

Abstract
Transition metal catalysis has traditionally relied on organometallic complexes that can cycle through a series of ground-state oxidation levels to achieve a series of discrete yet fundamental fragment-coupling steps. The viability of excited-state organometallic catalysis via direct photoexcitation has been demonstrated. Although the utility of triplet sensitization by energy transfer has long been known as a powerful activation mode in organic photochemistry, it is surprising to recognize that photosensitization mechanisms to access excited-state organometallic catalysts have lagged far behind. Here, we demonstrate excited-state organometallic catalysis via such an activation pathway: Energy transfer from an iridium sensitizer produces an excited-state nickel complex that couples aryl halides with carboxylic acids. Detailed mechanistic studies confirm the role of photosensitization via energy transfer.
Developments in transition metal catalysis over the past half-century (1, 2) have led to the invention of a vast number of molecular transformations, among which modern cross-coupling chemistry, enantioselective hydrogenation, and olefin metathesis have all been recognized as preeminent bond-forming reactions in organic chemistry. Note that all of these organometallic technologies employ catalysts that function exclusively within a range of ground-state oxidation levels to perform the fundamental fragment-coupling steps. Given the century-old history of organometallic excited-state (i.e., non–ground state) complexes that can be accessed via photoexcitation (3), it is remarkable to consider that the utility of excited-state metal catalysts remains largely unexploited in the realm of organic bond–forming reactions (Fig. 1). As a notable exception, the direct photoexcitation of transition-metal catalysts (without photosensitization) has been disclosed in elegant studies by Fu and Peters, Nocera, and Doyle and their colleagues. (4–7).
Fig. 1. Photosensitization and catalysis.
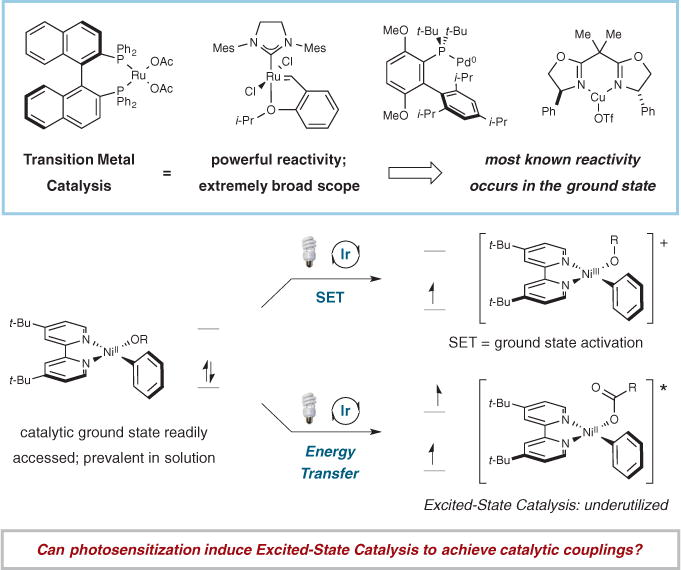
Photosensitization provides access to unexplored forms of catalysis in the excited state. Ph, phenyl; Ac, acetyl; Me, methyl; Mes, mesityl; Pr, propyl; i-Pr, isopropyl;_Bu, butyl; OTf, trifluoromethanesulfonate.
In the past 5 years, the field of metallaphotoredox catalysis has undergone widespread growth, in part, because of the high efficiency with which second- and third-row transition metals produce long-lived photoexcited states that can perform single-electron transfer (SET) with common organometallic catalysts (8–13). At the same time, these metallophotocatalysts have also found widespread use as photosensitizers to facilitate energy transfer with a diverse range of organic substrates (14–17). Indeed, these studies stand as a direct analogy to the abundance of organic excited-state reactions that can be accessed via organocatalytic photosensitization (e.g., with benzophenone), as popularized in part by Schenck, Turro, and Hammond and colleagues in the 1950s and 1960s (18–20). Given all of the above, it is surprising to consider that photoinduced energy transfer from photocatalysts to transition metals as a means to access organometallic excited states has not previously been exploited as a general activation pathway for reaction invention—especially in light of the historical success of energy transfer mechanisms in organic photochemistry (21–23) (Fig. 1). In this context, it is important to note that Molander and Doyle and co-workers have described the light-mediated arylation of α-oxy C–H bonds, wherein dual-catalysis photosensitization or direct transition metal photoexcitation were proposed, respectively, as the operative catalytic mechanism (7, 24).
We recognized that the application of photosensitization via energy transfer as a mechanism to switch on or facilitate excited-state organometallic catalysis might offer many opportunities in the field of organic cross coupling (11–14). Among many benefits, we anticipated that the use of energy-transfer photosensitization to access organometallic excited states would separate the roles of the light-harvesting complex from that of the transition-metal coupling catalyst. More specifically, protocols that operate via the direct photoexcitation of the cross-coupling catalyst are reliant on the visible light absorption cross section of the catalyst complex—a photophysical characteristic that is intrinsically disparate for each catalyst-substrate pair. In contrast, the use of a discrete photosensitization catalyst would effectively bypass the absorption cross-section requirement for the organometallic catalyst, thereby enabling a major improvement in tunability, efficiency, and, most important, generality in the application of excited-state organometallic catalysts for any given cross-coupling application.
Triplet sensitization is a long-established catalysis activation platform within the field of organic photochemistry. It is founded on the concept that organic substrates in their triplet excited states can participate in a range of valuable and unique bond-forming reactions. However, most organic molecules cannot readily access high-energy triplet states due to inefficient inter-system crossing from initially formed singlet excited states and/or vanishingly small absorption cross sections for direct singlet-triplet photoexcitation. To overcome this issue, photosensitizer catalysts are used that readily perform a light-harvesting role before a triplet-triplet energy transfer step with organic substrates—a pathway that allows efficient access to organic triplet excited states while bypassing the issues of direct photoexcitation. Quenching of the well-studied chromophore [Ru(bpy)3]2+ (where bpy is 2,2′-bipyridine) triplet excited state by transition-metal coordination compounds has long been established and often proceeds via energy transfer (25–28); however, this phenomenon has not been utilized in bond-forming catalysis. We noted that an analogous role-separation photosensitization concept might indeed be applied to organometallic catalysis. More specifically, using a strongly absorbing chromophore as a catalyst, we were able to gain entry to organometallic catalysts in their excited states via energy-transfer photosensitization and, thereafter, successfully access mechanistically distinct cross-coupling pathways. In this context, we report here the coupling of carboxylic acids with aryl halides to generate O-aryl ester products using a dtbbpy·Ni(0) complex (dtbbpy is 4,4′-di-tert-butyl-2,2′-bipyridine) as the cross-coupling catalyst and Ir(ppy)3 (1) (where ppy is ortho-metalated 2-phenylpyridine) as the optimal photosensitizer. Despite an extensive body of research into C–heteroatom bond formation over the past 40 years, an enduring challenge in this field has been the development of an efficient, mild, catalytic coupling between carboxylic acids and aryl halides to form O-aryl esters (29–33). As shown in Fig. 2A, our initial studies revealed that treatment of benzoic acid (3), with methyl p-bromobenzoate (4), enables the desired coupling in 85% yield using visible light and the catalyst combination outlined above. We further found that the organic sensitizer benzophenone (2) is also a moderately efficient photocatalyst in combination with the nickel catalyst described above (Fig. 2A and see the supplementary materials). Although in no way mechanistically definitive, we recognized that the successful use of benzophenone might implicate an energy transfer pathway in lieu of a SET process and that the efficiency of the Ni cycle most likely relied on access to an excited state of an intermediate Ni species. On this basis, we undertook a substantive mechanistic investigation that we here reveal is fully consistent with Ni excited-state catalysis, a mode of activation that is readily accessed using photosensitized energy transfer (14).
Fig. 2. Reaction development.
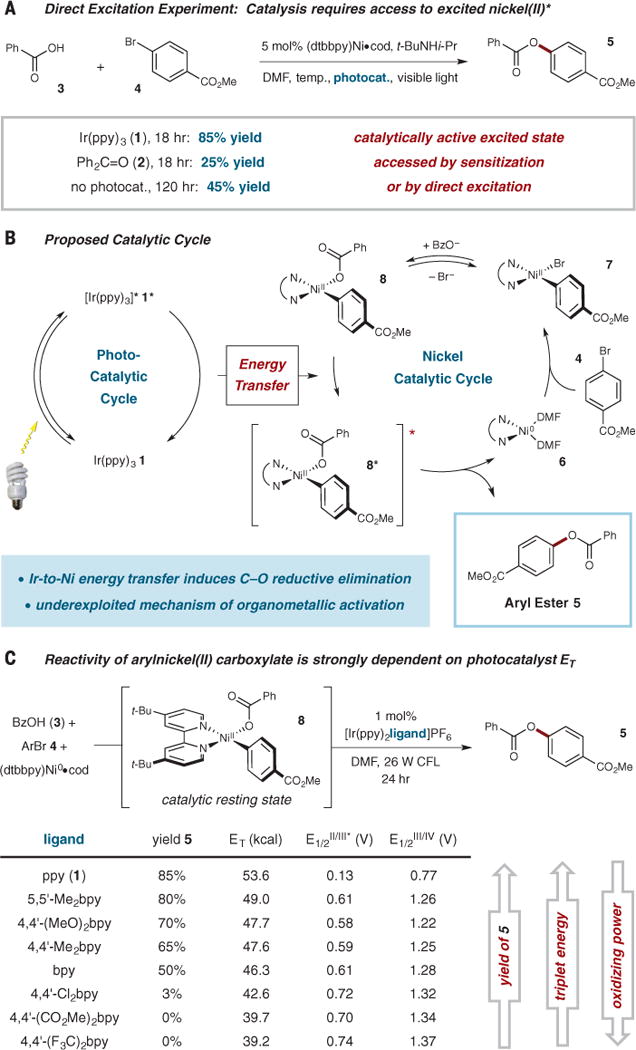
(A) Catalysis can be induced with visible light by using Ir(ppy)3 or benzophenone as photocatalyst; in the absence of a photocatalyst, extended reaction times and elevated temperatures are required. (B) Proposed catalytic cycle for the coupling of carboxylates with aryl bromides. (C) Photocatalysts with increasing triplet energy (ET) provide higher yields, whereas photocatalysts with increasing oxidation power provide lower yields; these findings support an energy transfer process and disfavor an electron transfer mechanism (oxidation potentials were measured from authentically synthesized or purchased materials: see figs. S1 to S8 and table S4; triplet energies were calculated from emission spectra, see fig. S9). Me, methyl; ppy, 2-phenylpyridine; Ph, phenyl; Ph2C=O, benzophenone; dtbbpy, 4,4′-di-tert-butyl-2,2′-bipyridine; Bu, butyl; Pr, propyl; CFL, compact fluorescent light, Bz, benzoyl; Ar, aryl; bpy, 2,2′-bipyridine.
A detailed description of the proposed energy transfer–driven catalytic cycle is shown in Fig. 2B. The cycle begins with oxidative addition of an aryl halide to dtbbpy·Ni(0) (6) to provide aryl–Ni(II) species 7. We believe that coordination of a carboxylate nucleophile, such as benzoate, should be rapid, yielding aryl–Ni(II) carboxylate 8. Meanwhile, Ir(ppy)3 (1) acts as an antenna, absorbing visible light to produce the characteristic triplet metal-to-ligand charge transfer (3MLCT) excited state 1*. At this stage, energy transfer can occur to produce electronically excited Ni(II) species 8* while simultaneously regenerating the ground state of 1. Reductive elimination from 8* generates the O-aryl ester product 5, regenerating Ni(0) species 6, and thus completing the catalytic cycle.
Despite the catalytic activity of benzophenone in our initial findings, we could not conclusively rule out the possibility of a SET oxidation mechanism on the basis of this information alone. Given our own experimental mechanistic studies (11) and those of Weix, Hu, and Fu and colleagues (34–36), we believed that all steps before reductive elimination would be facile, and as a corollary, we proposed that this was likely the photocatalyst-mediated step. Although computational studies have indicated that reductive elimination to form a C–O bond from Ni(II) is endothermic (37), our previous studies involving a nickel-catalyzed aryl etherification reaction had demonstrated that SET oxidation to Ni(III) is not only possible but is facile and can readily induce the key reductive elimination step.
A critical distinction between a photosensitization pathway and a SET process is that photosensitization creates an excited state of the Ni(II) species that should be directly accessible via excitation with visible light in the absence of the photocatalyst. Indeed, as shown in Fig. 2A, the coupling of benzoic acid (3) and methyl p-bromobenzoate (4) using our catalytic conditions but without a photocatalyst generated the desired adduct 5 in 45% yield after 120 hours. Furthermore, control experiments under identical conditions, with the exclusion of light, did not produce any detectable coupling product in the same time period. These experiments are fully consistent with the formation of a Ni excited-state complex that is an on-cycle intermediate and essential for productive bond formation.
We next turned our attention to studying the nature of the reactivity of the arylnickel(II) carboxylate complex subsequent to photosensitization. In this regard, it is important to underscore the fact that the electronic structure of a Ni(II) coordination complex is fundamentally different from those of more commonly used complexes of Ru(II), Ir(III), and Cu(I) insofar as the lowest energy excited state(s) of a Ni(II) system of the type we are using will be ligand-field (as opposed to charge-transfer) in nature. This class of excited states is far more likely to engage in bond-breaking and bond-forming reactions than the electron transfer chemistry typically observed for charge-transfer states. This point notwithstanding, a mechanistic assessment must still be made experimentally. Although time-resolved absorption spectroscopy can, in principle, differentiate between energy and electron transfer processes (14), the fact that the Ir sensitizer and the Ni(II) species both absorb in the same region, coupled with our desire to probe the reaction under catalytically relevant conditions, rendered mechanistic determination via this route very challenging (38). As an alternative, we designed a series of experiments to probe the capacity of photocatalysts to perform either energy transfer or electron transfer in the presence of arylnickel(II) compounds. The Ni(II) complexes are nonemissive; although this is consistent with the expected ligand-field nature of the lowest-energy excited state(s), it prevents a facile direct determination of excited–state energetics. A library of heteroleptic iridium photocatalysts of the type [Ir(ppy)2(ligand)](PF6), where “ligand” refers to a series of symmetrically substituted 2,2′-bipyridine ligands, was therefore synthesized. Systematically varying the electron-donating ability of this ligand allows for controlled tunability of the energy of the compound’s charge-transfer excited state, resulting in catalysts with a broad range of 3MLCT-state energies (Fig. 2C) (39, 40). Performing our catalytic reaction with this series of photocatalysts revealed two trends. First, the yield of 5 was observed to be strongly correlated with the energy of the Ir-based excited state and, in fact, exhibited a cut-off of ~40 kcal mol−1, below which no conversion occurred. This result thus defines a thermodynamic threshold for the reaction and a lower limit for the energy of the Ni(II)-based excited state responsible for the chemistry. Second, the yield of 5 was anticorrelated with the oxidizing power of the photocatalyst, a result that is wholly inconsistent with the necessary Ni(II) to Ni(III) change that would be involved in an SET process.
In order to isolate and study the reductive elimination event, we next prepared arylnickel(II) acetate species 9 (Fig. 3A) from the corresponding chloride complex (41, 42). Nickel complex 9 is stable for at least 1 week in dimethylformamide (DMF) solution, highlighting the thermally robust nature of this complex.
Fig. 3. Mechanism studies.
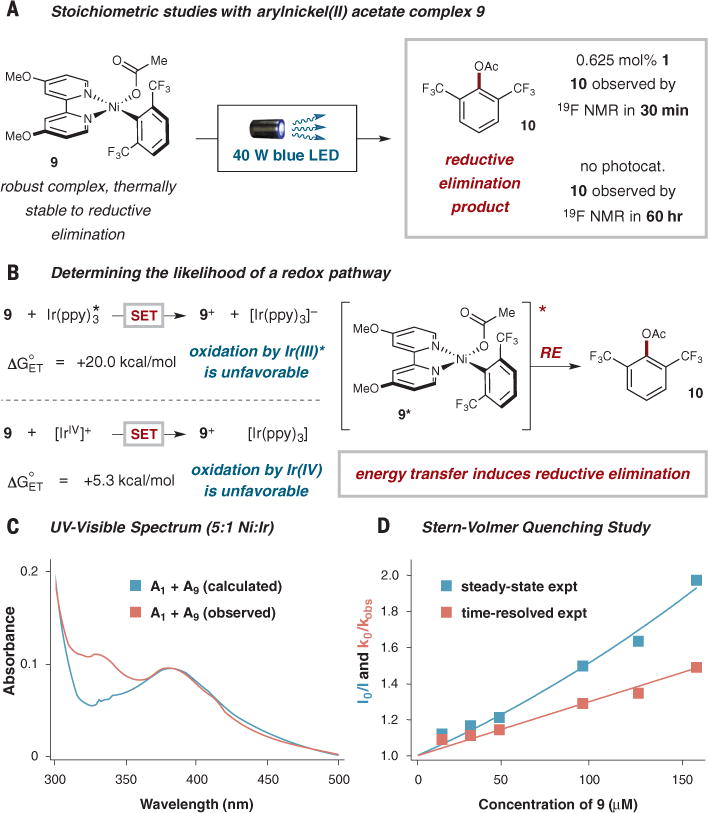
(A) Reductive elimination of 10 is induced with visible light and Ir(ppy)3 or with visible light alone. (B) The oxidation potentials of Ir(III)* (1*) and Ir(IV) indicate that SEToxidation is unfavorable, suggesting an alternate mechanism. (C) UV-visual absorption spectrum of a 5:1 mixture of 9:1 shows evidence of a ground-state association between the two. (D) Stern-Volmer quenching study shows evidence of both static and dynamic quenching. Me, methyl; Ac, acetyl; ppy, 2-phenylpyridine; SET, single electron transfer; RE, reductive elimination. ΔG°ET, ground-state Gibbs free energy of electron transfer.
We first sought to demonstrate that 9 could undergo the key reductive elimination step to form aryl ester 10. Indeed, irradiating complex 9 with a 40 W blue light-emitting diode lamp in the presence of Ir(ppy)3 (1) induced the formation of ester 10 after only 30 min as probed by 19F nuclear magnetic resonance (NMR) spectroscopy. More important, with the same light source, yet in the absence of photocatalyst, the same complex 9 was found to undergo reductive elimination to form appreciable quantities of 10 (albeit more slowly, in 60 hours). In both cases, control reactions in the absence of light did not yield any ester product. These observations conclusively demonstrate that a Ni(II) complex can undergo reductive elimination via the intermediacy of a Ni(II) excited state. The low-absorption cross section for this compound leads to the production of catalytically active excited-state Ni species in diminished quantities. As such, the paucity of active catalysts in solution that are available via direct excitation requires long reaction times with a relatively high intensity light source to produce similar levels of ester 10. Again, this highlights the advantages of separating the light-harvesting role from the catalyst being excited, as the number of catalytically active Ni species produced is far greater via an energy transfer.
Given that we could produce compound 9 as a single Ni species in solution, we elected to also study its electrochemistry (see fig. S11). More specifically, we calculated the driving forces for redox events between 1* and 9 using the method described by Rehm and Weller to determine the thermodynamic favorability of these processes (Fig. 3B) (43). We found that a SET oxidation of 9 by photoexcited 1* was strongly disfavored on thermodynamic grounds. Moreover, we recognized that [IrIV(ppy)3]+, which is a much stronger oxidant than photoexcited 1*, would be accessible via an initial oxidative quenching SET step. However, the corresponding oxidation potential of [IrIV(ppy)3]+ reveals that SET oxidation of 9 would also be unfavorable. We found that SET reduction of 9 by photoexcited 1* would be thermodynamically feasible (44). However, treatment of complex 9 with a series of single-electron reductants led only to the recovery of starting material (>75%) and in no case produced any detectable amount of reductive elimination product 10 (see table S6). These results further demonstrate that the critical reductive elimination step does not involve an electron transfer process, leaving energy transfer from the photoexcited Ir sensitizer to the arylnickel(II) species as the only possible mechanistic pathway that can account for all of our observations.
Additional information concerning the specific nature of the energy transfer process [i.e., Dexter (through-bond) versus Förster (dipolar)] was afforded from steady-state quenching measurements (14). At an early stage, we noted a strong deviation from the expected electronic absorption spectrum of a 5:1 mixture of complex 9 and photocatalyst 1 (the ratio used under the operative reaction conditions). As shown in Fig. 3C, an absorptive feature appeared around 330 nm that could not be accounted for on the basis of the calculated sum of the spectra of 9 and 1, suggesting that an association between the two compounds exists in solution. Furthermore, whereas Stern-Volmer luminescence quenching, as measured by time-resolved emission spectroscopy, revealed the expected linear correlation between the observed excited-state lifetime of the photosensitizer and the concentration of compound 9, the corresponding steady-state emission data (i.e., I0/I, where I0 and I are the integrated emission intensities of 1 in the absence and presence of a quencher, respectively) was quadratic (Fig. 3D) (14, 45). The latter observation indicates a major contribution from so-called static quenching, a likely consequence of the association inferred from the electronic absorption data shown in Fig. 3C. Given the relatively small degree of overlap between the emission spectrum of 1 and the absorption spectrum of 9 (see fig. S17), our data suggest that Dexter transfer—which can be extremely efficient at very short distances—is likely playing an important role in the reductive elimination step leading to C–O bond formation. In this context, it should be noted that coordination compounds are characterized by more complex electronic structures (both in terms of number and spin-state variability) than what is typically found in organic compounds. Accordingly, although we are confident that the lowest-energy excited state of the Ni(II) species responsible for the chemistry we are observing is ligand-field in nature, a more detailed understanding of the excited-state electronic structure of this and related systems is still under investigation (46).
In optimizing the reaction conditions (table S1) we found that NiBr2·diglyme was a superior pre-catalyst to bis(cyclooctadiene)nickel(0) [Ni(cod)2], coupling acid 3 and aryl halide 4 to yield ester 5 in 94% yield in only 6 hours (compare Fig. 2A, where a Ni(cod)2-derived catalyzed provided an 85% yield in 18 hours). Similarly, 4,4′-dOMebpy and dtbbpy ligands on the Ni(II) catalyst provided comparable efficiency. Moreover, secondary amines were found to be efficient bases for this protocol, with N-tert-butyl-isopropylamine proving optimal.
We were pleased to find that a wide array of carboxylic acids and aryl halides can be efficiently coupled using this energy transfer–driven mechanism. As shown in Fig. 4, this protocol is highly tolerant of a wide range of functional groups. The steric contribution of the acid component plays a minimal role in the coupling step, as primary, secondary, and tertiary acids, including the bulky adamantane system, can be used (11–14, 76 to 81% yield). Cyclopropyl rings, as well as ether-bearing substrates, do not inhibit catalysis (15–17, 76 to 83% yield). Moreover, benzoic acid derivatives are ideal substrates, regardless of the electronic nature of the aryl ring (entries 5, 19, 20, 93 to 95% yield). Protic functionality is tolerated without loss in efficiency, as revealed by the N–H-bearing amino acids N-Cbz-phenylalanine and N-Cbz-leucine (18 and 21, 86 and 81% yield, respectively). No racemization of these substrates was observed in this protocol (products were isolated in >99% enantiomeric excess).
Fig. 4. Coupling of carboxylic acids with aryl halides.
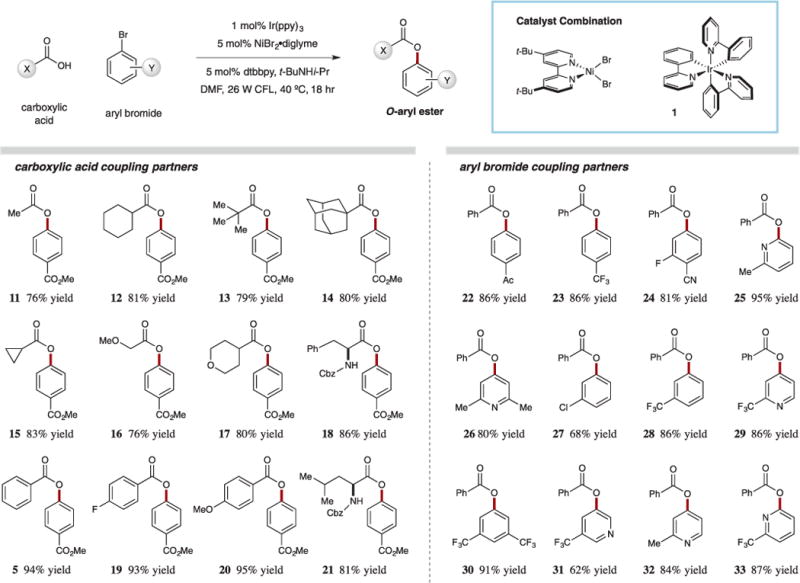
Substrate scope for the nickel-catalyzed coupling of aryl halides with carboxylic acids via excited-state catalysis. Cbz, benzyl carbamoyl.
With respect to the electrophilic coupling partner, we found that electron-deficient aryl bromides were preferred. Aryl bromides having ester, ketone, trifluoromethyl, and halide substitution function well in this C–O coupling (22–24, 27–28, 30, 68 to 91% yield). Also, a variety of substituted pyridine rings can be readily used, including 2-, 3-, and 4-bromopyridines (25–26, 29, 31–33, 62 to 95% yield). The use of these substrates is noteworthy, given the challenge of building similar products under classical conditions, because of the diminished nucleophilicity of the corresponding phenols.
This type of energy transfer–driven excited-state activation is likely to be broadly general, and we anticipate diverse applications across the many fields of chemistry that rely on organometallic coupling catalysis.
Supplementary Material
Acknowledgments
Research reported in this publication was supported by the National Institute of General Medical Sciences (NIGMS), NIH, under award number R01 GM078201-05 (D.W.C.M., E.R.W., and C.C.L.) and the NSF under the award number CHE-1300096 (J.K.M. and D.A.R.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIGMS or NSF. Additional data supporting the conclusions are available in supplementary material.
Footnotes
SUPPLEMENTARY MATERIAL
www.sciencemag.org/content/355/6323/380/suppl/DC1
Materials and Methods
References (47–61)
NMR Spectra
REFERENCES AND NOTES
- 1.Hegedus LS, Söderberg BCG. Transition Metals in the Synthesis of Complex Organic Molecules. 3. University Science Books; South Orange, NJ: 2010. [Google Scholar]
- 2.Johansson Seechurn CCC, Kitching MO, Colacot TJ, Snieckus V. Angew Chem Int Ed Engl. 2012;51:5062–5085. doi: 10.1002/anie.201107017. [DOI] [PubMed] [Google Scholar]
- 3.Bitterwolf TE. J Organomet Chem. 2004;689:3939–3952. [Google Scholar]
- 4.Creutz SE, Lotito KJ, Fu GC, Peters JC. Science. 2012;338:647–651. doi: 10.1126/science.1226458. [DOI] [PubMed] [Google Scholar]
- 5.Kainz QM, et al. Science. 2016;351:681–684. doi: 10.1126/science.aad8313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hwang SJ, et al. J Am Chem Soc. 2015;137:6472–6475. doi: 10.1021/jacs.5b03192. [DOI] [PubMed] [Google Scholar]
- 7.Shields BJ, Doyle AG. J Am Chem Soc. 2016;138:12719–12722. doi: 10.1021/jacs.6b08397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kalyani D, McMurtrey KB, Neufeldt SR, Sanford MS. J Am Chem Soc. 2011;133:18566–18569. doi: 10.1021/ja208068w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tellis JC, Primer DN, Molander GA. Science. 2014;345:433–436. doi: 10.1126/science.1253647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zuo Z, et al. Science. 2014;345:437–440. doi: 10.1126/science.1255525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Terrett JA, Cuthbertson JD, Shurtleff VW, MacMillan DWC. Nature. 2015;524:330–334. doi: 10.1038/nature14875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corcoran EB, et al. Science. 2016;353:279–283. doi: 10.1126/science.aag0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shaw MH, Twilton J, MacMillan DWC. J Org Chem. 2016;81:6898–6926. doi: 10.1021/acs.joc.6b01449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arias-Rotondo DM, McCusker JK. Chem Soc Rev. 2016;45:5803–5820. doi: 10.1039/c6cs00526h. [DOI] [PubMed] [Google Scholar]
- 15.Turro NJ. J Chem Educ. 1966;43:13–16. [Google Scholar]
- 16.Lu Z, Yoon TP. Angew Chem Int Ed Engl. 2012;51:10329–10332. doi: 10.1002/anie.201204835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singh K, Staig SJ, Weaver JD. J Am Chem Soc. 2014;136:5275–5278. doi: 10.1021/ja5019749. [DOI] [PubMed] [Google Scholar]
- 18.Schenck GO. Angew Chem. 1952;64:12–23. [Google Scholar]
- 19.Schenck GO, Hartmann WH, Mannsfeld SP, Metzner W, Krauch CH. Eur J Inorg Chem. 1962;95:1642–1647. [Google Scholar]
- 20.Hammond GS, Turro NJ, Fischer A. J Am Chem Soc. 1963;83:4674–4675. [Google Scholar]
- 21.Ikezawa H, Kutal C, Yasufuku K, Yamazaki H. J Am Chem Soc. 1986;108:1589–1594. [Google Scholar]
- 22.Islangulov RR, Castellano FN. Angew Chem Int Ed Engl. 2006;45:5957–5959. doi: 10.1002/anie.200601615. [DOI] [PubMed] [Google Scholar]
- 23.Müller C, Bauer A, Bach T. Angew Chem Int Ed Engl. 2009;48:6640–6642. doi: 10.1002/anie.200901603. [DOI] [PubMed] [Google Scholar]
- 24.Heitz DR, Tellis JC, Molander GA. J Am Chem Soc. 2016;138:12715–12718. doi: 10.1021/jacs.6b04789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Juris A, Gandolfi MT, Manfrin MF, Balzani V. J Am Chem Soc. 1976;98:1047–1048. [Google Scholar]
- 26.Demas JN, Addington JW. J Am Chem Soc. 1976;98:5800–5806. [Google Scholar]
- 27.Juris A, Manfrin MF, Maestri M, Serpone N. Inorg Chem. 1978;17:2258–2261. [Google Scholar]
- 28.Marciniak B, Buono-Core GE. Spectrosc Lett. 1990;23:149–160. [Google Scholar]
- 29.Muci AR, Buchwald SL. Top Curr Chem. 2002;219:131–209. [Google Scholar]
- 30.For the copper-catalyzed coupling of carboxylic acids with diaryliodonium salts, see (31). For examples of couplings between aryl halides and carboxylic acids mediated by super-stoichiometric transition metal catalysts at elevated temperatures, see (32, 33).
- 31.Bhattari B, Tay JH, Nagorny P. Chem Commun (Camb) 2015;51:5398–5401. doi: 10.1039/c4cc08604j. [DOI] [PubMed] [Google Scholar]
- 32.Thasana N, et al. J Org Chem. 2007;72:9379–9382. doi: 10.1021/jo701599g. [DOI] [PubMed] [Google Scholar]
- 33.Yoo K, Park H, Kim S, Kim M. Tetrahedron Lett. 2016;2016:781–783. [Google Scholar]
- 34.Biswas S, Weix DJ. J Am Chem Soc. 2013;135:16192–16197. doi: 10.1021/ja407589e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Breitenfeld J, Wodrich MD, Hu X. Organometallics. 2014;33:5708–5715. [Google Scholar]
- 36.Schley ND, Fu GC. J Am Chem Soc. 2014;136:16588–16593. doi: 10.1021/ja508718m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Macgregor SA, Neave GW, Smith C. Faraday Discuss. 2003;124:111–127. doi: 10.1039/b212309f. [DOI] [PubMed] [Google Scholar]
- 38.Although the molar absorptivity of arylnickel(II) carboxylate 9 is lower than that of Ir(ppy)3 (1) in the visible region, 9 is present in five times the concentration of 1 in a relevant reaction mixture. This translates to almost equal absorbances for the two compounds, which leads to an exceedingly small signal in a transient absorption experiment. Efforts are currently under way to circumvent these difficulties for future studies of this and related systems.
- 39.Dixon IM, et al. Chem Soc Rev. 2000;29:385–391. [Google Scholar]
- 40.Flamigni L, Barbieri A, Sabatini C, Ventura B, Barigelletti F. Top Curr Chem. 2007;281:143–203. [Google Scholar]
- 41.Standley EA, Smith SJ, Müller P, Jamison TF. Organometallics. 2014;33:2012–2018. doi: 10.1021/om500156q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.The 4,4′-dimethoxy-2,2′-dipyridyl (dOMebpy) ligand performs equally well in the coupling reaction as dtbbpy, and it was used here for ease of synthesis of 9.
- 43.Rehm D, Weller A. Isr J Chem. 1970;8:259–271. [Google Scholar]
- 44.It is highly unlikely that a SET reduction would lead to reductive elimination; however, this assumption cannot be guaranteed a priori.
- 45.Lakowicz JR. Principles of Fluorescence Spectroscopy. 3. Springer Science+Business Media; New York: 2006. [Google Scholar]
- 46.We thank a reviewer for an interesting discussion on the possibility of a disproportionation reaction subsequent to energy transfer that would lead to an active Ni(III) species responsible for reductive elimination. We can imagine two scenarios for this process, the first being bimolecular association of two photoexcited Ni(II)* species. The low light flux used in the reaction makes this scenario untenable. In addition, disproportionation between one photoexcited Ni(II)* and one ground-state Ni(II) species is possible; however, the low concentration of the Ni(II) species coupled with what is likely to be a sub-nanosecond lifetime of the Ni(II) excited state makes it unlikely that an excited Ni(II) complex can form an adduct with another Ni(II) complex before ground-state recovery of Ni(II)*.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.