

Significance
Antibiotic resistance leads to substantial mortality and morbidity and significant economic cost because it seriously undermines our ability to treat bacterial infections. Therefore, a better understanding of the effect of antibiotics on bacteria is needed to increase the effectiveness of treatments and slow the emergence of resistance. The bactericidal effects of antibiotics are triggered by target-specific interactions, but there is growing evidence that an important part of their cytotoxicity results from metabolic disturbances induced by treatment. In this article, we report that the perturbation of DNA replication by a wide-spectrum antibiotic, trimethoprim, affects bacterial metabolism, which provokes the production of genotoxic agents and DNA damage, whose processing ultimately contributes to cell death under both aerobic and anaerobic conditions.
Keywords: antibiotic, trimethoprim, oxidative stress, nitrate respiration, DNA repair
Abstract
The bactericidal effects of antibiotics are undoubtedly triggered by target-specific interactions, but there is growing evidence that an important aspect of cytotoxicity results from treatment-induced metabolic perturbations. In this study, we characterized molecular mechanisms whereby trimethoprim treatment results in cell death, using Escherichia coli as the model organism. E. coli cells grown in rich medium that contained all amino acids and low amounts of thymidine were treated with trimethoprim under aerobic and anaerobic conditions. Under these growth conditions, accelerated thymine depletion is the primary trigger of the processes leading to cell death. Thymine depletion-induced DNA replication stress leads to the production of reactive oxygen species under aerobic conditions and of the DNA-damaging byproducts of nitrate respiration under anaerobic conditions. Lowering the DNA replication initiation rate by introducing the dnaA(Sx) allele or by overexpressing Hda protein reduces the number of active replication forks, which reduces the consumption of thymidine and increases resistance to trimethoprim under both aerobic and anaerobic conditions. Analysis of the involvement of DNA repair enzymes in trimethoprim-induced cytotoxicity clearly indicates that different amounts and/or different types of DNA lesions are produced in the presence or absence of oxygen. Maladaptive processing of the DNA damage by DNA repair enzymes, in particular by MutM and MutY DNA glycosylases, ultimately contributes to cell death.
Tetrahydrofolate (THF) is required for the synthesis of several essential molecules (Fig. S1), including certain amino acids (methionine and glycine), purines (ATP and GTP), and thymidylate (1). Therefore, treatment with compounds that interfere with THF production leads to the starvation of molecules required for DNA replication, protein synthesis, and energy metabolism. For this reason, these compounds are used as antimicrobial, antimalarial, and anticancer drugs. Trimethoprim (TMP), a competitive inhibitor of bacterial dihydrofolate reductase, which mediates the reduction of dihydrofolate to THF, is commonly used as a wide-spectrum antibiotic (2–4). The effect of TMP treatment on bacteria depends on the composition of growth media. It is bacteriostatic in minimal medium, whereas it is bactericidal in rich medium and in minimal medium supplemented with amino acids and a purine source (5). The bactericidal effect of TMP results from thymidylate depletion, and therefore, the effect is assumed to be similar to that of a thymineless death (TLD) (6).
Fig. S1.

Schematic presentation of the folate cycle. TMP inhibits the reduction of dihydrofolate to THF by targeting dihydrofolate reductase, thus blocking the folate cycle.
The TLD phenomenon has been thoroughly studied using the Escherichia coli thyA mutant, which lacks thymidylate synthase (6). When thyA mutant cells are deprived of thymine, the DNA replication forks are rapidly stalled, but replication initiation continues, which results in massive DNA breakage, and ultimately cell death (6). Interestingly, although some DNA repair functions increase tolerance, others increased sensitivity to TLD. For example, recF mutants are more tolerant, whereas recB mutants are more sensitive to TLD (6). Although it is clear that the bactericidal effect of TMP, similar to TLD, is triggered by thymine depletion, the available data suggest that the mechanisms ultimately responsible for TLD and for the bactericidal effect of TMP may be different. For example, the screening of the E. coli Keio collection identified 31 mutants that showed significantly different sensitivity to TMP relative to the wild-type (wt) strain in the Luria Broth medium (LB) (5). Only one of those genes, recJ, which encodes a single-strand DNA exonuclease, was previously reported to contribute to TLD. It is likely that mechanisms responsible for TLD and for the bactericidal effect of TMP in LB medium are different because LB contains low amounts of thymidine, whereas TLD is always studied under conditions of total thymine deprivation.
In this study, we decided to use LB medium to study TMP cytotoxicity because thymidine is present in different mammalian tissues and body fluids (7, 8) and can, therefore, have an effect on the TMP treatment. We also tested an effect of oxygen availability on TMP cytotoxicity. Oxygen availability, which varies among different body compartments, has an important effect on bacterial cell metabolism, and therefore, on the antibiotic treatment outcome. To characterize how TMP treatment leads to bacterial cell death, we used a set of E. coli mutants with deficiencies in DNA replication, repair, and recombination ability, as well as several metabolic pathways. We grew these strains in LB supplemented with TMP under aerobic and anaerobic conditions. We found that bactericidal TMP effects are triggered by thymidine depletion under both aerobic and anaerobic conditions. Under both conditions, decreasing the DNA replication initiation rate and the inactivation of mutM and mutY genes, which code for DNA glycosylases, reduced TMP toxicity. In the presence of oxygen, TMP-induced cell death resulted from the increased production of reactive oxygen species (ROS). TMP-induced bactericidal potential under anaerobic conditions was boosted by nitrate respiration. Taken together, our data suggest that TMP-induced DNA replication stress leads to a perturbation of cell metabolism, which results in the production of the DNA-damaging agents. Maladaptive processing of the TMP-induced DNA damage by DNA repair enzymes ultimately contributes to cell death.
Results
TMP Treatment Provokes Thymidine Starvation and Induces SOS Response.
Because TMP affects several biosynthetic pathways (Fig. S1), we investigated which compound or compounds E. coli is starved for under our experimental conditions. We supplemented LB with one or a combination of the following molecules: thymidine, glycine, methionine, and inosine. Only thymidine was able, and sufficient, to completely inhibit TMP toxicity in liquid LB or on LB agar plates under aerobic and anaerobic conditions (Fig. 1 and Fig. S2A). Only half of a given dose of TMP was required to obtain the same effect on cell survival under anaerobic rather than aerobic conditions.
Fig. 1.

The bactericidal effect of TMP results from thymidine starvation. Overnight liquid cultures of the wt strain were serially diluted and plated on LB agar with or without TMP, and complemented, or not, with THF-derived molecules: thymidine (Thy), glycine (Gly), methionine (Met), and inosine (Ino). Plates were incubated under aerobic (A) or anaerobic (B) conditions. The presented results are representative of three independent experiments.
Fig. S2.

TMP induces thymidine starvation that triggers induction of stress responses. OD600 nm and fluorescence were measured during growth of the wt strain with different doses of TMP and with or without addition of thymidine. MIC of TMP for this strain is 0.64 μg/mL. (A) Effect of the TMP treatments on E. coli growth. (B–F) Induction of different stress response reporters: (B) PrecA-GFP, (C) PsodA-GFP, (D) PoxyR-GFP, (E) PkatG-GFP, and (F) Phda-GFP. Paraquat was used as a positive control for the induction of oxidative stress response reporters. Each graph represents the mean ± SEM of three independent replicates. MIC experiments were repeated at least three times. Gene expression data were analyzed using the algorithm published in ref. 42. TMP concentrations 0.08 μg/mL and 0.16 μg/mL significantly increased expression of all reporters relative to untreated controls.
It was previously reported that TMP induces the SOS response (5). We showed that this was also the case under our experimental conditions, using PrecA-GFP reporter fusion (Fig. S2B). Induction was abolished when thymidine was added to the media. Next, we examined the contribution of SOS induction to the survival of treated bacteria by using the lexA1 (Ind−) mutant, which cannot induce the SOS response. This mutant was more sensitive than wt to the higher doses of TMP in the presence of oxygen (Fig. 2A and Table S1). Under anaerobic conditions, it was more sensitive to low TMP doses, and it was as sensitive as the wt at the highest TMP dose (Fig. 2B and Table S1).
Fig. 2.

Survival of E. coli mutants treated with TMP under aerobic and anaerobic conditions. Overnight liquid cultures were serially diluted and plated on LB agar with or without TMP. Plates were incubated under aerobic (A) or anaerobic (B) conditions. The presented results are representative of at least three independent experiments.
Table S1.
Summary of all of the spot test-based susceptibility assays performed with different doses of TMP. Colors represent the relative susceptibility of the different strains to TMP. (A) Treated wild-type relative to nontreated wild-type strain. (B and C) Treated mutant strains relative to treated wild-type strain. This table represents the susceptibility of the different strains from at least three independent experiments
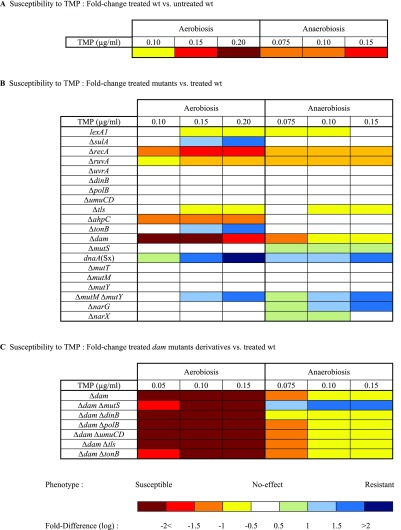 |
We also tested the involvement of several SOS-controlled functions in the survival of TMP-treated cells (Fig. 2 and Table S1). The deletion of the sulA gene, involved in the inhibition of cell division, increased the survival of cells treated with higher doses of TMP in the presence of oxygen, whereas it had no effect under anaerobic conditions. Both recA and ruvA mutants that have impaired homologous recombination showed high sensitivity to TMP under aerobic and anaerobic conditions. There was no significant difference in the survival of the nucleotide excision repair-deficient uvrA strain or of the single-deletion mutants of dinB, polB, and umuCD genes coding for the translesion synthesis (TLS) DNA polymerases under any experimental conditions. Triple TLS polymerases deletion mutant (referred to as tls) showed a decrease in survival at higher TMP doses in the presence and absence of oxygen. Therefore, as it was reported for TLD, some SOS functions contribute to the survival of TMP-treated cells, whereas others contribute to cell death.
ROS Production in TMP-Treated Cells.
Stalling of the replication forks and antibiotic treatments is known to increase the production of ROS, which contribute to cell death (9, 10). We investigated whether TMP treatment also induces ROS production. We first used reporter fusions of the promoters of soxS, sodA, oxyR, and katG genes and the gene coding for the fast-folding green fluorescent protein (GFP) (11). First, two genes are induced by increased intracellular superoxide levels, while two latter genes are induced by hydrogen peroxide (H2O2) (12). TMP treatment significantly increased expression of all reporter fusions in the wt background (Fig. 3 and Fig. S2 C–E). Induction of all reporters was abolished when thymidine was added to the media.
Fig. 3.

TMP induces oxidative stress response. OD600 nm and fluorescence were measured during growth of the wt strain, carrying a PsoxS-GFP reporter, in LB or LB TMP (0.08 µg/mL), with or without the addition of thymidine (0.3 mM). Paraquat (0.25 mM) was used as a positive control. After substraction of the autofluorescence background of the control strain without a PsoxS-GFP reporter, the fluorescence ratio between treated and nontreated cultures at OD600 nm = 0.4 was calculated. The mean ± SEM of three independent experiments is presented. *Significant difference of PsoxS expression in TMP-treated mutant cells compared with wt cells (t test, P values < 0.05). +Significant difference of PsoxS expression in thymidine-supplemented cultures compared with cultures without thymidine (t test, P values < 0.05).
Next, we showed that TMP treatment increased H2O2 production using Amplex Red assay (Fig. 4A). The increased H2O2 production in TMP-treated cells was further confirmed by the high sensitivity of the strain with a deleted ahpC gene, which codes for alkyl hydroperoxide reductase, which is the primary H2O2 scavenger, as well as of the Hpx− (ΔkatG ΔkatE ΔahpCF) strain, which is devoid of any catalase activity (13) (Figs. 2A and 4B and Table S1). Under anaerobic conditions, ahpC and Hpx− strains were no more sensitive than the wt strain (Fig. 2B and Fig. S3). The addition of catalase to the LB plates under aerobic conditions abolished sensitivity to TMP of wt, but also of ahpC and, to a great extent, of Hpx−. Importantly, catalase also abolished sensitivity of the recA and ruvA mutants, suggesting H2O2-induced damages require homologous recombination to be repaired (Fig. 4B).
Fig. 4.

H2O2 production in the TMP-treated cells and protective effect of catalase against TMP-induced cytotoxicity. (A) OD600 nm and fluorescence were measured during growth of the wt strain in LB or LB TMP (0.04 and 0.08 µg/mL). Paraquat (0.25 mM) was used as a positive control. Using Amplex Ultrared fluorescent dye, we followed intracellular H2O2 production. The mean ± SEM of six replicates from independent experiments is represented. (B and C) Overnight liquid cultures were serially diluted and plated on LB agar with or without TMP and with or without addition of catalase (3,000 units per plate). The presented results are representatives of at least three independent experiments.
Fig. S3.

Survival of E. coli mutants treated with TMP under aerobic and anaerobic conditions. Overnight liquid cultures were serially diluted and plated on LB agar with or without TMP. Plates were incubated under aerobic (A) or anaerobic (B) conditions. The presented results are representative of at least three independent experiments.
The hydroxyl radical is the most toxic ROS, which is produced through a Fenton reaction between H2O2 and iron. Therefore, diminishing the import of extracellular iron by tonB deletion should diminish hydroxyl radical production (12, 14). We found that the tonB mutant showed increased resistance to TMP compared with the wt under aerobic, but not under anaerobic, conditions (Fig. 2 and Table S1). Taken together, our data strongly suggest that TMP treatment increases ROS production that contributes to the TMP toxicity.
TMP-Induced Mutagenesis.
TMP treatment results in an imbalance in the dNTP pool and in ROS production, both of which result in increased mutagenesis (15, 16). We indeed observed that growth in liquid LB supplemented with 0.04 μg/mL TMP significantly induced mutagenesis in the wt (Fig. 5), as previously observed (17).
Fig. 5.

Mutagenesis in E. coli strains treated with sub-lethal concentration of TMP. Tetracycline resistant (TetR) mutant frequency in cultures grown in LB without (white bars) or with (black bars) 0.04 μg/mL TMP. The mean ± SEM of at least five independent experiments with three technical repeats each is presented. *Significant difference in TMP-induced mutagenesis between wt and mutant strains (Mann-Whitney, P value < 0.05).
As inactivation of the tonB gene increased survival after TMP treatment, we measured TMP-induced mutagenesis in the tonB mutant. TMP induced mutagenesis in this mutant to a significantly lower level than in the wt (Mann-Whitney, P value = 0.02; Fig. 5). This suggests hydroxyl radical production through the Fenton reaction contributes to the TMP-induced mutagenesis.
Because we observed that TLS polymerases contribute to the survival of the TMP-treated cells, and because it is known that they have lower DNA replication fidelity than the replicative DNA polymerase, we investigated their contribution to TMP-induced mutagenesis. However, deletions of genes coding for each or all three TLS polymerases did not have significant effects on mutagenesis compared with the wt (Mann-Whitney, P values > 0.05; Fig. 5). This indicates that TLS in the TMP-treated cells is mostly error-free and that replicative DNA polymerase may be responsible for most of the TMP-induced mutations.
DNA Methylation, Mismatch Repair, and Survival of the TMP-Treated Cells.
In E. coli, 99.99% of the errors made by DNA polymerases are corrected by mismatch repair (MMR) (18). MMR is targeted to a newly synthesized DNA strand, which carries the vast majority of the DNA replication errors. Discrimination between template and newly synthesized strands is based on the methylation of the adenines (A) in the 5′-GATC-3′ sequences by deoxyadenosine methylase (Dam). These As are methylated in the template strand and transiently unmethylated in the newly synthesized strand. In dam mutant, both DNA strands are unmethylated and MMR cuts them both indifferently, leading to DNA nicking and double-strand breaks (18). This confers high sensitivity to mutagens in dam mutants, which can be suppressed by inactivating the mutS gene. We showed that this is also the case with TMP treatment under aerobic and anaerobic conditions (Fig. 2 and Table S1). This confirms that TMP treatment is mutagenic.
We also verified whether TLS polymerases contribute to TMP-induced lethality in the dam mutant. The dam tls mutant was as sensitive as dam mutant under aerobic and anaerobic conditions (Table S1). This suggests TMP-induced mutations were not generated by the TLS polymerases when all three were present. Therefore, most of the TMP-induced mutations should be a consequence of replicative DNA polymerase errors. However, Pol IV polymerase may contribute to mutagenesis under anaerobic conditions because deletion of the dinB gene in the dam background attenuated toxicity of the low TMP dose (Fig. 2B and Table S1).
The dam mutant was more sensitive to TMP compared with the wt, under aerobic conditions. In addition, we found that PsoxS expression was more induced in TMP-treated dam than in wt, and that the induction was abolished by the addition of thymidine (Fig. 3). To evaluate how much ROS contribute to sensitivity of the dam mutant to TMP, we introduced tonB deletion in the dam mutant. Although dam tonB mutant grew rather poorly without treatment, it was more resistant at lower doses, but not at the higher TMP dose, under aerobic conditions. Under anaerobic conditions, dam and dam tonB had same sensitivity to TMP (Fig. 2 and Table S1). Finally, the addition of catalase to the LB plates under aerobic conditions attenuated to a great extent the sensitivity to TMP of the dam mutant, and totally abolished sensitivity to TMP of the dam mutS mutant (Fig. 4C).
Deletion of the mutS gene in the wt did not have any noticeable effect on E. coli survival in the presence of oxygen. Similarly, it was shown that MMR is not involved in TLD (19). Under anaerobic conditions, deletion of the mutS gene in the wt and dam backgrounds conferred resistance to TMP to a higher level than in wt (Fig. 2A and Table S1). The inactivation of the mutS gene increases survival of the TMP-treated dam mutant, but not to the wt level in the presence of oxygen, which suggests that some other mechanisms contribute to cell death of the dam mutant. Another known role of A methylation is the regulation of DNA replication initiation (20). The hemimethylated origin of replication, OriC, is sequestered by the SeqA protein, which prevents binding of the replication initiator protein, DnaA, thus inhibiting fast replication reinitiation. In the dam mutant background, replication initiation cannot be controlled by SeqA, which results in replication hyperinitiation and can contribute to sensitivity of the dam mutant to TMP.
DNA Replication Initiation and Sensitivity to TMP.
We examined whether the reduction of DNA replication initiation can increase resistance to TMP. We first introduced the dnaA(Sx) allele, which reduces DNA replication initiation rates (21), in wt strain. dnaA(Sx) mutant had higher resistance to all tested TMP doses than wt under aerobic and anaerobic conditions (Fig. 2 and Table S1). In addition, this mutant did not show any increase in expression of the PsoxS-GFP reporter on TMP treatment under aerobic conditions (Fig. 3). Next, we overproduced the hda gene, which codes for a protein that inhibits DNA replication reinitiation by binding to the DnaA protein (22), and found that it abolished TMP toxicity just as the dnaA(Sx) allele (Figs. 2 and 4B). Interestingly, we also found that TMP treatment induced expression of the hda gene (Fig. S2F), which suggests that reduction of the DNA replication initiation rates may be a cellular strategy to attenuate genotoxic stress.
Introduction of dnaA(Sx) in the dam mutant also increased resistance to TMP compared with the dam strain under aerobic and anaerobic conditions (Fig. 4C and Fig. S3). Although the dam dnaA(Sx) strain was more resistant than wt under anaerobic conditions, it was still more sensitive than wt under aerobic conditions. The addition of catalase diminished, but did not totally abolish, sensitivity of the dam dnaA(Sx) mutant to TMP under aerobic conditions relative to wt (Fig. 4C). This result further confirmed that ROS is a major cause of the lethality of TMP-treated aerobically growing cells.
Processing 8-Oxo-Deoxyguanosine DNA Damaged TMP-Treated Cells.
ROS can damage many cellular constituents including DNA. One of the most frequent oxidative DNA damages is 8-oxo-deoxyguanosine (8oxoG), which can be found both in the nucleotide pool and in the DNA (23, 24). High toxicity of the 8oxoG is illustrated by the fact that several DNA repair enzymes participate in its removal. MutT removes 8oxoG from the nucleotide pool. MutM (Fpg) DNA glycosylase removes 8oxoG from DNA, and MutY DNA glycosylase removes adenine bases when they are mispaired with guanine, cytosine, or 8oxoG (25). We investigated whether TMP treatment results in the production of 8oxoG by measuring sensitivity of the mutT, mutM, and mutY mutants to TMP. All three single mutants exhibited the same sensitivity to TMP as the wt under aerobic and anaerobic conditions. However, the double mutM mutY mutant was more resistant to TMP than the wt under aerobic and anaerobic conditions (Fig. 2 and Table S1). The hypothesis that the mutM mutY mutant, which has a high mutation rate (16), survives better than the wt because it generates mutations that confer resistance to TMP at a high rate can be ruled out because mutT and mutS mutants, which also have high mutation rates, do not survive better than wt.
TMP Cytotoxicity Under Anaerobic Conditions.
Increased ROS production, and processing of oxidative damages, are deleterious for TMP-treated cells under aerobic conditions. The question is, how are DNA lesions generated under anaerobic conditions where ROS cannot be produced. To test the effect of TMP on bacteria under anaerobic conditions, we used anaerobic jars. We are confident that ROS were not produced in the jars, or at least not to a level that can significantly affect cell survival, because tonB, ahpC, and Hpx− strains exhibited the same sensitivity to TMP as the wt under anaerobic conditions (Fig. 2B and Fig. S3B and Table S1). In the absence of oxygen, E. coli can use nitrogen as an electron acceptor, which is present in LB under the form of nitrates. Anaerobic nitrate respiration does not produce any ROS, but it does produce analogous byproducts; that is, reactive nitrogen species (RNS). RNS cause DNA damage, which are different from damage caused by ROS (26). To assess the role of nitrate respiration in TMP toxicity, we tested mutants with impaired nitrate respiration (Fig. 2B and Table S1). Inactivation of the narG gene that codes for the main anaerobic nitrate reductase greatly increased resistance to TMP treatment. Inactivation of the narX gene that participates in the signal transduction system, which controls anaerobic respiratory gene expression in response to nitrate and nitrite (27), also greatly increased resistance to TMP treatment, but to a lesser extent than inactivation of narG. Inactivation of the narG and narX genes had no effect on the sensitivity to TMP under aerobic conditions. Thus, nitrate respiration toxic byproducts are deleterious for TMP-treated cells under anaerobic conditions.
Discussion
In this study, we characterized molecular mechanisms whereby TMP treatment results in E. coli cell death. Cells were grown under aerobic and anaerobic conditions in the presence of low amounts of thymidine. We first found that thymidine depletion is the primary trigger of processes that lead to cell death under aerobic and anaerobic conditions. Next, we showed that TMP-treated cells increase production of ROS under aerobic conditions. Under anaerobic conditions, nitrate respiration of the TMP-treated cells produces toxic byproducts, most likely RNS. Finally, we showed that the processing of TMP-induced DNA damage by DNA repair enzymes is the ultimate contributor to the death of TMP-treated cells under both aerobic and anaerobic conditions.
Taken together, the following observations indicate that TMP-induced ROS production is a major contributor to cell death under aerobic conditions: TMP induces SoxS- and OxyR-regulated oxidative stress responses; using Amplex Red assay, we showed that TMP treatment increased H2O2 production; loss of genes coding for the H2O2 scavengers, katG, katE, and ahpCF, increases sensitivity to TMP only in the presence of oxygen; addition of the H2O2 scavenger, catalase, to growth medium greatly attenuates TMP toxicity for wt strain, as well as for the Hpx−, ahpC, dam, recA, and ruvA mutants; and inactivation of the tonB gene, responsible for iron intake, increases the resistance to TMP of the wt strain and dam mutant only in the presence of oxygen. The tonB mutant has also a significantly lower frequency of TMP-induced mutagenesis than wt.
In the presence of oxygen, the TMP dose that severely reduces the survival of wt cells barely affects the viability of the mutM mutY mutant. MutM removes 8oxoG from DNA, whereas MutY removes A from the 8oxoG:A mismatches. Deleterious consequences of the MutM and MutY activities result most likely from their lyase activity, which cleaves DNA at AP sites. If the resulting single-strand break is not processed with PolI before the arrival of the replication fork, it can provoke a double-strand break (28). The observation that recA and ruvA mutants are very sensitive to TMP suggests this treatment provokes DNA breaks. Because the addition of catalase eliminates sensitivity of recA and ruvA mutants to TMP, it indicates that ROS are involved in DNA breakage. It may be that homologous recombination can process double-strand breaks in the mutM mutY genetic background, but it is overwhelmed in the wt and single glycosylase mutants.
The inactivation of mutM and mutY genes also increased resistance to TMP treatment under anaerobic conditions. We found that TMP cytotoxicity under anaerobic conditions was dependent on the activity of enzymes involved in nitrate respiration. This dependency was previously observed for the toxicity of several antibiotics and silver nanoparticles (9, 29, 30). Most likely, toxic byproducts of the nitrate respiration are RNS such as nitric oxide and nitrous acid, which can damage different macromolecules, including DNA. Although MutM and MutY are known to process oxidative DNA damage, it cannot be excluded that they can also remove some RNS-damaged bases, which remain to be identified. We also found that TMP was more toxic in the absence than the presence of oxygen. This is in accordance with observations that the sensitivity of E. coli and Salmonella enterica to many antibiotics, including sulfamethoxazole/trimethoprim, was higher under anaerobic than under aerobic conditions (31).
The reduction of DNA replication initiation rates by introduction of the dnaA(Sx) allele, or by overproducing the hda gene, greatly attenuated TMP toxicity under aerobic and anaerobic conditions. Similarly, the impairment of DNA replication initiation by rifampicin or shifting the thermosensitive dnaA mutants to nonpermissive temperatures suppressed TLD (6). We also showed that hyperinitiation, in addition to MMR activity, contributes to sensitivity of the dam mutant to TMP. In addition, soxS was not induced in TMP-treated dnaA(Sx) cells. Attenuation of the TMP toxicity by diminishing DNA replication initiation rates can result from slower thymine depletion, resulting in a lower likelihood of blocking replication forks and, therefore, decreasing ROS production; and/or from a reduced probability that the advancing replication fork encounters either single-strand nicks generated by DNA glycosylase activity or previously blocked replication forks. The synergistic contribution of the DNA replication hyperinitiation, ROS and DNA glycosylases activity to the cell death is illustrated by the report that DNA replication hyperinitiation-induced lethality in E. coli cells can be attenuated with ROS scavengers and inactivation of the mutM gene (32).
The comparison of our results with other studies that examined the consequences of the blockage of DNA replication on the E. coli cell survival revealed many important similarities. Hence, blocking replication forks with hydroxyurea, or in the dnaEts mutant with shift to nonpermissive temperatures, induces ROS production that contributes to cell death (10, 33). Contribution of ROS to hydroxyurea-induced lethality is illustrated by the observation that it can be attenuated by inactivation of the major respiratory cytochrome oxidase, by treating cells with antioxidants, and by interrupting iron uptake (10).
Several research groups have provided independent lines of evidence that the interactions of antibiotics with their targets cannot completely account for the antibiotic toxicity (ref. 9 and references therein). It was proposed that interactions of antibiotics with their targets cause metabolic perturbations, alter the cellular redox state, and induce formation of ROS, which contribute to cell death. This hypothesis was challenged by a proposition that ROS production is a consequence and not a causal contributor to the cell death (34, 35). However, an important support for this hypothesis can be found in the existence of clinical isolates whose antibiotic tolerance involves mutations in oxidative stress response genes. Finally, our study also shows that the bactericidal effect of TMP is triggered by target-specific interactions, which results in thymine starvation, but that an important aspect of cytotoxicity results from the processing of the ROS-induced lesions.
Interestingly, ROS production also greatly contributes to the cytotoxic effects of antifolate drugs, such as methotrexate and pemetrexed, which are used as anticancer chemotherapy agents (36, 37). Our observation that nitrosative stress contributes to TMP-induced cytotoxicity under anaerobic conditions was not examined for the other abovementioned DNA replication-blocking treatments. However, it is likely that this may be the case because nitrosative stress also plays a critical role in methotrexate-induced intestinal damage in rats (38). Therefore, it can be concluded that in prokaryotes and eukaryotes, DNA synthesis-inhibiting drugs primarily interact with their different cellular targets, which then provoke ROS and RNS production and DNA damage, the processing of which ultimately contributes to cell death.
Materials and Methods
Bacterial Strains and Growth Conditions.
Bacterial strains used in this study are described in Tables S2 and S3.
Table S2.
List of the strains used in this study
| Strain | Relevant genotype* | Source |
| MG1655 (parental strain) | Wild-type | Laboratory strain collection |
| MG1655 lexA1(Ind−) | ∆ins::FRT† lexA1 | This work (43) |
| MG1655 ∆sulA | ∆sulA::FRT | This work |
| MG1655 ∆uvrA | ∆uvrA::Kan | This work |
| MG1655 ∆dinB | ∆dinB::Kan | This work |
| MG1655 ∆polB | ∆polB::Kan | This work |
| MG1655 ∆umuCD | ∆umuCD::Kan | This work |
| MG1655 ∆recA | ∆recA::Kan | This work |
| MG1655 ∆ruvA | ∆ruvA::Kan | This work |
| MG1655 ∆mutS | ∆mutS::Spec/Strep | This work |
| MG1655 ∆dam | ∆dam::Kan | This work |
| MG1655 ∆dam ∆mutS | ∆dam::Kan ∆mutS::Spec/Strep | This work |
| MG1655 ∆dam ∆dinB | ∆dinB::Kan ∆dam::Cm | This work |
| MG1655 ∆dam ∆polB | ∆polB::Kan ∆dam::Cm | This work |
| MG1655 ∆dam ∆umuCD | ∆umuCD::Kan ∆dam::Cm | This work |
| MG1655 ∆dam ∆tonB | ∆tonB::Kan ∆dam::Cm | This work |
| MG1655 dnaA(Sx) | dnaA(Sx)721 ∆zib::Tn10 | Ref. 44 |
| MG1655 ∆dam dnaA(Sx) | ∆dam::Cm dnaA(Sx)721 ∆zib::Tn10 | This work |
| MG1655 ∆mutT | ∆mutT::Cm | This work |
| MG1655 ∆mutM | ∆mutM::Cm | This work |
| MG1655 ∆mutY | ∆mutY::Tn10 | This work |
| MG1655 ∆mutM ∆mutY | ∆mutM::Cm ∆mutY::Tn10 | This work |
| MG1655 ∆ahpC | ∆ahpC::Kan | This work |
| MG1655 ∆tonB | ∆tonB::Kan | This work |
| MG1655 ∆narG | ∆narG::Kan | This work |
| MG1655 ∆narX | ∆narX::Kan | This work |
| MG1655 Hpx− | ∆katE::Tet ∆katG::Cm ∆ahpCF::Kan | Ref. 45 |
| MG1655pro‡ | SpecR lacR tetR | Ref. 9 |
| IBDS1§ | MG1655 attλ::cI (Ind−) λpR tetA Δara::FRT Δmeter::FRT | Ref. 39 |
| IBDS1 ∆dinB | ∆dinB::FRT | Ref. 39 |
| IBDS1 ∆polB | ∆polB::FRT | Ref. 39 |
| IBDS1 ∆umuCD | ∆umuCD::FRT | Ref. 39 |
| IBDS1 ∆tls | ∆dinB::FRT ∆polB::FRT ∆umuCD::FRT | Ref. 39 |
| IBDS1 ∆dam ∆tls | ∆dinB::FRT ∆polB::FRT ∆umuCD::FRT ∆dam::cm | This work |
Because all strains are derivatives of parental strain, only differences compared with the genotype of parental strain are indicated.
FRT indicates a scar that was left on elimination of the antibiotic resistance cassette using the FLP recombinase (47).
MG1655pro strain carrying pZE21-APX was used for H2O2 measurements.
IBDS1 strains were used for the measurements of TetR mutant frequency.
Table S3.
List of the plasmids used in this study
| Plasmid name | Relevant features | Source |
| pWSK29 | AmpR, pSC101 origin | Ref. 46 |
| pJBC200 | pWSK29 with hda allele under Plac control | Ref. 46 |
| pUA66 | KanR, promoter-less and gfp-less | Ref. 11 |
| pUA66-PrecA-GFP | pUA66 with recA promoter fused to gfpmut2 gene | Ref. 11 |
| pUA66-PsoxS-GFP | pUA66 with soxS promoter fused to gfpmut2 gene | Ref. 11 |
| pUA66-PoxyR-GFP | pUA66 with oxyR promoter fused to gfpmut2 gene | Ref. 11 |
| pUA66-PsodA-GFP | pUA66 with sodA promoter fused to gfpmut2 gene | Ref. 11 |
| pUA66-PkatG-GFP | pUA66 with katG promoter fused to gfpmut2 gene | Ref. 11 |
| pUA66-Phda-GFP | pUA66 with hda promoter fused to gfpmut2 gene | Ref. 11 |
| pZE21-APX‡ | KanR, pZE21-mcs origin, APX (ascorbate peroxidase) reporter gene under pL(tetO) control | Ref. 9 |
Overnight liquid cultures were grown in 5 mL LB (Difco) at 37 °C with 150 rpm shaking. When needed, medium was supplemented with antibiotics (Sigma Aldrich) at the following concentrations: 30 µg/mL chloramphenicol (Cm), 100 µg/mL ampicillin, 75 µg/mL spectinomycin, 50 µg/mL streptomycin, 50 µg/mL kanamycin, and 12.5 µg/mL tetracycline.
Measuring Expression of the GFP-Based Transcription Reporters.
Fresh overnight cultures were diluted to a final ratio of 1/10,000 vol/vol and inoculated in 96-well microtiter plates containing LB with or without TMP (0.04, 0.08, and 0.16 µg/mL) and with or without 0.3 mM thymidine. As a control, cells were treated with 0.25 mM paraquat that has the same effect on growth as 0.08 µg/mL TMP. Microtiter plates were incubated at 37 °C in a microplate reader incubator (Infinite M200 Pro from TECAN). Optical density (600 nm) and fluorescence (GFP 480/510) were read every 20 min for 16 h. The strain with the control plasmid without reporter fusion was used as an autofluorescence control for all experiments. Values obtained for the autofluorescence control cultures were subtracted from the corresponding cultures.
H2O2 Measurement.
Intracellular H2O2 concentration was measured using Amplex Red assay as described in ref. 9, with some modifications (SI Materials and Methods).
Mutation Assay.
To study the effect of TMP on mutagenesis, we used a previously described mutation assay (39). For more details see SI Materials and Methods.
Spot Test.
Fresh overnight cultures were serially diluted in 10 mM MgSO4. A 7-µL volume of each dilution was spotted on LB agar plates with different amounts of TMP (from 0.05 to 0.2 µg/mL). When needed, LB medium was supplemented with: 0.3 mM thymidine, 0.3 mM glycine, 0.3 mM methionine, 0.3 mM inosine, or 3,000 U/plate catalase (Sigma). After 16 h of incubation at 37 °C, growth of the spots was verified and compared with the spots on the control LB agar plates. Alternatively, plates were incubated at 37 °C in anaerobic jars with anaerobic atmosphere generation bags (Sigma-Aldrich) for 40 h. Anaerobic conditions were verified using an anaerobe indicator test (Sigma).
Statistical Analyses.
All statistical analyses were performed using the software Graphpad Prism 7 (https://www.graphpad.com).
All data, protocols, and materials are available upon request (ivan.matic@inserm.fr).
SI Materials and Methods
Bacterial Strains.
Bacterial strains used in this study are described in Table S2. All strains were derivatives of the E. coli K12 MG1655 strain, which is referred to as wt in the text. All mutants were constructed by P1 allelic transduction (40) from the Keio collection (41) or INSERM U1001 laboratory strain collection to the wt strain. Transductants were verified using polymerase chain reaction amplification, with primers described in ref. 41.
H2O2 Measurement.
The Amplex Red assay used to monitor peroxide generation is based the pea ascorbate peroxidase (APX) activity that catalyzes the H2O2-dependent oxidation of a fluorogenic substrate, which results in the production of the fluorescent product. Tester cells contain pZE21-APX plasmid encoding the APX W41F mutant. Experiments were performed as described in ref. 9, with some modifications. Briefly, fresh overnight cultures of MG1655pro cells with the pZE21-APX plasmid were diluted to a final ratio of 1/10,000 vol/vol in 50 mL LB in a 250-mL Erlenmeyer flask. Cultures were grown to an optical density at 600 nm (OD600 nm) of 0.1 and then were diluted 1/10 vol/vol in LB with anhydrotetracycline (30 ng/mL) to induce APX expression. At this step, TMP or paraquat was added to the culture. Cultures were then grown to an OD600 nm of 0.2 and 5-mL aliquots were dispensed into 15-mL polypropylene tubes. Amplex Ultrared reagent (Molecular Probes) was added to a final concentration of 50 µM in each tube. To quantify Amplex Ultrared fluorescence, 100 µL of cultures was transferred to a 96-well microtiter plate. OD600 nm and fluorescence (530/595) of the control cultures containing untreated cells and cultures containing treated cells were measured every 2.5 min for 1 h at 37 °C, using the SPARK 10M (TECAN) microplate reader. It should be noted that optical density values measured in the microplate reader are different from values obtained in the spectrophotometer because of the different width of the 1-mL cuvettes and the microplates.
Mutation Assay.
To study the effect of TMP on mutagenesis, we used mutation assay, which scores mutations in the bacteriophage Lambda cI (Ind−) repressor gene (39). This mutant repressor, which cannot undergo self-cleavage stimulated by the coprotease activity of the RecA protein, represses the tetA gene whose native promoter was replaced by the Lambda pR promoter. Any mutation that inactivates cI derepresses the tetA gene, thus conferring resistance to tetracycline, which can be selected for.
Fresh overnight cultures were diluted 1/1,000,000 vol/vol in 5 mL LB with or without a sub-MIC of TMP (0.04 µg/mL). After a 24-h incubation at 37 °C with 150 rpm shaking, cultures were serially diluted in 10 mM MgSO4. Tetracycline resistant (TetR) mutants were scored by plating 100 µL nondiluted and 10−1 diluted cultures on LB + Tet agar plates. The total number of colony-forming units was determined by plating 100 µL of the 10−5 and 10−6 diluted cultures on LB agar plates. Colony-forming units were counted after 16 h of growth at 37 °C. For each strain, at least five independent experiments with three technical repeats were performed. Mutation frequency was calculated as the ratio between TetR mutants and the total number of colony-forming units. For each pair of cultures (treated and nontreated), the TMP-induced mutagenesis increase was calculated as the ratio of TetR mutant frequencies between the treated and nontreated cultures.
Supplementary Material
Acknowledgments
We thank Arnaud Gutierrez and Sébastien Fleurier for critical reading of the manuscript. This work was supported by grants from Idex ANR-11-IDEX-0005-01/ANR-11-LABX-0071, Idex-Sorbonne Paris Cité, AXA Research Fund, and FRM Grant DBF20160635736.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1706236114/-/DCSupplemental.
References
- 1.Harvey RJ, Dev IK. Regulation in the folate pathway of Escherichia coli. Adv Enzyme Regul. 1975;13:99–124. doi: 10.1016/0065-2571(75)90010-2. [DOI] [PubMed] [Google Scholar]
- 2.Burchall JJ, Hitchings GH. Inhibitor binding analysis of dihydrofolate reductases from various species. Mol Pharmacol. 1965;1:126–136. [PubMed] [Google Scholar]
- 3.Bushby SRM, Hitchings GH. Trimethoprim, a sulphonamide potentiator. Br Pharmacol Chemother. 1968;33:72–90. doi: 10.1111/j.1476-5381.1968.tb00475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burchall JJ. Mechanism of action of trimethoprim-sulfamethoxazole. II. J Infect Dis. 1973;128(Suppl):437–441. doi: 10.1093/infdis/128.supplement_3.s437. [DOI] [PubMed] [Google Scholar]
- 5.Sangurdekar DP, Zhang Z, Khodursky AB. The association of DNA damage response and nucleotide level modulation with the antibacterial mechanism of the anti-folate drug trimethoprim. BMC Genomics. 2011;12:583. doi: 10.1186/1471-2164-12-583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khodursky A, Guzmán EC, Hanawalt PC. Thymineless death lives on: New insights into a classic phenomenon. Annu Rev Microbiol. 2015;69:247–263. doi: 10.1146/annurev-micro-092412-155749. [DOI] [PubMed] [Google Scholar]
- 7.Indiveri MC, Hirsh DC. Tissues and exudates contain sufficient thymidine for growth of anaerobic bacteria in the presence of inhibitory levels of trimethoprim-sulfamethoxazole. Vet Microbiol. 1992;31:235–242. doi: 10.1016/0378-1135(92)90081-4. [DOI] [PubMed] [Google Scholar]
- 8.Stokes A, Lacey RW. Effect of thymidine on activity of trimethoprim and sulphamethoxazole. J Clin Pathol. 1978;31:165–171. doi: 10.1136/jcp.31.2.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dwyer DJ, et al. Antibiotics induce redox-related physiological alterations as part of their lethality. Proc Natl Acad Sci USA. 2014;111:E2100–E2109. doi: 10.1073/pnas.1401876111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davies BW, et al. Hydroxyurea induces hydroxyl radical-mediated cell death in Escherichia coli. Mol Cell. 2009;36:845–860. doi: 10.1016/j.molcel.2009.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zaslaver A, et al. A comprehensive library of fluorescent transcriptional reporters for Escherichia coli. Nat Methods. 2006;3:623–628. doi: 10.1038/nmeth895. [DOI] [PubMed] [Google Scholar]
- 12.Imlay JA. The molecular mechanisms and physiological consequences of oxidative stress: Lessons from a model bacterium. Nat Rev Microbiol. 2013;11:443–454. doi: 10.1038/nrmicro3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jacobson FS, Morgan RW, Christman MF, Ames BN. An alkyl hydroperoxide reductase from Salmonella typhimurium involved in the defense of DNA against oxidative damage. Purification and properties. J Biol Chem. 1989;264:1488–1496. [PubMed] [Google Scholar]
- 14.Jakubovics NS, Jenkinson HF. Out of the iron age: New insights into the critical role of manganese homeostasis in bacteria. Microbiology. 2001;147:1709–1718. doi: 10.1099/00221287-147-7-1709. [DOI] [PubMed] [Google Scholar]
- 15.Schaaper RM, Mathews CK. Mutational consequences of dNTP pool imbalances in E. coli. DNA Repair (Amst) 2013;12:73–79. doi: 10.1016/j.dnarep.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fowler RG, et al. Interactions among the Escherichia coli mutT, mutM, and mutY damage prevention pathways. DNA Repair (Amst) 2003;2:159–173. doi: 10.1016/s1568-7864(02)00193-3. [DOI] [PubMed] [Google Scholar]
- 17.Baharoglu Z, Mazel D. Vibrio cholerae triggers SOS and mutagenesis in response to a wide range of antibiotics: A route towards multiresistance. Antimicrob Agents Chemother. 2011;55:2438–2441. doi: 10.1128/AAC.01549-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marinus MG. DNA mismatch repair. EcoSal Plus. 2012;5 doi: 10.1128/ecosalplus.7.2.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuong KJ, Kuzminov A. Stalled replication fork repair and misrepair during thymineless death in Escherichia coli. Genes Cells. 2010;15:619–634. doi: 10.1111/j.1365-2443.2010.01405.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taghbalout A, et al. Competition between the replication initiator DnaA and the sequestration factor SeqA for binding to the hemimethylated chromosomal origin of E. coli in vitro. Genes Cells. 2000;5:873–884. doi: 10.1046/j.1365-2443.2000.00380.x. [DOI] [PubMed] [Google Scholar]
- 21.Ginés-Candelaria E, Blinkova A, Walker JR. Mutations in Escherichia coli dnaA which suppress a dnaX(Ts) polymerization mutation and are dominant when located in the chromosomal allele and recessive on plasmids. J Bacteriol. 1995;177:705–715. doi: 10.1128/jb.177.3.705-715.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Riber L, et al. Hda-mediated inactivation of the DnaA protein and dnaA gene autoregulation act in concert to ensure homeostatic maintenance of the Escherichia coli chromosome. Genes Dev. 2006;20:2121–2134. doi: 10.1101/gad.379506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neeley WL, Essigmann JM. Mechanisms of formation, genotoxicity, and mutation of guanine oxidation products. Chem Res Toxicol. 2006;19:491–505. doi: 10.1021/tx0600043. [DOI] [PubMed] [Google Scholar]
- 24.Haghdoost S, Sjölander L, Czene S, Harms-Ringdahl M. The nucleotide pool is a significant target for oxidative stress. Free Radic Biol Med. 2006;41:620–626. doi: 10.1016/j.freeradbiomed.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 25.Lu A-L, Li X, Gu Y, Wright PM, Chang D-Y. Repair of oxidative DNA damage: Mechanisms and functions. Cell Biochem Biophys. 2001;35:141–170. doi: 10.1385/CBB:35:2:141. [DOI] [PubMed] [Google Scholar]
- 26.Burney S, Caulfield JL, Niles JC, Wishnok JS, Tannenbaum SR. The chemistry of DNA damage from nitric oxide and peroxynitrite. Mutat Res. 1999;424:37–49. doi: 10.1016/s0027-5107(99)00006-8. [DOI] [PubMed] [Google Scholar]
- 27.Bonnefoy V, Demoss JA. Nitrate reductases in Escherichia coli. Antonie van Leeuwenhoek. 1994;66:47–56. doi: 10.1007/BF00871632. [DOI] [PubMed] [Google Scholar]
- 28.Foti JJ, Devadoss B, Winkler JA, Collins JJ, Walker GC. Oxidation of the guanine nucleotide pool underlies cell death by bactericidal antibiotics. Science. 2012;336:315–319. doi: 10.1126/science.1219192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma Y, Guo C, Li H, Peng X-X. Low abundance of respiratory nitrate reductase is essential for Escherichia coli in resistance to aminoglycoside and cephalosporin. J Proteomics. 2013;87:78–88. doi: 10.1016/j.jprot.2013.05.019. [DOI] [PubMed] [Google Scholar]
- 30.Du H, Lo T-M, Sitompul J, Chang MW. Systems-level analysis of Escherichia coli response to silver nanoparticles: The roles of anaerobic respiration in microbial resistance. Biochem Biophys Res Commun. 2012;424:657–662. doi: 10.1016/j.bbrc.2012.06.134. [DOI] [PubMed] [Google Scholar]
- 31.DeMars Z, Biswas S, Amachawadi RG, Renter DG, Volkova VV. Antimicrobial susceptibility of enteric gram negative facultative anaerobe bacilli in aerobic versus anaerobic conditions. PLoS One. 2016;11:e0155599. doi: 10.1371/journal.pone.0155599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Charbon G, Bjørn L, Mendoza-Chamizo B, Frimodt-Møller J, Løbner-Olesen A. Oxidative DNA damage is instrumental in hyperreplication stress-induced inviability of Escherichia coli. Nucleic Acids Res. 2014;42:13228–13241. doi: 10.1093/nar/gku1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakayashiki T, Mori H. Genome-wide screening with hydroxyurea reveals a link between nonessential ribosomal proteins and reactive oxygen species production. J Bacteriol. 2013;195:1226–1235. doi: 10.1128/JB.02145-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Keren I, Wu Y, Inocencio J, Mulcahy LR, Lewis K. Killing by bactericidal antibiotics does not depend on reactive oxygen species. Science. 2013;339:1213–1216. doi: 10.1126/science.1232688. [DOI] [PubMed] [Google Scholar]
- 35.Liu Y, Imlay JA. Cell death from antibiotics without the involvement of reactive oxygen species. Science. 2013;339:1210–1213. doi: 10.1126/science.1232751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buqué A, et al. Molecular mechanism implicated in Pemetrexed-induced apoptosis in human melanoma cells. Mol Cancer. 2012;11:25. doi: 10.1186/1476-4598-11-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang C-C, et al. Ornithine decarboxylase prevents methotrexate-induced apoptosis by reducing intracellular reactive oxygen species production. Apoptosis. 2005;10:895–907. doi: 10.1007/s10495-005-2947-z. [DOI] [PubMed] [Google Scholar]
- 38.Kolli VK, Abraham P, Rabi S. Methotrexate-induced nitrosative stress may play a critical role in small intestinal damage in the rat. Arch Toxicol. 2008;82:763–770. doi: 10.1007/s00204-008-0287-9. [DOI] [PubMed] [Google Scholar]
- 39.Bjedov I, et al. Involvement of Escherichia coli DNA polymerase IV in tolerance of cytotoxic alkylating DNA lesions in vivo. Genetics. 2007;176:1431–1440. doi: 10.1534/genetics.107.072405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moore SD. Assembling new Escherichia coli strains by transduction using phage P1. Methods Mol Biol. 2011;765:155–169. doi: 10.1007/978-1-61779-197-0_10. [DOI] [PubMed] [Google Scholar]
- 41.Baba T, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: The Keio collection. Mol Syst Biol. 2006;2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mathieu A, et al. Discovery and function of a general core hormetic stress response in E. coli induced by sublethal concentrations of antibiotics. Cell Reports. 2016;17:46–57. doi: 10.1016/j.celrep.2016.09.001. [DOI] [PubMed] [Google Scholar]
- 43.Mount DW. Isolation and genetic analysis of a strain of Escherichia coli K-12 with an amber recA mutation. J Bacteriol. 1971;107:388–389. doi: 10.1128/jb.107.1.388-389.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sutera VA, Jr, Lovett ST. The role of replication initiation control in promoting survival of replication fork damage. Mol Microbiol. 2006;60:229–239. doi: 10.1111/j.1365-2958.2006.05093.x. [DOI] [PubMed] [Google Scholar]
- 45.Hébrard M, Viala JPM, Méresse S, Barras F, Aussel L. Redundant hydrogen peroxide scavengers contribute to Salmonella virulence and oxidative stress resistance. J Bacteriol. 2009;191:4605–4614. doi: 10.1128/JB.00144-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baxter JC, Sutton MD. Evidence for roles of the Escherichia coli Hda protein beyond regulatory inactivation of DnaA. Mol Microbiol. 2012;85:648–668. doi: 10.1111/j.1365-2958.2012.08129.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]