

Constitutive activation of the FMS-related tyrosine kinase-3 (FLT3) by mutations or high expression levels is common in acute leukemias.1 Aberrant FLT3 signaling is mediated via the RAS/MAPK and PI-3-Kinase/AKT pathways leading to proliferation, survival, and therapy resistance.1 Infants and children with KMT2A (MLL)-rearranged acute lymphoblastic leukemia (KMT2Ar-ALL) have an unfavorable prognosis. Activating FLT3 tyrosine kinase domain mutations (TKDs) and N/KRAS mutations are frequently identified in KMT2Ar-ALL.2–6 This subgroup is also characterized by exceptionally high FLT3 expression associated with ligand-independent signaling.7,8 In order to investigate the prognostic impact of a constitutive activation of FLT3 in infants and children, we analyzed FLT3 transcription levels and common mutations in FLT3 and NRAS/KRAS. We show that in infants, high FLT3 activation correlates with a superior prognosis. Moreover, we show that RAS mutations alter the positive effect of constitutive FLT3 activation and increase relapse risk. High FLT3 activation surprisingly inhibits proliferation of human KMT2Ar-ALL samples in patient-derived xenografts (PDX). Small molecule FLT3 inhibition was efficient in FLT3high PDX without an activating FLT3 mutation suggesting that FLT3 expression, regardless of mutations, may play a role for novel therapies in the future.
Analyses were performed on samples from infants and children with KMT2Ar-ALL from trials ALL-BFM 86, 90, 95, 2000, AEIOP-BFM ALL 2009 and German patients enrolled in Interfant-99/-06 (Online Supplementary Methods, Online Supplementary Table S1). We detected mutations in the TKD or the juxtamembrane domain (JMD) of FLT3 in 15/167 (9%) patients (Table 1). In children (patients >1 year, n=72), only one FLT3 aberration (TKD-D835H) was found (1/72, 1.4%). In infants (n=95), FLT3-TKD mutations were identified in 12/95 (12.6%) patients. Seven of these had D835 substitutions and 4 had I836 deletions, all of which are activating.9,10 One infant had a novel 12-base pair (bp) deletion and 3-bp insertion involving codons D835 to S838 similar to the mutation reported by Taketani et al.3 Notably, of the 12 infants with a FLT3-TKD mutation, only 3 suffered from relapse; 3-year cumulative incidence of relapse (CIR) 26±14%. In only 2/95 (2.1%) infant patients, alterations in the JMD of FLT3 were detected, namely one uncharacterized duplication, and one novel Y589_F594 deletion. Both patients relapsed.
Table 1.
Clinical features and FLT3 transcription level of infants and children with MLL-rearranged ALL and mutations in the FLT3, NRAS, or KRAS genes.
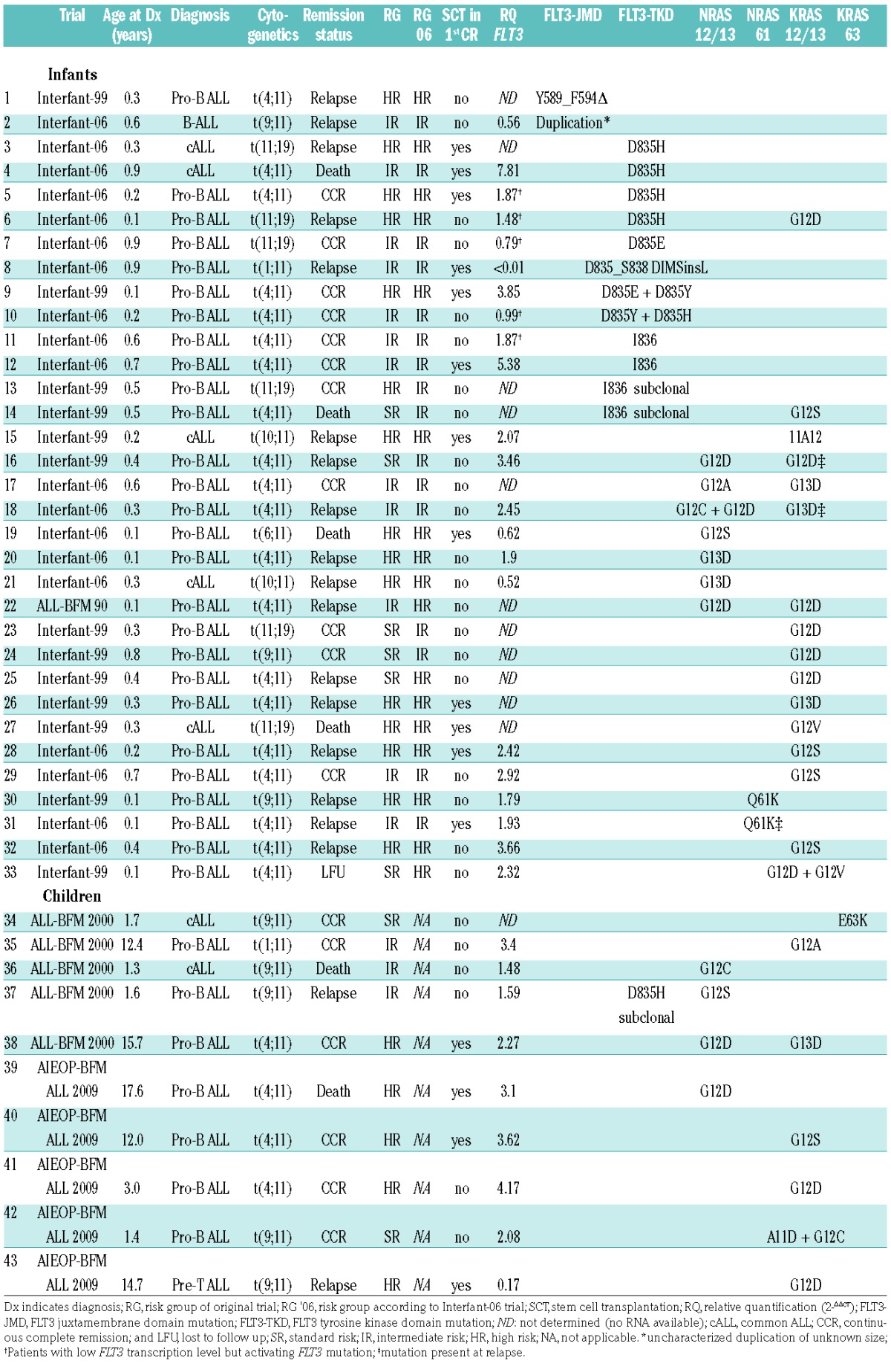
We further identified 39 non-synonymous N/KRAS mutations in 21/95 (22.1%) infants and 10/72 (13.9%) children (Table 1). N/KRAS 12/13 mutations were detected in 28/167 patients. Seven patients had more than one N/KRAS 12/13 mutation. One of these had a KRAS A11D and a G12C mutation. Another patient had a 3-bp insertion leading to a KRAS 10A11 mutation. NRAS 61 mutations were found in 2 patients. No KRAS 61, but one KRAS E63K mutation, previously described in Philadelphia chromosome-like ALL,11 was identified. Of 8 available relapse samples, 3 had RAS mutations present at diagnosis. Of the 5 relapse specimens that did not have RAS mutations at relapse, only 1 had a RAS mutation at diagnosis, suggesting a subclonal mutation. None of the 8 patients had a FLT3 mutation, neither at diagnosis nor at relapse. In infants, the presence of RAS mutations correlated with a lower 3-year probability of event-free survival (3y-pEFS mut 16±8% vs. wt 43±6%, P=0.04), and a higher 3y-CIR (mut 69±11% vs. wt 40±6%, P=0.01).
Next, we analyzed FLT3 transcription by qRT-PCR in 124 patients (69 infants, 55 children). When we separated the infant cohort into 2 groups according to the median RQ value (< or ≥2, Figure 1A), a low FLT3 transcription level significantly correlated with a low pEFS (FLT3low 16±7% vs. FLT3high 57±9%, P=0.009) and a high CIR (FLT3low 69±8% vs. FLT3high 25±8%, P=0.001) (Figure 1B–C). Moreover, the median FLT3 transcription level was low in all 8 relapse samples analyzed (initial 1.85 vs. relapse 1.46, P=0.263). It is important that median FLT3 transcription levels in the FLT3low group of infants was 16-fold higher than in bone marrow of healthy controls (FLT3low n=35 vs. healthy n=8, P<0.0001) and 4.6-fold higher than in BCR-ABL-positive ALLs (FLT3low n=35 vs. BCR-ABL n=15, P=0.009). In multivariate analysis, the prognostic impact of FLT3 transcription levels in infants was independent of age (< vs. ≥6 months), initial white blood cell count (< vs. ≥300,000/μl), prednisone response (poor vs. good), and non-remission on day 33: in a Cox regression analysis for EFS and relapse incidence with these covariables, FLT3low resulted in a hazard ratio of 3.80 (95% confidence interval (CI) 1.71–8.44, P=0.001) for EFS and 6.68 (95% CI 2.37–18.8, P<0.0001) for relapse incidence (Online Supplementary Table S2). No prognostic relevance of FLT3 transcription was observed for children with B- or biphenotypic ALL (3y-pEFS FLT3low 71±11% vs. FLT3high 65±10%, P=0.73, and 3y-CIR FLT3low 12±8% vs. FLT3high 12±7%, v=0.93). Children with T-ALL were excluded as they showed very low FLT3 transcription (B-cell/biphenotypic ALL, n=46, median 3.17 vs. T-ALL, n=9, median 0.17, P<0.00001). Of the 6 infants with low FLT3 transcription, but with a FLT3-TKD mutation, two had a relapse. Importantly, in one of these two patients (patient 8), a FLT3-TKD mutation (new del/ins with unknown effects) was present, but the FLT3 gene was not transcribed at all. We next analyzed infants with a presumed high FLT3 activation (FLT3high/mut, FLT3high/non-mut, FLT3low/mut) compared to those with a low FLT3 activation (FLT3low/non-mut/FLT3mut but no transcription) and found the CIR to be further reduced and the pEFS to be further increased in the group with high FLT3 activation (Figure 1D–E). The other relapsed patient with a FLT3-TKD mutation (patient 6) had, in addition, a RAS mutation (Table 1). Likewise, most patients with a high FLT3 transcription level that suffered from relapse also had RAS mutations, suggesting that RAS mutations alter the positive effect of FLT3 activation. A Cox regression analysis of relapse incidence in infants with the same variables as above plus FLT3low vs. FLT3high, RAS mutation vs. wildtype and the interaction of FLT3high and RAS wildtype resulted in a hazard ratio of 0.04 (95% CI 0.01–0.37, P=0.005) for the interaction (Online Supplementary Table S3).
Figure 1.

Prognostic impact of FLT3 transcription and FLT3 activation in infant KMT2Ar-ALL. A: Box plot with FLT3 expression levels in FLT3high, FLT3low, BCR-ABL1 positive patients and in healthy bone marrow. B–E: Kaplan-Meier estimates for probability of event-free survival (pEFS) (B and D) and for cumulative incidence of relapse (CIR) (C and E) at 3 years. Plots for low vs. high levels of FLT3 transcription (RQ < or ≥2) are shown in B and C, and plots for low vs. high FLT3 activation are shown in D and E. High or low activation of FLT3 was derived from FLT3 transcription level (RQ < or ≥2) and FLT3 mutational status (FLT3high/mut, FLT3high/non-mut, FLT3low/mut vs. FLT3low/non-mut/FLT3mut but no transcription).
In order to clarify the protective mechanism of high FLT3 activation, we performed xenotransplantation assays of 19 KMT2Ar samples into NOD.CgPrkdcscid Il2rgtm1Wjl/SzJ mice. When these patients were separated according to FLT3 activation (FLT3high/mut, FLT3high/non-mut, FLT3low/mut vs. FLT3low/non-mut/FLT3mut but no transcription), a high FLT3 activation correlated with prolonged PDX survival, confirming the clinical data (P=0.0054, Figure 2A). In vivo bromodeoxyuridine (BrdU) assays in a subset of 3 FLT3high and 3 FLT3low PDX bearing different patients revealed significantly slower proliferation rates in human leukemic cells from FLT3high mice (Figure 2B–C). Human PDX cells from six situations (FLT3high/mut, FLT3high/non-mut, FLT3low/mut, FLT3low/non-mut, FLT3high/Ras-mut, FLT3low/Ras-mut) revealed an activation of p-Erk in FLT3high, FLT3mut and RAS-mutated, but not in FLT3low/non-mut samples (Figure 2D) supporting that FLT3high/mut, FLT3high/non-mut and FLT3low/mut correspond to high FLT3 activation. High p-Akt was detected in FLT3high, but not in FLT3low samples without RAS-mutations. No changes were detected in RAS-mutated samples, implying a minor role of p-Akt in that situation. Interestingly, CDKN1A/p21 indicating cell cycle arrest was higher in FLT3high patients (Figure 2D), supporting our BrdU data. In order to test if FLT3high ALLs are targetable by FLT3 inhibitors irrespective of FLT3 mutations, we performed experiments in FLT3high/non-mut compared to FLT3low/non-mut PDX. Treatment with Lestaurtinib resulted in a small but significant prolongation of median survival in FLT3high/non-mut xenografts (57 vs. 54 days, P<0.0001) but not in FLT3low/non-mut animals (Figure 2E). When Lestaurtinib was combined with a regimen mimicking standard induction chemotherapy in humans (vincristine, dexamethasone, PEG-asparaginase), survival could be increased by ≈30% as compared to chemotherapy only in FLT3high/non-mut animals (113 vs. 86 days, P=0.0003), whereas this effect was only marginal in the FLT3low/non-mut setting (63 vs. 60 days, P<0.0001) (Figure 2E). These data support the relevance of FLT3 expression irrespective of FLT3 mutations.
Figure 2.

High FLT3 activation inhibits the growth of KMT2Ar-ALL cells in vivo. Cells from patients with a high and a low FLT3 activation status as indicated were injected into NSG mice and mice were sacrificed upon detection of leukemic symptoms. A: Survival prolongation in mice bearing patients with a high FLT3 activation status (FLT3high/mut, FLT3high/non-mut, FLT3low/mut), Kaplan-Meier log-rank test. B: A subgroup of 3 mice from each group, were fed with BrdU before euthanasia. Human leukemic cells were recovered from the spleens (upper panel) and the bone marrow (lower panel), stained with an anti-BrdU antibody and 7-AAD and FACS analysis was performed. The percentages of cells in the different cell cycle phases are depicted. H = high FLT3 activation status; L = low FLT3 activation status; *P<0.05; **P<0.01; ***P<0.001. C: Representative plots of cell cycle distributions of two samples from both groups as indicated and schematic overview. D: Human leukemic cells were recovered from fully engrafted xenograft spleens. Cells were lysed and subjected to Western Blotting experiments for the markers indicated. H = high FLT3 expression; L = low FLT3 expression. Quantification of the p-Erk/Erk ratio was determined using Image J software. E: Xenograft mice (FLT3high/non-mut, upper panel and FLT3low/non-mut, lower panel) were subjected to treatments with vehicle only, Lestaurtinib, chemotherapy only (dexamethasone, vincristine and PEG-asparaginase), or a combination of chemotherapy and Lestaurtinib, as indicated. Survival curves, Kaplan-Meier log-rank test.
In summary, we report that FLT3 activation is associated with a good prognosis in KMT2Ar infant ALL, which is in contrast to several studies.12–14 Chillon et al.13 analyzed a cohort of 17 KMT2A-AF4-positive ALL cases including only 4 infants. Kang et al.14 used FLT3 transcription levels to separate infant KMT2Ar and KMT2A-germline ALL. The latter were characterized by low FLT3 transcription and a good prognosis. Stam et al.12 analyzed a smaller cohort of KMT2Ar infants and identified a small group of FLT3high patients with a low 1y-EFS, but the level of significance was also low. In accordance with a previous study, we confirmed that RAS mutations correlate with a poor prognosis in KMT2Ar-ALL,4 abolishing the positive effect of high FLT3. Conversely, low FLT3 expression is associated with an inferior prognosis, regardless of RAS. Our in vivo studies confirm our clinical data as high FLT3 inhibits cell cycle and delays leukemic outgrowth in mice, suggesting pro-survival functions. Distinction of high/low FLT3 levels remains artificial, and the lack of a validation cohort and small sample sizes should be addressed in the future. Also, we cannot exclude that mutations in other genes may interfere with our findings. A recent clinical trial was unable to show a benefit from adding Lestaurtinib to chemotherapy in KMT2Ar infant ALL patients, even though 95% confidence intervals were very large.15 Further studies are needed to confirm potential effects in subgroups, depending on FLT3 expression and FLT3 and RAS mutational status.
Supplementary Material
Acknowledgments
We thank Birthe Fedders, Katrin Timm-Richert and Katrin Neumann for excellent technical assistance. We thank the Institute of Clinical Molecular Biology in Kiel for providing Sanger sequencing as supported in part by the DFG Cluster of Excellence “Inflammation at Interfaces” and “Future Ocean”. We thank Sandra Greve, Tanja Henke and Stefanie Arndt for technical support.
Footnotes
Funding: this work was supported by the European Union’s Seventh Framework Program (FP7/2007-2013) under the project European Network for Cancer Research in Children and Adolescents (ENCCA), grant agreement HEALTH-F2-2011-261474. J. S. is supported by the Madeleine Schickedanz-Kinderkrebs-Stiftung. D. M. S. is supported by a Max-Eder Junior Research Grant by the Deutsche Krebshilfe e. V. and by the Wilhelm-Sander Stiftung.
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Annesley CE, Brown P. The biology and targeting of FLT3 in pediatric leukemia. Front Oncol. 2014;4:263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Armstrong SA, Kung AL, Mabon ME, et al. Inhibition of FLT3 in MLL. Validation of a therapeutic target identified by gene expression based classification. Cancer Cell. 2003;3(2):173–183. [DOI] [PubMed] [Google Scholar]
- 3.Taketani T, Taki T, Sugita K, et al. FLT3 mutations in the activation loop of tyrosine kinase domain are frequently found in infant ALL with MLL rearrangements and pediatric ALL with hyperdiploidy. Blood. 2004;103(3):1085–1088. [DOI] [PubMed] [Google Scholar]
- 4.Driessen EM, van Roon EH, Spijkers-Hagelstein JA, et al. Frequencies and prognostic impact of RAS mutations in MLL-rearranged acute lymphoblastic leukemia in infants. Haematologica. 2013;98(6):937–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liang DC, Shih LY, Fu JF, et al. K-Ras mutations and N-Ras mutations in childhood acute leukemias with or without mixed-lineage leukemia gene rearrangements. Cancer. 2006;106(4):950–956. [DOI] [PubMed] [Google Scholar]
- 6.Andersson AK, Ma J, Wang J, et al. The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat Genet. 2015;47(4):330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Armstrong SA, Staunton JE, Silverman LB, et al. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet. 2002;30(1):41–47. [DOI] [PubMed] [Google Scholar]
- 8.Stam RW, den Boer ML, Schneider P, et al. Targeting FLT3 in primary MLL-gene-rearranged infant acute lymphoblastic leukemia. Blood. 2005;106(7):2484–2490. [DOI] [PubMed] [Google Scholar]
- 9.Yamamoto Y, Kiyoi H, Nakano Y, et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood. 2001;97(8):2434–2439. [DOI] [PubMed] [Google Scholar]
- 10.Grundler R, Thiede C, Miething C, Steudel C, Peschel C, Duyster J. Sensitivity toward tyrosine kinase inhibitors varies between different activating mutations of the FLT3 receptor. Blood. 2003;102(2):646–651. [DOI] [PubMed] [Google Scholar]
- 11.Roberts KG, Li Y, Payne-Turner D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. 2014;371(11):1005–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stam RW, Schneider P, de Lorenzo P, Valsecchi MG, den Boer ML, Pieters R. Prognostic significance of high-level FLT3 expression in MLL-rearranged infant acute lymphoblastic leukemia. Blood. 2007;110(7):2774–2775. [DOI] [PubMed] [Google Scholar]
- 13.Chillon MC, Gomez-Casares MT, Lopez-Jorge CE, et al. Prognostic significance of FLT3 mutational status and expression levels in MLL-AF4+ and MLL-germline acute lymphoblastic leukemia. Leukemia. 2012;26(11):2360–2366. [DOI] [PubMed] [Google Scholar]
- 14.Kang H, Wilson CS, Harvey RC, et al. Gene expression profiles predictive of outcome and age in infant acute lymphoblastic leukemia: a Children’s Oncology Group study. Blood. 2012;119(8):1872–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown PK, Wang C, Dreyer Z, et al. Addition of FLT3 inhibitor lestaurtinib to post-induction chemotherapy does not improve outcomes in Mll-rearranged infant acute lymphoblastic leukemia (ALL): AALL0631, a Children’s Oncology Group Study. Pediatr Blood Cancer. 2016;63(S5):S321. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.