

 The α-ketoacid–hydroxylamine (KAHA) ligation enables the direct cyclization of unprotected peptides upon cleavage, without coupling reagents or purification of precursors. We report the synthesis of a library of 24 cyclic peptides and a detailed mechanistic study.
The α-ketoacid–hydroxylamine (KAHA) ligation enables the direct cyclization of unprotected peptides upon cleavage, without coupling reagents or purification of precursors. We report the synthesis of a library of 24 cyclic peptides and a detailed mechanistic study.
Abstract
The α-ketoacid–hydroxylamine (KAHA) ligation with 5-oxaproline enables the direct cyclization of peptides upon cleavage from a solid support, without coupling reagents, protecting groups, or purification of the linear precursors. This Fmoc SPPS-based method was applied to the synthesis of a library of 24 homoserine-containing cyclic peptides and was compared side-by-side with the synthesis of the same products using a standard method for cyclizing side-chain protected substrates. A detailed mechanistic study including 2H and 18O labeling experiments and the characterization of reaction intermediates by NMR and mass spectrometry is reported.
Introduction
Macrocyclic peptides are an important class of molecules for drug discovery and development.1 The conformational restriction induced by cyclization confers on peptides a higher selectivity2 for binding, an improved proteolytic stability3 and a defined structural organization, which enable them to be used as a preferred platform for mimicking protein epitopes4 and disrupting protein–protein interactions.5 The cyclization of macrocycles is often a critical step and different approaches6 including on-resin cyclization,7 chemical ligation,8 copper-catalyzed azide–alkyne cycloaddition,9 ring-closing metathesis10 and electrostatically-controlled macro-cyclizations11 have been developed. However, the synthesis of this class of molecules for drug discovery screening programs or on a preparative scale remains a formidable challenge.
The most common method of preparing backbone cyclized peptides remains the solution-phase cyclization of side-chain protected peptides, often prepared on 2-Cl trityl resin followed by a head-to-tail cyclization and global deprotection.12 While the overall yields of cyclic products can be satisfactory – particularly for substrates with elements that favor cyclization – a considerable number of steps and purifications are required.
Depending on the sequence and the ring size, cyclization can be accompanied by substantial epimerization, even with mild C-terminal activation.13 With increasing size, the solubility of linear protected peptides becomes an issue and has triggered the development of minimal-protected cyclization strategies to increase solubility at the expense of unwanted side reactions.14 These issues are particularly relevant in the preparation of libraries, as complicated workflows and substrate specific optimization are often required. Alternative cyclizations, including native chemical ligation (NCL) using C-terminal thioesters and N-terminal cysteines, have been advanced but the preparation of the linear precursors remains a bottleneck.
Despite elegant advances in the synthesis of thioester surrogates, Fmoc-SPPS based methods delivering unprotected cyclization substrates upon resin-cleavage, without the need for subsequent transformation to a thioester remain underdeveloped.15
In our own efforts, we have previously reported the cyclization of unprotected linear peptides16 by KAHA ligations.17 This method makes possible the cyclization of peptides bearing a C-terminal α-ketoacid and a N-terminal hydroxylamine in the presence of unprotected side chain functional groups and without the need for coupling reagents or other additives. Unfortunately, the methods for introducing the α-ketoacid and hydroxylamine functional groups were cumbersome and detracted from the overall ease of preparing cyclic peptides. An ideal method would allow cyclization to occur simply upon cleavage of the linear peptide from the resin followed by in situ cyclization and purification.18 With this in mind, we sought to develop a method especially suited for library synthesis of cyclic peptide with a minimum of steps required for manual handling.
In this report, we document the spontaneous cyclization of linear, unprotected peptides to form macrocyclic peptides and depsipeptides by KAHA ligation with (S)-5-oxaproline19 as a stable, easily handled hydroxylamine and acetal protected α-ketoacids (Scheme 1). The linear peptides prepared by standard Fmoc solid-phase peptide synthesis20 (SPPS) are cyclized in CH3CN/H2O directly after resin cleavage and side-chain deprotection, resulting in a homoserine residue at the ligation site. The immediate products of the cyclization are the synthetically valuable depsipeptides, which can be either isolated or rearranged to the corresponding amide product.21 The optimized KAHA cyclization conditions were tested on a library of 24 sequences, ranging from 8 to 20 amino acids and the results were compared head-to-head against the standard synthetic method involving preparation of peptides on 2-Cl trityl resin, cyclization, and global deprotection.
Scheme 1. One step cleavage and cyclization of unprotected peptides with the KAHA ligation.

Results and discussion
Initial studies
In order to establish that the 5-oxaproline hydroxylamine and the new precursors to α-ketoacids were suitable substrates for peptide cyclization with the KAHA ligation, we prepared the 10-residue peptide22 3 by Fmoc SPPS. The synthesis began with our recently reported resin for forming C-terminal Leu α-ketoacid residues directly upon cleavage of the peptide from resin under acidic conditions.23 These resins can be easily prepared on multi-gram scale and can be stored without decomposition. By incorporating (S)-5-oxaproline at the N-terminus, bifunctional cyclization substrate 3 could be prepared and isolated by preparative HPLC without premature cyclization or oligomerization. We were pleased to find that 3 converted fully to cyclic peptide 4 within 2 h upon warming to 60 °C in 7 : 3 DMSO/H2O in the presence of 0.1 M oxalic acid at a substrate concentration of 1 mM (Scheme 2). Two distinct products were formed: amide-4 as a minor component and the depsipeptide depsi-4 as the major product. The isolated depsi-4 was characterized by extensive NMR studies (see ESI†) and could be cleanly converted to amide-4 in pH 11 phosphate buffer in CH3CN/H2O within 18 h.
Scheme 2. Cyclization of 3 in 7 : 3 DMSO/H2O with 0.1 M oxalic acid at 60 °C after (a) 0 h (b) 2 h. (c) Purified depsi-4. (d) O,N-Acyl shift of depsi-4 at pH 11 in 2 : 1 CH3CN/H2O (200 mM phosphate buffer) after 4 h and (e) after 18 h.

Optimization
These initial findings encouraged us to further optimize and generalize the cyclization conditions, with the aim of establishing a process for resin cleavage, cyclization, and O,N-acyl shift without isolation of any intermediates.
We first examined the effect of the reaction conditions on the cyclization. As shown in Fig. 1a, the reaction was faster in CH3CN/H2O mixtures than in DMSO, NMP and DMF. A higher CH3CN content further improved the reaction rate. For solubility reasons we chose 2 : 1 CH3CN/H2O as our standard solvent mixture. The KAHA cyclization proceeded slowly at room temperature but reached >50% conversion after 30 min at 60 °C (Fig. 1b).
Fig. 1. Cyclization of 3: conversion determined by HPLC at 220 nm as relative area% of starting material 3 and products depsi-4 and amide-4 dependent on (a) solvents after 30 min at 60 °C in the presence of 0.1 M oxalic acid and (b) temperature in 2 : 1 CH3CN/H2O (0.1 M oxalic acid).

In order to reduce the amount of solvent needed for the cyclizations, we investigated the formation of oligomeric species as a function of the concentration of the linear precursor 3. As shown in Fig. 2, only 5% of dimers were formed at a concentration of 5 mM and less than 10% at 10 mM. For library preparation we employed a substrate concentration of 6.25 mM, which is suitable for preparative work and is higher than the usual concentration employed for peptide cyclization. In order to minimize the number of manipulation steps, we planned to bypass purifications after resin cleavage and cyclization with the goal of only isolating the final amide products after the O,N-acyl shift. In fact, direct cyclization of the cleaved peptide without prior purification proved to positively affect the process as we found that typically the HPLC peak shapes of the cyclized products were much sharper than the linear α-ketoacid precursors and facilitated purification.
Fig. 2. Influence of substrate concentration on the formation of cyclodimers during the cyclization of 3 in 2 : 1 CH3CN/H2O (0.1 M oxalic acid) at 60 °C. The ratio was determined by HPLC at 220 nm as relative area% of depsi and amide products.

To eliminate the second purification step, we performed the O,N-acyl shift in a one-pot reaction directly after cyclization. Basic aqueous buffers have been used to perform the O,N-acyl shift but the risk of salt-precipitation in organic co-solvents prompted us to search for an easier alternative. We were pleased to find that the addition of aqueous ammonia at room temperature offered an operationally simple protocol that facilitated the overall process and dramatically reduced the time needed for the acyl shift. For most of the cyclic peptides examined, the acyl shift was completed within 1 h. We did not observe ammonolysis of the ester bond even after 3 h; the cyclic depsipeptides were fully converted to the desired homodetic cyclic peptides, which were isolated by preparative HPLC. Table 2 shows the optimized workflow for the KAHA cyclization.
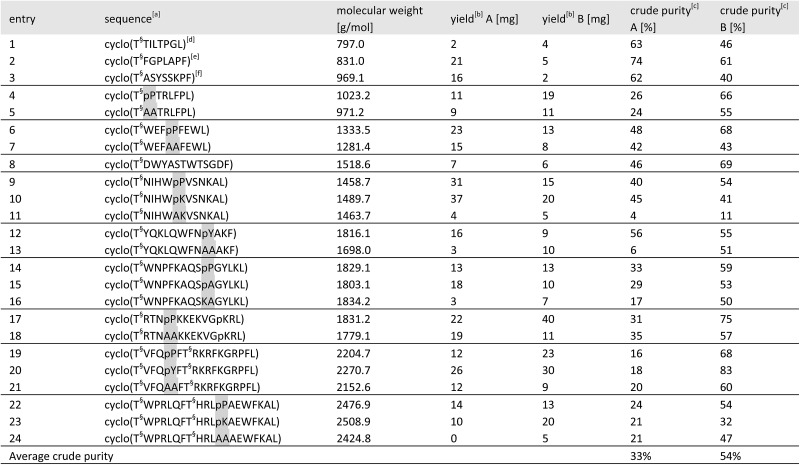
Table 2. Results of the cyclic peptide library synthesis by protected cyclization in solution (method A) and by KAHA cyclization (method B).
|
|
aHighlighted residues indicate the distinctive position of similar sequences with and without a turn inducing template. The last position indicates the α-ketoacid used for cyclization (Leu or Phe). The peptide concentration for cyclization was 6.25 mM for both methods. [T§ = (S)-homoserine].
bIsolated yields on 50 μmol scale after RP-HPLC purification.
cDetermined by HPLC as relative area% at 220 nm.
dGypsophin B25 analogue.
ePseudostellarin G25 analogue.
fSegetalin F25 analogue.
Epimerization analysis
After optimizing the cyclization conditions we investigated possible epimerization at the cyclization site. Since separation of the d-Leu and l-Leu epimers of amide-4 and depsi-4 was not possible by HPLC, the cyclic peptides were hydrolysed and converted into volatile derivatives. This allowed analysis of single amino acid derivatives by chiral GC/MS. Analysis of the final product amide-4 after cyclization and one-pot rearrangement with ammonia showed 8% of d-Leu (Table 1, entry 4). This rather high, but still acceptable, level of epimerization prompted us to perform a more detailed study on the origin of epimerization (Table 1, entries 1–5), especially as the initially formed amide-4 showed a significant amount of d-Leu (13%, entry 2), while the initially formed depsi-4 showed only 5.6% of d-Leu. The cleavage of the α-ketoacid protecting group and the rearrangement of depsi-4 to amide-4 under basic conditions (entry 1 and 3) do not cause any significant epimerization.
Table 1. Epimerization analysis by chiral GC/MS.
| Entry | Compound | Description | Content of d-Le a |
| 1 | Depsi-4 | From reaction mixture b | 5.6% |
| 2 | Amide-4 | 13% | |
| 3 | Amide-4 | After rearrangement of isolated depsi-4 c | 4.7 e % |
| 4 | Amide-4 | After rearrangement of crude reaction mixture c | 8.1% |
| 5 | Amide-4 | From reaction in 1 : 1 (CH3)3COH/H2O d | 18% |
aIsolated peptides were hydrolyzed in 6 N DCl in D2O, converted into volatile derivatives and analyzed by chiral GC/MS.
b2 : 1 CH3CN/0.05 M oxalic acid in H2O, 50 °C, 15 h.
c2 : 1 CH3CN/0.05 M oxalic acid in H2O, 1.6 M NH3, 23 °C, 3 h.
d1 : 1 (CH3)3COH/H2O, 0.1 M oxalic acid, 60 °C, 15 h.
eThe slightly lower content of d-Leu after rearrangement is probably a result of the isolation by preparative HPLC.
Mechanistic investigation
Although unexpected, the small amount of epimerization sheds light on the mechanism of the KAHA ligation with 5-oxaproline. The different extents of epimerization of the depsi and the amide products suggest that epimerization occurs not from a common intermediate but rather via two distinct pathways. We have proposed two possible pathways for the formation of depsi and amide product, starting from the common nitrilium intermediate 9 (Scheme 3).21 The epimerization most likely occurs after the nitrilium intermediate 9, otherwise the same level of epimerization would be expected for both products.
Scheme 3. Proposed mechanisms for the formation of 18 by KAHA ligation with 5-oxaproline.

We hypothesized that both the epimerized amide and ester product arise from a planar intermediate formed by enolization and reprotonation of a transient cyclization product. If deuterium instead of a proton would be incorporated during this step, 50% of the incorporated deuterium would correspond to the amount of epimerized product. To test this hypothesis, we performed the cyclization reaction of 3 in a 2 : 1 CH3CN/D2O mixture and isolated the products by HPLC. The isolated HPLC fractions were incubated for 3 h at 60 °C to ensure complete back exchange of reversibly incorporated deuterium. Based on the obtained isotope pattern by MALDI HR-MS the percentage of deuterium incorporation was calculated to be 6% for depsi-4 and 25% for amide-4 corresponding to 3% and 12.5% d-Leu. Within a certain error, these values are in accordance with the values obtained by GC/MS (5.6% and 13%) and verified our hypothesis.
Our previously reported mechanism involves the formation of iminoether 19, although we had been unsuccessful in the attempt to observe or isolate it (Scheme 4A). During the mechanistic investigations of the epimerization we found that if 3 is warmed under anhydrous conditions the crude MALDI-MS spectrum shows exclusively the mass of the iminoether intermediate 19. Subsequent addition of 18OH2 or HPLC analysis (with CH3CN/H2O) leads to the almost exclusive formation of (18O)-depsi-4 or depsi-4 respectively. We were able to reduce the iminoether by addition of NaBH4 to amine 21 which was formed along with depsi-4. As further evidence, we sought to isolate the iminoether intermediate on a small model system. In order to increase the hydrolytic stability of the postulated iminoether by conjugation we decided to use benzoylformic acid 22 as α-ketoacid. Indeed, the reaction with 5-oxaproline dipeptide 23 under anhydrous conditions afforded iminoether 24 as major product, which was isolated by preparative HPLC. The structure was confirmed by NMR and MALDI HR-MS.
Scheme 4. Formation of iminoethers. (A) During the cyclization of 3 in dry CH3OH (B) isolation and characterization of a stable iminoether formed by the reaction between 5-oxaproline containing peptide 23 and α-ketoacid 22.

Since anhydrous conditions strongly favored the formation of the depsi product – which showed less epimerization – we attempted to decrease the overall level of epimerization by favoring the depsi formation. We unfortunately found, using our deuterium incorporation assay with 3, that anhydrous d4-methanol/CH3CN as solvent gave almost completely epimerized (36% d-Leu after 45 min and 45% d-Leu after 4 h; see ESI† for details) depsi-4. The mechanistic implication is that the imino ether intermediate 19 – which is the stable resting state under anhydrous conditions – is configurationally unstable; the epimerization of the depsi product occurs via enamine intermediate 13 (Scheme 3, path A2).
In contrast, the epimerization of the amide product is likely to proceed via another pathway. It is known that peptides with highly electrophilic C-terminal carbonyls are prone to epimerize via 5-exo-trig attack of the next amide carbonyl oxygen followed by the loss of chiral information trough oxazol formation.24 In the case of the nitrilium intermediate 9, also a highly electrophilic carbon is present which could react analogously by 5-exo-dig attack leading to 10 with subsequent formation of oxazol 11 (path C). Also direct epimerization by tautomerization of nitrilium 9 followed by hydrolysis cannot be ruled out.
Library synthesis
Although the cyclization by KAHA ligation with 5-oxaproline is not completely stereo retentive, we found that its potential for facile library synthesis outweighed the small amounts of epimerization observed. Using the optimized process, we confirmed the generality of our method by synthesizing a 24-membered library of cyclic peptides. We chose for our study three analogues of natural products25 as well as random sequences covering a wide range of ring sizes and amino acid composition, with only methionine and cysteine residues excluded in the present study.26 For the α-ketoacid, we used our recently reported acetal protected leucine α-ketoacid resin 25, as well as the corresponding phenylalanine derivative. As a comparison of this new method, we synthesized in parallel, the same library of cyclic peptides by the established method of side chain-protected cyclization in solution.
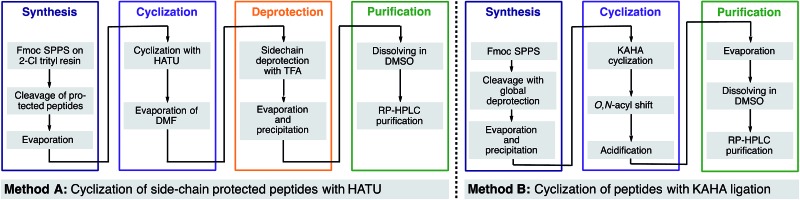
For cyclization with traditional coupling reagents, the side-chain protected peptides were synthesized on 2-Cl trityl resin with N-terminal homoserine, in order to keep the same cyclization points as in the KAHA cyclization. The side chain-protected peptides were cleaved from the resin with 1% TFA in CH2Cl2, cyclized with HATU (1-[bis(dimethylamino)-methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexa-fluorophosphate) and DIPEA (N,N-diisopropylethylamine) in DMF solution27 and deprotected with the deprotection cocktail TFA/TIS/H2O 95 : 2.5 : 2.5 (see Table 2 and ESI† for details).
A representative example by KAHA ligation is shown in Scheme 5 for the synthesis of the 13 residue cyclic peptide amide-28 (Table 2, entry 9). The linear peptide 27 was assembled by Fmoc-SPPS using aminomethyl polystyrene resin 25 preloaded with the protected leucine α-ketoacid. After the standard coupling of Fmoc-5-oxaproline to the N-terminus, the peptide was globally deprotected and cleaved from the resin with the deprotection cocktail TFA/DODT/H2O 95 : 2.5 : 2.5.
Scheme 5. Synthesis of cyclic peptide amide-28 using the optimized KAHA cyclization process. Analytical HPLC traces at 220 nm: (a) crude linear peptide 27 after cleavage from resin. (b) Crude cyclic peptide depsi-28 after 18 h at 50 °C. (c) Crude cyclic peptide amide-28 3 h after the addition of 1.6 M aq NH3. [DODT = 3,6-dioxa-1,8-octanedithiol].28 .

The crude peptide was dissolved in 2 : 1 CH3CN/H2O (0.05 M oxalic acid) and cyclized at 50 °C overnight. Typically the reactions were complete within a few hours, but we chose to elongate the reaction times to 18 h for convenience in parallel synthesis. After cyclization, the crude reaction mixture was treated with aqueous ammonia for 3 h at room temperature to convert depsi-28 into amide-28. This rearrangement proceeded cleanly for all peptides and we did not observe any ammonolysis of the ester bond or reactions of the side-chain functional groups. The mixture was acidified with TFA and the solvent removed by evaporation. The crude product was dissolved in DMSO and purified by RP-HPLC. In most cases, a single purification of the crude peptide macrocycle provided material of >95% purity. As shown in Table 2, we chose random sequences with and without turn inducing motifs such as d-Pro-Pro, which is known to favor the head-to-tail cyclization with standard chemistry.29 To evaluate the influence of these residues on the KAHA cyclization we also included the same sequences in which the turn inducing residues were replaced by l-alanine or l-lysine.
For the 24 sequences synthesized, both methods afforded the desired peptides in comparable overall yields. A significant difference was noticed in the average crude purity of the cyclized peptides where we found that the macrocycles prepared by KAHA ligation were formed in higher purity, greatly facilitating purification.
In sequences without turn-inducing residues, the crude purity was found to be up to 8 times higher in the case of KAHA cyclization (see Table 2 entries 13, 16 and 24). Despite the limited number of synthesized peptides, the data suggests that KAHA cyclization is less dependent on the presence of turn-inducing moieties. The ability to cyclize fully unprotected peptides and the initial formation of ester-linked depsipeptides followed by O,N-acyl shift – which has been reported30 as a strategy to synthesize constrained cyclic peptides – may play a beneficial role on the KAHA cyclizations.
Conclusions
We have established a protocol for the cyclization of fully unprotected peptides, obtained by Fmoc-SPPS, to homoserine-containing macrocycles. The initial ester-linked cyclic depsipeptides can be isolated or rearranged in a one-pot procedure to afford the corresponding native cyclic peptides in good yields and excellent purity. The cyclization of longer peptides can be challenging by standard methods but is facilitated by use of the unprotected linear peptides in the case of the KAHA cyclization. Although our method does not overcome one of the notorious problems with cyclic peptide synthesis, epimerization, the ease of the process and the facile access to valuable cyclic depsipeptides make KAHA cyclizations a valuable tool for the preparation of macrocyclic peptide libraries. These studies also provide further mechanistic insights into the highly unusual chemoselective ester formation, which is the primary product of the KAHA ligation with 5-oxaproline residues.
The protocol presented here is fully compatible with automated, parallel Fmoc SPPS and requires significantly less time, solvents, and manual operation steps than other methods for head-to-tail cyclization, which establishes the KAHA cyclization as a viable synthetic process for the generation of cyclic peptide libraries. Currently, it delivers homoserine-containing products – which have advantages in terms of novelty and intellectual property – due to the use of N-terminal 5-oxaproline as the hydroxylamine partner for the α-ketoacid. By developing alternative N-terminal hydroxylamines31 and preparing new acetal protected α-ketoacids, the variability and generality of this method will be further improved.
Acknowledgments
This work was supported by ETH Zürich and the Swiss National Science Foundation (200020_150073). We thank the LOC MS Service and the LOC NMR Service for analyses, Marc-Olivier Ebert, Vijaya R. Pattabiraman, Thomas G. Wucherpfennig and Christopher J. White for helpful discussions, Stéphane Gable and Véronique Saussin for parallel purification and isolation of the cyclic peptides.
Footnotes
References
- For reviews about this topic see: ; (a) Driggers E. M., Hale S. P., Lee J., Terret N. K. Nat. Rev. Drug Discovery. 2008;7:608–624. doi: 10.1038/nrd2590. [DOI] [PubMed] [Google Scholar]; (b) Marsault E., Peterson M. L. J. Med. Chem. 2011;54:1961–2004. doi: 10.1021/jm1012374. [DOI] [PubMed] [Google Scholar]; (c) Bockus A. T., McEwen C. M., Lokey S. Curr. Top. Med. Chem. 2013;13:821–836. doi: 10.2174/1568026611313070005. [DOI] [PubMed] [Google Scholar]; (d) Yudin A. K. Chem. Sci. 2015;6:30–49. doi: 10.1039/c4sc03089c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Dias R. L. A., Fasan R., Moehle K., Renard A., Obrecht D., Robinson J. A. J. Am. Chem. Soc. 2006;128:2726–2732. doi: 10.1021/ja057513w. [DOI] [PubMed] [Google Scholar]; (b) Henchey L. K., Porter J. R., Ghosh I., Arora P. S. ChemBioChem. 2010;11:2104–2107. doi: 10.1002/cbic.201000378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairlie D. P., Tyndall J. D. A., Reid R. C., Wong A. K., Abbenante G., Scanlon M. J., March D. R., Bergman D. A., Chai C. L. L., Burkett B. A. J. Med. Chem. 2000;43:1271–1281. doi: 10.1021/jm990315t. [DOI] [PubMed] [Google Scholar]
- (a) Obrecht D., Chevalier E., Moehle K., Robinson J. A. Drug Discovery Today: Technol. 2012;9:e63–e69. [Google Scholar]; (b) Obrecht D., Robinson J. A., Bernardini F., Bisang C., DeMarco S. J., Moehle K., Gombert F. O. Curr. Med. Chem. 2009;16:42–65. doi: 10.2174/092986709787002844. [DOI] [PubMed] [Google Scholar]; (c) Giordanetto F., Kihlberg J. J. Med. Chem. 2014;57:278–295. doi: 10.1021/jm400887j. [DOI] [PubMed] [Google Scholar]
- (a) Khakshoor O., Demeler B., Nowick J. S. J. Am. Chem. Soc. 2007;129:5558–5569. doi: 10.1021/ja068511u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Liu C., Sawaya M. R., Cheng P.-N., Zheng J., Nowick J. S., Eisenberg D. J. Am. Chem. Soc. 2011;133:6736–6744. doi: 10.1021/ja200222n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Villar E. A., Beglov D., Chennamadhavuni S., Porco Jr J. A., Kozakov D., Vajda S., Whitty A. Nat. Chem. Biol. 2014;10:723–731. doi: 10.1038/nchembio.1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For a recent review about this topic see: White C. J., Yudin A. K., Nat. Chem., 2011, 3 , 509 –524 . [DOI] [PubMed] [Google Scholar]
- (a) Kates S. A., Solé N. A., Johnson C. R., Hudson D., Barany G., Albericio F. Tetrahedron Lett. 1993;34:1549–1552. [Google Scholar]; (b) Rosenbaum C., Waldmann H. Tetrahedron Lett. 2001;42:5677–5680. [Google Scholar]; (c) Yang L., Morriello G. Tetrahedron Lett. 1999;40:8197–8200. [Google Scholar]
- (a) Clark R. J., Craik D. J. Biopolymers. 2010;94:414–422. doi: 10.1002/bip.21372. [DOI] [PubMed] [Google Scholar]; (b) Camarero J. A., Muir T. W. Chem. Commun. 1997:1369–1370. [Google Scholar]; (c) Shao Y., Lu W., Kent S. B. H. Tetrahedron Lett. 1998;39:3911–3914. [Google Scholar]; (d) Wong C. T. T., Rowlands D. K., Wong C.-H., Lo T. W. C., Nguyen G. K. T., Li H.-Y., Tam J. P. Angew. Chem., Int. Ed. 2012;51:5620–5624. doi: 10.1002/anie.201200984. [DOI] [PubMed] [Google Scholar]; (e) Zheng J.-S., Tang S., Guo Y., Chang H.-N., Liu L. ChemBioChem. 2012;13:542–546. doi: 10.1002/cbic.201100580. [DOI] [PubMed] [Google Scholar]; (f) Lam H. Y., Zhang Y., Liu H., Xu J., Wong C. T. T., Xu C., Li X. J. Am. Chem. Soc. 2013;135:6272–6279. doi: 10.1021/ja4012468. [DOI] [PubMed] [Google Scholar]; (g) Kleineweischede R., Hackenberger C. P. R. Angew. Chem., Int. Ed. 2008;47:5984–5988. doi: 10.1002/anie.200801514. [DOI] [PubMed] [Google Scholar]
- Bock V. D., Perciaccante R., Jansen T. P., Hiemstra H., van Maarseveen J. A. Org. Lett. 2006;8:919–922. doi: 10.1021/ol053095o. [DOI] [PubMed] [Google Scholar]
- Miller S. J., Blackwell H. E., Grubbs R. H. J. Am. Chem. Soc. 1996;118:9606–9614. [Google Scholar]
- (a) Londregan A. T., Farley K. A., Limberakis C., Mullins P. B., Piotrowski D. W. Org. Lett. 2012;14:2890–2893. doi: 10.1021/ol301173m. [DOI] [PubMed] [Google Scholar]; (b) Hili R., Rai V., Yudin A. K. J. Am. Chem. Soc. 2010;132:2289–2891. doi: 10.1021/ja910544p. [DOI] [PubMed] [Google Scholar]
- Barlos K. and Adermann K., in Amino Acids, Peptides and Proteins in Organic Chemistry Vol. 3-Building Blocks, Catalysis and Coupling Chemistry, ed. A. B. Hughes, Wiley-VCH, Weinheim, 2011, pp. 392–394. [Google Scholar]
- Schmidt U., Langer J. J. Pept. Res. 1997;49:67–73. doi: 10.1111/j.1399-3011.1997.tb01122.x. [DOI] [PubMed] [Google Scholar]
- Camarero J. A., Cairó J. J., Giralt E., Andreu D. J. Pept. Sci. 2004;1:241–250. doi: 10.1002/psc.310010405. [DOI] [PubMed] [Google Scholar]
- (a) Harris P. W. R., Brimble M. A. Biopolymers. 2013;100:356–365. doi: 10.1002/bip.22223. [DOI] [PubMed] [Google Scholar]; (b) Taichi M., Hemu X., Qiu Y., Tam J. P. Org. Lett. 2013;15:2620–2623. doi: 10.1021/ol400801k. [DOI] [PubMed] [Google Scholar]; (c) Fang G.-M., Li Y.-M., Shen F., Huang Y.-C., Li J.-B., Lin Y., Cui H.-K., Liu L. Angew. Chem., Int. Ed. 2011;50:7645–7649. doi: 10.1002/anie.201100996. [DOI] [PubMed] [Google Scholar]; (d) Ingenito R., Bianchi E., Fattori D., Pessi A. J. Am. Chem. Soc. 1999;121:11369–11374. [Google Scholar]; (e) Shin Y., Winans K. A., Backes B. J., Kent S. B. H., Ellman J. A., Bertozzi C. R. J. Am. Chem. Soc. 1999;121:11684–11689. [Google Scholar]; (f) Boll E., Ebran J.-P., Drobecq H., El-Mahdi O., Raibaut L., Ollivier N., Melnyk O. Org. Lett. 2015;17:130–133. doi: 10.1021/ol503359w. [DOI] [PubMed] [Google Scholar]
- Fukuzumi T., Ju L., Bode J. W. Org. Biomol. Chem. 2012;10:5837–5844. doi: 10.1039/c2ob25129a. [DOI] [PubMed] [Google Scholar]
- Bode J. W., Fox R. M., Baucom K. D. Angew. Chem., Int. Ed. 2006;45:1248–1252. doi: 10.1002/anie.200503991. [DOI] [PubMed] [Google Scholar]
- Chandra K., Roy T. K., Shalev D. E., Loyter A., Gilon C., Gerber R. B., Friedler A. Angew. Chem., Int. Ed. 2014;53:9450–9455. doi: 10.1002/anie.201402789. [DOI] [PubMed] [Google Scholar]
- (a) Pattabiraman V. R., Ogunkoya A. O., Bode J. W. Angew. Chem., Int. Ed. 2012;51:5114–5118. doi: 10.1002/anie.201200907. [DOI] [PubMed] [Google Scholar]; (b) Ogunkoya A. O., Pattabiraman V. R., Bode J. W. Angew. Chem., Int. Ed. 2012;51:9693–9697. doi: 10.1002/anie.201204144. [DOI] [PubMed] [Google Scholar]
- Merrifield R. B. J. Am. Chem. Soc. 1963;85:2149–2154. [Google Scholar]
- Wucherpfennig T. G., Rohrbacher F., Pattabiraman V. R., Bode J. W. Angew. Chem., Int. Ed. 2014;53:12244–12247. doi: 10.1002/anie.201406097. [DOI] [PubMed] [Google Scholar]
- Fasan R. R., Dias L. A., Moehle K., Zerbe O., Vrijbloed J. W., Obrecht D., Robinson J. A. Angew. Chem., Int. Ed. 2004;43:2109–2112. doi: 10.1002/anie.200353242. [DOI] [PubMed] [Google Scholar]
- Wucherpfennig T. G., Pattabiraman V. R., Limberg F. R. P., Ruiz-Rodríguez J., Bode J. W. Angew. Chem., Int. Ed. 2014;53:12248–12252. doi: 10.1002/anie.201407014. [DOI] [PubMed] [Google Scholar]
- El-Faham A., Albericio F. Chem. Rev. 2011;111:6557–6602. doi: 10.1021/cr100048w. [DOI] [PubMed] [Google Scholar]
- (a) Morita H., Kayashita T., Kobata H., Gonda A., Takeya K., Itokawa A. Tetrahedron. 1994;50:9975–9982. [Google Scholar]; (b) Morita A., Eda A., Iizuka T., Hirasawa Y., Sekiguchi M., Yun Y. S., Itokawa H., Takeya K. Bioorg. Med. Chem. Lett. 2006;16:4458–4461. doi: 10.1016/j.bmcl.2006.06.083. [DOI] [PubMed] [Google Scholar]; (c) Wang G., Luo J.-G., Yang M.-H., Wang X.-B., Kong L.-Y. Chem. Pharm. Bull. 2013;61:489–495. doi: 10.1248/cpb.c12-01094. [DOI] [PubMed] [Google Scholar]
- Cysteines and methiones are well tolerated by the KAHA ligation but were not desired for the macrocyclic peptides prepared in the present study
- Ehrlich A., Heyne H.-U., Winter R., Beyermann M., Haber H., Carpino L. A., Bienert M. J. Org. Chem. 1996;61:8831–8838. doi: 10.1021/jo951108d. [DOI] [PubMed] [Google Scholar]
- Teixeira A., Benckhuijsen W. E., de Koning P. E., Valentijn A. R. P. M., Drijfhout J. W. Protein Pept. Lett. 2002;9:379–385. doi: 10.2174/0929866023408481. [DOI] [PubMed] [Google Scholar]
- Favre M., Moehle K., Jiang L., Pfieffer B., Robinson J. A. J. Am. Chem. Soc. 1999;121:2679–2685. [Google Scholar]
- (a) Meutermans W. D. F., Golding S. W., Bourne G. T., Miranda L. P., Dooley M. J., Alewood P. F., Smythe M. L. J. Am. Chem. Soc. 1999;121:9790–9796. [Google Scholar]; (b) Lécaillon J., Gilles P., Subra G., Martines J., Amblard M. Tetrahedron Lett. 2008;49:4674–4676. [Google Scholar]
- Pusterla I., Bode J. W. Nat. Chem. 2015 doi: 10.1038/NCHEM.2282. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.