qiu SH, et al. Solution structure of a ubiquitin-like domain from tubulin-binding cofactor B. J Biol Chem 2004;279:46787–46793. [DOI] [PubMed] [Google Scholar]
- 26.Sato R, Takanashi J, Tsuyusaki Y, et al. Association between invisible basal ganglia and ZNF335 mutations: a case report. Pediatrics 2016;138:e20160897. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.