

Abstract
A major concept to sensitize cancer cells to DNA damaging agents is by inhibiting proteins in the DNA repair pathways. X-family DNA polymerases play critical roles in both base excision repair (BER) and nonhomologous end joining (NHEJ). In this study, we examined the effectiveness of honokiol to inhibit human DNA polymerase β (pol β), which is involved in BER, and DNA polymerase λ (pol λ), which is involved in NHEJ. Kinetic analysis with purified polymerases showed that honokiol inhibited DNA polymerase activity. The inhibition mode for the polymerases was a mixed-function noncompetitive inhibition with respect to the substrate, dCTP. The X-family polymerases, pol β and pol λ, were slightly more sensitive to inhibition by honokiol based on the Ki value of 4.0 μM for pol β, and 8.3 μM for pol λ, while the Ki values for pol η and Kf were 20 and 26 μM, respectively. Next we extended our studies to determine the effect of honokiol on the cytotoxicity of bleomycin and temozolomide in human cancer cell lines A549, MCF7, PANC-1, UACC903, and normal blood lymphocytes (GM12878). Bleomycin causes both single strand DNA damage that is repaired by BER and double strand breaks that are repaired by NHEJ, while temozolomide causes methylation damage repaired by BER and O6-alkylguanine-DNA alkyltransferase. The greatest effects were found with the honokiol and bleomycin combination in MCF7, PANC-1, and UACC903 cells, in which the EC50 values were decreased 10-fold. The temozolomide and honokiol combination was less effective; the EC50 values decreased three-fold due to the combination. It is hypothesized that the greater effect of honokiol on bleomycin is due to inhibition of the repair of the single strand and double strand damage. The synergistic activity shown by the combination of bleomycin and honokiol suggests that they can be used as combination therapy for treatment of cancer, which will decrease the therapeutic dosage and side effects of bleomycin.
Graphical Abstract
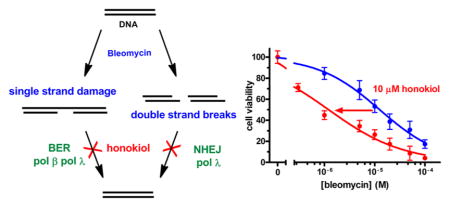
INTRODUCTION
Recent studies show that novel plant-derived compounds act as antitumor agents through modulation of biological pathways.1 Honokiol (2-(4-hydroxy-3-prop-2-enyl-phenyl)-4-prop-2-enyl-phenol (Figure 1), a biologically active biphenolic compound isolated from the Magnolia officinalis/grandiflora, has received significant attention due to its potent antineoplastic and antiangiogenic properties.2,3 It has yielded promising results against skin, colon, lung, and breast cancers.2,4–7 Furthermore, honokiol (HNL) is less toxic to normal cells than tumor cells, it has preferential activity against patient-derived chronic lymphocytic leukemia cells versus normal lymphocytes,8 and HNL killed myeloma cells but not peripheral blood mononuclear cells from relapsed patients.9 HNL and analogs are attractive therapeutic agents because HNL is nontoxic with no side effects in rats treated with a dose of 100 mg/kg.10 In addition, HNL crosses the blood–brain and blood–cerebrospinal fluid barriers3 and is effective in prolonging life in mice xenografted with HL60 promyelocytic leukemia cells.10,11 HNL has been shown to produce its anticancer effects via multiple mechanisms including NF-κB, STAT3, EGFR, mTOR HDAC, and caspase-mediated common pathways.12–14 The typical concentrations used to observe these effects have been in the micromolar range. However, it is still unclear if there is a single therapeutic target or if the effective in vivo target has been found. Since HNL is relatively nontoxic, a potentially useful application of HNL may be its ability to potentiate the actions of chemotherapeutic agents. In this regard, we evaluated the potentiation of two chemotherapeutic DNA damaging agents, bleomycin and temozolomide, and examined two novel targets of HNL, the X-family polymerases β and λ.
Figure 1.
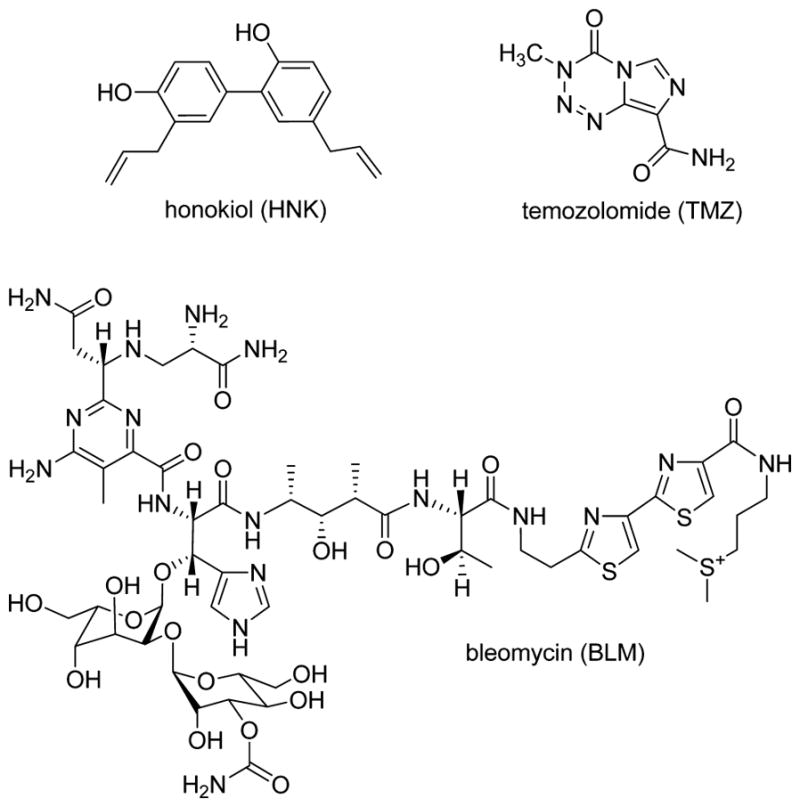
Structures of molecules used in this study.
Bleomycins are a family of glycopeptide antibiotics that bind to Fe(II) and DNA. Upon binding oxygen and undergoing a one electron reduction, the activated bleomycin initially produces 4′-oxidized abasic sites, or single-strand breaks containing 3′-phosphoglycolate/5′-phosphate ends.15 Subsequent reactivation of the DNA-bound bleomycin and a second reaction can lead to double strand breaks (DSBs). While the ssDNA breaks can be repaired by the short-patch base-excision repair pathway that utilizes DNA polymerase β (pol β),16,17 the oxidized abasic sites react with the lyase domain of pol β to form a DNA–protein adduct during short-patch BER.18 Even though the protein–DNA cross-links can be repaired by a pathway involving the proteasome, the protein adducts are toxic.19 However, the oxidized abasic sites can be effectively repaired by long-patch BER,20 a process that involves pol β21–23 and perhaps δ or ε.24 Bleomycin-induced DSBs can be repaired by nonhomologous end joining (NHEJ), a process that utilizes the X-family polymerases λ and μ.25,26 Thus, the X-family polymerases β and λ are integral in attenuating the toxicity of BLM. In fact, inhibition of pol β sensitizes the cells to bleomycin damage,27–29 while increased expression of pol β attenuates BLM toxicity.30
Temozolomide (TMZ) is a chemotherapeutic agent that produces methyl diazonium ions that react with DNA to form adducts such as 7-methylguanine (7mG), 3-methyladenine (3mA), and O6-methylguanine (O6mG). Each of these adducts contributes to the toxicity of TMZ. While O6mG is repaired by O6-alkylguanine-DNA alkyltransferase, 7mG and 3mA, which comprise over 80% of the TMZ DNA adducts, are repaired by BER. BER inhibition, via PARP1 or pol β inhibition, increases the toxicity of TMZ, while pol β activity decreases the cytotoxicity of TMZ.31–33
While BLM and TMZ produce different types of DNA damage, the X-family polymerases, pol β and λ, are involved in the repair their damage. Pol β is the primary polymerase involved in BER, while pol λ functions as a backup.34 Pol λ is also involved in NHEJ. In this manuscript, we evaluated the inhibitory activity of HNL toward pol β and pol λ, and two other polymerases, pol η and E. coli DNA polymerase I (Klenow fragment) with the proofreading activity inactivated (KF(exo-)), to evaluate the selectivity of the inhibition. In addition, we studied the effect that HNL has on the cytotoxicity of BLM and TMZ. Our results show that HNL inhibits eukaryotic pol β, pol λ, and pol η activities but has lesser inhibition of prokaryotic Kf(exo-). In addition, we found that HNL chemosensitizes the cancer cell lines to cytotoxic effects of BLM to a greater extent than does TMZ.
MATERIALS AND METHODS
Caution: These chemicals are dangerous
The following chemicals are hazardous and should be handled carefully: bleomycin, temozolomide, and honokiol.
Chemicals and Reagents
T4 polynucleotide kinase was purchased from Epicenter (Lexington, KY) and [γ-32P]ATP (6000 Ci/mmol) from PerkinElmer (Waltham, MA). DNA oligomers were purchased from IDT (Coralville, IA). The concentrations of the dNTPs (Promega, Madison WI) were each determined by UV absorbance.35 HNK, BLM, and TMZ, purchased from Sigma-Aldrich (St. Louis, MO), were freshly prepared in DMSO, stored at −80 °C, and diluted in buffer or complete medium just before use. 3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) was purchased from Promega (Madison, WI, USA), and phenazine metho-sulfate (PMS) was purchased from Sigma-Aldrich (St. Louis, MO, USA). MTS/PMS solution was prepared at a concentration of 2 mg/mL of MTS and 0.92 mg/mL of PMS in phosphate-buffered saline (PBS) and stored in dark bottle at 4 °C. Fetal bovine serum was purchased from Atlanta Biologicals Inc. The human cancer cell lines, MCF7, A549, PANC-1, UACC903, were purchased from American Type Culture Collection (ATCC), and GM12878 cells were purchased from Coriell Institute for Medical Research.
DNA Substrates
The single gapped DNA substrate for pol β and pol λ contained a template, a primer, and a downstream blocking oligodeoxynucleotide as illustrated in Table 1. The primer strand was 5′-end-labeled with T4 polynucleotide kinase and [γ-32P]ATP (6000 Ci/mmol) as previously described.36 The blocking strands were phosphorylated on the 5′-end with nonradioactive ATP. The unreacted [γ-32P]ATP and ATP were removed using a Sephadex G-25 spin column and annealed at a primer/blocker/template ratio of 1:3:1. The DNA substrates for pol η and KF(exo-) were prepared by mixing a 32P labeled 15-mer primer with the 24-mer template at a molar ratio of 1:1.2.
Table 1.
DNA Substrates for Polymerases
| pol β and λ | ||||||||||||||||||||||||||||||||||||||||
| 5′- | G | A | A | G | A | C | T | G | G | T | G | A | A | G | A | C | T | T | G | A | G | T | A | C | A | G | G | T | C | G | A | T | T | C | A | T | G | G | -3′ | |
| 3′- | C | T | T | C | T | G | A | C | C | A | C | T | T | C | T | G | A | A | C | T | C | G | A | T | G | T | C | C | A | G | C | T | A | A | G | T | A | C | C | -5′ |
| KF(exo-) and pol η | ||||||||||||||||||||||||||||||||||||||||
| 5′- | G | C | A | C | C | G | C | A | G | A | C | G | C | A | G | -3′ | ||||||||||||||||||||||||
| 3′- | C | G | T | G | G | C | G | T | C | T | G | C | G | T | C | G | A | C | A | G | C | G | T | C | -5′ | |||||||||||||||
Cell Lines and Culture Conditions
The human cancer cell lines MCF7, A549, PANC-1, and UACC903 were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (v/v). The normal blood lymphoblasts (GM12878) were cultured in RPMI 1640 medium containing 15% fetal bovine serum. All cell lines were cultured at 37 °C in a humidified incubator with 5% CO2 (v/v).
DNA Polymerases
DNA polymerase I from E. coli (Klenow fragment) with the proofreading activity inactivated (Kf(exo-)) was purchased from USB scientific. His-tagged human DNA polymerases β,37 η,38 and λ39 were expressed and purified as previously described. The polymerase concentrations were determined by analyzing the burst intensity as described previously.38
Steady-State Primer Extension Assay
The DNA polymerase reaction was initiated by adding equal volumes of DNA polymerase, DNA substrate, and the appropriate amount of honokiol in buffer with dNTP and MgCl2 at 37 °C. The final buffer concentrations depended on the polymerase: KF, 50 mM Tris-HCl (pH 7.8), 5 mM MgCl2, 0.1 mM EDTA, 3 mM DTT, and 100 μg/mL of BSA; pol η, 40 mM Tris-HCl (pH 8.0), 5 mM MgCl2, 0.1 mM EDTA, 3 mM DTT, and 100 μg/mL of BSA with 2.5% glycerol (v/v); pol β and λ, 50 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 100 mM NaCl, 100 μg/mL of BSA, 1 mM DTT. The concentrations of the polymerases varied from 0.01–0.2 nM, and the DNA concentration was 10 nM during the reactions. The gapped DNA substrate was used for pol β and λ, while the simple primer template was used for Kf and pol η (Table 1). The reaction was quenched by addition of an equal volume of STOP solution (95% formamide, 20 mM Na2EDTA, 0.025% bromphenol blue (w/v), and 0.025% xylene cyanol (w/v)).
Polymerase in Excess Primer Extension Assay
The reaction was initiated by adding equal volumes of DNA polymerase, DNA substrate, and the appropriate amount of honokiol in buffer (as above) with dNTP and MgCl2 at 37 °C with a rapid quench instrument (RQF-3, KinTek Corp). The concentrations of the polymerase and DNA during the reaction were 100 nM and 15 nM, respectively. The reaction was quenched by addition of 0.3 M EDTA (pH 8.0).
Analysis of Reactions
The reaction products were separated by electrophoresis on a 15% (w/v) polyacrylamide (19:1 (w/w), acrylamide/bis-acrylamide) gel containing 8 M urea in TBE buffer (89 mM Tris-HCl, 89 mM boric acid, and 2 mM EDTA, pH 8.3). The amounts of radioactivity in the reactant and product bands were quantified using a Typhoon 9200 and ImageQuant software (GE Healthcare).
Polymerase–DNA Interaction
The binding affinities of DNA and the polymerases were examined with an electrophoretic mobility shift assay.40 The DNA, polymerase, and honokiol were incubated for 20 min in the correct DNA polymerase buffer at 37 °C. Samples were loaded onto a 6% native polyacrylamide gel (0.5 × TBE) and run at 100 V for 2 h. Bound protein was quantified using ImageQuant software after the gel was scanned using a Typhoon 9200 and ImageQuant software (GE Healthcare). Protein bound to DNA resulted in a shift of the DNA on the gel when compared to DNA without bound protein.
MTS Assay of Cell Proliferation
A colorimetric cell proliferation assay7,10 was used to assess the effect on cell proliferation of potentiation of the cytotoxicity of BLM with HNL in cancer cell lines A549, MCF7, PANC-1, UACC903, and the immortalized normal cell line GM12878. For these studies, 5 × 103 cells were plated and grown for 24 h in 100 μL of growth medium in 96-well microtiter plates at 37 °C in a humidified 5% CO2 atmosphere. For treatments, stock solution of HNL (10 mM), BLM (10 mM), or TMZ (100 mM) in DMSO were diluted with fresh complete medium immediately before use. Cells were treated for 24 and 72 h in 200 μL of fresh media containing various concentrations of HNL, BLM, or TMZ alone or in combination. Control cells were treated with equivalent concentrations of DMSO. In all cases, final concentration of DMSO was 0.2%, well below the concentrations that interfere with proliferation in the above cell lines. After a 24 or 72 h incubation period, the number of viable cells was determined by measuring the bioreduction by intracellular dehydrogenases of the tetrazolium compound MTS in the presence of the electron coupling reagent PMS. To perform the assay, 20 μL of combined MTS/PMS solution containing 2 mg/mL MTS and 0.92 mg of PMS in PBS, pH 7.2, was added to each well, and the mixture was incubated for 4 h at 37 °C in a humidified 5% CO2 atmosphere. Absorbance at 492 nm was measured using an ELISA microplate reader (Flexstation 3 Molecular Devices, Softmax Pro 5). Background absorbance of the medium was measured in wells that contained medium and the MTS/PMS solution without added cells.
Data Analysis
IC50 values for the inhibition of the polymerases were obtained by fitting the data to eq 1, in which Y was the amount of product, A was the amount of product with no honokiol, and X was the concentration of honokiol and IC50, the concentration of honokiol that reduces the amount of product to 50%. IC50 values for the inhibition of cell viability were also obtained by fitting the data of eq 1, in which Y was the normalized absorbance value, A was 100, and X was the concentration of the test compound and IC50, the concentration that reduces the normalized absorbance to 50%.
| (1) |
Vmaxapp and Kmapp values were obtained by fitting the initial rates (v0) versus [dNTP] to eq 2, where [E]0 and [S]0 are the initial concentrations of the polymerase and dNTP. Eqs 1 and 2 were fitted using GraphPad Prism version 4 for Windows (GraphPad Software, San Diego, CA):
| (2) |
The kcat, Km, and Ki were determined by fitting v0 directly to the hyperbolic multitype inhibition eq 3 using Matlab 2015b:
| (3) |
The time course experiments were fit to the burst eq 3 where P is the total product, A the burst amplitude, kb the burst, and kss the steady-state rate constants:
| (4) |
The Kd for honokiol was evaluated from the burst amplitudes by fitting the amplitudes to eq 5 in which A represents the burst amplitude, Amin and Amax the minimum and maximum burst amplitudes, and [I], the HNL concentration:
| (5) |
RESULTS
Effects of HNL on the Activities of Select Eukaryotic and Prokaryotic DNA Polymerases
We initially examined the inhibitory activity of honokiol on selected polymerases using the primer extension assay with 1 nM polymerase, 10 nM DNA, and 50 μM dCTP and various concentrations of HNL. As illustrated in Figure 2, HNL inhibited the X-family polymerases (β and λ) to a greater degree than all of the polymerases studied with IC50 values of 0.6 μM for pol β, 0.8 μM for pol λ, 10 μM for pol η, and 80 μM for Kf(exo-). The low IC50 values for pol β and λ lead us to evaluate the mechanism underlying the inhibition. The inhibition could be at several stages including inhibition of DNA or dNTP binding or by allosteric inhibition of the catalytic activity.
Figure 2.
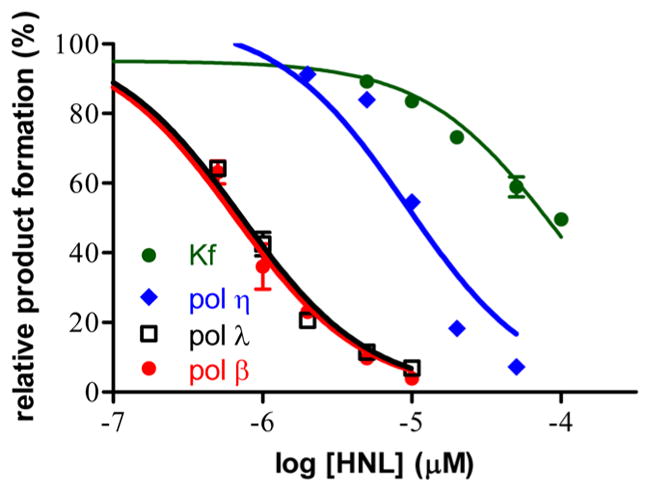
Dose–response curves of honokiol with DNA polymerases. Each polymerase (1 nM) enzyme was preincubated with honokiol and 10 nM DNA substrate for 10 min prior to initiation of the reaction by addition of dCTP and MgCl2. After a 15 min incubation at 37 °C for 15 min. The DNA polymerase activity in the absence of HNL was taken as 100% activity.
Honokiol Does Not Inhibit the Binding of Polymerases to DNA
Since the honokiol effects were greatest with pols β and λ, we examined whether honokiol inhibited the binding of the DNA substrates to these enzymes. As shown in Figure 3, we used the EMSA to determine a KdDNA of 10.0 ± 1.5 nM for pol β and 11.4 ± 3.0 nM for pol λ, values consistent with previous results for pol β.41–43 We then incubated honokiol with the polymerases and the appropriate DNA substrate and evaluated DNA binding by EMSA. Figure 3 shows experiments with polymerase in excess of DNA so that the amount of unbound radiolabeled DNA is minimal and consequently we would be able to detect unbound DNA. Up to its solubility limit of 200 μM, honokiol did not inhibit the binding of the DNA to the polymerase. On the basis of equilibria equations, a conservative estimate for the KiHNL would be greater than 500 μM. Since we observed polymerase inhibition at lower concentrations of honokiol, we concluded that competitive inhibition DNA binding is not the mechanism by which honokiol inhibits these DNA polymerases.
Figure 3.

Honokiol does not inhibit DNA binding to polymerases. EMSA showing native PAGE of of (A) pol β, (B) pol λ, (C) pol η, and (D) KF(exo-).
Honokiol Inhibits Polymerases by Mixed-type Inhibition
To determine the kinetic mechanism by which honokiol inhibits polymerase activity, the initial rates were determined over a range of dCTP and honokiol concentrations. The data were fitted to the Michaelis–Menten eq 2 by nonlinear least-squares analysis. The plots for pol β and λ are shown in Figure 4. These plots clearly show that the Vmaxapp decreases as honokiol concentrations increase. As shown in Figures S1 and S2, both Vmaxapp and Kmapp decrease for all four polymerases with increasing HNL concentrations. The data were visualized with Lineweaver–Burk plots as shown in Figure 4C and D (also Figure S4). The observation that the lines intersect to the left of the y-axis and below the x-axis is consistent with hyperbolic mixed type inhibition, illustrated in Scheme 1 with kinetic eq 3 in which α < 1 and β < 1.44 The experiment was conducted in triplicate, the data were fit directly to the hyperbolic mixed type inhibition eq 3, and the results are presented in Table 2. The β values for pol β, λ, and Kf(exo-) are small, but greater than 0, indicating that a small amount of catalysis occurs with HNL bound to the polymerase complex. The β value for pol η has 95% confidence limits that includes 0. With β = 0, the mechanism would be described as linear mixed-type inhibition. The α values are approximately 0.5, which indicate that binding of dCTP or HNL increases the affinity of the other ligand.
Figure 4.

Inhibition of the pol β and pol λ catalyzed incorporation of dCTP into DNA by honokiol. Initial rate versus dNTP plot for (A) pol β and (B) pol λ. The lines are the best fit to eq 2. Lineweaver–Burk plots for (C) pol β and (D) pol λ. The lines are the best fit to a linear equation. The HNL concentrations are 0 (open circle), 25 (diamond), 50 (down triangle), 100 (up triangle), 250 (square), and 500 (filled circle) μM. Each point represents the mean ± SEM of three experiments.
Scheme 1.
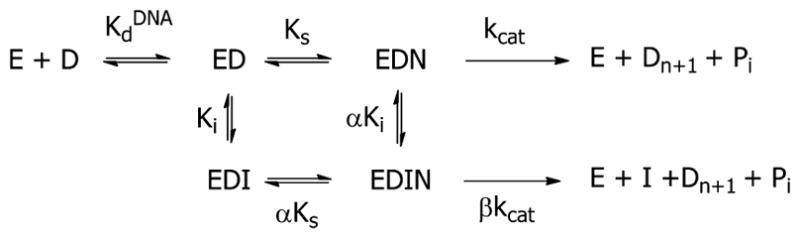
Hyperbolic Mixed-type Inhibition of DNA Polymerases by HNL
Table 2.
Steady State Kinetic Parameters for the Hyperbolic Mixed-type Inhibition of DNA Polymerase by HNLa
| pol | kcat (min−1) | Km (nM) | Ki (μM) | α | β |
|---|---|---|---|---|---|
| β | 28 (26, 30) | 220 (180, 260) | 4.0 (2.4, 5.6) | 0.55 (0.31, 0.79) | 0.20 (0.15, 0.25) |
| λ | 5.2 (4.8, 5.5) | 274 (230, 310) | 8.3 (4.8, 11.9) | 0.51 (0.24, 0.78) | 0.07 (0.02, 0.12) |
| η | 7.4 (6.7, 8.0) | 390 (330, 450) | 20 (7, 34) | 0.49 (0.12, 0.85) | 0.03 (–0.10, 0.15) |
| Kf | 18 (17, 20) | 76 (63, 88) | 26 (14, 38) | 0.48 (0.23, 0.74) | 0.16 (0.09, 0.23) |
Inhibition experiments were conducted in triplicate. All data points were used to fit the data to eq 3. The data are reported as the values with the lower and upper 95% confidence limits.
Presteady State Burst Kinetics of Pol β and Pol λ in the Presence of HNL
Presteady-state kinetic analysis of enzyme inhibition is a powerful approach for determining which step or steps are affected by a given inhibitor. This technique has been applied to study a number of systems including D-3-phosphoglycerate dehydrogenase,45 beta-site amyloid precursor protein-cleaving enzyme,46 ATP-phosphoribosyl-transferase, 47 DNA polymerase γ,48 and HIV-RT.49,50
Presteady-state analysis was carried out using a rapid quench apparatus to further characterize the mechanism of inhibition of pol β and pol λ. We initially examined the reaction with no inhibitor and found kpol = 18 ± 1 s−1 and Kd dCTP = 20 ± 3 μM for pol β (data not shown), values similar literature values.41,42 Preincubated gapped DNA (15 nM), pol β (100 nM), and honokiol were added to dCTP (100 μM) with Mg2+. The reactions were quenched at various times and the data presented in Figure 5A. The data were fit to the burst eq 4. With no inhibitor, the burst amplitude is close to the polymerase concentration. The addition of HNL decreased the burst amplitude but neither the burst (kb) nor the steady-state (kss) rate constants. The burst amplitude was fitted to the hyperbolic eq 5 to obtain KdHNL = 20 ± 4 μM. The minimum burst amplitude is close to zero. A decrease in burst amplitude indicates that honokiol inhibits pol β by forming a catalytically inactive quaternary species that reverses slowly. The off rate for HNL would have to be slower than the kcat. This type of inhibition was observed in the non-nucleoside inhibitors of HIV-RT.49,50 We found similar results with pol λ (Figure S4).
Figure 5.
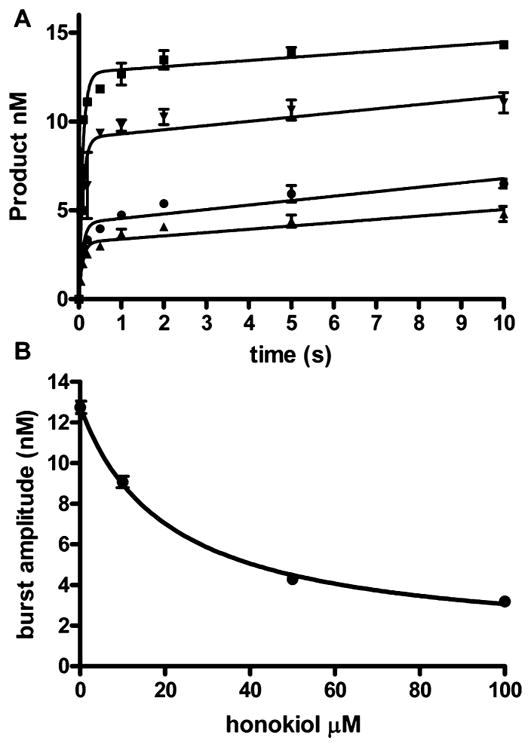
Reduced pol β burst amplitudes in the presence of honokiol. (A) Time course for the insertion of 200 μM dCTP opposite dG in the presence of 0 (square), 10 (down triangle), 50 (circle), and 100 (up triangle) μM honokiol. The lines are the best fit to the burst equation. The kb and kss were unchanged with increasing honokiol concentrations at 11.7 ± 1.0 s−1 and 0.2 ± 0.6 nM−1 s−1, respectively. (B) Plot of burst amplitude versus honokiol concentration. The line is the best fit to the hyperbolic equation with Amax = 12.8 ± 0.3 nM, Amin = 1.0 ± 0.5, K = 21 ± 4 μM. The data points are the mean ± SEM of three experiments.
Honokiol Chemosensitizes the Cancer Cells for BLM Toxicity
The inhibition of pol β and λ may also reduce the activity of the BER pathway in cells. If this occurs, then cytotoxic DNA damaging agents that are repaired by BER may become more toxic with coadministration of HNL. Thus, we examined the ability of HNL to potentiate the cytotoxicity of BLM and TMZ in human cancer cell lines A549, MCF7, PANC1, UACC903, and GM12878. The cells were treated with various concentrations of honokiol, BLM, and TMZ to determine EC50 values. Figure 6 shows the effect on the EC50 values with PANC1 cells. The open markers represent treatment with a single compound, while the filled symbols represent dual treatment in which 10 μM BLM or 250 μM TMZ was added to HNL, or 10 μM HNL was added to BLM or TMZ. Panels A and B show the effect of HNL on TMZ. The small leftward shift of the curves indicates that HNL and TMZ weakly enhance each other’s cytotoxicity. The EC50 values decreased up to three-fold. Panels C and D illustrate that HNL and BLM more strongly enhance each other’s cytotoxicity. In these graphs, larger leftward shifts in the curves are more apparent. The EC50 values decreased 10-fold. The logEC50 values for all cell lines are presented in Table 3, and the plots for the other cell lines are presented in Figure S5–S9. The effect on the logEC50 of the dual administration of compounds is graphically illustrated in Figure 6. The data indicate that the BLM and HNL combination increased cellular toxicity to a greater extent than TMZ and HNL in the MCF7, PANC1, and UCAA cell lines.
Figure 6.

Changes in EC50 due to combination of HNL with BLM and TMZ in PANC1 cells. The cell viability was determined at different concentrations of HNL, TMZ, or BLM alone (represented by the open symbols) and in combination (represented by solid symbols). The data are the mean ± SE of threee experiments.
Table 3.
Toxicity of HNL, TMZ, and BLM with Select Combinations with Cancer Cell Linesa
| tested compound | supplemented with | −log(IC50) (95% CI) | |
|---|---|---|---|
|
| |||
| 24 h | 72 h | ||
| GM12878 | |||
| TMZ | >3 | 3.5 (3.4, 3.7) | |
| TMZ | HNL | >3 | 3.9 (3.8, 4.1) |
| HNL | >3 | 4.7 (4.6, 4.9) | |
| HNL | TMZ | >3 | 4.9 (4.8, 5.1) |
| BLM | 4.1 (3.91, 4.3) | 4.1 (4.0, 4.3) | |
| BLM | HNL | 4.2 (4.0, 4.3) | 4.20 (4.0, 4.4) |
| HNL | 3.9 (3.7, 4.0) | 4.0 (3.8, 4.1) | |
| HNL | BLM | 3.9 (3.8, 4.1) | 4.0 (3.9, 4.1) |
| A549 | |||
| TMZ | 3.4 (3.3, 3.5) | 3.6 (3.4, 3.7) | |
| TMZ | HNL | 3.6 (3.5 3.7) | 4.1 (4.0, 4.2) |
| HNL | 4.6 (4.4, 4.7) | 4.0 (3.9, 4.0) | |
| HNL | TMZ | 4.9 (4.7, 5.0) | 4.1 (4.0, 4.2) |
| BLM | 4.8 (4.7, 5.0) | 5.2 (5.0, 5.3) | |
| BLM | HNL (10 μM) | 5.30 (5.18, 5.43) | 5.6 (5.5, 5.7) |
| HNL | 4.6 (4.5, 4.8) | 4.8 (4.6, 4.9) | |
| HNL | BLM (10 μM) | 5.0 (4.8, 5.1) | 5.3 (5.2, 5.4) |
| MCF7 | |||
| TMZ | 3.3 (3.2, 3.4) | 3.7 (3.6, 3.8) | |
| TMZ | HNL (10 μM) | 3.7 (3.6, 3.8) | 3.9 (3.7, 4.0) |
| HNL | 4.7 (4.6, 4.8) | 4.8 (4.7, 4.9) | |
| HNL | TMZ (250 μM) | 4.9 (4. 8, 5.0) | 5.0 (4.9, 5.1) |
| BLM | 4.9 (4.8, 5.0) | 5.3 (5.2, 5.4) | |
| BLM | HNL (10 μM) | 6.0 (5.7, 6.2) | 6.3 (6.1, 6.6) |
| HNL | 4.8 (4.7, 4.8) | 4.9 (4.8, 5.0) | |
| HNL | BLM (10 μM) | 5.6 (5.4, 5. 7) | 5.6 (5.4, 5.8) |
| PANC1 | |||
| TMZ | 3.5 (3.4, 3.7) | 3.8 (3.7, 3.9) | |
| TMZ | HNL (10 μM) | 3.9 (3.8, 4.0) | 4.3 (4.2 4.4) |
| HNL | 4.7 (4.5, 4.9) | 5.1 (5.0, 5.3) | |
| HNL | TMZ(250 μM) | 5.0 (4.9, 5.1) | 5.3 (5.2, 5.4) |
| BLM | 4.9 (4.7, 5.1) | 5.2 (5.0, 5.3) | |
| BLM | HNL (10 μM) | 5.9 (5.7, 6.2) | 6.09 (5.9, 6.3) |
| HNL | 4.6 (4.5, 4.8) | 4.8 (4.7, 4.9) | |
| HNL | BLM (10 μM) | 5.5 (5.3, 5.7) | 5.6 (5.4, 5. 8) |
| UACC903 | |||
| TMZ | 3.2 (3.1, 3.3) | 3.5 (3.4, 3.7) | |
| TMZ | HNL (10 μM) | 3.6 (3.5, 3.8) | 3.9 (3.8, 4.1) |
| HNL | 4.6 (4.5 4.7) | 4.8 (4.7, 4.9) | |
| HNL | TMZ(250 μM) | 4.8 (4.7, 4.9) | 5.0 (4.9, 5.1) |
| BLM | 4.9 (4.7, 5.0) | 5.22 (5.1, 5.4) | |
| BLM | HNL (10 μM) | 5.5 (5.4, 5.7) | 6.3 (6.0, 6.5) |
| HNL | 4.6 (4.47, 4.8) | 4.8 (4.65, 4.9) | |
| HNL | BLM (10 μM) | 5.1 (5.0 5.2) | 5.4 (5.2, 5.5) |
The −log(EC50) values were determined for the tested compound alone and in the presence of 10 μM HNL, 10 μM BLM, or 250 μM TMZ. The experiments were conducted in triplicate. All data points were used to fit the data to eq 3. The data are reported as the value with the lower and upper 95% confidence limits.
DISCUSSION
Chemotherapeutic agents have a concentration window where they kill tumor cells but not normal cells. The idea of chemotherapeutic cocktails is to use low nontoxic doses of two or more compounds that take advantage of the altered protein expression levels in tumor cells to selectively kill these cells. Many tumor cells have upregulated DNA polymerase β levels,51,52 perhaps to compensate for a higher oxidative stress levels in the tumors. Our strategy is to stress these tumor cells by simultaneously inhibiting DNA polymerase β while stressing the cells with a DNA damaging agent that would need the BER pathway. In this, we hope to enlarge the therapeutic window between toxicity for tumor cells and normal cells. This strategy has been successful with many pol β inhibitors that have been identified in the past two decades. For example, a modified benzenesulfonamide inhibits the binding of the APC-protein to pol β and thus inhibits long-patch BER activity.32 This action sensitizes colon cancer cells to TMZ. Similarly, triterpenoids and fatty acid-like compounds have increased the cytotoxic activity of BLM27–29 and methylmethanesulfonate.32
Several triterpenes, such as koetjapic acid, inhibit pol β via a linear mixed-mode inhibition mechanism with Ki values in the low micromolar range.53 Here we show that HNL also inhibits both pol β and λ with Ki values as low as these natural products. Steady-state kinetic analysis indicated that HNL inhibition versus dCTP is mixed-type noncompetitive for pol β, pol λ, and KF(exo-), and linear mixed-type noncompetitive for pol η. While the formal nomenclatures of the inhibition mechanisms are different, essentially the inhibition is of a noncompetitive nature with a kinetic mechanism illustrated in Scheme 1.
NMR studies show that koetjapic acid and related triterpenoids bind in a cleft on the N-terminal 8 kDa domain of pol β, which is responsible for the deoxyribose phosphate lyase activity of the enzyme.54,55 Binding to the N-terminal 8 kDa domain of pol β occurs with other large fatty acid-like compounds.55,56 HNL can potentially bind to the same region. These compounds are similar to HNL in that they contain large hydrophobic hydrocarbon regions punctuated with oxygen to provide some hydrophobicity. However, HNL is smaller than these compounds and does not contain a carboxylic acid moiety. In addition, while this mechanism may also occur with the X-family pol λ, pol η and KF(exo-) do not have an analog to the N-terminal 8 kDa lyase domain, so inhibition of these polymerases must occur via a different mechanism. Thus, the molecular mechanism by which honokiol inhibits DNA polymerases is unknown.
Triterpenes require preincubation with the polymerase for successful inhibition; an indication that the binding of the inhibitor to the polymerase is slow or that inhibition requires a slow conformational change to form an inactive complex. Using presteady-state kinetics, we have found that HNL inhibits the reaction by decreasing the burst amplitude of the reaction. This type of kinetic plot would be observed only if the inhibitor complexes (EDI and EDNI) are slow to dissociate. If the binding and dissociation of HNL were rapid, then addition of HNL would decrease the rate constant, not the amplitude. Modeling the reaction with a noncompetitive inhibition scheme results in a honokiol off rate constant of 0.16 s−1 (Figure S10). If HNL and triterpenes inhibit pol β by similar mechanisms, then the need for preincubation and the slow off rates are consistent. This type of inhibition was observed previously with non-nucleoside inhibitions and HIV-RT.49
In summary, HNL is an inhibitor of pol β with a Ki < 10 μM. This Ki value, while not very low, is similar to IC50 and Ki values of other compounds.27,29,55–66 The kinetic mechanism involves HNL sequestering the polymerase–DNA–dNTP complex in an unreactive configuration that decomposes slowly. To evaluate if HNL can be utilized to chemosensitize damage DNA, which is repaired by BER, we examined two reagents TMZ and BLM.
TMZ is a methylating agent approved for use with anaplastic astrocytoma and glioblastoma,67 has clinical activity in metastatic melanoma, and is under clinical evaluation for use in other cancers including leukemia, lymphoma, aerodigestive tract, pancreatic, and neuroendocrine tumors. TMZ causes cancer cell cytotoxicity by methylating genomic DNA, producing cytotoxic or mutagenic abnormal DNA bases.68 The major DNA adducts studied include 7mG, 3mA, and O6mG. 7mG and 3mA are cytotoxic because they depurinate and form abasic sites. 3mA is also block to DNA polymerases.69 These adducts are repaired by BER which utilizes pol β as the primary polymerase. The inhibition of this repair system with PARP or pol β inhibitors leads to increased efficacy of TMZ.31–33,70,71 O6mG is mutagenic, and its cytotoxicity is derived from the futile cycling of the mismatch repair system.72 It is not repaired by BER, but by O6-alkylguanine DNA alkyltransferase. Low activity of AGT also increases the efficacy of TMZ.73–75 The net toxicity of TMZ is determined through the interaction of at least two DNA repair pathways.
Bleomycin produces 4′-oxidized abasic sites, single-strand breaks containing 3′-phosphoglycolate/5′-phosphate ends as well as double stranded breaks.15 Pol β is involved in the repair of the single-strand breaks via the BER pathway.16,17 While short-patch BER of oxidized abasic sites produces a DNA-pol β cross-link, the adduct can be repaired via long patch BER. Inhibition of pol β sensitizes the cells to bleomycin damage.27–29 Links between DNA pol β expression and sensitivity to BLM also support the ability of pol β to attenuate BLM toxicity.30 BLM also caused double-strand breaks that may be the primary source of toxicity. Double-strand breaks are repaired by homologous recombination (HR), nonhomologous end joining (NHEJ), and alternative NHEJ. NHEJ operates during all phases of the cell cycle and is predominant during G0/G1.76 HR is absent in G1 and most active in S and G2.26 Alternative NHEJ appears to operate as a backup repair system for HR and NHEJ.77 Each of these pathways involves many proteins, but relevant to our study, NHEJ utilizes the X-family polymerases λ and μ.76
HNL is relatively nontoxic to normal cells and may be an alternative to stronger inhibitors that have side effects. The EC50 values for a 24 h incubation in our GM12878 cells were >1 mM in one study and >100 μM in another. We evaluated the toxicity of HNL, BLM, and TMZ in five cell lines, GM12878, a normal cell line immortalized with the Epstein Bar virus, with four tumor derived lines A549, MCF7, PANC1, and UACC903. As shown in Figure 7, the EC50 values for TMZ and BLM are decreased in the presence of HNL (Figure 7A,C), and the EC50 for HNL is decreased in the presence of both TMZ and BLM (Figure 7B,D). Importantly, in the nontumor derived GM12878 cells, HNL does not increase the toxicity of TMZ and has a very small effect with BLM. This property would be useful in therapy.
Figure 7.

Changes in log EC50 due to dual administration of HNL and either BLM or TMZ. Change in log EC50 of (A) TMZ caused by 10 μM HNL; (B) HNL caused by 250 μM HNL; (C) BLM cause by 10 μM HNL; and (D) HNL caused by 10 μM BLM.
HNL increases the toxicity of both BLM and TMZ, but the effect is greater for BLM. The EC50 values decreased up to 10-fold in MCF7, PANC1, and UACC903 cells due to the NHL/BLM combination (Figure 7C). The effect with TMZ is less, with the EC50 values being decreased up to three-fold with HNL (Figure 7A). We speculate that the increased activity with BLM is because HNL inhibits repair of both single-strand damage and DSBs caused by BLM. As discussed above, both BLM and TMZ produce various types of DNA damage. TMZ produces 3mA and 7mG, adducts that are repaired by BER, and O6mG, which is repaired by O6-alkylguanine-DNA alkyltransferase. BLM produces both single-strand damage (single-strand breaks and oxidized abasic sites) and DSBs. The single-strand breaks are repaired by short patch BER, the oxidized abasic sites can be repaired by long patch BER, and the DSBs are repaired by NHEJ. In this work, we found that HNL inhibits both pol β, which is involved in BER, and pol λ, which is involved in both BER and NHEJ. Thus, HNL can inhibit the repair of the major DNA damage produced by BLM. In contrast, HNL inhibits the BER repair of 7mG and 3mA but not the repair of O6mG.
The effects of NHL on modulating the cytotoxicity of TMZ and BLM are similar for the four cancer cell lines. In Figure 7A, the effect of HNL on TMZ toxicity was much less than the other cell lines at the 72 h time point. However, this effect was not observed in the 24 h time point nor in Figure 7B, so we do not want to draw any conclusion based upon this result. In the BLM studies, the A549 cell line is more resistant to the effects of HNL than the other cell lines tested (Figure 7C,D). HNL has multiple possible targets for its anticancer effects such as the NF-κB, STAT3, EGFR, mTOR HDAC, and caspases.12–14 We have identified pol β and λ as possible HNL targets. The effect that HNL has in different cell lines will be affected by the interplay between these pathways. More work needs to be done to understand how the different cell lines respond to HNL treatment.
In summary, HNL is an inhibitor for X-family polymerases β and λ. The mechanism is noncompetitive inhibition in which the complex between HNL/polymerase/DNA/dNTP, while not extremely stable, is slow to dissociate. HNL modulates the cytotoxicity of TMZ and BLM, possibly through the inhibition of pol β and λ. More work needs to be done to test this mechanism. The approach of combining HNL with BLM shows promise as HNL can inhibit polymerases involved in the repair of single strand damage and double strand breaks. Thus, by developing a target defined strategy of chemotherapy and with the appropriate understanding of the mechanism of action, the strategy of combination of HNL with BLM may provide a foundation for both clinical and scientific development of cancer treatment.
Supplementary Material
Acknowledgments
Funding
The work was supported by the National Institutes of Health Grant No. ES021762 to T.E.S.
ABBREVIATIONS
- BML
bleomycin
- BSA
bovine serum albumin
- DMEM
Dulbecco’s modified Eagle’s medium
- FBS
fetal bovine serum
- HNL
honokiol
- Kf(exo-)
Klenow fragment of E. coli DNA polymerase I with the proofreading exonuclease activity inactivated
- MTS
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
- PMS
phenazine metho-sulfate
- PBMCs
peripheral blood mononuclear cells
- pol β
DNA polymerase β
- pol η
DNA polymerase η
- pol λ
DNA polymerase λ
- TBE
Tris-HCl, boric acid, and EDTA buffer
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.chemrestox.6b00451.
Figures of steady-state kinetics for inhibition of DNA polymerases by HNL and EC50 plots for all cell lines (PDF)
References
- 1.Gupta SC, Kim JH, Prasad S, Aggarwal BB. Regulation of survival, proliferation, invasion, angiogenesis, and metastasis of tumor cells through modulation of inflammatory pathways by nutraceuticals. Cancer Metastasis Rev. 2010;29:405–434. doi: 10.1007/s10555-010-9235-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang X, Duan X, Yang G, Zhang X, Deng L, Zheng H, Deng C, Wen J, Wang N, Peng C, Zhao X, Wei Y, Chen L. Honokiol crosses BBB and BCSFB, and inhibits brain tumor growth in rat 9L intracerebral gliosarcoma model and human U251 xenograft glioma model. PLoS One. 2011;6:e18490. doi: 10.1371/journal.pone.0018490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen F, Wang T, Wu YF, Gu Y, Xu XL, Zheng S, Hu X. Honokiol: a potent chemotherapy candidate for human colorectal carcinoma. World J Gastroenterol. 2004;10:3459–3463. doi: 10.3748/wjg.v10.i23.3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chilampalli S, Zhang X, Fahmy H, Kaushik RS, Zeman D, Hildreth MB, Dwivedi C. Chemopreventive effects of honokiol on UVB-induced skin cancer development. Anticancer Res. 2010;30:777–783. [PubMed] [Google Scholar]
- 5.Avtanski DB, Nagalingam A, Bonner MY, Arbiser JL, Saxena NK, Sharma D. Honokiol activates LKB1-miR-34a axis and antagonizes the oncogenic actions of leptin in breast cancer. Oncotarget. 2015;6:29947. doi: 10.18632/oncotarget.4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prasad R, Katiyar SK. Honokiol, an active compound of magnolia plant, inhibits growth, and progression of cancers of different organs. In: Gupta SC, Prasad S, Aggarwal BB, editors. Anti-inflammatory Nutraceuticals and Chronic Diseases. Springer International Publishing; Cham: 2016. pp. 245–265. [DOI] [PubMed] [Google Scholar]
- 7.Lin JM, Prakasha Gowda AS, Sharma AK, Amin S. In vitro growth inhibition of human cancer cells by novel honokiol analogs. Bioorg Med Chem. 2012;20:3202–3211. doi: 10.1016/j.bmc.2012.03.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Battle TE, Arbiser J, Frank DA. The natural product honokiol induces caspase-dependent apoptosis in B-cell chronic lymphocytic leukemia (B-CLL) cells. Blood. 2005;106:690–697. doi: 10.1182/blood-2004-11-4273. [DOI] [PubMed] [Google Scholar]
- 9.Ishitsuka K, Hideshima T, Hamasaki M, Raje N, Kumar S, Hideshima H, Shiraishi N, Yasui H, Roccaro AM, Richardson P, Podar K, Le Gouill S, Chauhan D, Tamura K, Arbiser J, Anderson KC. Honokiol overcomes conventional drug resistance in human multiple myeloma by induction of caspase-dependent and -independent apoptosis. Blood. 2005;106:1794–1800. doi: 10.1182/blood-2005-01-0346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arora S, Bhardwaj A, Srivastava SK, Singh S, McClellan S, Wang B, Singh AP. Honokiol arrests cell cycle, induces apoptosis, and potentiates the cytotoxic effect of gemcitabine in human pancreatic cancer cells. PLoS One. 2011;6:e21573. doi: 10.1371/journal.pone.0021573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li L, Han W, Gu Y, Qiu S, Lu Q, Jin J, Luo J, Hu X. Honokiol induces a necrotic cell death through the mitochondrial permeability transition pore. Cancer Res. 2007;67:4894–4903. doi: 10.1158/0008-5472.CAN-06-3818. [DOI] [PubMed] [Google Scholar]
- 12.Kumar A, Kumar Singh U, Chaudhary A. Honokiol analogs: a novel class of anticancer agents targeting cell signaling pathways and other bioactivities. Future Med Chem. 2013;5:809–829. doi: 10.4155/fmc.13.32. [DOI] [PubMed] [Google Scholar]
- 13.Bai X, Cerimele F, Ushio-Fukai M, Waqas M, Campbell PM, Govindarajan B, Der CJ, Battle T, Frank DA, Ye K, Murad E, Dubiel W, Soff G, Arbiser JL. Honokiol, a small molecular weight natural product, inhibits angiogenesis in vitro and tumor growth in vivo. J Biol Chem. 2003;278:35501–35507. doi: 10.1074/jbc.M302967200. [DOI] [PubMed] [Google Scholar]
- 14.Singh T, Prasad R, Katiyar SK. Inhibition of class I histone deacetylases in non-small cell lung cancer by honokiol leads to suppression of cancer cell growth and induction of cell death in vitro and in vivo. Epigenetics. 2013;8:54–65. doi: 10.4161/epi.23078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen J, Stubbe J. Bleomycins: towards better therapeutics. Nat Rev Cancer. 2005;5:102–112. doi: 10.1038/nrc1547. [DOI] [PubMed] [Google Scholar]
- 16.Xu Y-j, Kim EY, Demple B. Excision of C-4′-oxidized deoxyribose lesions from double-stranded DNA by human apurinic/apyrimidinic endonuclease (Ape1 protein) and DNA polymerase β. J Biol Chem. 1998;273:28837–28844. doi: 10.1074/jbc.273.44.28837. [DOI] [PubMed] [Google Scholar]
- 17.Parsons JL, Dianova II, Dianov GL. APE1 is the major 3′-phosphoglycolate activity in human cell extracts. Nucleic Acids Res. 2004;32:3531–3536. doi: 10.1093/nar/gkh676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DeMott MS, Beyret E, Wong D, Bales BC, Hwang JT, Greenberg MM, Demple B. Covalent trapping of human DNA polymerase β by the oxidative DNA lesion 2-deoxyribonolactone. J Biol Chem. 2002;277:7637–7640. doi: 10.1074/jbc.C100577200. [DOI] [PubMed] [Google Scholar]
- 19.Quiñones JL, Thapar U, Yu K, Fang Q, Sobol RW, Demple B. Enzyme mechanism-based, oxidative DNA–protein cross-links formed with DNA polymerase β in vivo. Proc Natl Acad Sci U S A. 2015;112:8602–8607. doi: 10.1073/pnas.1501101112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sung JS, Demple B. Roles of base excision repair subpathways in correcting oxidized abasic sites in DNA. FEBS J. 2006;273:1620–1629. doi: 10.1111/j.1742-4658.2006.05192.x. [DOI] [PubMed] [Google Scholar]
- 21.Dianov GL, Prasad R, Wilson SH, Bohr VA. Role of DNA polymerase beta in the excision step of long patch mammalian base excision repair. J Biol Chem. 1999;274:13741–13743. doi: 10.1074/jbc.274.20.13741. [DOI] [PubMed] [Google Scholar]
- 22.Prasad R, Lavrik OI, Kim SJ, Kedar P, Yang XP, Vande Berg BJ, Wilson SH. DNA polymerase β-mediated long patch base excision repair: POLY(ADP-RIBOSE) POLYMERASE-1 STIMULATES STRAND DISPLACEMENT DNA SYNTHESIS. J Biol Chem. 2001;276:32411–32414. doi: 10.1074/jbc.C100292200. [DOI] [PubMed] [Google Scholar]
- 23.Liu Y, Beard WA, Shock DD, Prasad R, Hou EW, Wilson SH. DNA polymerase β and flap endonuclease 1 enzymatic specificities sustain DNA synthesis for long patch base excision repair. J Biol Chem. 2005;280:3665–3674. doi: 10.1074/jbc.M412922200. [DOI] [PubMed] [Google Scholar]
- 24.Stucki M, Pascucci B, Parlanti E, Fortini P, Wilson SH, Hubscher U, Dogliotti E. Mammalian base excision repair by DNA polymerases delta and epsilon. Oncogene. 1998;17:835–843. doi: 10.1038/sj.onc.1202001. [DOI] [PubMed] [Google Scholar]
- 25.Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mao Z, Bozzella M, Seluanov A, Gorbunova V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle. 2008;7:2902–2906. doi: 10.4161/cc.7.18.6679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li SS, Gao Z, Feng X, Jones SH, Hecht SM. Plant sterols as selective DNA polymerase beta lyase inhibitors and potentiators of bleomycin cytotoxicity. Bioorg Med Chem. 2004;12:4253–4258. doi: 10.1016/j.bmc.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 28.Sun DA, Deng JZ, Starck SR, Hecht SM. Mispyric acid, a new monocyclic triterpenoid with a novel skeleton from Mischocarpus pyriformis that inhibits DNA polymerase β. J Am Chem Soc. 1999;121:6120–6124. [Google Scholar]
- 29.Gao Z, Maloney DJ, Dedkova LM, Hecht SM. Inhibitors of DNA polymerase β: Activity and mechanism. Bioorg Med Chem. 2008;16:4331–4340. doi: 10.1016/j.bmc.2008.02.071. [DOI] [PubMed] [Google Scholar]
- 30.Liu S, Lai Y, Zhao W, Wu M, Zhang Z. Links between DNA polymerase beta expression and sensitivity to bleomycin. Toxicology. 2011;281:63–69. doi: 10.1016/j.tox.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 31.Tentori L, Ricci-Vitiani L, Muzi A, Ciccarone F, Pelacchi F, Calabrese R, Runci D, Pallini R, Caiafa P, Graziani G. Pharmacological inhibition of poly(ADP-ribose) polymerase-1 modulates resistance of human glioblastoma stem cells to Temozolomide. BMC Cancer. 2014;14:151. doi: 10.1186/1471-2407-14-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jaiswal AS, Banerjee S, Panda H, Bulkin CD, Izumi T, Sarkar FH, Ostrov DA, Narayan S. A novel Inhibitor of DNA polymerase β enhances the ability of Temozolomide to impair the growth of colon cancer cells. Mol Cancer Res. 2009;7:1973–1983. doi: 10.1158/1541-7786.MCR-09-0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tang JB, Svilar D, Trivedi RN, Wang XH, Goellner EM, Moore B, Hamilton RL, Banze LA, Brown AR, Sobol RW. N-methylpurine DNA glycosylase and DNA polymerase beta modulate BER inhibitor potentiation of glioma cells to Temozolomide. Neuro Oncol. 2011;13:471–486. doi: 10.1093/neuonc/nor011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Braithwaite EK, Kedar PS, Stumpo DJ, Bertocci B, Freedman JH, Samson LD, Wilson SH. DNA polymerases β and λ mediate overlapping and independent roles in base excision repair in mouse embryonic fibroblasts. PLoS One. 2010;5:e12229. doi: 10.1371/journal.pone.0012229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Borer P. Optical properties of nucleic acids, absorption, and circular dichroism spectra. In: Fasman GD, editor. Handbook of Biochemistry and Molecular Biology. CRC Press; Boca Raton, FL: 1977. p. 589. [Google Scholar]
- 36.Kretulskie AM, Spratt TE. Structure of purine-purine mispairs during misincorporation and extension by E. coli DNA polymerase I. Biochemistry. 2006;45:3740–3746. doi: 10.1021/bi052306u. [DOI] [PubMed] [Google Scholar]
- 37.Prakasha Gowda AS, Polizzi JM, Eckert KA, Spratt TE. Incorporation of gemcitabine and cytarabine into DNA by DNA polymerase beta and ligase III/XRCC1. Biochemistry. 2010;49:4833–4840. doi: 10.1021/bi100200c. [DOI] [PubMed] [Google Scholar]
- 38.Gowda ASP, Spratt TE. DNA polymerases η and ζ combine to bypass O2-[4-(3-pyridyl)-4-oxobutyl]thymine, a DNA adduct formed from tobacco carcinogens. Chem Res Toxicol. 2016;29:303–316. doi: 10.1021/acs.chemrestox.5b00468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brown JA, Fiala KA, Fowler JD, Sherrer SM, Newmister SA, Duym WW, Suo Z. A novel mechanism of sugar selection utilized by a human X-family DNA polymerase. J Mol Biol. 2010;395:282–290. doi: 10.1016/j.jmb.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carey J. Gel retardation. Methods Enzymol. 1991;208:103–117. doi: 10.1016/0076-6879(91)08010-f. [DOI] [PubMed] [Google Scholar]
- 41.Vande Berg BJ, Beard WA, Wilson SH. DNA structure and aspartate 276 influence nucleotide binding to human DNA polymerase beta. Implication for the identity of the rate-limiting conformational change. J Biol Chem. 2001;276:3408–3416. doi: 10.1074/jbc.M002884200. [DOI] [PubMed] [Google Scholar]
- 42.Ahn J, Kraynov VS, Zhong X, Werneburg BG, Tsai MD. DNA polymerase beta: effects of gapped substrates on dNTP specificity, fidelity, processivity and conformational changes. Biochem J. 1998;331:79–87. doi: 10.1042/bj3310079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Werneburg BG, Ahn J, Zhong X, Hondal RJ, Kraynov VS, Tsai MD. DNA polymerase beta: pre-steady-state kinetic analysis and roles of arginine-283 in catalysis and fidelity. Biochemistry. 1996;35:7041–7050. doi: 10.1021/bi9527202. [DOI] [PubMed] [Google Scholar]
- 44.Segel IH. Enzyme Kinetics. Wiley-Interscience; New York: 1975. Rapid Equilibrium Partial and Mixed-Type Inhibition; pp. 161–226. [Google Scholar]
- 45.Burton RL, Chen S, Xu XL, Grant GA. Transient kinetic snalysis of the interaction of L-serine with Escherichia coli D-3-phosphoglycerate dehydrogenase reveals the mechanism of V-type regulation and the order of effector binding. Biochemistry. 2009;48:12242–12251. doi: 10.1021/bi901489n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao L, Pence MG, Eoff RL, Yuan S, Fercu CA, Guengerich FP. Elucidation of kinetic mechanisms of human translesion DNA polymerase kappa using tryptophan mutants. FEBS J. 2014;281:4394–4410. doi: 10.1111/febs.12947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kamath-Loeb AS, Balakrishna S, Whittington D, Shen JC, Emond MJ, Okabe T, Masutani C, Hanaoka F, Nishimura S, Loeb LA. Sphingosine, a modulator of human translesion DNA polymerase activity. J Biol Chem. 2014;289:21663–21672. doi: 10.1074/jbc.M114.570242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fowler JD, Brown JA, Johnson KA, Suo Z. Kinetic investigation of the inhibitory effect of gemcitabine on DNA polymerization catalyzed by human mitochondrial DNA polymerase gamma. J Biol Chem. 2008;283:15339–15348. doi: 10.1074/jbc.M800310200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Spence RA, Kati WM, Anderson KS, Johnson KA. Mechanism of inhibition of HIV-1 reverse transcriptase by nonnucleoside inhibitors. Science. 1995;267:988–993. doi: 10.1126/science.7532321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Johnson KA. Transient-state kinetic analysis of enzyme reaction pathways. In: Sigman DS, editor. The Enzymes (Amsterdam) Academic Press; 1992. pp. 1–61. [Google Scholar]
- 51.Lange SS, Takata K, Wood RD. DNA polymerases and cancer. Nat Rev Cancer. 2011;11:96–110. doi: 10.1038/nrc2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Loeb LA, Monnat RJ. DNA polymerases and human disease. Nat Rev Genet. 2008;9:594–604. doi: 10.1038/nrg2345. [DOI] [PubMed] [Google Scholar]
- 53.Sun DA, Starck SR, Locke EP, Hecht SM. DNA Polymerase β Inhibitors from Sandoricum koetjape. J Nat Prod. 1999;62:1110–1113. doi: 10.1021/np990104r. [DOI] [PubMed] [Google Scholar]
- 54.Mizushina Y, Ohkubo T, Date T, Yamaguchi T, Saneyoshi M, Sugawara F, Sakaguchi K. Mode Analysis of a Fatty Acid Molecule Binding to the N-terminal 8-kDa Domain of DNA Polymerase β: A 1:1 COMPLEX AND BINDING SURFACE. J Biol Chem. 1999;274:25599–25607. doi: 10.1074/jbc.274.36.25599. [DOI] [PubMed] [Google Scholar]
- 55.Hu HY, Horton JK, Gryk MR, Prasad R, Naron JM, Sun DA, Hecht SM, Wilson SH, Mullen GP. Identification of small molecule synthetic inhibitors of DNA polymerase beta by NMR chemical shift mapping. J Biol Chem. 2004;279:39736–39744. doi: 10.1074/jbc.M402842200. [DOI] [PubMed] [Google Scholar]
- 56.Barakat KH, Gajewski MM, Tuszynski JA. DNA polymerase beta (pol beta) inhibitors: a comprehensive overview. Drug Discovery Today. 2012;17:913–920. doi: 10.1016/j.drudis.2012.04.008. [DOI] [PubMed] [Google Scholar]
- 57.Strittmatter T, Brockmann A, Pott M, Hantusch A, Brunner T, Marx A. Expanding the scope of human DNA polymerase lambda and beta inhibitors. ACS Chem Biol. 2014;9:282–290. doi: 10.1021/cb4007562. [DOI] [PubMed] [Google Scholar]
- 58.Arian D, Hedayati M, Zhou H, Bilis Z, Chen K, DeWeese TL, Greenberg MM. Irreversible inhibition of DNA polymerase beta by small-molecule mimics of a DNA lesion. J Am Chem Soc. 2014;136:3176–3183. doi: 10.1021/ja411733s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maloney DJ, Deng JZ, Starck SR, Gao Z, Hecht SM. (+)-Myristinin A, a naturally occurring DNA polymerase beta inhibitor and potent DNA-damaging agent. J Am Chem Soc. 2005;127:4140–4141. doi: 10.1021/ja042727j. [DOI] [PubMed] [Google Scholar]
- 60.Deng JZ, Starck SR, Hecht SM. Pentacyclic triterpenoids from Freziera sp. that inhibit DNA polymerase beta. Bioorg Med Chem. 2000;8:247–250. doi: 10.1016/s0968-0896(99)00276-x. [DOI] [PubMed] [Google Scholar]
- 61.Ma J, Starck SR, Hecht SM. DNA polymerase beta inhibitors from Tetracera boiviniana. J Nat Prod. 1999;62:1660–1663. doi: 10.1021/np990326p. [DOI] [PubMed] [Google Scholar]
- 62.Li SS, Gao Z, Feng X, Hecht SM. Biscoumarin derivatives from Edgeworthia gardneri that inhibit the lyase activity of DNA polymerase beta. J Nat Prod. 2004;67:1608–1610. doi: 10.1021/np040127s. [DOI] [PubMed] [Google Scholar]
- 63.Feng X, Gao Z, Li S, Jones SH, Hecht SM. DNA polymerase beta lyase inhibitors from Maytenus putterlickoides. J Nat Prod. 2004;67:1744–1747. doi: 10.1021/np040057p. [DOI] [PubMed] [Google Scholar]
- 64.Chaturvedula VS, Zhou BN, Gao Z, Thomas SJ, Hecht SM, Kingston DG. New lupane triterpenoids from Solidago canadensis that inhibit the lyase activity of DNA polymerase beta. Bioorg Med Chem. 2004;12:6271–6275. doi: 10.1016/j.bmc.2004.08.048. [DOI] [PubMed] [Google Scholar]
- 65.Barakat K, Gajewski M, Tuszynski J. DNA repair inhibitors: our last disposal to improve cancer therapy. Curr Top Med Chem. 2012;12:1376. [PubMed] [Google Scholar]
- 66.Goellner EM, Svilar D, Almeida KH, Sobol RW. Targeting DNA polymerase β for therapeutic intervention. Curr Mol Pharmacol. 2012;5:68–87. [PMC free article] [PubMed] [Google Scholar]
- 67.Mrugala MM, Chamberlain MC. Mechanisms of disease: Temozolomide and glioblastoma–look to the future. Nat Clin Pract Oncol. 2008;5:476–486. doi: 10.1038/ncponc1155. [DOI] [PubMed] [Google Scholar]
- 68.Sobol RW. Temozolomide. In: Schwab M, editor. Encyclopedia of Cancer. Springer; Berlin Heidelberg: 2012. pp. 3637–3642. [Google Scholar]
- 69.Larson K, Sahm J, Shenkar R, Strauss B. Methylation-induced blocks to in vitro DNA replication. Mutat Res, Fundam Mol Mech Mutagen. 1985;150:77–84. doi: 10.1016/0027-5107(85)90103-4. [DOI] [PubMed] [Google Scholar]
- 70.Horton TM, Jenkins G, Pati D, Zhang L, Dolan ME, Ribes-Zamora A, Bertuch AA, Blaney SM, Delaney SL, Hegde M, Berg SL. Poly(ADP-ribose) polymerase inhibitor ABT-888 potentiates the cytotoxic activity of Temozolomide in leukemia cells: influence of mismatch repair status and O6-methylguanine-DNA methyltransferase activity. Mol Cancer Ther. 2009;8:2232–2242. doi: 10.1158/1535-7163.MCT-09-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bobola MS, Kolstoe DD, Blank A, Chamberlain MC, Silber JR. Repair of 3-methyladenine and abasic sites by base excision repair mediates glioblastoma resistance to Temozolomide. Front Oncol. 2012;2:176. doi: 10.3389/fonc.2012.00176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Marchesi F, Turriziani M, Tortorelli G, Avvisati G, Torino F, De Vecchis L. Triazene compounds: Mechanism of action and related DNA repair systems. Pharmacol Res. 2007;56:275–287. doi: 10.1016/j.phrs.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 73.Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, Bromberg JEC, Hau P, Mirimanoff RO, Cairncross JG, Janzer RC, Stupp R. MGMT gene silencing and benefit from Temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 74.Quinn JA, Jiang SX, Reardon DA, Desjardins A, Vredenburgh JJ, Rich JN, Gururangan S, Friedman AH, Bigner DD, Sampson JH, McLendon RE, Herndon JE, 2nd, Walker A, Friedman HS. Phase II trial of Temozolomide plus O6-benzylguanine in adults with recurrent, Temozolomide-resistant malignant glioma. J Clin Oncol. 2009;27:1262–1267. doi: 10.1200/JCO.2008.18.8417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stephen ZR, Kievit FM, Veiseh O, Chiarelli PA, Fang C, Wang K, Hatzinger SJ, Ellenbogen RG, Silber JR, Zhang M. Redox-responsive magnetic nanoparticle for targeted convection-enhanced delivery of O6-benzylguanine to brain tumors. ACS Nano. 2014;8:10383–10395. doi: 10.1021/nn503735w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lieber MR, Ma Y, Pannicke U, Schwarz K. Mechanism and regulation of human non-homologous DNA end-joining. Nat Rev Mol Cell Biol. 2003;4:712–720. doi: 10.1038/nrm1202. [DOI] [PubMed] [Google Scholar]
- 77.Iliakis G, Murmann T, Soni A. Alternative end-joining repair pathways are the ultimate backup for abrogated classical non-homologous end-joining and homologous recombination repair: Implications for the formation of chromosome translocations. Mutat Res, Genet Toxicol Environ Mutagen. 2015;793:166–175. doi: 10.1016/j.mrgentox.2015.07.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.