

Abstract
In B-cell precursor acute lymphoblastic leukemia, the initial leukemic cells share the same antigen receptor gene rearrangements. However, due to ongoing rearrangement processes, leukemic cells with different gene rearrangement patterns can develop, resulting in subclone formation. We studied leukemic subclones and their distribution in the bone marrow and peripheral blood at diagnosis. Antigen receptor gene rearrangements (IGH, IGK, TRG, TRD, TRB) were analyzed by next-generation sequencing in seven paired bone marrow samples and five paired bone marrow-peripheral blood samples. Background-thresholds were defined, which enabled identification of leukemic gene rearrangements down to very low levels. Paired bone marrow analysis showed oligoclonality in all 7 patients and up to 34 leukemic clones per patient. Additional analysis of evolutionary-related IGH gene rearrangements revealed up to 171 leukemic clones per patient. Interestingly, overall 86% of all leukemic gene rearrangements, including small subclones, were present in both bone marrow samples (range per patient: 72–100%). Paired bone marrow-peripheral blood analysis showed that 83% of all leukemic gene rearrangements in bone marrow were also found in peripheral blood (range per patient: 81–100%). Remarkably, in the paired bone marrow samples and paired bone marrow-peripheral blood samples the vast majority of leukemic gene rearrangements had a similar frequency (<5-fold frequency difference) (96% and 96% of leukemic rearrangements, respectively). Together, these results indicate that B-cell precursor acute lymphoblastic leukemia is generally highly oligoclonal. Nevertheless, the vast majority of leukemic clones, even the minor antigen receptor-defined subclones, are homogeneously distributed throughout the bone marrow and peripheral blood compartment.
Introduction
During B-cell and T-cell differentiation, genes that encode the B-cell receptor (Immunoglobulin; IG) and the T-cell receptor (TR) are rearranged, respectively. This process of IG and TR gene rearrangement includes the assembly of Variable (V), Diversity (D) and Joining (J) gene segments and the random insertion and deletion of nucleotides between these gene segments,1,2 resulting in unique V-(D)-J junctions. In normal B cells and T cells, productive V-(D)-J exons encode the antigen-binding domains of the IG and TR molecules. The large diversity of V-(D)-J junctions contributes significantly to the large diversity of IG and TR molecules, thereby providing the B and T lymphocytes with a wide variety of antigen specificities.
B-cell precursor acute lymphoblastic leukemia (BCP-ALL) arises from a malignantly transformed B-cell precursor (BCP).3 Therefore, BCP-ALL cells contain IG gene rearrangements in the vast majority of cases.4 In addition, most BCP-ALL cells also harbor certain TR gene rearrangements, which are uncommon in normal BCPs.5–7 Since BCP-ALL is thought to arise from a single cell, a BCP-ALL population is expected to be monoclonal with regard to IG/TR gene rearrangements, i.e. to consist of cells which all share the same IG/TR gene rearrangements. However, continuing rearrangement processes and V-segment replacements that occur in normal BCP cells can also occur during the development of BCP-ALL.8–10 As a result, a BCP-ALL population can be oligoclonal, i.e. can contain subclones with IG/TR gene rearrangements that deviate from those in the initial leukemic cell.
Several previous studies had analyzed IG/TR gene rearrangements in BCP-ALL diagnosis samples by using PCR-based methods in combination with Southern blot (SB), which allowed identification of clonal IG/TR gene rearrangements down to the level of 1–5%.11,12 In these studies, oligoclonality was detected in 40% of patients when analyzing the IGH and TRD loci and in 10–20% of patients when analyzing the IGK and TRB loci.11–13 With regard to complete IGH gene rearrangements, a maximum of 9 clonal rearrangements could be found per patient.14 More recently, IGH gene rearrangements in BCP-ALL at diagnosis were analyzed with highly sensitive next-generation sequencing (NGS), which showed that many more clonal IGH gene rearrangements (up to 4024) can be present in a single patient.15 However, in NGS studies of IG/TR gene rearrangements in BCP-ALL patients, it is difficult to discriminate leukemic rearrangements at low-frequency levels from the background of normal lymphocyte-derived rearrangements and from technical artifacts. Furthermore, NGS-based analysis of gene rearrangements other than complete IGH gene rearrangements has, to the best of our knowledge, so far not been performed in patients with BCP-ALL. Therefore, the degree of oligoclonality in BCP-ALL patients, especially at low-frequency level, remains unclear.
BCP-ALL subclones may develop at different locations in the body, after which they may disseminate throughout the bone marrow (BM) and peripheral blood (PB) compartment. Whereas minimal residual disease (MRD) levels were shown to be comparable between paired BM samples during follow up,16 limited data are available on the distribution of BCP-ALL clones at the time of diagnosis. Our early SB study on IG/TR gene rearrangements in paired BM-PB samples at diagnosis reported that in 5 out of 10 oligoclonal patients, subclonal IGH gene rearrangements were found in the BM sample, but not in the corresponding PB sample.17 However, the relative frequency of these subclonal IGH gene rearrangements in PB might have been too low to be detected with SB. Therefore, it is still unknown to what extent BCP-ALL clones are distributed throughout the BM and PB compartments.
Here, we studied the distribution of leukemic clones in BCP-ALL patients by performing NGS of IG/TR gene rearrangements on paired BM-BM samples as well as paired BM-PB samples.
Methods
Patient and control samples
Bone marrow and PB samples from a total of 12 children with newly diagnosed BCP-ALL were collected (Online Supplementary Table S1). These samples included seven left BM-right BM pairs, i.e. BM from the left and right pelvic bone of the same patient, and five BM-PB pairs, all taken at initial diagnosis. One BM sample from a healthy child (residual material from a donor for allogeneic BM transplantation), two BM samples from BCP-ALL patients at one year after end of therapy, and five regenerating BM samples from T-ALL patients at day 79 and month 5 during therapy were collected to be used as control samples. All control samples were MRD negative according to allele-specific oligonucleotide PCR assays, as described previously.4,18 All samples were obtained according to the guidelines of the local Medical Ethics Committees (MEC 2004-203 and MEC 2012-287) and in line with the Declaration of Helsinki Protocol.
Next-generation sequencing by the Adaptive Biotechnologies method
Genomic DNA was isolated from BM and PB mononuclear cells (MNCs) by the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). A detailed description of the NGS-based immunosequencing method has been published elsewhere.19 Briefly, a DNA quantity of 420–480 ng, corresponding to 70,000–80,000 MNCs, was used for the amplification of complete IGH gene rearrangements (Vh-DJh; three separate PCRs) and a DNA quantity of 120–180 ng, corresponding to 20,000–30,000 MNCs, was used for the amplification of incomplete IGH gene rearrangements (Dh-Jh), IGK gene rearrangements (Vκ-Jκ, Vκ-KDE, intronRSS-KDE), TRG gene rearrangements (Vγ-Jγ), TRD gene rearrangements (Vδ-Dδ, Dδ-Dδ) and TRB gene rearrangements (Vβ-Jβ) (all single PCRs). These rearrangements were amplified in a first PCR reaction of 25 cycles, using locus-specific primer sets.20 Next, 1:100 of these PCR products was further amplified in a second PCR reaction of 14 cycles, using universal primers complementary to the adaptors that were linked to the locus-specific primers with sample-identifiers. The final PCR products were sequenced using the Illumina HiSEQ platform. Low-quality reads were filtered out and sequences with a single read were excluded.19 For each sequence, the absolute read count, i.e. the read count corrected for PCR amplification by the spike-in method, was calculated (Online Supplementary Methods). Also the frequency for each sequence was determined. Importantly, the frequency represents a fraction of the rearranged genes of the involved locus, rather than a fraction of all, including germline, IG and TR genes, as is the case in SB and PCR-based IG/TR gene rearrangement analysis (Online Supplementary Methods).
NGS clonal variant comparison pipeline and data analysis
We developed a new analysis tool (PRISCA: PRecISe Clonal Analysis) in Galaxy21 to compare sequences from paired and triplicate samples (Online Supplementary Methods). The graphical output from this tool was used for further data analysis. The IG/TR gene rearrangements at diagnosis were subsequently analyzed according to the criteria described in the Online Supplementary Methods. In line with previous reports,19,22–24 an index clone was arbitrarily defined as a clonal IG/TR gene rearrangement with a frequency >5%.
Results
Normal and regenerating BM samples define thresholds for the exclusion of normal lymphocyte-derived IG/TR gene rearrangements
For in-depth analysis of IG/TR gene rearrangements in BCP-ALL patients, it is necessary to discriminate the IG/TR gene rearrangements derived from leukemic clones from the background that also contains IG/TR gene rearrangements derived from normal B-cell and T-cell clones. We therefore aimed to define read count thresholds (RCTs) above which we can fairly assume normal lymphocyte-derived IG/TR gene rearrangements to be absent. To define these RCTs, we determined the maximum of the absolute read counts of normal lymphocyte-derived IG/TR gene rearrangements detected in eight non-leukemic BM samples, i.e. a normal BM sample from a healthy child, two normal BM samples from BCP-ALL patients one year after end of therapy, and five regenerating BM samples from T-ALL patients during therapy intervals. Importantly, regenerating BM samples did not contain residual leukemic IG/TR gene rearrangements (as determined by the absence of leukemic IG/TR gene rearrangements which were found by NGS in the corresponding diagnosis sample) (data not shown). We particularly included regenerating BM samples (and only limited healthy BM samples) as controls since we expected the highest background of IG/TR rearrangements in such samples with increased numbers of early B-cell precursors. Analysis of complete IGH gene rearrangements showed that the maximum read count of complete IGH gene rearrangements in the non-leukemic BM samples was 26 (Figure 1). The IGH Vh-DJh RCT was therefore set at 30 reads. The same strategy was applied to the other types of IG/TR gene rearrangements, resulting in an RCT of 30 reads for Dh-Jh rearrangements, 70 reads for Vκ-Jκ rearrangements, 40 reads for Vκ-KDE rearrangements, 170 reads for intronRSS-KDE rearrangements, 50 reads for Vγ-Jγ rearrangements, 20 reads for Vδ-Dδ rearrangements, 30 reads for Dδ-Dδ rearrangements, and 30 reads for Vγ-Jβ rearrangements (Online Supplementary Figure S1). The relatively high maximum read count of intronRSS-KDE rearrangements was probably due to the limited junctional variability of intronRSS-KDE rearrangements. Importantly, these RCTs are dependent on DNA input and are therefore not universal, but are specific for our data.
Figure 1.

Read counts (RC) of Vh-DJh rearrangements in bone marrow (BM) from a healthy child, a T- cell acute lymphoblastic leukemia (TALL) patient at a therapy interval (regenerating BM) and a B-cell precursor (BCP)-acute lymphoblastic leukemia (ALL) patient at one year after end of therapy (representative examples of all non-leukemic BM samples; total n=8). Vh-DJh with a read count of <1 (caused by amplification-correction) were displayed as rearrangements with a read count of 1. Black line indicates the established threshold for Vh-DJh rearrangements. Regenerating BM showed structurally lower numbers of unique Vh-DJh rearrangements compared to healthy control BM and BM one year after end of therapy. This was also true for Vκ-Jκ, Vκ-KDE and intronRSS-KDE rearrangements, but not Dh-Jh rearrangements (Online Supplementary Figure S1). This was expected, since the B-cell population in regenerating BM mainly consists of pre-B-I stage cells.25,26
Samples run in triplicate show that our thresholds also exclude IG/TR gene rearrangements derived from technical artifacts
We aimed to evaluate whether IG/TR gene rearrangements with a read count above the RCTs were truly clonal, i.e. were derived from leukemic clones and not from technical artifacts (non-clonal rearrangements whose read counts were insufficiently corrected after PCR amplification). We therefore prepared three independent libraries of the same sample and performed three independent NGS-runs, based on the fair assumption that IG/TR gene rearrangements can be regarded as clonal if present in all three samples or if present in two of the three samples (since small clones can coincidently be absent in a sampling volume). Analysis of complete IGH gene rearrangements in a BM sample at diagnosis showed that all complete IGH gene rearrangements with a read count above the previously defined RCT, and thus considered to be derived from leukemic clones, were present in all three replicates, indicating that these were not technical arte-facts (Figure 2). Analysis of the other IG/TR gene rearrangement types in this diagnosis sample showed the same (Online Supplementary Figure S2). Thus, by using these RCTs, we ensured that the included rearrangements (above the RCT) were very likely to be derived from leukemic clones (as opposed to normal lymphocytes or technical artefacts), thereby accepting that leukemic rearrangements with a read count below the RCT will be ignored. Analysis of IG/TR gene rearrangements in a regenerating BM sample of a T-ALL patient showed, as expected, that none of the rearrangements had a read count above the RCT. Nevertheless, part of the IG/TR gene rearrangements with a read count below the RCT were present in two or three of the replicates, and could thus be derived from normal B-cell or T-cell clones (Online Supplementary Figure S3).
Figure 2.

Read counts of Vh-DJh rearrangements in bone marrow (BM) replicates from a B-cell precursor (BCP)-acute lymphoblastic leukemia (ALL) patient at diagnosis. This BM was sequenced in triplicate, resulting in IGH gene rearrangements that were found in one, in two or in all three replicates. Vh-DJh rearrangements with a read count of <1 (caused by amplification-correction) were displayed as rearrangements with a read count of 1. Black line indicates the previously defined threshold for Vh-DJh rearrangements (30 reads).
Paired BM samples show homogeneous distribution in BCP-ALL, also for small subclones
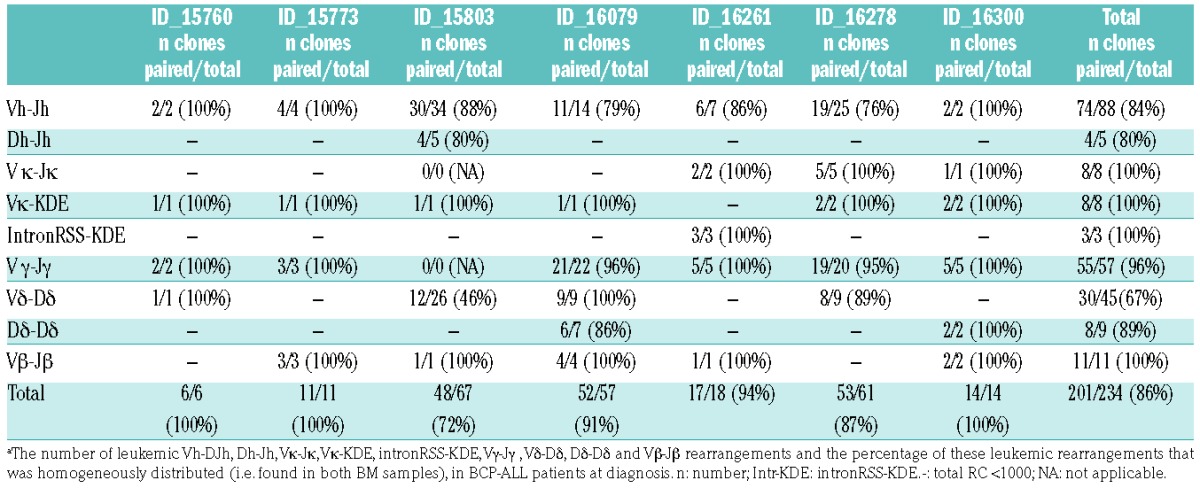
Next, we aimed to analyze the number and particularly the tissue distribution of clones in BCP-ALL patients. Therefore, we performed NGS of IG/TR gene rearrangements in paired BM samples (from the left and the right pelvic bone) from newly diagnosed BCP-ALL patients (n=7). By applying the previously described thresholds (now expressed as frequency) and analysis criteria (Online Supplementary Methods) to complete IGH rearrangements, oligoclonality was detected in 6 out of 7 BCP-ALL patients (Figure 3). The number of index clones (frequency of >5%) ranged from 1 to 5 (median: 1), whereas the total number of (sub)clones ranged from 2 to 34. With regard to distribution of the clones, 84% (74 out of 88) of all leukemic Vh-DJh rearrangements were paired (i.e. present in both BM samples), ranging from 76% to 100% per patient (Figure 3). Importantly, 61 out of the 74 (82%) paired leukemic Vh-DJh rearrangements had a comparable frequency (<5-fold difference) in the two BM samples. Within the 13 paired leukemic Vh-DJh index clones, all Vh-DJh rearrangements had a comparable frequency in the two BM samples. The leukemic Vh-DJh rearrangements which were present in only one of the two samples all had a very low frequency (below 0.2%) and often shared a common Dh-Jh stem with a paired leukemic Vh-DJh rearrangement (see below). Analysis of the other types of IG/TR gene rearrangements also showed that the vast majority of the leukemic rearrangements were paired: 4 out of 5 (80%) Dh-Jh rearrangements, 8 out of 8 (100%) Vκ-Jκ rearrangements, 8 out of 8 (100%) Vκ-KDE rearrangements, 3 out of 3 (100%) intronRSS-KDE rearrangements, 55 out of 57 (96%) Vγ-Jγ rearrangements, 30 out of 45 (67%) Vδ-Dδ rearrangements, 8 out of 9 (89%) Dδ-Dδ rearrangements, and 11 out of 11 (100%) Vβ-Jβ rearrangements (Online Supplementary Figure S4). Within all paired leukemic IG/TR gene rearrangements, 171 out of all 199 (86%) showed a comparable frequency in the two BM samples. The overall results per patient are summarized in Table 1.
Figure 3.

Frequencies of leukemic Vh-DJh rearrangements in paired bone marrow (BM) samples [left (L) and right (R) pelvic bone] from B-cell precursor (BCP)-acute lymphoblastic leukemia (ALL) patients at diagnosis. For each leukemic Vh-DJh rearrangement, the presence in both BM samples (L+R) or in only one of the two BM samples (L or R) is indicated. The background area, which also contains Vh-DJh rearrangements derived from normal B-cell clones, is indicated for each sample (gray). *Vh-DJh rearrangement sharing a common Dh-Jh stem with a paired leukemic Vh-DJh rearrangement.
Table 1.
Number and distribution of leukemic clones at B-cell precursor acute lymphoblastic leukemia (BCP-ALL) diagnosis in paired bone marrow (BM) samples.a
Analysis of leukemic Dh-Jh stems at very low frequency levels suggests even higher numbers of homogeneously distributed subclones
We subsequently aimed to estimate the number of leukemic IG/TR gene rearrangements that were present at very low frequencies and that were, based on the background-threshold, excluded from previous analyses. Therefore, we visualized all paired complete IGH gene rearrangements, irrespective of their frequency (i.e. also with a frequency below the threshold). Only paired IGH gene rearrangements (i.e. present in both BM samples) were visualized in order to exclude potential technical artifacts. The resulting graph showed that, besides the previously detected paired IGH gene rearrangements, many more paired IGH gene rearrangements were present below the threshold (Figure 4). However, a paired IGH gene rearrangement below the threshold can either be derived from a leukemic clone or derived from a normal mature B-cell clone which had been distributed throughout the BM compartment. To discriminate, at sub-threshold level, leukemia-derived IGH gene rearrangements from normal B-cell-derived IGH gene rearrangements, we searched for common Dh-Jh stems. An IGH gene rearrangement below the threshold that shared a common Dh-Jh stem with a leukemic IGH gene rearrangement above the threshold was also considered leukemic (since both were derived from the same ancestral clone). This analysis showed that, on top of the leukemic IGH gene rearrangements found in the previous analyses (above the threshold), even more leukemic IGH gene rearrangements are present at very low frequency levels (below the threshold) (summarized in Figure 5). The exact number of leukemic IGH gene rearrangement per patient remains unknown, since the origin (leukemia- or mature B-cell-derived) of the IGH gene rearrangements with a frequency below the threshold and without a common Dh-Jh stem could not be determined. Of note, the IGH gene rearrangements with a common Dh-Jh stem also often had a common V-D junction, indicating that V to D-J joining as well as Vh-replacement had taken place.
Figure 4.

Frequencies of Vh-DJh rearrangements in paired bone marrow (BM) samples [left (L) and right (R) pelvic bone] from B-cell precursor (BCP)-acute lymphoblastic leukemia (ALL) patients at diagnosis. All paired Vh-DJh rearrangements are shown, i.e. with a frequency above as well as below the threshold. Background area of each sample is indicated in gray.
Figure 5.

Schematic representation of the relations between Vh-DJh rearrangements, based on common Dh-Jh stems, in bone marrow (BM) samples from B-cell precursor (BCP)-acute lymphoblastic leukemia (ALL) patients at diagnosis. The size of the circle represents the frequency of the Vh-DJh rearrangement. Blue or red indicates a leukemic origin (blue: based on frequency; red: based on a common Dh-Jh stem), whereas green indicates small clones of unknown origin (no leukemic Dh-Jh stem).
Paired BM-PB samples confirm the homogeneous distribution of BCP-ALL clones via PB
The above data indicate that BCP-ALL clones are distributed homogeneously over the BM compartment, implying that migration via PB is required. Therefore, we investigated whether leukemia-derived IG/TR gene rearrangements detected in BM can also be found in PB. To do so, we performed NGS of IG/TR gene rearrangements in paired BM-PB samples from 5 newly diagnosed BCP-ALL patients. Leukemic IG/TR gene rearrangements found in the BM sample (i.e. with a frequency above the previously established thresholds) were searched in the corresponding PB sample and classified as ‘paired’ when also present in the PB sample. Analysis of complete IGH gene rearrangements showed that 98% (43 out of 44) of all leukemic Vh-DJh rearrangements were paired (Figure 6). Forty-two of the 43 (98%) paired leukemic Vh-DJh rearrangements had a comparable frequency (<5-fold difference) in the two corresponding samples. The only leukemic Vh-DJh rearrangement present in the BM sample but not in the PB sample, had a frequency of 0.04% and shared a common Dh-Jh stem with a paired leukemic Vh-DJh rearrangement. Analysis of the other IG and TR gene rearrangement types also showed that the majority of the leukemic rearrangements were paired: 30 out of 32 (94%) Dh-Jh rearrangements, 2 out of 2 (100%) Vκ-Jκ rearrangements, 1 out of 1 (100%) Vκ-KDE rearrangements, 9 out of 9 (100%) Vγ-Jγ rearrangements, 320 out of 403 (79%) Vδ-Dδ rearrangements, 17 out of 17 (100%) Dδ-Dδ rearrangements, and 4 out of 4 (100%) Vβ-Jβ rearrangements (no clonal intronRSS-KDE rearrangements were found) (Online Supplementary Figure S5). Within all paired leukemic IG/TR gene rearrangements, 411 out of all 426 (96%) showed a comparable frequency between the BM and the corresponding PB sample.
Figure 6.

Frequencies of leukemic Vh-DJh rearrangements in paired bone marrow (BM)-peripheral (PB) samples from B-cell precursor (BCP)-acute lymphoblastic leukemia (ALL) patients at diagnosis. For each leukemic Vh-DJh rearrangement in the BM sample (above the background threshold), the presence in both samples (BM+PB) or in only the BM sample (BM) is indicated. Diamonds represent Vh-DJh rearrangements found in BM, triangles represent Vh-DJh rearrangements found in PB. Background area of each sample is indicated in gray. *Vh-DJh rearrangement sharing a common Dh-Jh stem with a paired leukemic Vh-DJh rearrangement.
Discussion
To study the number of clones and their distribution in patients with BCP-ALL, we performed NGS of IG/TR gene rearrangements in paired BM-BM and BM-PB samples at diagnosis. First, we aimed to carefully define background RCTs, which exclude normal lymphocyte-derived IG/TR gene rearrangements when using an identical amount of DNA per sample. Therefore, we determined the maximum read count of these normal lymphocyte-derived IG/TR gene rearrangements in non-leukemic BM, i.e. healthy control BM, regenerating BM, and BM one year after end of therapy. Second, by analyzing BCP-ALL samples at diagnosis in triplicate, we confirmed that all IG/TR gene rearrangements with a read count above the pre-set RCT represented clonal IG/TR gene rearrangements, and not technical artifacts. Of note, RCTs could not exclude IG/TR gene rearrangements derived from normal lymphocyte expansions directed against leukemic cells, since these expansions were not present in the studied follow-up samples (or in the healthy control BM sample). However, since many Vh-DJh rearrangements shared a common stem with an index clone and since, besides clonal TR gene rearrangements, also clonal IG gene rearrangements were observed (whereas reactive expansions against leukemic cells would be mainly of T-cell origin), it is likely that most IG/TR gene rearrangements with read counts above the RCTs are of leukemic origin.
Analysis of IG/TR gene rearrangements in paired BM-BM diagnosis samples showed that all 7 patients were oligoclonal. Remarkably high numbers of leukemic Vh-DJh, Vγ-Jγ and Vδ-Dδ rearrangements per patient could be detected (e.g. up to 34 leukemic Vh-DJh rearrangements). Even higher numbers of leukemic rearrangements per patient were detected when common Dh-Jh stem (junction sequence) analysis was used to also include Vh-DJh rearrangements below the background threshold (up to 171 leukemic Vh-DJh rearrangements). Previous studies that had used SB to analyze IG/TR gene rearrangements reported that only 40% of the ALL-patients were oligoclonal, with maximally 9 leukemic rearrangements per patient.11,14 The discrepancies between these previous findings and our current results can be explained by the difference in sensitivity of the applied methods. A more recent study that used NGS to analyze IGH gene rearrangements, showed, in line with our results, that far more IGH gene rearrangements per patient can be found when analysis down to very low frequency levels is allowed.15 However, in contrast with our results, this study also reported on a few patients (4 out of the 51 studied patients) in which more than 1000 leukemic IGH gene rearrangements were found. Patients with such exceptionally high numbers of IGH gene rearrangements were not present in our study, which is probably related to the lower number of patients analyzed. In any case, we showed that the number of leukemic IG/TR gene rearrangements per BCP-ALL patient is considerably higher than previously observed.
IG/TR gene rearrangements are used as PCR targets for MRD detection.27–31 Insight into subclone formation and distribution of these subclones might improve selection of MRD targets. Our current findings indicate that MRD targets have so far been selected from only a small part of the total number of leukemic IG/TR gene rearrangements present at diagnosis, i.e. from those that were sufficiently large to be detected by PCR-based methods (generally index clones, i.e. with a frequency >5%). Still, more than 90% of the relapsed cases had maintained the IG and TR targets selected at diagnosis.30 Apparently, monitoring of only the index clones might be sufficient to predict relapse in most BCP-ALL patients. Furthermore, our observation that the vast majority of leukemic clones could be detected both in BM and PB implies that also PB samples may reliably be used for NGS-based selection of MRD targets.
By analyzing paired BM samples, we showed that the vast majority of the leukemic rearrangements were present in both BM samples and that their frequency was generally similar in both samples. The few leukemic rearrangements that were present in only one of the two samples generally had a low frequency (<2%). Probably, also these low-frequent clones were present in the whole BM compartment, but were absent in one of the two samples based on statistical chance. By analyzing paired BM-PB samples (from other patients), we showed that almost all leukemic rearrangements that were present in BM, were also present in PB, generally at a similar frequency. As an exception to the above discussed results, relatively many leukemic Vδ-Dδ rearrangements in patient 15803 (child with a TCF3-PBX1 translocation) and in patient 15507 (infant with an MLL-ENL translocation) were present in only one of the paired BM samples (54% and 22%, respectively). Since infants with a MLL-R translocation are known to be highly oligoclonal,32,33 the specific genetic aberrancies might be associated with the deviating results on Vδ-Dδ rearrangements.34 It is important to note that we analyzed diagnosis samples in which tumor percentages were generally high, and that the situation during treatment (with lower numbers of ALL cells) might be different. Nevertheless, our previous study16 suggests that also in an MRD setting the ALL clones are homogeneously distributed over the BM compartment.
Next-generation sequencing-based IG and TR gene rearrangement analysis in paired BM-BM and BM-PB samples from newly diagnosed BCP-ALL patients has, to our knowledge, not been performed previously. Our early SB study on IG gene rearrangements in paired BM-PB samples of newly diagnosed BCP-ALL patients showed that a relatively high number of leukemic IGH gene rearrangements were detected in the BM sample, but not or in a lower frequency in the corresponding PB sample,17 which seems to be in contrast with our current results. However, this discrepancy is probably due to technical differences between Southern blot and NGS analytical methods (see Methods section).
Together, our paired BM-BM and paired BM-PB NGS analyses showed that almost all leukemic IG/TR gene rearrangements are present in comparable frequencies in the BM and PB compartment. We did not study leukemic IG/TR gene rearrangements in tissues other than BM or PB. However, it might be speculated that extramedullary dissemination is different from distribution within the BM and PB compartment, since only clones with specific characteristics (e.g. homing receptors) are able to migrate to extramedullary sites.35 Furthermore, it should be noted that we defined subclones based on IG/TR gene rearrangements. It may well be that subclones with different somatic mutations, related to leukemogenesis and prone to positive selection, are distributed in a less homogeneous way. Exome sequencing of paired BM samples may answer this question.
In conclusion, by using NGS data in combination with carefully defined thresholds, we were able to study leukemic IG/TR gene rearrangements down to a low level (0.01–0.1%), without the interference of background rearrangements. By studying paired BM-BM and paired BM-PB samples, we showed that the relative quantity of IG/TR gene rearrangements within the total (rearranged) BCP-ALL population is generally similar throughout the BM compartment and between BM and PB. Apparently, after their development at different BM sites in the body, almost all IG/TR-defined clones within a BCP-ALL population, even the small subclones, disseminate homogeneously via PB throughout the BM compartment.
Supplementary Material
Acknowledgments
We gratefully thank Arjan Lankester and the involved technicians of the Laboratory for Medical Immunology, in particular Rianne Noordijk, Patricia Hoogeveen and Maaike de Bie. We acknowledge Auke Beishuizen for providing BM samples from the BCP-ALL patients.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/102/11/1869
Funding
The research for this manuscript was (in part) performed within the framework of the Erasmus Postgraduate School Molecular Medicine and was financially supported by PrioMedChild, project 40-41800-98-027.
References
- 1.Tonegawa S. Somatic generation of antibody diversity. Nature. 1983;302(5909):575–581. [DOI] [PubMed] [Google Scholar]
- 2.Alt FW, Oltz EM, Young F, Gorman J, Taccioli G, Chen J. VDJ recombination. Immunol Today. 1992;13(8):306–314. [DOI] [PubMed] [Google Scholar]
- 3.Pui CH, Campana D, Evans WE. Childhood acute lymphoblastic leukaemia–current status and future perspectives. Lancet Oncol. 2001;2(10):597–607. [DOI] [PubMed] [Google Scholar]
- 4.van der Velden VH, van Dongen JJ. MRD detection in acute lymphoblastic leukemia patients using Ig/TCR gene rearrangements as targets for real-time quantitative PCR. Methods Mol Biol. 2009;538:115–150. [DOI] [PubMed] [Google Scholar]
- 5.Chen Z, Le Paslier D, Dausset J, et al. Human T cell gamma genes are frequently rearranged in B-lineage acute lymphoblastic leukemias but not in chronic B cell proliferations. J Exp Med. 1987;165(4):1000–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Szczepanski T, Beishuizen A, Pongers-Willemse MJ, et al. Cross-lineage T cell receptor gene rearrangements occur in more than ninety percent of childhood precursor-B acute lymphoblastic leukemias: alternative PCR targets for detection of minimal residual disease. Leukemia. 1999;13(2):196–205. [DOI] [PubMed] [Google Scholar]
- 7.Fronkova E, Krejci O, Kalina T, Horvath O, Trka J, Hrusak O. Lymphoid differentiation pathways can be traced by TCR delta rearrangements. J Immunol. 2005;175(4):2495–2500. [DOI] [PubMed] [Google Scholar]
- 8.Choi Y, Greenberg SJ, Du TL, et al. Clonal evolution in B-lineage acute lymphoblastic leukemia by contemporaneous VH-VH gene replacements and VH-DJH gene rearrangements. Blood. 1996;87(6):2506–2512. [PubMed] [Google Scholar]
- 9.Moreira I, Papaioannou M, Mortuza FY, et al. Heterogeneity of VH-JH gene rearrangement patterns: an insight into the biology of B cell precursor ALL. Leukemia. 2001;15(10):1527–1536. [DOI] [PubMed] [Google Scholar]
- 10.Stankovic T, Weston V, McConville CM, et al. Clonal diversity of Ig and T-cell receptor gene rearrangements in childhood B-precursor acute lymphoblastic leukaemia. Leuk Lymphoma. 2000;36(3–4):213–224. [DOI] [PubMed] [Google Scholar]
- 11.Beishuizen A, Hahlen K, Hagemeijer A, et al. Multiple rearranged immunoglobulin genes in childhood acute lymphoblastic leukemia of precursor B-cell origin. Leukemia. 1991;5(8):657–667. [PubMed] [Google Scholar]
- 12.van der Velden VH, Bruggemann M, Hoogeveen PG, et al. TCRB gene rearrangements in childhood and adult precursor-BALL: frequency, applicability as MRD-PCR target, and stability between diagnosis and relapse. Leukemia. 2004;18(12):1971–1980. [DOI] [PubMed] [Google Scholar]
- 13.van der Velden VH, Szczepanski T, Wijkhuijs JM, et al. Age-related patterns of immunoglobulin and T-cell receptor gene rearrangements in precursor-B-ALL: implications for detection of minimal residual disease. Leukemia. 2003;17(9):1834–1844. [DOI] [PubMed] [Google Scholar]
- 14.Beishuizen A, Verhoeven MA, van Wering ER, Hahlen K, Hooijkaas H, van Dongen JJ. Analysis of Ig and T-cell receptor genes in 40 childhood acute lymphoblastic leukemias at diagnosis and subsequent relapse: implications for the detection of minimal residual disease by polymerase chain reaction analysis. Blood. 1994;83(8):2238–2247. [PubMed] [Google Scholar]
- 15.Gawad C, Pepin F, Carlton VE, et al. Massive evolution of the immunoglobulin heavy chain locus in children with B precursor acute lymphoblastic leukemia. Blood. 2012;120(22):4407–4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van der Velden VH, Hoogeveen PG, Pieters R, van Dongen JJ. Impact of two independent bone marrow samples on minimal residual disease monitoring in childhood acute lymphoblastic leukaemia. Br J Haematol. 2006;133(4):382–388. [DOI] [PubMed] [Google Scholar]
- 17.Beishuizen A, Verhoeven MA, Hahlen K, van Wering ER, van Dongen JJ. Differences in immunoglobulin heavy chain gene rear-rangmeent patterns between bone marrow and blood samples in childhood precursor B-acute lymphoblastic leaukemia at diagnosis. Leukemia. 1993;7(6):60–63. [PubMed] [Google Scholar]
- 18.van der Velden VH, Cazzaniga G, Schrauder A, et al. Analysis of minimal residual disease by Ig/TCR gene rearrangements: guidelines for interpretation of real-time quantitative PCR data. Leukemia. 2007;21(4):604–611. [DOI] [PubMed] [Google Scholar]
- 19.Faham M, Zheng J, Moorhead M, et al. Deep-sequencing approach for minimal residual disease detection in acute lymphoblastic leukemia. Blood. 2012;120(26):5173–5180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Faham M. Methods of monitoring conditions by sequence analysis. 2011. Available from: http://www.google.sr/patents/US9228232
- 21.Afgan E, Baker D, van den Beek M, et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 2016;44(W1):W3–W10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sekiya Y, Xu Y, Muramatsu H, et al. Clinical utility of next-generation sequencing-based minimal residual disease in paediatric B-cell acute lymphoblastic leukaemia. Br J Haematol. 2017;176(2):248–257. [DOI] [PubMed] [Google Scholar]
- 23.Pulsipher MA, Carlson C, Langholz B, et al. IgH-V(D)J NGS-MRD measurement pre- and early post-allotransplant defines very low- and very high-risk ALL patients. Blood. 2015;125(22):3501–3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ladetto M, Bruggemann M, Monitillo L, et al. Next-generation sequencing and real-time quantitative PCR for minimal residual disease detection in B-cell disorders. Leukemia. 2014;28(6):1299–1307. [DOI] [PubMed] [Google Scholar]
- 25.van Lochem EG, Wiegers YM, van den Beemd R, Hahlen K, van Dongen JJ, Hooijkaas H. Regeneration pattern of precursor-B-cells in bone marrow of acute lymphoblastic leukemia patients depends on the type of preceding chemotherapy. Leukemia. 2000;14(4):688–695. [DOI] [PubMed] [Google Scholar]
- 26.van Wering ER, van der Linden-Schrever BE, Szczepanski T, et al. Regenerating normal B-cell precursors during and after treatment of acute lymphoblastic leukaemia: implications for monitoring of minimal residual disease. Br J Haematol. 2000;110(1):139–146. [DOI] [PubMed] [Google Scholar]
- 27.Szczepanski T, Orfao A, van der Velden VH, San Miguel JF, van Dongen JJ. Minimal residual disease in leukaemia patients. Lancet Oncol. 2001;2(7):409–417. [DOI] [PubMed] [Google Scholar]
- 28.van Dongen JJ, Seriu T, Panzer-Grumayer ER, et al. Prognostic value of minimal residual disease in acute lymphoblastic leukaemia in childhood. Lancet. 1998;352(9142):1731–1738. [DOI] [PubMed] [Google Scholar]
- 29.van der Velden VH, Hochhaus A, Cazzaniga G, Szczepanski T, Gabert J, van Dongen JJ. Detection of minimal residual disease in hematologic malignancies by real-time quantitative PCR: principles, approaches, and laboratory aspects. Leukemia. 2003;17(6):1013–1034. [DOI] [PubMed] [Google Scholar]
- 30.Pieters R, de Groot-Kruseman H, Van der Velden V, et al. Successful Therapy Reduction and Intensification for Childhood Acute Lymphoblastic Leukemia Based on Minimal Residual Disease Monitoring: Study ALL10 From the Dutch Childhood Oncology Group. J Clin Oncol. 2016;34(22):2591–601. [DOI] [PubMed] [Google Scholar]
- 31.van Dongen JJ, van der Velden VH, Bruggemann M, Orfao A. Minimal residual disease diagnostics in acute lymphoblastic leukemia: need for sensitive, fast, and standardized technologies. Blood. 2015;125(26):3996–4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bardini M, Woll PS, Corral L, et al. Clonal variegation and dynamic competition of leukemia-initiating cells in infant acute lymphoblastic leukemia with MLL rearrangement. Leukemia. 2015;29(1):38–50. [DOI] [PubMed] [Google Scholar]
- 33.Jansen MW, Corral L, van der Velden VH, et al. Immunobiological diversity in infant acute lymphoblastic leukemia is related to the occurrence and type of MLL gene rearrangement. Leukemia. 2007;21(4):633–641. [DOI] [PubMed] [Google Scholar]
- 34.Brumpt C, Delabesse E, Beldjord K, et al. The incidence of clonal T-cell receptor rearrangements in B-cell precursor acute lymphoblastic leukemia varies with age and genotype. Blood. 2000;96(6):2254–2261. [PubMed] [Google Scholar]
- 35.van der Velden VH, de Launaij D, de Vries JF, et al. New cellular markers at diagnosis are associated with isolated central nervous system relapse in paediatric B-cell precursor acute lymphoblastic leukaemia. Br J Haematol. 2016;172(5):769–781. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.