

Abstract
Objective:
To determine the association between synaptic functioning as measured via neurogranin in CSF and cognition relative to established Alzheimer disease (AD) biomarkers in neurologically healthy older adults.
Methods:
We analyzed CSF concentrations of neurogranin, β-amyloid (Aβ42), phosphorylated tau (p-tau), and total tau (t-tau) among 132 neurologically normal older adults (mean 64.5, range 55–85), along with bilateral hippocampal volumes and a measure of episodic memory (Auditory Verbal Learning Test, delayed recall). Univariable analyses examined the relationship between neurogranin and the other AD-related biomarkers. Multivariable regression models examined the relationship between neurogranin and delayed recall, adjusting for age and sex, and interaction terms (neurogranin × AD biomarkers).
Results:
Higher neurogranin concentrations were associated with older age (ρ = 0.20, p = 0.02), lower levels of p-tau and t-tau, and smaller hippocampal volumes (p < 0.03), but not with CSF Aβ42 (p = 0.18). In addition, CSF neurogranin demonstrated a significant relationship with memory performance independent of the AD-related biomarkers; individuals with the lowest CSF neurogranin concentrations performed better on delayed recall than those with medium or high CSF neurogranin concentrations (p < 0.01). Notably, CSF p-tau, t-tau, and Aβ42 and hippocampal volumes were not significantly associated with delayed recall scores (p > 0.40), and did not interact with neurogranin to predict memory (p > 0.10).
Conclusions:
Synaptic dysfunction (assessed via neurogranin) may be an early pathologic process in age-related neurodegeneration, and a sensitive marker of age-related cognitive abilities, potentially preceding or even acting independently from AD pathogenesis. Synaptic functioning may be a useful early marker of cognitive aging and possibly a target for future brain aging interventions.
Synapses are critical for long-term potentiation (LTP) and cognition, decline early in aging1 and Alzheimer disease (AD), and are more strongly related to cognitive deficits than amyloid or tau pathology.2,3
Neurogranin is a neural-specific postsynaptic protein involved in LTP signaling.4–6 Concentrated on dendritic spines in the hippocampus and basal forebrain, neurogranin binds to calmodulin to regulate calcium influxes.7 In animals, neurogranin knockdown models inhibit LTP and cognition,8 while upregulation promotes LTP and improves cognitive performance.9
Neurogranin concentrations decrease in hippocampi with increasing age and are related to CNS dysfunction in AD, though their relationship with AD biomarkers in asymptomatic or normal aging remains unknown.7,10 In AD, neurogranin is depleted in the brain, elevated in CSF, and associated with tau and (to a lesser degree) amyloid pathology and poorer cognitive performance.11–15 Higher baseline CSF neurogranin is associated with faster declines in cognition, cortical glucose metabolism, and hippocampal volumes in individuals with mild cognitive impairment and AD.16 While there are differences in neurogranin concentrations between healthy individuals and patients with AD, no clinical studies have examined neurogranin's relationship to cognition and markers of AD development in aging.
We aimed to address this major gap by characterizing the association between neurogranin and memory abilities in relation to established AD biomarkers among clinically normal older adults. We hypothesized that higher CSF neurogranin would be associated with older age and poorer memory performance. In addition, given that AD neuropathology develops early in aging, we hypothesized that AD-related biomarkers would interact with neurogranin to confer greater risk of reduced memory performance.
METHODS
Participants.
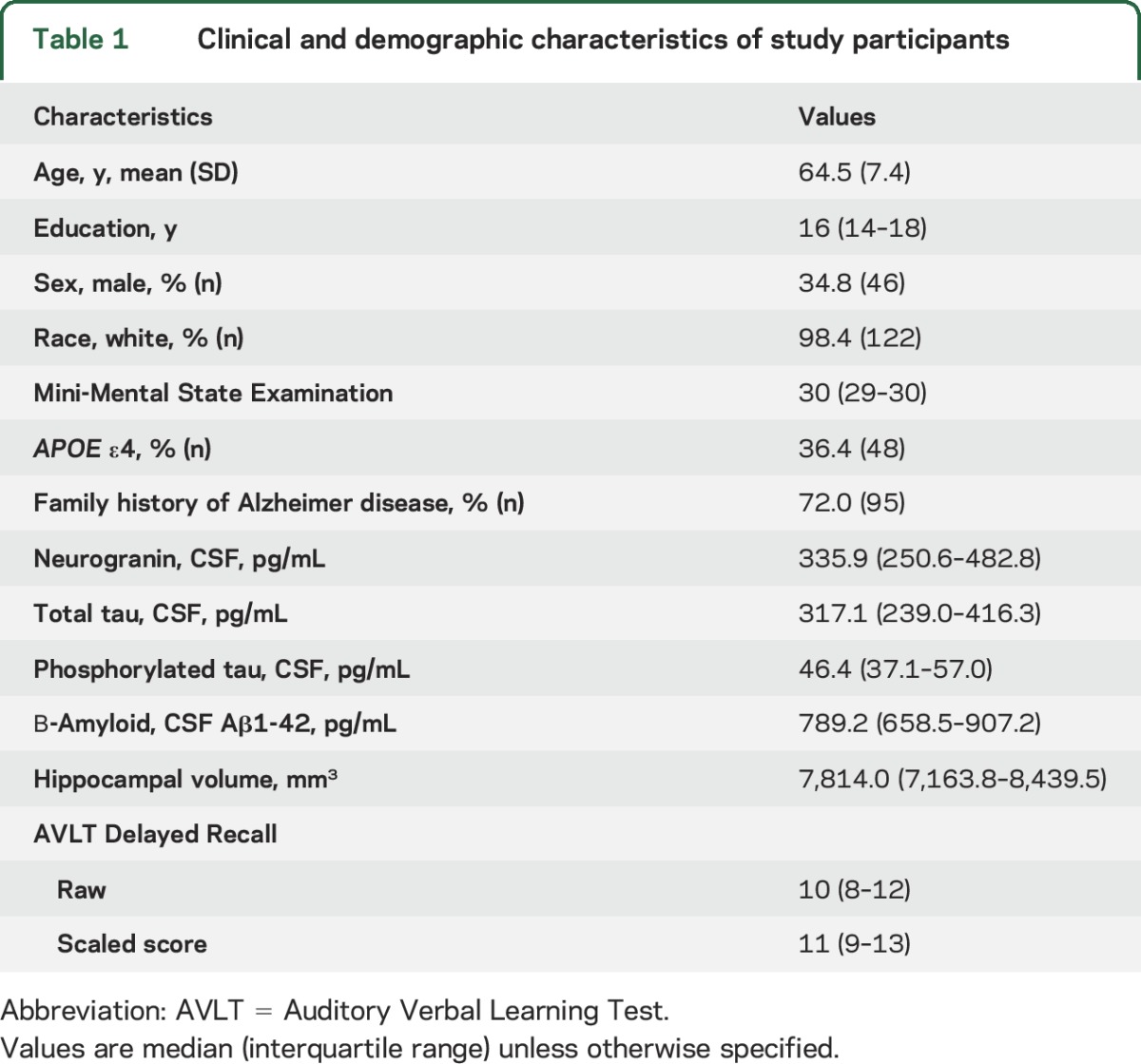
We studied 132 community-dwelling older adults cross-sectionally from the Wisconsin Registry for Alzheimer's Prevention study and the Wisconsin Alzheimer's Disease Research Center who completed neuroimaging, lumbar puncture, blood draw, and memory testing. Inclusion criteria included (1) neuropsychological functioning within normative standards determined via a comprehensive battery and consensus review,17 (2) no history of neurologic or major psychiatric disease, (3) no history of major medical illness, and (4) no MRI contraindications. All participants with available data were included in analyses; all participants had complete data on the major variable of interest (CSF neurogranin). We selected the study group to represent healthy middle-aged to older adults with and without parents with late-onset AD. Therefore, the current sample was enriched for family history of AD and the genetic AD risk factor APOE ε4 (table 1). We defined a parental history of AD by a validated interview18 or by confirmed AD at autopsy as reviewed by a multidisciplinary diagnostic consensus. Participants were determined to have a negative parental history of AD if the participant's father survived to >70 years and mother to >75 years without a diagnosis of dementia or cognitive decline17 per comprehensive medical interviews.
Table 1.
Clinical and demographic characteristics of study participants
Standard protocol approvals, registrations, and patient consents.
All study procedures were approved by the University of Wisconsin Health Sciences institutional review board in accordance with US federal regulations and the Helsinki declaration. All participants provided written informed consent.
CSF collection and analysis.
CSF samples were collected in the morning after a 12-hour fasting period. Approximately 22 mL of CSF were tapped and inverted to avoid possible gradients, gently mixed, and centrifuged at 2,000g for 10 minutes before being frozen in 0.5 mL aliquots and stored at −80°C.
CSF samples were analyzed for phosphorylated tau (p-tau), total tau (t-tau), and the 42 amino acid form of β-amyloid (Aβ42) via standard commercially available ELISA (INNOTEST assays; Fujiurebio, Ghent, Belgium).19 Details regarding CSF neurogranin analysis are given elsewhere5,13; in brief, we utilized the monoclonal anti-Ng7 antibody Ng7 and the primary rabbit anti-Ng antibody (Upstate) in a sandwich ELISA format. Board-certified laboratory technicians blinded to participant clinical history performed analyses per protocols approved by the Swedish Board of Accreditation and Conformity Assessment using one batch of reagents (intra-assay coefficient of variation <10%).
MRI.
Participants underwent imaging on a GE (Fairfield, CT) 3T MR750 system equipped with an 8-channel head coil and parallel imaging (array coil spatial sensitivity encoding [ASSET]). T1-weighted brain volumes were acquired in axial plane with a 3D fast spoiled gradient-echo sequence using the following measures: inversion time 450 ms; repetition time 8.1 ms; echo time 3.2 ms; flip angle 12o; acquisition matrix 256 × 256 mm; field of view 256 mm; slice thickness 1.0 mm.
FMRIB Software Library's FIRST Tool was used to calculate bilateral hippocampal volumes (fmrib.ox.ac.uk/fsl/). The shape/appearance models used in FIRST are constructed via manually segmented images provided by the Center for Morphometric Analysis, Massachusetts General Hospital, Boston.
Neuropsychological testing.
Memory.
Given that neurogranin shows increased concentrations in the hippocampi, our primary outcome of interest was delayed recall performances assessed via the Rey Auditory Verbal Learning Test (AVLT).20 The AVLT consists of a 15-item word list that participants are asked to immediately recall across 5 learning trials. Participants are then presented an interference trial of a novel 15-item word list before freely recalling the original list. After a 20-minute delay, participants are again asked to recall the original word list; this delayed recall score was our outcome of interest.
Other cognitive domains.
We additionally examined other cognitive domains to determine the specificity of the relationship between neurogranin and memory. The Wechsler Adult Intelligence Scale, 3rd edition (WAIS-III), Symbol Digit Coding, was given to measure processing speed.21 WAIS-III Digit Span forward and backward performances (longest span) were used as measures of echoic attention and working memory. Finally, the Boston Naming Test, a 60-item confrontation naming task, was administered to assess semantic retrieval.22
Statistical analyses.
We conducted Spearman rho (ρ) rank correlations or Wilcoxon/Kruskal-Wallis rank sum tests to characterize relationships between neurogranin, demographics, AD-related biomarkers, and cognition. Given that prior published works examining neurogranin have primarily utilized Spearman rho associations, we opted to report these to be comparable with previous results. Plotting the relationship between neurogranin and delayed recall evidenced nonlinear curvature. Therefore, we additionally conducted regression models testing for polynomic effects (e.g., quadratic [x + x2] and cubic [x + x2 + x3]). Models examining relationships with memory performances were additionally adjusted for sex (utilizing age-corrected scores) and hippocampal models for age, sex, and total gray matter volume. To increase interpretation of polynomic effects, independent variables in the models were centered to the mean; therefore, the linear measure represents the linear change in neurogranin at the means of all other predictors on the outcome, while the polynomic parameter represents curvature of the regression line. In all analyses, missing data were minimal and excluded pairwise when necessary.
We additionally examined the independent association between neurogranin and memory performance in relation to the AD-related biomarkers and AD risk factors (APOE ε4 status, AD family history), and tested whether the magnitude of the association between neurogranin with memory depended on the level of AD-related biomarkers (interaction terms). Finally, to increase interpretation and help describe the clinical significance of the observed polynomic effects between neurogranin and delayed recall, we converted neurogranin into tertiles.
RESULTS
Table 1 presents descriptive statistics of our study sample. Notably, 72% of participants had a first-degree relative with AD and about one-third carried the APOE ε4 risk allele, though participants were intact on a global cognitive screener (median Mini-Mental State Examination 30) and performed above average on a measure of delayed recall (AVLT scaled score = 11). CSF neurogranin concentrations were slightly skewed (Shapiro-Wilk W = 0.86, p < 0.01). Higher levels of neurogranin were associated with older age (ρ = 0.20, p = 0.02), though no polynomic relationships with age were observed (neurogranin2 p = 0.87). Neurogranin was not significantly associated with sex (z = −0.90, p = 0.37), education (ρ = −0.06, p = 0.51), or race (z = −0.87, p = 0.38).
Neurogranin and AD biomarkers.
Higher CSF neurogranin was associated with higher CSF p-tau (ρ = 0.68, p < 0.001) and t-tau (ρ = 0.75, p < 0.001). Specifically, neurogranin demonstrated quadratic relationships with both p-tau (neurogranin2 p < 0.01) and t-tau (neurogranin2 p = 0.02; table 2).
Table 2.
CSF neurogranin is associated with phosphorylated and total tau and hippocampal volumes but not amyloid in neurologically normal older adults
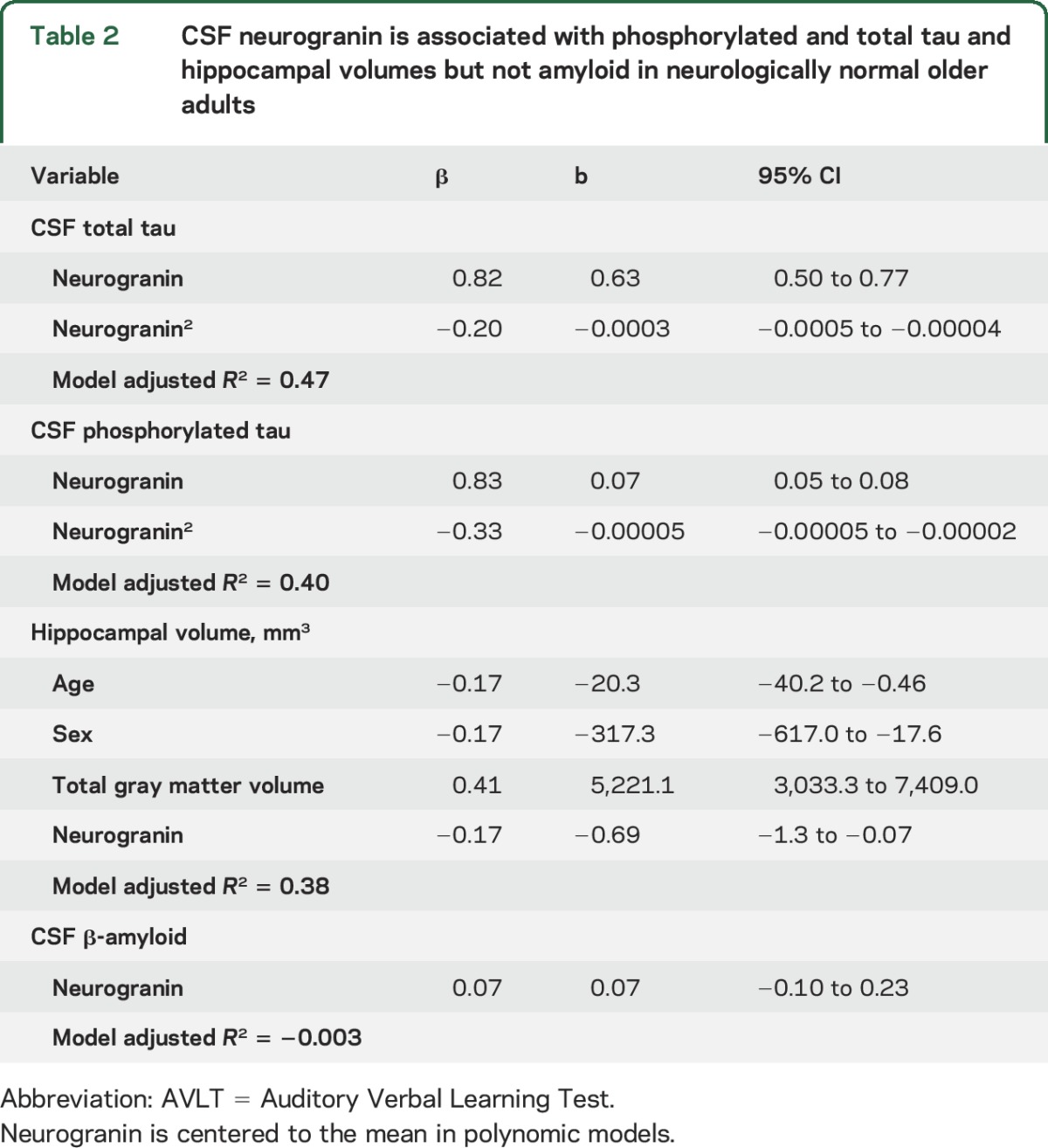
Higher concentrations of neurogranin were also associated with smaller hippocampal volumes, after adjusting for age, sex, and total gray matter volumes (p = 0.03), though no significant polynomic relationships were evidenced (neurogranin2 p = 0.72).
Neurogranin was not significantly associated with CSF Aβ42 concentrations (ρ = 0.12, p = 0.18; neurogranin2: β = −0.01, p = 0.97), APOE ε4 status (z = −0.13, p = 0.90), or AD family history (z = 0.29, p = 0.77).
Neurogranin is associated with delayed recall.
Higher levels of CSF neurogranin were associated with poorer delayed recall performances (ρ = −0.21, p = 0.02) such that there was a quadratic relationship between neurogranin and memory (neurogranin: b = −0.005, β = −0.36, p = 0.002; neurogranin2: b = 0.00001, β = 0.38, p = 0.001). In contrast, no other AD biomarker was significantly associated with memory scores via either linear or polynomic modeling (figure 1): t-tau (ρ = −0.17, p = 0.06), p-tau (ρ = −0.14, p = 0.13), Aβ42 (ρ = −0.02, p = 0.87), or hippocampal volume (β = −0.02, p = 0.90) (polynomial model ps > 0.10).
Figure 1. CSF neurogranin is the only CNS marker associated with delayed recall performance among aging adults.

Analyses adjusted for age. 95% Confidence intervals (CIs) calculated via bootstrapping (100 samples). β-Amyloid, total tau, phosphorylated tau (p-tau), and neurogranin represent CSF concentrations.
The quadratic relationship between neurogranin and memory performance remained significant in individual models or a combined model adjusting for Aβ42, t-tau, p-tau, hippocampal volumes, APOE ε4 status, or family history of AD, and none of the AD-related markers reached significance in these models (combined model: neurogranin2 β = 0.41, p = 0.002; individual models: neurogranin2 β range 0.38–0.42, ps < 0.002; AD-related markers: β range −0.04 to 0.04, ps > 0.50). In addition, the quadratic relationship between neurogranin and memory performance was not significantly moderated by any of the CNS AD biomarkers (interaction measures β range 0.05–0.17, ps > 0.16). Finally, neurogranin remained significantly associated with delayed recall performance after excluding 8 participants with a Clinical Dementia Rating of 0.5 (neurogranin2 p = 0.003).
Ultimately, the scatterplot visualizing the relationship between neurogranin and delayed recall (figure 2) evidenced 5 data points of individuals with high CSF neurogranin (>1,000 pg/mL) and high memory performance that appeared to be contributing to the quadratic line curvature. Given that neurogranin is a novel in vivo clinical biomarker and polynomic effects have not been reported previously, we further explored this relationship. We excluded the 5, refit the model, and found that the quadratic relationship was no longer significant, though a significant negative, linear effect between CSF neurogranin and delayed recall remained (adjusting for age, sex, AD-related biomarkers, APOE ε4, and AD family history) (neurogranin2: b = 0.00001, β = 0.06, p = 0.54; neurogranin: b = −0.005, β = −0.27, p = 0.04).
Figure 2. Scatterplot illustrates the relationship between CSF neurogranin and delayed recall performance.

n = 5 data points with CSF neurogranin >1,000 pg/mL driving the quadratic effects. Open markers denote individuals with Clinical Dementia Rating of 0.5; delayed recall scaled score is age-adjusted (mean 10, SD 3).
Nonetheless, when all participants were categorized into tertiles, neurogranin tertiles were associated with delayed recall adjusting for age and sex (F = 5.8, p = 0.004), such that individuals with the lowest concentrations of CSF neurogranin demonstrated significantly better delayed recall performances compared to those with medium or high levels (ps < 0.03), the latter of which did not differ from one another (p = 0.99) (figure 3).
Figure 3. Low CSF neurogranin is associated with better delayed recall performance in aging adults.
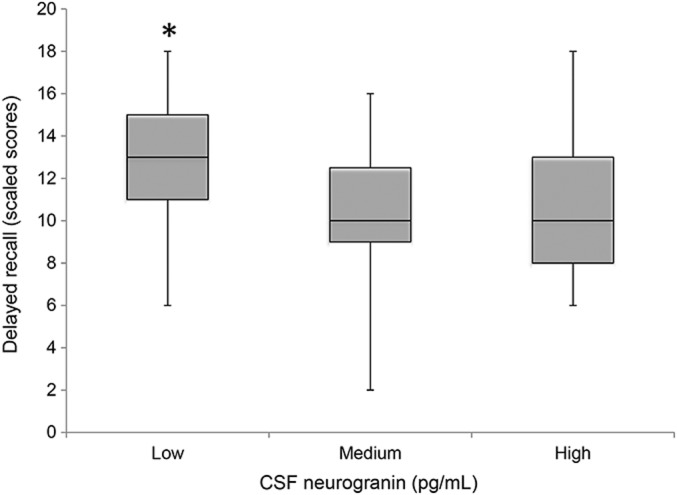
Delayed recall scaled score is age-adjusted (mean 10, SD 3).
Neurogranin and other cognitive domains.
Higher concentrations of CSF neurogranin were additionally associated with slower processing speed in univariate analyses (ρ = −0.33) and after adjusting for age and sex (β = −0.19, p = 0.02), though the quadratic effect was not significant (neurogranin2 β = 0.11, p = 0.28). However, slower processing speed was also associated with some AD biomarkers, including t-tau (ρ = −0.43, p < 0.001; adjusting for age and sex β = −0.21, p = 0.01) and p-tau (ρ = −0.31, p = 0.001; adjusting for age and sex β = −0.16, p = 0.06), but not Aβ42 (ρ = −0.03, p = 0.71) or hippocampal volumes (β = 0.02, p = 0.82). Adjusting for t-tau or p-tau, neurogranin was no longer significantly associated with processing speed (ps < 0.05).
CSF neurogranin was not associated with echoic attention (ρ = 0.005), working memory (ρ = 0.008), or semantic retrieval (ρ = −0.17) (ps > 0.05).
DISCUSSION
As an indicator of synaptic physiology, CSF neurogranin was associated with age, hippocampal volumes, and tau, but was the only CNS marker linked to memory performance in clinically normal older adults. In addition, though processing speed was associated with neurogranin, this relationship was not independent of other CNS markers (i.e., t-tau and p-tau). Given that processing speed is a highly sensitive but nonspecific measure of brain functioning, the relationship between neurogranin and speed may reflect more global abilities that are also detected by the other CNS markers. These findings suggest that (1) age-related synaptic dynamics are highly sensitive to cognitive functioning (especially subtle memory abilities); (2) synaptic dysfunction may potentially precede, act synergistically, or even independently from early AD pathogenesis; and (3) monitoring synaptic status may be valid marker of brain functioning in clinically normal older adults. Though previous works have demonstrated alterations in neurogranin concentrations in patients with AD compared to healthy individuals,14 ours is among the first to closely characterize neurogranin in cognitively normal older adults, including the polynomic nature of the relationship, and extend its association with cognition beyond brief screeners. Our findings are in adults with elevated risk for AD, limiting some generalizability of the effects; however, the independence of the association between neurogranin and memory from AD-specific biomarkers supports the potential application of synaptic protein markers to objectively monitor brain wellness even in clinically normal aging.
Though neurogranin was the most sensitive marker of delayed recall, the nature of the effect may be nuanced. We found a quadratic relationship between neurogranin and memory that was driven by a small subset of individuals (n = 5) with high CSF neurogranin and high memory scores. Yet, even when these outlying individuals were included in analyses, overall low concentrations of CSF neurogranin were associated with better memory performances. Although the study design does not allow for conclusions regarding causality, it is tempting to speculate that low CSF concentrations of neurogranin reflect higher neurogranin availability in synapses. In the presence of NMDA-related calcium influxes, neurogranin releases calmodulin, freeing it to activate downstream protein kinase C pathways. Ultimately, these pathways increase AMPA receptor insertion, subsequently enhancing LTP. That is, higher concentrations of neurogranin sequester larger amounts of available calmodulin in the synapse, lowering the basal neuronal activity threshold needed to induce LTP and learning.23 Conversely, a subset of individuals with the highest CSF neurogranin concentrations also demonstrated high memory performance. Given that this was not necessarily expected and the novelty of neurogranin as an in vivo clinical biomarker, interpretation of this finding would be speculative at this point. The small subset of outlying neurogranin concentrations (>1,000 pg/mL) may represent spurious assay-related variability or biologically relevant information. Future studies are needed to replicate and clarify the polynomic nature of our findings.
Nonetheless, CSF neurogranin was the only marker sensitive to clinical memory performance. Given that neurogranin is concentrated in the hippocampi and the hippocampi are strongly linked to memory abilities,7 CSF neurogranin may reflect hippocampal-related plasticity processes important to learning and recall in our older adults. In contrast, hippocampal volumes were not associated with memory scores, suggesting that more nuanced hippocampal functioning subtleties may be captured by neurogranin.
Regarding its association with other markers of CNS function, we found that neurogranin was more strongly associated with CSF tau proteins and hippocampal structure than amyloid concentrations, which is consistent with recent clinical neurogranin literature in AD. Though some studies evidence a significant link between neurogranin and amyloid levels in both the brain and CSF among individuals with AD-related impairment, these relationships are consistently smaller than those observed between neurogranin and tau.12,13,24 In addition, several studies, particularly those examining earlier stages of AD development, did not identify a significant relationship between neurogranin and amyloid concentrations at all.11,25 Similarly, we found that neurogranin was not associated with and independently predicted memory beyond APOE ε4 status or family history of AD, both of which are associated with greater amyloid deposition.26 Though longitudinal studies are needed, taken together these findings suggest that the relationship between neurogranin and amyloid may be distinct and only become meaningful later in disease or at an elevated threshold of amyloid aggregation. That is, though amyloid accumulates with age, it may only interfere with or relate to neurogranin functioning once it has reached pathologic levels. This may suggest that synaptic proteins represent the broader neurologic aging spectrum not necessarily specific to AD development (i.e., aging does not equate AD).
On the other hand, neurogranin was strongly associated with p-tau and t-tau, as well as hippocampal volumes, though interactions with these markers did not moderate its relationship with memory performance. These results are also consistent with the previous studies demonstrating moderate to large effect sizes between neurogranin and tau, and hippocampal-specific changes in neurogranin with age, and may point to early concurrent aging processes.6,11–13,16,25 Though commonly considered markers of AD-related pathogenesis, tau aggregation and hippocampal atrophy manifest in early aging (i.e., 20s–30s).27 Neurogranin may also be such a fluid metric of the broader neural aging process. These data are also complementary to recent work by Portelius et al.16 demonstrating a relationship between high baseline CSF neurogranin and increased rapidity of cognitive, hippocampal, and cortical metabolic declines in healthy individuals and those with AD-related impairment across time, suggesting that the neurogranin relationships observed here have high clinical relevance for subsequent disease development. However, Portelius et al. did not find an association between baseline neurogranin and performance on global cognitive screeners. Our data suggest that the relationship between neurogranin and cognition is more nuanced and perhaps specific to memory abilities. We also demonstrate that the relationship between cognition and neurogranin extends into asymptomatic individuals, though perhaps disrupted baseline neurogranin affects cognition globally given time. Continued characterization of how neurogranin and other age-sensitive markers develop, potentially interact, and change with time will be critical to our understanding of age-related cognitive decline and neurodegeneration.
One major limitation of our work is the cross-sectional design. We cannot discern age-related changes in synapse-related memory abilities vs premorbid relationships. Given that we posit these synaptic functions to be dynamic with aging and to directly affect cognitive and behavioral abilities, future work utilizing longitudinal designs and experimental behavioral manipulation are needed to determine directionality of these relationships. Nonetheless, ours are the first data beginning to parse out the role of neurogranin and cognition in clinically normal older adults and lay a foundation for future synaptic aging work. In addition, our participants have overrepresented family histories of AD; therefore, the generalizability of our results may be attenuated. However, it is notable that even given the increased risk of AD, we did not observe significant relationships between neurogranin and amyloid, AD family history, or APOE ε4, suggesting that preclinical AD was at least not the sole driver of our observed effects.
Markers sensitive to early clinical change in age-related brain health are needed to track and identify at-risk individuals who warrant preventative and early intervention. Integrity of synaptic functioning, and neurogranin specifically, may represent one such marker that can be used to objectively monitor clinically meaningful brain development. Given that synaptic dynamics are plastic with age and experience, further work characterizing synapse development under normal and pathologic conditions may help identify targets for intervention and be a relevant metric of interest for future clinical trials.
GLOSSARY
- Aβ42
42 amino acid form of β-amyloid
- AD
Alzheimer disease
- AVLT
Rey Auditory Verbal Learning Test
- LTP
long-term potentiation
- p-tau
phosphorylated tau
- t-tau
total tau
- WAIS-III
Wechsler Adult Intelligence Scale, 3rd edition
AUTHOR CONTRIBUTIONS
Kaitlin Casaletto: project conceptualization, statistical analyses and interpretation, manuscript preparation and writing. Fanny Elahi: statistical interpretation, manuscript preparation and writing. Brianne Bettcher: project conceptualization, critical revision of manuscript. John Neuhaus: statistical analyses. Barbara Bendlin: critical revision of manuscript, study coordination. Sterling Johnson: critical revision of manuscript, study coordination, obtaining funding. Sanjay Asthana: critical revision of manuscript, study coordination. Kristine Yaffe: critical revision of manuscript. Cynthia Carlsson: critical revision of manuscript. Kaj Blennow: critical revision of manuscript and CSF analysis. Henrik Zetterberg: critical revision of manuscript and CSF analysis. Joel Kramer: project conceptualization, interpretation of data, critical revision of manuscript, study supervision/coordination, obtaining funding.
STUDY FUNDING
Supported by NIH-NIA grants NIA 1R01AG032289 (PI: Kramer), R01AG048234 (PI: Kramer), UCSF ADRC P50 AG023501, UW ADRC P50 AG033514 (PI: Asthana), R01AG037639 (PI: Bendlin), R01AG021155, and R01AG027161 (PI: Johnson); Larry L. Hillblom Network Grant for the Prevention of Age-Associated Cognitive Decline 2014-A-004-NET (PI: Kramer); and work at the University of Gothenburg was supported by the Swedish Alzheimer Foundation, the Swedish Brain Foundation, and the Torsten Söderberg Foundation.
DISCLOSURE
K. Casaletto, F. Elahi, B. Bettcher, B. Bendlin, S. Johnson, S. Asthana, K. Yaffe, and C. Carlsson report no disclosures relevant to the manuscript. K. Blennow is a co-founder of Brain Biomarker Solutions in Gothenburg AB, a GU Venture-based platform company at the University of Gothenburg, and has served at advisory boards or as a consultant for Eli Lilly, Fujirebio Europe, IBL International, and Roche Diagnostics. H. Zetterberg is a co-founder of Brain Biomarker Solutions in Gothenburg AB, a GU Venture-based platform company at the University of Gothenburg, and has served at advisory boards for Roche Diagnostics, Eli Lilly, and Pharmasum Therapeutics. J. Kramer reports no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Morrison JH, Baxter MG. The ageing cortical synapse: hallmarks and implications for cognitive decline. Nat Rev Neurosci 2012;13:240–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Terry RD, Masliah E, Salmon DP, et al. Physical basis of cognitive alterations in Alzheimers disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 1991;30:572–580. [DOI] [PubMed] [Google Scholar]
- 3.Blennow K, Bogdanovic N, Alafuzoff I, Ekman R, Davidsson P. Synaptic pathology in Alzheimer's disease: relation to severity of dementia, but not to senile plaques, neurofibrillary tangles, or the ApoE4 allele. J Neural Transm 1996;103:603–618. [DOI] [PubMed] [Google Scholar]
- 4.Zhong L, Gerges NZ. Neurogranin and synaptic plasticity balance. Commun Integr Biol 2010;3:340–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kvartsberg H, Duits FH, Ingelsson M, et al. Cerebrospinal fluid levels of the synaptic protein neurogranin correlates with cognitive decline in prodromal Alzheimer's disease. Alzheimers Dement 2015;11:1180–1190. [DOI] [PubMed] [Google Scholar]
- 6.Zetterberg H, Blennow K. Neurogranin levels in cerebrospinal fluid a new addition to the Alzheimer disease diagnostic toolbox. JAMA Neurol 2015;72:1237–1238. [DOI] [PubMed] [Google Scholar]
- 7.Mons N, Enderlin V, Jaffard R, Higueret P. Selective age-related changes in the PKC-sensitive, calmodulin-binding protein, neurogranin, in the mouse brain. J Neurochem 2001;79:859–867. [DOI] [PubMed] [Google Scholar]
- 8.Hayashi Y. Long-term potentiation: two pathways meet at neurogranin. EMBO J 2009;28:2859–2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhong L, Cherry T, Bies CE, Florence MA, Gerges NZ. Neurogranin enhances synaptic strength through its interaction with calmodulin. Embo J 2009;28:3027–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Masliah E, Mallory M, Hansen L, Deteresa R, Terry RD. Quantitative synaptic alterations in the human neocortex during normal aging. Neurology 1993;43:192–197. [DOI] [PubMed] [Google Scholar]
- 11.Kester MI, Teunissen CE, Crimmins DL, et al. Neurogranin as a cerebrospinal fluid biomarker for synaptic loss in symptomatic Alzheimer disease. JAMA Neurol 2015;72:1275–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mattsson N, Insel PS, Palmqvist S, et al. Cerebrospinal fluid tau, neurogranin, and neurofilament light in Alzheimer's disease. Embo Mol Med 2016;8:1184–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wellington H, Paterson RW, Portelius E, et al. Increased CSF neurogranin concentration is specific to Alzheimer disease. Neurol 2016;86:829–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thorsell A, Bjerke M, Gobom J, et al. Neurogranin in cerebrospinal fluid as a marker of synaptic degeneration in Alzheimer's disease. Brain Res 2010;1362:13–22. [DOI] [PubMed] [Google Scholar]
- 15.Davidsson P, Blennow K. Neurochemical dissection of synaptic pathology in Alzheimer's disease. Int Psychogeriatr 1998;10:11–23. [DOI] [PubMed] [Google Scholar]
- 16.Portelius E, Zetterberg H, Skillback T, et al. Cerebrospinal fluid neurogranin: relation to cognition and neurodegeneration in Alzheimer's disease. Brain 2015;138:3373–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 2011;7:263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kawas C, Segal J, Stewart WF, Corrada M, Thai LJ. A validation study of the dementia questionnaire. Arch Neurol 1994;51:901–906. [DOI] [PubMed] [Google Scholar]
- 19.Palmqvist S, Zetterberg H, Blennow K, et al. Accuracy of brain amyloid detection in clinical practice using cerebrospinal fluid beta-amyloid 42 a cross-validation study against amyloid positron emission tomography. JAMA Neurol 2014;71:1282–1289. [DOI] [PubMed] [Google Scholar]
- 20.Lezak MD. Neuropsychological Assessment. 2nd ed. New York: Oxford University Press; 1983. [Google Scholar]
- 21.Wechsler D. WAIS-III: Wechsler Adult Intelligence Scale. San Antonio: Psychological Corporation; 1997. [Google Scholar]
- 22.Kaplan E, Goodglass H, Weintraub S. Boston Naming Test. Austin: Pro-Ed; 2001. [Google Scholar]
- 23.Huang KP, Huang FL, Jager T, Li JF, Reymann KG, Balschun D. Neurogranin/RC3 enhances long-term potentiation and learning by promoting calcium-mediated signaling. J Neurosci 2004;24:10660–10669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hellwig K, Kvartsberg H, Portelius E, et al. Neurogranin and YKL-40: independent markers of synaptic degeneration and neuroinflammation in Alzheimer's disease. Alzheimers Res Ther 2015;7:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Vos A, Jacobs D, Struyfs H, et al. C-terminal neurogranin is increased in cerebrospinal fluid but unchanged in plasma in Alzheimer's disease. Alzheimers Dement 2015;11:1461–1469. [DOI] [PubMed] [Google Scholar]
- 26.Ossenkoppele R, Jansen WJ, Rabinovici GD, et al. Prevalence of amyloid PET positivity in dementia syndromes a meta-analysis. JAMA 2015;313:1939–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 Years. J Neuropathol Exp Neurol 2011;70:960–969. [DOI] [PubMed] [Google Scholar]