

Abstract
Introduction
Infantile nystagmus has many causes, some life threatening. We determined the most common diagnoses in order to develop a testing algorithm.
Methods
Retrospective chart review. Exclusion criteria were no nystagmus, acquired after 6 months, or lack of examination. Data collected: pediatric eye examination findings, ancillary testing, order of testing, referral, and final diagnoses. Final diagnosis was defined as meeting published clinical criteria and/or confirmed by diagnostic testing. Patients with a diagnosis not meeting the definition were “unknown.” Patients with incomplete testing were “incomplete.” Patients with multiple plausible etiologies were “multifactorial.” Patients with negative complete workup were “motor.”
Results
284 charts were identified; 202 met inclusion criteria. The 3 most common causes were Albinism(19%), Leber Congenital Amaurosis(LCA)(14%) and Non-LCA retinal dystrophy (13%). Anatomic retinal disorders comprised 10%, motor another 10%. The most common first test was MRI (74/202) with a diagnostic yield of 16%. For 28 MRI-first patients, nystagmus alone was the indication; for 46 MRI-first patients other neurologic signs were present. 0/28 nystagmus-only patients had a diagnostic MRI while 14/46 (30%) with neurologic signs did. Yield of ERG as first test was 56%, OCT 55%, and molecular genetic testing 47%. 90% of patients had an etiology identified.
Conclusion
The most common causes of infantile nystagmus were retinal disorders (56%), however the most common first test was brain MRI. For patients without other neurologic stigmata complete pediatric eye examination, ERG, OCT and molecular genetic testing had a higher yield than MRI scan. If MRI is not diagnostic, a complete ophthalmologic workup should be pursued.
Keywords: Electroretinogram, etiology, nystagmus
Introduction
Infantile nystagmus syndrome, an involuntary eye movement disorder, may be considered a diagnosis in itself with various underlying causes, or a clinical sign requiring a systematic evaluation. Nystagmus waveforms have been extensively studied (1) and various nomenclatures have been proposed over the years. The CEMAS nomenclature (2) is the most exhaustive. In our paper Infantile Nystagmus will be used to signify any involuntary oscillatory eye movement disorder that occurs in the first 6 months of life, not associated with medication or other causes of acquired nystagmus. We will consider Infantile Nystagmus a sign rather than a final diagnosis, and therefore we include those entities listed in the CEMAS classification as infantile nystagmus syndrome (INS), fusion maldevelopment syndrome or latent/manifest latent nystagmus (FMNS), spasmus nutans (SNS), vestibular nystagmus, gaze holding deficiency nystagmus, vision loss nystagmus, other pendular nystagmus, and ocular bobbing, as our purpose is to develop an algorithm for arriving at the etiology of the many different types of “shaking eyes” that present to the pediatric ophthalmologist. A major deviation from the CEMAS nomenclature is that in the CEMAS schema “motor nystagmus” is folded into INS. In our study we are grouping patients by etiology rather than nystagmus waveform type and are therefore using the term “motor nystagmus” to signify a diagnosis of exclusion category in which sensory and neurologic etiologies have been ruled out, resulting in an oculomotor diagnosis with stable, relatively good visual acuity. This has been referred to as Idiopathic Infantile Nystagmus Syndrome in some publications (3).
Infantile nystagmus is a common reason for referral to pediatric, retina and genetic eye disorder specialists as well as to neurologists and neuro-ophthalmologists, and yet there is no agreed upon protocol for the evaluation of these patients. In the clinical experience of this author, children often have brain magnetic resonance imaging (MRI) performed as the first study by the first provider they encounter, and may receive a diagnosis of “motor nystagmus” (or Idiopathic Infantile Nystagmus Syndrome), without further workup if the MRI is negative and visual acuity appears grossly normal for age. Sometime later, either because of decreased vision or school assessments, another consultation is requested and decisions must be made about how to proceed. Because of the uncertainty about which diagnoses are most common and which tests are most useful, we undertook a retrospective chart review of 202 consecutive patients referred to either pediatric ophthalmology or the pediatric genetic eye disease service with infantile nystagmus. Highlights of this study, combined with a review of the literature, will be summarized in this paper along with an algorithm for the workup of infantile nystagmus based on this data.
Methods
An IRB approved retrospective chart review was performed using nystagmus and diagnoses associated with nystagmus as key words in the electronic medical record and in a pediatric genetic eye disease database for patients seen from 2008–2016 by one physician (AVD). Charts were excluded if nystagmus was not documented to be present, was acquired after 6 months of age, or if there was no eye examination recorded. Clinical data was collected including results from complete pediatric eye examination and all testing completed both before and after referral, with documentation of the order in which testing was performed. Referring diagnosis, initial diagnosis and final diagnosis were recorded. Final diagnosis was considered definitive if it met published clinical criteria for a disorder (e.g. albinism with the triad of nystagmus, iris transillumination, and foveal hypoplasia), if it was confirmed by diagnostic testing such as electroretinogram (ERG) or optical coherence tomography (OCT) to be localized to a specific site in the eye (e.g. retinal dystrophy or degeneration, foveal or optic nerve hypoplasia) or brain by MRI (e.g. septo-optic dysplasia, brain malformation), and/or if it was confirmed with molecular genetic testing (e.g. the appropriate number of disease-causing mutations in a gene known to cause a specific disorder that fit the clinical presentation). Patients in whom a clinical or molecular diagnosis did not meet the rigorous definitions above after all testing was complete were called “unknown.” Patients who were lost to follow up, declined testing, or had investigation ongoing at the time of chart review were called “incomplete.” Patients in whom more than one plausible etiology of nystagmus was discovered were called “multifactorial.” If retinal, optic nerve, and central nervous system were normal and vision was 20/200 or better, the patient was classified as “motor nystagmus” as a diagnosis of exclusion. If vision was worse than 20/200, they were placed in the unknown category as this is atypical for purely oculomotor nystagmus.
Results
284 charts were identified, of which 202 met inclusion criteria. 119 were male and 83 were female. The 3 most common causes of nystagmus in this population were Albinism (19%), Leber Congenital Amaurosis (14%) and Non-Leber Congenital Amaurosis retinal dystrophy/degeneration (13%). Anatomic retinopathies such as foveal hypoplasia, familial exudative vitreoretinopathy and coloboma comprised an additional 10%, with motor nystagmus (Idiopathic Infantile Nystagmus Syndrome) another 10% (see figure 1).
Figure 1.

Causes of infantile nystagmus in 202 patients.
74/202 patients had MRI performed as their first test, with the majority performed before referral. The diagnostic yield for these 74 patients was 16%, or 3.5% of the total 202 nystagmus patients. 28 of the 74 had MRI as their first test with nystagmus alone as the indication; 46 had MRI performed first because of neurologic signs in addition to nystagmus. 0/28 nystagmus-only patients had a diagnostic first MRI while 14/46 (30%) with other neurologic signs did (see figure 2). When considering all patients who had MRI, either as initial test or as a subsequent test, 100/202 patients had MRI scans and 24% of the total had findings related to nystagmus. The MRI findings were 9 septo-optic dysplasia (SOD) spectrum, 1 coloboma with intraconal cyst, 2 cerebellar dysplasia (Joubert syndrome), 1 encephalocele, 1 crowded foramen magnum/mild Arnold Chiari malformation, 1 hamartomas on optic nerves.
Figure 2.

Yield of MRI as first test in patients with no neurologic signs, vs. those with neurologic signs. The Y axis represents number of patients. The non-neurologic signs group had solely ocular findings at pediatric eye examination. The neurologic group had abnormal optic nerve appearance, abnormal head circumference, developmental delay, or other neurologic signs at the time of pediatric eye examination.
All patients had a complete pediatric eye examination. This examination was able to detect the cause of nystagmus in 67% of cases, later confirmed by other testing as necessary. After MRI, the most common test to be utilized first was genetic testing when karyotype and chromosome microarray are included, and was ERG when only specific ocular genetic testing is considered. ERG as the first test had a 56% diagnostic yield, OCT 55%, and molecular genetic testing 47% (see figure 3a). The choice of first ocular test was driven by clinical signs noted in the pediatric eye examination, specifically presence or absence of iris transillumination, presence or absence of optic nerve anomaly, presence or absence of systemic neurologic signs, presence or absence of abnormal retinal pigmentation, and presence or absence of positive family history for a disorder associated with nystagmus. Overall as a first test, MRI had the lowest yield and ERG the highest, but all ocular tests had higher yields than MRI. In most cases if the first test was not diagnostic, other testing was pursued. When we calculated the percent positive yield of each test for ever using that test, not only as a first test, Genetic testing and OCT were tied for the highest yield at 58%, ERG was next at 47% and MRI was positive in 16% of cases (see Figure 3b). The complete pediatric eye exam actually had the highest yield for narrowing down the diagnosis to a specific category of etiology that could then be confirmed through other testing.
Figure 3.

(a) First test results. (b). All tests results. The Y axis represents number of patients in each category. The number of patients receiving each test ever is shown by the blue bar, and number of patients who had an abnormality found that was related to nystagmus is on the orange bar. MRI = brain magnetic resonance imaging; OCT = optical coherence tomography; ERG = electroretinogram; Genetic testing = all molecular genetic testing including chromosome microarray and karytype; Genetic Eye Tests = specific testing of eye-related genes by gene panels, exome sequencing, and del/dup testing; Eye Examination = complete pediatric eye examination.
Categories of Nystagmus Etiology
We found 3 broad categories of nystagmus etiology that can help to guide workup: neurologic, ocular, motor. Below are exemplar cases from each of these categories to demonstrate the decision making process.
I. Neurologic causes of nystagmus
Exemplar case 1: A 5 month old male was referred for evaluation of nystagmus. It was described as intermittent and his vision appeared to be normal. Growth and development had been proceeding normally. On examination, there was high frequency horizontal nystagmus of both eyes with brief periods of cessation. Vision was fix and follow with each eye. Anterior segment examination revealed no abnormalities, including no iris transillumination. Dilated fundus examination revealed normal retina and macula, but markedly small, flat optic nerves (see figure 4). MRI scan of brain was ordered due to small optic nerves: the septum pellucidum was noted to be absent, however the pituitary gland appeared normal. The child was referred for vision services, and for endocrine evaluation. At the age of 7 years visual acuity is 20/70 right and 20/60 left, and large amplitude horizontal nystagmus is present. He has not developed hypopituitarism or diabetes insipidus.
Exemplar case 2. A three year old boy was referred from neurosurgery for ocular examination due to nystagmus and concern for encephalocoele. Visual acuity was 20/80 right eye, less than 20/400 left eye with bilateral horizontal jerk nystagmus. There was a frontonasal mass, soft to palpation, and epiphora on the left. Anterior segment and fundus examinations were unremarkable. There was a marked anisometropia. MRI confirmed a left frontal encephalocoele (figure 5), along with cerebellar tonsillar ectopia. Pituitary gland was normal. One year later following repair, spectacle correction and patching therapy, visual acuity was 20/25 right eye, 20/60 left eye with fine pendular horizontal nystagmus.
Discussion of cases 1 and 2: Neurologic disorders associated with nystagmus in children may be apparent, such as encephalocoele, or very subtle. During the history portion of the examination it may become clear that a child is not meeting developmental milestones, has a seizure disorder, or has other signs of a neurologic disorder. In some children only mild neurologic signs can be elicited by history, but upon examination the head circumference is noted to be large or small (it is wise to obtain standardized growth and head circumference curves from children’s primary care doctor, or to measure the head and plot it on a standard curve chart in the office). Other signs of a primarily neurologic disorder may be unusual nystagmus such as see-saw or spasmus nutans (shimmering and asymmetric) (4,5), co-occurrence of ocular motor apraxia (6), ataxia, pale or small optic nerves, or swollen optic nerves. Chorioretinal colobomas may also be associated with midline brain defects, while iris-only colobomas, when in an unusual position away from the inferonasal region, may signify Gillespie syndrome due to mutations in PAX6 or other genes (7,8). Small optic nerves may be due to primary optic nerve hypoplasia (ONH), with or without abnormalities of the septum pellucidum, pituitary gland and posterior pituitary bright spot. Isolated ONH may be associated with pituitary dysfunction especially if the gland is anatomically abnormal on MRI. Life threatening deficiency of cortisol as well as low growth and thyroid hormone may be present making recognition and treatment of great importance. Analysis of a child’s growth cures may reveal “falling off” a percentile curve over time as a sign of growth hormone failure. There are risk factors for ONH such as young maternal age (9), and there are also genetic types, for example due to mutations in HESX1 (10). At this writing the yield with genetic testing for ONH is not very high, but it should be considered in familial cases and discussed with parents in all cases. Many patients with albinism also have small, grey optic nerves. When associated with albinism there is no need for MRI as the association with pituitary and septum pellucidum abnormalities has not been reported. Some patients with partial aniridia also have optic nerve hypoplasia, and in these cases genetic testing of PAX6 rather than MRI is diagnostic. This makes differentiation between primary optic nerve hypoplasia and albinism or aniridia especially important. Rarely, congenital brain tumors or other brain anomalies may present as infantile nystagmus, especially with shimmering, spasmus nutans type (11). Arnold Chiari malformation and other malpositioning of the cerebellar tonsils have been associated with nystagmus (12). In the second case described above it is interesting that the nystagmus changed but did not resolve after brain surgery, and that vision in the non-amblyopic eye is now in the normal range.
Figure 4.

Right eye with severe optic nerve hypoplasia, left with mild in a child with vertical and horizontal nystagmus and septo-optic dysplasia.
Figure 5.
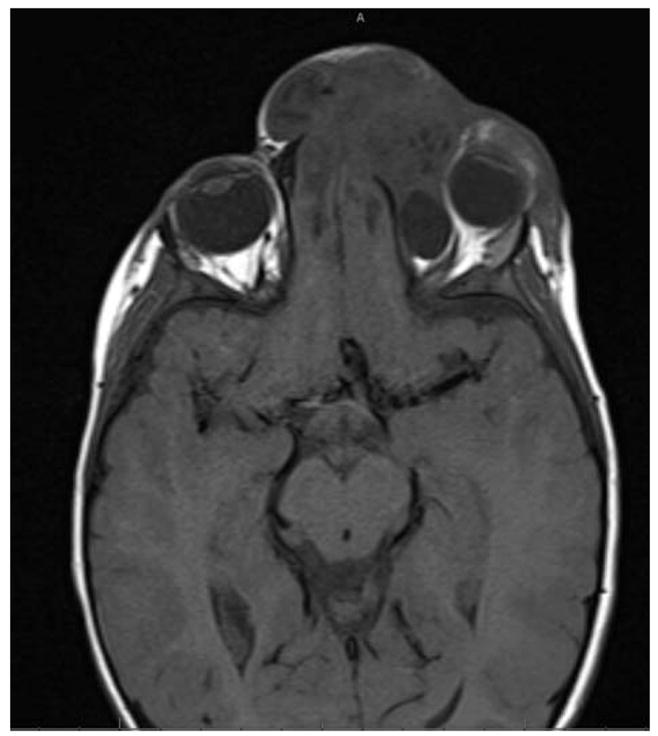
Frontal encephalocoele presenting with infantile nystagmus. Cerebellar tonsillar ectopia was also present.
2. Ocular Causes of Nystagmus
Exemplar case 3: A 7 year old boy presented with a lifelong history of “spasmus nutans.” He had been noted to have shimmering, asymmetric nystagmus by 6 months of age and was seen by several doctors between the ages of 1 year and 6.5 years. He had 4 brain MRI scans, all of them normal, the first ordered because of the known association of spasmus nutans with intracranial pathology, with subsequent scans ordered due to motion artifact obscuring some regions, a new sign (esotropia) developing, and decreased vision being documented. Because the parents had been told that spasmus nutans was self-limited and associated with normal vision, they were surprised when teachers began to notice poor vision. His acuity improved with a myopic correction, but could not be corrected to better than 20/60. Upon referral to the genetic eye disease service ERG was performed (indication: nystagmus, no iris transillumination, decreased best corrected vision, moderate photophobia, and normal MRI) and showed markedly reduced amplitudes in all conditions with electronegative standard combined response (see figure 6). A tentative diagnosis of Leber Congenital Amaurosis/Severe Cone Rod Dystrophy vs. Congenital Stationary Night Blindness (CSNB) was made; the ERG fit CSNB but was unusual in the setting of photophobia and lack of night blindness. A mutation in CACNA1F was discovered, confirming this extremely variable form of “CSNB,” which may also present with cone dysfunction. An extended family history was obtained and multiple male relatives related through females were discovered with a range of signs and symptoms consistent with CACNA1F mutation. At the age of 12 years vision is 20/70 right and 20/40 left. Nystagmus is less prominent but has persisted and is still asymmetric and very fine.
Figure 6.

Electroretinogram of patient with CACNA1F mutation and spasmus-nutans-like nystagmus, compared to normal ERG.
Discussion of case 3: Spasmus-nutans-like nystagmus is a valid reason for obtaining a brain MRI because this type of asymmetric, shimmering nystagmus has been associated with diencephalic and optic nerve tumors (4). However, if the MRI is normal, and the nystagmus does not resolve over the course of 2–4 years as is typical of benign spasmus nutans, the evaluation should shift to looking for ocular causes. Spasmus-nutans-like, asymmetric, shimmering nystagmus can also be caused by retinal disorders (13,14). Other clues that could have prompted earlier referral include photophobia, and decreased vision.
Examplar case 4: A 16 week old male was referred for infantile nystagmus. The child was very blond, as was the entire family. Nystagmus had been noted at about 2 weeks of age. Visual responses had been essentially absent, but the child was beginning to grossly follow faces. On examination iris transillumination was present and there was no apparent fovea on fundus examination. Optic nerves were small and grey. Albinism could be diagnosed clinically, but parents were interested in genetic testing for greater understanding of the syndrome and for family planning. Two mutations in OCA1 were discovered.
Discussion of case 4: Oculocutaneous albinism can be autosomal recessive or X-linked. In the past only patients with complete albinism, lacking all or most pigment, could be diagnosed. The advent of genetic testing has taught us that some people have partial enzyme activity and can have quite normal appearing pigmentation despite having foveal hypoplasia and other signs of albinism. In blond families light pigmentation is not a helpful sign and even normal blue irides may have a few iris transillumination defects. Genetic testing for the 16 plus genes now known to cause albinism can allow for family planning and can rule in or out the rare but real forms of albinism that are associated with other morbidity such as Hermansky Pudlak Syndrome (bleeding diathesis, pulmonary fibrosis) and Chediak Higashi Syndrome (immune deficiency). In infants and older patients with diffuse iris transillumination this sign can be easily seen with a penlight or other illuminator. In more subtle cases iris transillumination can only be seen at the slit lamp. In the case of young children it is often useful to have a slit lamp photograph taken with the light positioned to show the red reflex through the pupil. This may reveal subtle but diffuse iris transillumination not readily apparent on the fleeting examination otherwise possible (see figure 7).
Figure 7.
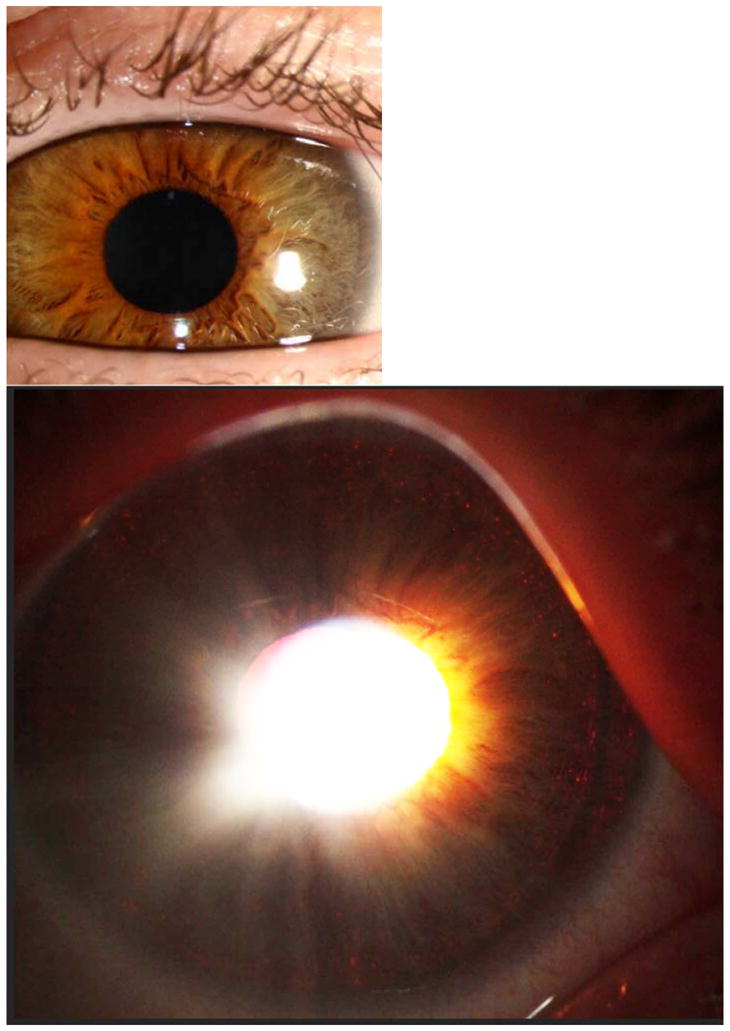
Hazel iris in oblique illumination on the top, with pupillary illumination on the bottom. Note the edge of the crystalline lens visible with surrounding iris transillumination temporally.
Exemplar case 5: A 20 month old female was referred with infantile nystagmus and possible achromatopsia due to intense photophobia since a few months of age. Vision was central, unsteady and maintained at distance and near in each eye. There was photophobia to room light. Nystagmus was fine, horizontal, pendular. Anterior segment and fundus examination were unremarkable and low hyperopia was present. Because she was at an age when electroretinogram must be obtained under anesthesia, molecular genetic testing was the first test obtained with the idea that if positive, anesthesia could be avoided. Testing was sent for achromatopsia with reflex to LCA since there is significant overlap in the presentation during infancy. Achromatopsia testing was negative, but the LCA panel revealed two pathologic mutations in RPGRIP1.
Exemplar case 6: An 11 month old boy was referred with infantile nystagmus and possible optic nerve hypoplasia. Vision was fix and follow each eye at near but there was no distance fixation. Horizontal nystagmus was present. Anterior segment was normal without transillumination of iris. Fundus exam was normal with borderline small optic nerves. Cycloplegic refraction revealed high hyperopia. Because nerves were only borderline small, visual acuity was worse than nerves would suggest, and high hyperopia was present, LCA was suspected. Molecular genetic testing was ordered (LCA panel) and ERG under anesthesia was scheduled for a future date in case genetic testing was non-diagnostic. Genetic testing revealed 2 disease causing mutations in CEP290, obviating the need for ERG. Visual acuity was 20/400 by Teller cards at age one year, however at age 8 years vision had decreased to light perception.
Discussion of cases 5 and 6: Because commercial testing for relatively common conditions like LCA and achromatopsia is advanced and rapid, molecular genetic testing is often the most efficient first test for young children who present with classic symptoms and signs. ERG is difficult to perform in an awake child between the ages of approximately 1 to 5 years, so genetic testing is a good first option in these patients. Infants with achromatopsia, LCA, and albinism can present with almost identical findings, especially if the child is blond. It is important to note that photoaversion occurs in some patients with LCA (15), which is less well known than the photoaversion seen in albinism and achromatopsia (16). If ERG cannot easily be obtained, molecular genetic testing starting with the most likely of the 3 disorders can be used to rapidly distinguish between them. Everyone carries multiple single copies of mutated recessive genes. When exome sequencing or large panels of genes are queried without a diagnosis in mind, it is common to find single mutated alleles for unrelated disorders that can inaccurately sway the diagnosis. An hypothesis should guide genetic testing based on the clinical findings and the narrowest molecular genetic “question” should be asked to avoid misleading results (17).
3. Motor Nystagmus (Idiopathic Infantile Nystagmus)
Examplar case 7: A 6 month old boy was referred to evaluate infantile nystagmus present since shortly after birth. He was blond, as was most of his family. His mother, brother and maternal uncle also have infantile nystagmus. On examination he had horizontal left beating jerk nystagmus of moderate amplitude with good fixation and following visual responses. There was no iris transillumination. Retinas, optic nerves and foveas appeared normal. Mother’s vision, with horizontal jerk nystagmus, was 20/30 each eye. OCT was performed on mother and showed a normal fovea. There was no night blindness or photophobia present in any affected family member. A pedigree was obtained of XL inheritance with manifesting females and good visual acuity. FRMD7 gene sequencing was obtained and was normal. Intragenic deletion testing was obtained and an intragenic deletion was detected in FRMD7 that segregates with the nystagmus in the family.
Discussion of case 7: X-linked motor/idiopathic infantile nystagmus has been described in many publications (18,19) and mutations in FRMD7 are common (20,21,22) and intragenic deletions have also been reported (23,24). This type of nystagmus appears very similar to the nystagmus seen in albinism, however on nystagmography there are subtle differences (25). Patients with FRMD7 motor nystagmus have no iris transillumination defects and a fovea is present on OCT, although at least one report states the foveas may be thinner than normal (26). Both X Linked ocular albinism and FRMD7 nystagmus are inherited as X linked traits, so either OCT or molecular genetic testing is vital to differentiate the two (27); patients with FRMD7 mutations will likely have significantly better visual acuity in adulthood. In infants, the hand held OCT may be very useful to differentiate these disorders (28), although sedation or anesthesia is often required during the toddler years. Molecular genetic testing may be a good first step in some patients.
4. Ocular versus Neurologic: you decide
Examplar case 8: A baby boy was seen at 4 months of age for infantile nystagmus, present since birth. He was noted to have developmental delays and had two MRI scans which were read as normal. His eye examination which unremarkable and the cause of the nystagmus was suspected to be neurologic, related to developmental delay but without a localizing MRI. He was followed for many years and at the age of 14 years he was noted to have optic nerve pallor. He had walked late and had balance problems his entire life. A repeat MRI was obtained, also read as normal, and he was referred for ocular re-evaluation. An ERG was performed, which was essentially nonrecordable. Because of the combination of infantile nystagmus and non-recordable rod and cone ERG responses, LCA genetic testing was obtained but was negative. Testing was broadened to a retinal exome panel, and 2 pathologic mutations were discovered in AHI1, known to be associated with Joubert syndrome. Oculomotor apraxia was not present. A repeat reading of his most recent MRI after genetic testing results were shared with the radiologist led to detection of a subtle molar tooth sign. At age 16 years with his myopic correction visual acuity is 20/70 each eye.
Discussion of case 8: In this patient neurologic signs led appropriately to an early MRI, then repeat MRIs, as his findings progressed, however radiologists are best able to diagnose subtle findings when they know where to look. In this case the molar tooth sign was not detected until the molecular genetic diagnosis of Joubert syndrome was made. Joubert syndrome was not initially suspected due to the negative MRI scan reports but became part of the differential diagnosis after ERG results were obtained. This case demonstrates the need to periodically reassess patients with nystagmus who do not have a definite diagnosis, and to communicate with other specialties in cases of complex patients. New technologies can diagnose many patients with subtle findings who could not be diagnosed in the past.
Discussion
Based on our data, a child presenting with infantile nystagmus and no other neurologic signs or symptoms is far more likely to have an ocular sensory cause of nystagmus than a neurologic cause. However, despite falling into several broad groups, the etiologies of infantile nystagmus are many and varied (see table 1), making the diagnostic workup a challenge. It is best described to patients and families as a process in which a logical stepwise evaluation will be performed. Despite best efforts, about 4% of patients will have an unknown cause of infantile nystagmus, and another 10% will be grouped as motor nystagmus or idiopathic, a diagnosis of exclusion which nonetheless has a good prognosis for stable, near normal vision.
Table 1.
Etiology of Nystagmus in 202 patients.
| Diagnosis | Molecular | Clinical | Likely | Total | Percent of All Patients |
|---|---|---|---|---|---|
| Albinism | 14 | 22 | 2 | 38 | 18.81 |
| LCA | 23 | 3 | 2 | 28 | 13.86 |
| Motor | 3 | 15 | 2 | 20 | 9.90 |
| Incomplete Workup | 13 | 6.44 | |||
| ONH with or without SOD | 11 | 11 | 5.45 | ||
| CSNB | 7 | 1 | 1 | 9 | 4.46 |
| PAX6 | 8 | 8 | 3.96 | ||
| Achromatopsia | 6 | 2 | 8 | 3.96 | |
| Multifactorial | 8 | 3.96 | |||
| Unknown | 8 | 8 | 3.96 | ||
| Foveal Dysplasia | *7 | 7 | 3.47 | ||
| Down Syndrome | 3 | **4 | 7 | 3.47 | |
| Joubert Syndrome | 4 | 1 | 5 | 2.48 | |
| Retinal Dystrophy | 5 | 5 | 2.48 | ||
| Retinal Dystrophy plus Syndrome | 4 | 4 | 1.98 | ||
| Neurologic | 4 | 4 | 1.98 | ||
| Coloboma | 1 | 2 | 3 | 1.49 | |
| FEVR | 2 | 1 | 3 | 1.49 | |
| Chromosomal Deletion Syndromes | 3 | 3 | 1.49 | ||
| Optic Nerve Atrophy | 2 | 2 | 0.99 | ||
| Maculopathy | 2 | 2 | 0.99 | ||
| BBS | 1 | 1 | 0.50 | ||
| Visual Deprivation Nystagmus | ***1 | 1 | 0.50 | ||
| Blue Cone Monochromatism | 1 | 1 | 0.50 | ||
| Donnai-Barrow Syndrome | 1 | 1 | 0.50 | ||
| Zellweger syndrome | 1 | 1 | 0.50 | ||
| Tuberous Sclerosis | ****1 | 1 | 0.50 |
This includes patients with isolated abnormal fovea on OCT including former severely premature babies and one child with Waardenburg syndrome.
Some patients have no karyotype data in their charts, however it is assumed that trisomy 21 was confirmed by prior providers.
Dense congenital cataracts referred at 3 months of age.
Tuberous sclerosis with bilateral optic nerve hamartomas.
Molecular = molecular genetic diagnosis confirmed with 2 disease causing alleles in trans or hemizygous disease causing allele; Clinical = all clinical signs of the disorder +/− one allele found in a disease causing gene; Likely = most signs of the disorder without genetic confirmation; LCA = Leber Congenital Amaurosis; Motor= diagnosis of exclusion after all testing has been completed and visual acuity is 20/200 or better; Incomplete workup = workup in progress or lost to follow up or declined further testing; ONH = Optic nerve hypoplasia; SOD = septo-optic dysplasia; CSNB = congenital stationary night blindness; PAX6 = mutations in PAX6 gene leading to aniridia or other manifestations; FEVR = familial exudative vitreoretinopathy; BBS = Bardet Biedl Syndrome.
Pediatric ophthalmologists and genetic eye disease specialists have a vested interest in developing an algorithm for the evaluation of these patients. The CEMAS classification of nystagmus (2), while helpful for categorizing the waveforms of nystagmus, has not been proven useful in diagnosing the etiologies. Because nystagmography is not routinely available in pediatric ophthalmology offices, and because the utility of such recordings for diagnosis in a large clinical sample have not been studied, this would be a fertile area for further research. A study comparing the nystagmus of albinism patients and FRMD7-related patients found that while there were group differences the differences between individuals were not diagnostic (25). In our study population we found essentially the same phenomenon (Figure 8). For example, roving nystagmus was over represented in LCA, but was also found in many other disorders. While nystagmus waveform is not currently helpful in diagnosis to most practitioners, many other current technological and molecular genetic resources are.
Figure 8.

Nystagmus waveform type versus diagnosis by percentage. The y-axis represents the nystagmus waveform types. The x-axis represents a selection of the most common diagnoses. The waveform types may add up to more than 100% per diagnosis due to some patients having more than one waveform type mentioned in the clinical description of their nystagmus in the chart.
We have found it helpful to think about the differential diagnosis of infantile nystagmus by first dividing it into 3 broad categories: Neurologic causes, Vision/Ocular related causes, and Oculomotor/Eye Movement disorder causes. For brevity we refer to these groups as the Neurologic, Ocular and Motor groups. The first dividing point is based on the patient’s birth and family history, growth and development. If there is no relevant family history, and there are any signs of neurologic issues, a brain MRI is the first test. If there is no relevant family history and there are NO neurologic signs, the findings on a complete pediatric eye examination are used to direct ancillary testing with the most likely tests being ordered first. Is the vision very poor and accompanied by high hyperopia? This best fits LCA and molecular genetic testing would be ordered first. Are there iris transillumination defects? Macula OCT can be obtained, or if the child is too young, molecular genetic testing for albinism can be considered, especially if the child has easy bruising or bleeding, and/or if the family would use the information for family planning. Hand held OCT is especially useful in young infants, in whom it may be accomplished while awake (28), or in toddlers it may be performed under anesthesia. Is the pupil ectopic or oval? PAX6 testing should be considered. If there are no obvious findings to direct the testing, ERG is often the best first test. This test can divide the causes broadly into genetic retinal dystrophies versus all others (neurologic, anatomic, motor).
Asymmetric VEP has been reported to be a specific feature of albinism. This is due to anomalous optic chiasm crossing of fibers leading to a larger potential in the contralateral visual cortex. The asymmetry between potentials varies by age as well as by patient (29). In the molecular era, we have found VEP to be far more difficult to interpret than other tests that are available. For that reason, we have not incorporated VEP into our algorithm, however, some practitioners who are very experienced in its use with albinism patients may choose to incorporate it into their personal work-up for infantile nystagmus patients. Figure 9 represents one possible algorithm, which is a modification of a flow chart submitted to the American Academy of Ophthalmology Knights Templar Pediatric Ophthalmology Education Site(www.aao.org).
Figure 9.

Flow chart algorithm for the workup of infantile nystagmus. Used with permission from American Academy of Ophthalmology Knights Templar Pediatric Ophthalmology Education Site(www.aao.org).
Key: MRI = magnetic resonance imaging; TIDs= transillumination defects; OCT = optical coherence tomography; LCA=Leber Congenital Amaurosis; ONH= optic nerve hypoplasia; CVI=cortical vision impairment; CSNB=congenital stationary night blindness; JXLR=juvenile Xlinked retinoschisis; Abnl=abnormal; achroma=achromatopsia; RP=retinitis pigmentosa; PAX6=PAX6 gene, responsible for aniridia and related syndromes; FRMD7=FRMD7 gene, an X linked gene associated with motor (idiopathic infantile) nystagmus.
The first step is always a complete pediatric eye examination including cycloplegic refraction with suspicion for, and use of techniques necessary to detect, very high refractive errors. Very high refractive error alone can cause nystagmus (generally greater than −15D myopia or +10D hyperopia) and can be easily missed in a fussy infant without very careful cycloplegic retinoscopy. If the retinoscopy reflex appears to be plano, i.e. no movement with the usual working distance offset, high plus and high minus lenses (−10, +10 etc.) should be used to assess whether one of these gets the reflex to move, signifying extremely high refractive error.
Nystagmus of any type and in any direction may be referred to pediatric ophthalmologists and genetic eye disease specialists. Two patients in our series had shimmering, asymmetric, fine nystagmus that is typical of spasmus nutans. Spasmus nutans is defined as a self-limited benign nystagmus of childhood, often thought to be related to neurologic immaturity. This type of nystagmus can also be present with infantile brain tumors, so physicians are acutely aware of the risk of missing a lesion that can be seen on MRI. But although this pattern of nystagmus is typical of spasmus nutans, or, more ominously, diencephalic or optic chiasm tumors, it is not specific for those disorders. If the MRI is negative, further workup must be explored. Several patients in our series had a delay of years between obtaining a negative MRI scan and receiving a full ophthalmic workup. Of interest, the two patients with an initial diagnosis of spasmus nutans in our series both had an electronegative ERG with a final diagnosis of CSNB due to CACNA1F in one patient and TRPM1 in another. Neither of these patients complained of night blindness and both had lessening of nystagmus over time, though it still persisted in early adolescence. Nystagmus is known to resolve in some CSNB patients. There are many reports in the literature of spasmus nutans-like nystagmus being caused by retinal dystrophies (30,31), and even an excellent article entitled, “Electroretinography is necessary for spasmas nutans workup” because of these diagnoses (32). Still, many spasmus nutans patients may never have a full workup if their vision is near normal and the nystagmus resolves, and because many CSNB patients have very protean manifestations of their genetic mutations, it is possible that many more patients with “spasmus nutans” have CSNB than we know.
These cases demonstrate that a normal or negative MRI in a child with nystagmus should never be the last step—ophthalmologic workup should follow a negative MRI. By the same token, if an ophthalmologic workup is performed and there is an ocular diagnosis, if there are changes, or if the diagnosis does not fit the clinical picture, MRI should still be performed. We have seen a patient with classic negative wave ERG and nystagmus and visual acuity consistent with CSNB who later started losing vision. An MRI revealed a midline brain tumor. While unusual, some patients will have two disorders. Clinical acumen plays an important role in the workup of these complex patients.
Careful slit lamp exam must be done to examine irides for transillumination, which can be seen in albinism, aniridia, and PAX6 disease without complete aniridia, all of which are associated with foveal hypoplasia and nystagmus. PAX6 disease may present with complete aniridia or with cataract, ellipsoid iris or other mild iris anomalies (33,34). Waardenburg syndrome patients may also have iris transillumination and may possibly have foveal hypoplasia and nystagmus as well; one patient in our foveal dysplasia category has Waardenburg syndrome with an MITF mutation, and foveal hypoplasia. There is a report in the literature of a Waardenburg patient with MITF mutation and nystagmus which the authors attribute to digenic inheritance with an OCA1 mutation (35), however it is more likely that Waardenburg syndrome alone may be associated with foveal hypoplasia and nystagmus since MITF is in the pigmentation cascade. More study on this is needed. A condition called FHONDA has been reported, which is foveal hypoplasia without decreased pigmentation (36,37). We have included the gene for FHONDA in our albinism gene panel. Slit lamp photographs taken specifically to capture iris transillumination may demonstrate defects that were not visible at the slit lamp in a young, moving child. Lisch nodules or other anomalies of the iris may offer clues to tumors and syndromes associated with nystagmus, such as neurofibromatosis. Another important part of the slit lamp examination is to view the anterior vitreous under high magnification and assess the presence or absence of cells. Vitritis may be a primary immune condition, or can be secondary to some, but not all, pediatric onset retinal degenerations (38).
The discovery of FRMD7 as a “motor” nystagmus gene is fascinating and the function of this gene and protein should pave the way to better understanding of the mechanisms behind nystagmus and of treatment targets (39). Likewise, while Down syndrome has long been known to be a predisposition for nystagmus, only recently have the ocular or neurologic aberrations responsible for nystagmus in trisomy 21 patients been studied and reported (40).
In our study as in other studies of infantile nystagmus, there are more affected males than females. This likely represents the importance of X linked disorders such as XL Albinism, XL FEVR, XLRP and FRMD7.
Fundus examination is vital and often holds the key to diagnosis. As with slit lamp examination, if a child is not able to fully cooperate it may be easier to get a quick fundus photograph than to get a full look with the indirect ophthalmoscope. The retina must be examined for the bone-spicule-like pigmentation in the periphery seen in various types of retinitis pigmentosa (however this is not usually present in early childhood even if it will develop as time goes on), nummular pigment, or narrowed arterioles. The macula must be examined for pigmentary changes, and the fovea for blunting, or for absence as is seen in foveal hypoplasia. In its most severe form, vessels may course directly over the area that should be the fovea. OCT has revolutionized our ability to diagnose two causes of nystagmus: foveal hypoplasia and cystoid macular edema (CME). Foveal hypoplasia may be seen in several conditions and is very commonly associated with nystagmus. CME is much less commonly a cause of nystagmus, however if it is severe and of early onset, such as is seen in some cases of Usher Type 1, or if it is not true CME but the foveal cysts seen in Juvenile X-linked retinoschisis with a severe early onset, nystagmus can be present.
It is clear that patients with an abnormal retinal examination should have ERG, OCT or genetic testing for retinal disorders. The the other group of patients who need this workup, paradoxically, are those with a completely normal appearing retina. Many types of LCA, neuronal ceroid lipofuscinosis, CSNB, achromatopsia and other disorders have no retinal signs on fundoscopy early in life.
Optic nerve hypoplasia is fairly common. It has been found to be more common in offspring of very young mothers with their first pregnancy (41). If it is mild and subtle, optic nerve OCT may be helpful to confirm the size of the disc and thickness of the retinal nerve fiber layer. If it is suspected, brain MRI must be performed to evaluate the pituitary and the septum pellucidum. If the nerves are pale there may have been pre- or peri-natal insult causing optic atrophy, which may be confirmed with MRI. This may also be associated with extremely premature birth.
A careful family history, especially asking about family members with poor vision who developed neurologic signs, kidney disease or other associated findings should be taken. The patient’s history is important as well, even if they are very young. Premature birth, expecially extreme prematurity of 28 weeks gestational age or less, may be associated with sequellae of retinopathy of prematurity, abnormalities of the fovea and macula, and neurologic sequellae. Several children with very premature birth are in our series spread between the foveal dysplasia, neurologic and multifactorial categories, demonstrating how complex the etiology may be.
Growth charts for height, weight, and head circumference should be obtained from the primary care doctor as well as ages at which children met developmental milestones. Watch the child walk across the room, reach for toys, sit unassisted. Any deviation in these neurologic parameters should suggest the utility of an MRI scan.
The findings on the pediatric eye examination as well as the history should direct the next step in the workup. If there are optic nerve anomalies, an MRI may be the appropriate first step. However, patients with albinism and PAX6 related disorders often have small, grey or abnormal optic nerves. In order to spare them an unnecessary MRI, the search for iris trans illumination and for foveal hypoplasia must be avid. Hand held OCT is now available and while it is difficult to obtain in an awake child, it can be done more quickly under anesthesia than an MRI. Another option is to consider genetic testing for albinism or PAX6 very early if there signs that the iris and fovea are not normal, yet definitive slit lamp and OCT cannot be obtained. In these cases if 2 definite disease causing mutations are found in a known albinism gene, or one definite mutation is found in PAX6, further workup may be unnecessary.
In an infant with nystagmus, especially if roving with very poor vision and no other ocular abnormalities, the most likely diagnosis is Leber Congenital Amaurosis. A non-recordable electroretinogram can make the diagnosis of this category, however it cannot specify which of the 19 known genes is causing it. There have been successful clinical trials of subretinal gene replacement for one type of LCA, RPE65-associated, and patients with mutations in this gene may benefit from treatment (42,43,44). In addition, it is possible for parents to do in vitro fertilization (IVF) with pre-implantation genetic testing for future children if they know the mutations in their affected child. Because genetic testing for LCA is now standard and commercially available, a molecular genetic diagnosis is the most accurate one for these children. In addition, ERG in an awake child is a challenge for both parent and child (and whomever performs the ERG) while doing ERG under anesthesia carries risks of anesthesia. If the genetic testing is not diagnostic, the algorithm reflexes back to ERG since this may provide a surprise, such as a pattern more suggestive of CSNB or achromatopsia than LCA, which can guide further genetic testing. There are likely more than the 19 known genes for LCA since some typical patients still have no mutations found; these patients can be offered enrollment in a research protocol designed to find these unknown genes.
Another benefit of molecular genetic testing in children with a clinical diagnosis of LCA, either before or after ERG, is prognosis for kidney failure. Senior-Loken syndrome, or nephronophthisis in the setting of retinal degeneration, used to be a black box with no way of knowing which LCA patients were at risk. We now know that mutations in the NPHP genes can cause LCA alone, LCA with renal failure, or renal failure with later onset retinitis pigmentosa (45,46). Identifying these patients early can get them to renal providers before kidney failure occurs.
In our series, even when MRI was the correct first test, it sometimes turned out not to be the most helpful first test. Four children in two unrelated families presented with nystagmus, ataxic gait, developmental delay and delayed speech to their primary care doctors. These children had MRI scans which were read as normal. Several years later they were referred for genetic eye disease evaluation and ERG was performed for the indication of nystagmus and nightblindness. The electroretinograms were remarkably abnormal. This combined with their other symptoms and signs put Joubert syndrome in the differential diagnosis, and retinal exome sequencing panel revealed 2 mutations in a gene known to cause Joubert syndrome in each family. After these ERG and molecular genetic diagnoses were made, we asked to have the scans re-read and a “molar tooth” sign was noticed in one child of each family. This is yet another example of why children with infantile nystagmus deserve a full pediatric eye evaluation early in life, with a complete workup, even if neurologic signs are also present.
A weakness of our study is that patients with a neurologic cause for infantile nystagmus may have been missed because these patients were referred to pediatric ophthalmology or genetic eye disease services. There may be other patients who present elsewhere with nystagmus alone, have an MRI with a diagnostic abnormality found for their nystagmus, and are never referred. This is possible and it would be interesting to see a study of diagnoses of infantile nystagmus in children presenting to other types of physicians to do a comparison. It would be necessary, however, only to include patients with a complete workup. The more patients without a full workup in a series, the higher the percentage of the “motor” or idiopathic category. For example, a study of 62 patients with nystagmus at a school for the blind in Sweden found that about 43 had an obvious underlying condition, and of 19 with isolated “congenital nystagmus” as their only diagnosis; upon workup 2 had albinism, 4 apparently isolated foveal hypoplasia, 3 achromatopsia, 1 rod cone dystrophy, and 1 high myopia (47). In a paper by Fu et al. (3), the visual acuity in 214 patients with Infantile Nystagmus Syndrome was assessed. It was concluded that visual acuity correlated with underlying etiology of nystagmus, which was garnered from records as being Idiopathic INS in 84, albinism in 71, ONH in 23, congenital retinal disorder 36 (including achromatopsia/blue cone monochromacy in 13), LCA in 8, cone rod or cone degeneration in 9. Foveal hypoplasia was diagnosed in 6. Thus is a series similar in size to our own, the most common diagnosis was idiopathic, similar to our “motor” classification. Congenital Idiopathic Nystagmus (CIN), equivalent to our “motor nystagmus” and Idiopathic Infantile Nystagmus, can be inherited as an autosomal dominant, recessive or X linked disorder, but because the only gene known so far is FRMD7, it must still be a diagnosis of exclusion for most patients (48). Without a standard workup, however, one cannot be sure how many of the patients in this category have another, more specific, diagnosis.
Many patients in our series who have an underlying neurologic diagnosis were first seen by neurologists, were diagnosed with MRI, and then were referred for pediatric eye evaluation because of the understanding that nystagmus might be affecting their vision, or might be a sign of vision problems in addition to the primary cause of the nystagmus. Despite this referral pattern, only 2% of patients in this series had nystagmus due to a purely neurologic cause.
Conclusion
The most common causes of infantile nystagmus in this pediatric ophthalmology cohort are retinal disorders, totaling 56% of all cases. Conversely, the most common first test in the nystagmus work-up was brain MRI. MRI is not the best first test for patients with infantile nystagmus in the absence of other neurologic stigmata. Complete pediatric eye examination, ERG, OCT and molecular genetic testing in an order determined by clinical findings should be performed early in patients with infantile nystagmus who do not have other neurologic signs. In patients with neurologic signs MRI should be obtained, and if it is non-diagnostic, a complete workup considering ERG, OCT and molecular genetic testing should be pursued.
Acknowledgments
Thanks to Edwin Stone, MD, PhD, and the Carver Lab for molecular genetic testing. Thanks to Frank Bertsch, BA, MS, for assistance with data analysis.
Funding
Support for this project came from the Carver College of Medicine, NIH T35 HL007485 (Bertsch), and The Ronald Keech Associate Professorship in Pediatric Genetic Eye Disease Research, Vision for Tomorrow Foundation, Foundation Fighting Blindness, Research to Prevent Blindness, and the Wynn Institute for Vision Research (Drack).
Footnotes
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article. Arlene V. Drack, MD, is a co-investigator in the Phase III RPE65 gene therapy trial which is funded by a grant from Spark Therapeutics.
References
- 1.Hertle RW, Maldanado VK, Maybodi M, Yang D. Clinical and ocular motor analysis of the infantile nystagmus syndrome in the first 6 months of life. Br J Ophthalmol. 2002 Jun;86(6):670–5. doi: 10.1136/bjo.86.6.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.CEMAS Working Group. The National Eye Institute Publications. Bethesda, MD: The National Eye Institute, The National Institutes of Health; 2001. A National Eye Institute Sponsored Workshop and Publication on The Classification Of Eye Movement Abnormalities and Strabismus (CEMAS) ( www.nei.nih.gov). The National Institutes of Health. [Google Scholar]
- 3.Fu VL, Bilonick RA, Felius J, Hertle RW, Birch EE. Visual acuity development of children with infantile nystagmus syndrome. Invest Ophthalmol Vis Sci. 2011 Mar 14;52(3):1404–11. doi: 10.1167/iovs.09-4686. Print 2011 Mar. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brodsky MC, Keating GF. Chiasmal glioma in spasmus nutans: a cautionary note. J Neuroophthalmol. 2014 Sep;34(3):274–5. doi: 10.1097/WNO.0000000000000121. [DOI] [PubMed] [Google Scholar]
- 5.Delorme C, Gras D, Roze E. Spasmus Nutans: More Than Meets the Eye. Pediatr Neurol. 2015 Oct;53(4):367–8. doi: 10.1016/j.pediatrneurol.2015.06.011. Epub 2015 Jun 20. [DOI] [PubMed] [Google Scholar]
- 6.Weiss AH1, Doherty D, Parisi M, Shaw D, Glass I, Phillips JO. Eye movement abnormalities in Joubert syndrome. Invest Ophthalmol Vis Sci. 2009 Oct;50(10):4669–77. doi: 10.1167/iovs.08-3299. Epub 2009 May 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McEntagart M, Williamson KA, Rainger JK, Wheeler A, Seawright A, De Baere E, Verdin H, Bergendahl LT, Quigley A, Rainger J, Dixit A, Sarkar A, López Laso E, Sanchez-Carpintero R, Barrio J, Bitoun P, Prescott T, Riise R, McKee S, Cook J, McKie L, Ceulemans B, Meire F, Temple IK, Prieur F, Williams J, Clouston P, Németh AH, Banka S, Bengani H, Handley M, Freyer E, Ross A, van Heyningen V, Marsh JA, Elmslie F, FitzPatrick DR DDD Study. A Restricted Repertoire of De Novo Mutations in ITPR1 Cause Gillespie Syndrome with Evidence for Dominant-Negative Effect. Am J Hum Genet. 2016 May 5;98(5):981–92. doi: 10.1016/j.ajhg.2016.03.018. Epub 2016 Apr 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ansari M, Rainger J, Hanson IM, Williamson KA, Sharkey F, Harewood L, Sandilands A, Clayton-Smith J, Dollfus H, Bitoun P, Meire F, Fantes J, Franco B, Lorenz B, Taylor DS, Stewart F, Willoughby CE, McEntagart M, Khaw PT, Clericuzio C, Van Maldergem L, Williams D, Newbury-Ecob R, Traboulsi EI, Silva ED, Madlom MM, Goudie DR, Fleck BW, Wieczorek D, Kohlhase J, McTrusty AD, Gardiner C, Yale C, Moore AT, Russell-Eggitt I, Islam L, Lees M, Beales PL, Tuft SJ, Solano JB, Splitt M, Hertz JM, Prescott TE, Shears DJ, Nischal KK, Doco-Fenzy M, Prieur F, Temple IK, Lachlan KL, Damante G, Morrison DA, van Heyningen V, FitzPatrick DR. Genetic Analysis of ‘PAX6-Negative’ Individuals with Aniridia or Gillespie Syndrome. PLoS One. 2016 Apr 28;11(4):e0153757. doi: 10.1371/journal.pone.0153757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tornqvist K1, Ericsson A, Källén B. Optic nerve hypoplasia: Risk factors and epidemiology. Acta Ophthalmol Scand. 2002 Jun;80(3):300–4. doi: 10.1034/j.1600-0420.2002.800313.x. [DOI] [PubMed] [Google Scholar]
- 10.McNay DE, Turton JP, Kelberman D, Woods KS, Brauner R, Papadimitriou A, Keller E, Keller A, Haufs N, Krude H, Shalet SM, Dattani MT. HESX1 mutations are an uncommon cause of septooptic dysplasia and hypopituitarism. J Clin Endocrinol Metab. 2007 Feb;92(2):691–7. doi: 10.1210/jc.2006-1609. Epub 2006 Dec 5. [DOI] [PubMed] [Google Scholar]
- 11.Gottlob I1, Wizov SS, Reinecke RD. Spasmus nutans. A long-term follow-up. Invest Ophthalmol Vis Sci. 1995 Dec;36(13):2768–71. [PubMed] [Google Scholar]
- 12.Tubbs RS1, Soleau S, Custis J, Wellons JC, Blount JP, Oakes WJ. Degree of tectal beaking correlates to the presence of nystagmus in children with Chiari II malformation. Childs Nerv Syst. 2004 Jul;20(7):459–61. doi: 10.1007/s00381-004-0948-9. Epub 2004 Apr 2. [DOI] [PubMed] [Google Scholar]
- 13.Papageorgiou E, McLean RJ, Gottlob I. Nystagmus in childhood. Pediatr Neonatol. 2014 Oct;55(5):341–51. doi: 10.1016/j.pedneo.2014.02.007. Epub 2014 Jul 31. [DOI] [PubMed] [Google Scholar]
- 14.Richards MD, Wong A. Infantile nystagmus syndrome: clinical characteristics, current theories of pathogenesis, diagnosis, and management. Can J Ophthalmol. 2015 Dec;50(6):400–8. doi: 10.1016/j.jcjo.2015.07.010. [DOI] [PubMed] [Google Scholar]
- 15.Traboulsi EI, Maumenee IH. Photoaversion in Leber’s congenital amaurosis. Ophthalmic Genet. 1995 Mar;16(1):27–30. doi: 10.3109/13816819509057851. [DOI] [PubMed] [Google Scholar]
- 16.Sundin OH, Yang JM, Li Y, Zhu D, Hurd JN, Mitchell TN, Silva ED, Maumenee IH. Genetic basis of total colourblindness among the Pingelapese islanders. Nat Genet. 2000 Jul;25(3):289–93. doi: 10.1038/77162. [DOI] [PubMed] [Google Scholar]
- 17.Stone EM. Leber congenital amaurosis - a model for efficient genetic testing of heterogeneous disorders: LXIV Edward Jackson Memorial Lecture. Am J Ophthalmol. 2007 Dec;144(6):791–811. doi: 10.1016/j.ajo.2007.08.022. Epub 2007 Oct 26. [DOI] [PubMed] [Google Scholar]
- 18.Kerrison JB, Vagefi MR, Barmada MM, Maumenee IH. Congenital motor nystagmus linked to Xq26-q27. Am J Hum Genet. 1999 Feb;64(2):600–7. doi: 10.1086/302244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kerrison JB, Giorda R, Lenart TD, Drack AV, Maumenee IH. Clinical and genetic analysis of a family with X-linked congenital nystagmus (NYS1) Ophthalmic Genet. 2001 Dec;22(4):241–8. doi: 10.1076/opge.22.4.241.2216. [DOI] [PubMed] [Google Scholar]
- 20.Tarpey P, Thomas S, Sarvananthan N, Mallya U, Lisgo S, Talbot CJ, Roberts EO, Awan M, Surendran M, McLean RJ, Reinecke RD, Langmann A, Lindner S, Koch M, Jain S, Woodruff G, Gale RP, Bastawrous A, Degg C, Droutsas K, Asproudis I, Zubcov AA, Pieh C, Veal CD, Machado RD, Backhouse OC, Baumber L, Constantinescu CS, Brodsky MC, Hunter DG, Hertle RW, Read RJ, Edkins S, O’Meara S, Parker A, Stevens C, Teague J, Wooster R, Futreal PA, Trembath RC, Stratton MR, Raymond FL, Gottlob I. Mutations in FRMD7, a newly identified member of the FERM family, cause X-linked idiopathic congenital nystagmus. Nat Genet. 2006 Nov;38(11):1242–4. doi: 10.1038/ng1893. Epub 2006 Oct 1. Erratum in: Nat Genet. 2011 Jul;43(7):720. Bastawrous, Andrew [added] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thomas MG, Thomas S, Kumar A, Proudlock FA, Gottlob I. FRMD7-Related Infantile Nystagmus. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC, Smith RJH, Stephens K, editors. F GeneReviews® [Internet] Seattle (WA): University of Washington, Seattle; 2009. Feb 12, pp. 1993–2016. [updated 2011 Sep 29] [Google Scholar]
- 22.Watkins RJ, Thomas MG, Talbot CJ, Gottlob I, Shackleton S. The Role of FRMD7 in Idiopathic Infantile Nystagmus. J Ophthalmol. 2012;2012:460956. doi: 10.1155/2012/460956. Epub 2011 Aug 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fingert JH, Roos B, Eyestone ME, Pham JD, Mellot ML, Stone E. Novel intragenic FRMD7 deletion in a pedigree with congenital X-linked nystagmus. Ophthalmic Genet. 2010 Jun;31(2):77–80. doi: 10.3109/13816810903584989. [DOI] [PubMed] [Google Scholar]
- 24.AlMoallem B, Bauwens M, Walraedt S, Delbeke P, De Zaeytijd J, Kestelyn P, Meire F, Janssens S, van Cauwenbergh C, Verdin H, Hooghe S, Kumar Thakur P, Coppieters F, De Leeneer K, Devriendt K, Leroy BP, De Baere E. Novel FRMD7 Mutations and Genomic Rearrangement Expand the Molecular Pathogenesis of X-Linked Idiopathic Infantile Nystagmus. Invest Ophthalmol Vis Sci. 2015 Feb 12;56(3):1701–10. doi: 10.1167/iovs.14-15938. [DOI] [PubMed] [Google Scholar]
- 25.Clinical and oculomotor characteristics of albinism compared to FRMD7 associated infantile nystagmus. Kumar A, Gottlob I, McLean RJ, Thomas S, Thomas MG, Proudlock FA. Invest Ophthalmol Vis Sci. 2011 Apr 8;52(5):2306–13. doi: 10.1167/iovs.10-5685. [DOI] [PubMed] [Google Scholar]
- 26.Thomas MG, Crosier M, Lindsay S, Kumar A, Araki M, Leroy BP, McLean RJ, Sheth V, Maconachie G, Thomas S, Moore AT, Gottlob I. Abnormal retinal development associated with FRMD7 mutations. Hum Mol Genet. 2014 Aug 1;23(15):4086–93. doi: 10.1093/hmg/ddu122. Epub 2014 Mar 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Han R, Wang X, Wang D, Wang L, Yuan Z, Ying M, Li N. GPR143 Gene Mutations in Five Chinese Families with X-linked Congenital Nystagmus. Sci Rep. 2015 Jul 10;5:12031. doi: 10.1038/srep12031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee H, Sheth V, Bibi M, Maconachie G, Patel A, McLean RJ, Michaelides M, Thomas MG, Proudlock FA, Gottlob I. Potential of handheld optical coherence tomography to determine cause of infantile nystagmus in children by using foveal morphology. Ophthalmology. 2013 Dec;120(12):2714–24. doi: 10.1016/j.ophtha.2013.07.018. Epub 2013 Oct 22. [DOI] [PubMed] [Google Scholar]
- 29.Neveu MM, Jeffery G, Burton LC, Sloper JJ, Holder GE. Age-related changes in the dynamics of human albino visual pathways. European Journal of Neuroscience. 2003;18:1939–1949. doi: 10.1046/j.1460-9568.2003.02929.x. [DOI] [PubMed] [Google Scholar]
- 30.Gottlob I, Helbling A. Nystagmus mimicking spasmus nutans as the presenting sign of Bardet-Biedl syndrome. Am J Ophthalmol. 1999 Dec;128(6):770–2. doi: 10.1016/s0002-9394(99)00293-7. [DOI] [PubMed] [Google Scholar]
- 31.Kiblinger GD, Wallace BS, Hines M, Siatkowski RM. Spasmus nutans-like nystagmus is often associated with underlying ocular, intracranial, or systemic abnormalities. J Neuroophthalmol. 2007 Jun;27(2):118–22. doi: 10.1097/WNO.0b013e318067b59f. [DOI] [PubMed] [Google Scholar]
- 32.Smith DE, Fitzgerald K, Stass-Isern M, Cibis GW. Electroretinography is necessary for spasmus nutans diagnosis. Pediatr Neurol. 2000 Jul;23(1):33–6. doi: 10.1016/s0887-8994(00)00134-x. [DOI] [PubMed] [Google Scholar]
- 33.Thomas S, Thomas MG, Andrews C, Chan WM, Proudlock FA, McLean RJ, Pradeep A, Engle EC, Gottlob I. Autosomal-dominant nystagmus, foveal hypoplasia and presenile cataract associated with a novel PAX6 mutation. Eur J Hum Genet. 2014 Mar;22(3):344–9. doi: 10.1038/ejhg.2013.162. Epub 2013 Aug 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sharan S, Mirzayans F, Footz T, Walter M, Levin AV. Elliptical anterior iris stromal defects associated with PAX6 gene sequence changes. J AAPOS. 2008 Aug;12(4):340–3. doi: 10.1016/j.jaapos.2007.11.021. Epub 2008 Apr 25. [DOI] [PubMed] [Google Scholar]
- 35.Chiang PW1, Spector E, McGregor TL. Evidence suggesting digenic inheritance of Waardenburg syndrome type II with ocular albinism. Am J Med Genet A. 2009 Dec;149A(12):2739–44. doi: 10.1002/ajmg.a.33128. [DOI] [PubMed] [Google Scholar]
- 36.Al-Araimi M, Pal B, Poulter JA, van Genderen MM, Carr I, Cudrnak T, Brown L, Sheridan E, Mohamed MD, Bradbury J, Ali M, Inglehearn CF, Toomes C. A new recessively inherited disorder composed of foveal hypoplasia, optic nerve decussation defects and anterior segment dysgenesis maps to chromosome 16q23.3–24.1. Mol Vis. 2013 Nov 1;19:2165–72. eCollection 2013. [PMC free article] [PubMed] [Google Scholar]
- 37.Poulter JA, Al-Araimi M, Conte I, van Genderen MM, Sheridan E, Carr IM, Parry DA, Shires M, Carrella S, Bradbury J, Khan K, Lakeman P, Serfgouniotis PI, Wester AR, Moore AT, Bishwanath P, Mohamed MD, Venkataramana A, Ramprasad V, Shetty R, Saktival M, Kumaramanickavel G, Tan A, Mackey DA, Hewitt AW, Banfi S, Ali M, Inglehearn CR, Toomes C. Recessive mutations in SLC38A8 cause foveal hypoplasia and optic nerve misrouting without albinism. Am J Hum Genet. 2013;93:1143–1150. doi: 10.1016/j.ajhg.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stunkel M, Bhattarai S, Kemerley A, Stone EM, Wang K, Mullins RF, Drack AV. Vitritis in pediatric genetic retinal disorders. Ophthalmology. 2015 Jan;122(1):192–9. doi: 10.1016/j.ophtha.2014.07.037. Epub 2014 Sep 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thomas S, Proudlock FA, Sarvananthan N, Roberts EO, Awan M, McLean R, Surendran M, Kumar AS, Farooq SJ, Degg C, Gale RP, Reinecke RD, Woodruff G, Langmann A, Lindner S, Jain S, Tarpey P, Raymond FL, Gottlob I. Phenotypical characteristics of idiopathic infantile nystagmus with and without mutations in FRMD7. Brain. 2008 May;131(Pt 5):1259–67. doi: 10.1093/brain/awn046. Epub 2008 Mar 27. [DOI] [PubMed] [Google Scholar]
- 40.Weiss AH, Kelly JP, Phillips JO. Infantile Nystagmus and Abnormalities of Conjugate Eye Movements in Down Syndrome. Invest Ophthalmol Vis Sci. 2016 Mar;57(3):1301–9. doi: 10.1167/iovs.15-18532. [DOI] [PubMed] [Google Scholar]
- 41.Garcia-Filion P, Borchert M. Prenatal determinants of optic nerve hypoplasia: review of suggested correlates and future focus. Surv Ophthalmol. 2013 Nov-Dec;58(6):610–9. doi: 10.1016/j.survophthal.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maguire AM, Simonelli F, Pierce EA, Pugh EN, Jr, Mingozzi F, Bennicelli J, Banfi S, Marshall KA, Testa F, Surace EM, Rossi S, Lyubarsky A, Arruda VR, Konkle B, Stone E, Sun J, Jacobs J, Dell’Osso L, Hertle R, Ma JX, Redmond TM, Zhu X, Hauck B, Zelenaia O, Shindler KS, Maguire MG, Wright JF, Volpe NJ, McDonnell JW, Auricchio A, High KA, Bennett J. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N Engl J Med. 2008 May 22;358(21):2240–8. doi: 10.1056/NEJMoa0802315. Epub 2008 Apr 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bainbridge JW, Smith AJ, Barker SS, Robbie S, Henderson R, Balaggan K, Viswanathan A, Holder GE, Stockman A, Tyler N, Petersen-Jones S, Bhattacharya SS, Thrasher AJ, Fitzke FW, Carter BJ, Rubin GS, Moore AT, Ali RR. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N Engl J Med. 2008 May 22;358(21):2231–9. doi: 10.1056/NEJMoa0802268. Epub 2008 Apr 27. [DOI] [PubMed] [Google Scholar]
- 44.Hauswirth WW, Aleman TS, Kaushal S, Cideciyan AV, Schwartz SB, Wang L, Conlon TJ, Boye SL, Flotte TR, Byrne BJ, Jacobson SG. Treatment of leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a phase I trial. Hum Gene Ther. 2008 Oct;19(10):979–90. doi: 10.1089/hum.2008.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ronquillo CC1, Bernstein PS, Baehr W. Senior-Løken syndrome: a syndromic form of retinal dystrophy associated with nephronophthisis. Vision Res. 2012 Dec 15;75:88–97. doi: 10.1016/j.visres.2012.07.003. Epub 2012 Jul 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stone EM1, Cideciyan AV, Aleman TS, Scheetz TE, Sumaroka A, Ehlinger MA, Schwartz SB, Fishman GA, Traboulsi EI, Lam BL, Fulton AB, Mullins RF, Sheffield VC, Jacobson SG. Variations in NPHP5 in patients with nonsyndromic leber congenital amaurosis and Senior-Loken syndrome. Arch Ophthalmol. 2011 Jan;129(1):81–7. doi: 10.1001/archophthalmol.2010.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Holmström G1, Bondeson ML, Eriksson U, Åkerblom H, Larsson E. ‘Congenital’ nystagmus may hide various ophthalmic diagnoses. Acta Ophthalmol. 2014 Aug;92(5):412–6. doi: 10.1111/aos.12250. Epub 2013 Jul 29. [DOI] [PubMed] [Google Scholar]
- 48.Self J, Lotery A. A review of the molecular genetics of congenital Idiopathic Nystagmus (CIN) Ophthalmic Genet. 2007 Dec;28(4):187–91. doi: 10.1080/13816810701651233. Review. [DOI] [PubMed] [Google Scholar]